")
Back to Journals » Journal of Asthma and Allergy » Volume 15
Pathobiology of Airway Remodeling in Asthma: The Emerging Role of Integrins
Received 20 December 2021
Accepted for publication 25 March 2022
Published 11 May 2022 Volume 2022:15 Pages 595—610
DOI https://doi.org/10.2147/JAA.S267222
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Amrita Dosanjh
Chitra Joseph, Amanda L Tatler
Centre for Respiratory Research, National Institute for Health Research Biomedical Research Centre, School of Medicine, University of Nottingham, Nottingham, UK
Correspondence: Amanda L Tatler, Centre for Respiratory Research, National Institute for Health Research Biomedical Research Centre, School of Medicine, University of Nottingham, Hucknall Road, Nottingham, NG5 1PB, UK, Tel +44 11 5823 1683, Email [email protected]
Abstract: Airway remodeling is a complex clinical feature of asthma that involves long-term disruption and modification of airway architecture, which contributes significantly to airway hyperresponsiveness (AHR) and lung function decline. It is characterized by thickening of the airway smooth muscle layer, deposition of a matrix below the airway epithelium, resulting in subepithelial fibrosis, changes within the airway epithelium, leading to disruption of the barrier, and excessive mucous production and angiogenesis within the airway wall. Airway remodeling contributes to stiffer and less compliant airways in asthma and leads to persistent, irreversible airflow obstruction. Current asthma treatments aim to reduce airway inflammation and exacerbations but none are targeted towards airway remodeling. Inhibiting the development of airway remodeling or reversing established remodeling has the potential to dramatically improve symptoms and disease burden in asthmatic patients. Integrins are a family of transmembrane heterodimeric proteins that serve as the primary receptors for extracellular matrix (ECM) components, mediating cell–cell and cell–ECM interactions to initiate intracellular signaling cascades. Cells present within the lungs, including structural and inflammatory cells, express a wide and varying range of integrin heterodimer combinations and permutations. Integrins are emerging as an important regulator of inflammation, repair, remodeling, and fibrosis in the lung, particularly in chronic lung diseases such as asthma. Here, we provide a comprehensive summary of the current state of knowledge on integrins in the asthmatic airway and how these integrins promote the remodeling process, and emphasize their potential involvement in airway disease.
Keywords: asthma, airway remodeling, integrins, matrix, fibrosis, biomechanics
Introduction
Asthma is a heterogeneous chronic inflammatory disease of the airways characterized by airway hyper-responsiveness, bronchoconstriction, and airway remodeling. Asthma affects over 260 million people worldwide and was responsible for over 21 million Disability-adjusted Life Years (DALYs) in 2019,1 representing significant morbidity and economic burden. The prevalence of asthma is highest in countries with the highest socio-demographic index (SDI); however, death rates are highest in countries with low–middle SDI.1
 |
Table 1 Overview of Integrins Involved in Asthmatic Airway Remodeling |
The symptoms of asthma include wheezing, shortness of breath, chest tightness, and cough that fluctuate in frequency and intensity, as well as variable expiratory airflow restriction.2 Treatment includes targeting bronchoconstriction through the use of β2 adrenergic agonists, or some cases muscarinic receptor antagonists, and reducing airway inflammation via inhaled or oral corticosteroids. Such an approach is sufficient to control symptoms in most patients, however, some patients suffer from difficult-to-treat asthma, with uncontrolled symptoms despite good adherence to treatment. Severe asthma, defined as uncontrolled symptoms despite treatment with highest doses of inhaled corticosteroids in combination with an additional controller medication (eg, long-acting β2 agonist), affects approximately 5–10% of patients and is associated with frequent and uncontrolled exacerbations, and a long-term decrease in lung function.3–5
Remodeling of the airways contributes to airway wall thickening and has a detrimental effect on asthma. It is associated with accelerated decline in lung function, an increased rate of exacerbation in asthmatic patients, and irreversible airflow obstruction.6–8 Thickening of the airways is not limited to patients suffering from the severest forms of the disease and can be evident even in mild forms of asthma; however, the degree of thickening is associated with increased disease severity and degree of airflow obstruction.9,10 Airway remodeling is thought to play a vital role in the uncontrolled symptoms and disease burden observed in severe asthmatics. Over recent years many studies have implicated a family of cell surface receptors known as integrins in the development and progression of airway remodeling. This review aims to bring together our current knowledge of how integrins may either drive or inhibit airway remodeling in asthma, and discuss the potential utility of targeting integrins as a therapeutic strategy in severe asthma.
Airway Remodeling in Asthma
Airway remodeling is the collective term given to the structural changes that occur within the asthmatic airway. These changes include sub-epithelial fibrosis, thickening of the airway smooth muscle (ASM) layer, mucous gland hyperplasia, angiogenesis, and loss of epithelial layer integrity, all of which contribute to a thickened and stiffened airway wall. The development of airway remodeling begins early in the disease course, with structural changes being evident in preschool children with clinically confirmed wheeze, even prior to an asthma diagnosis.11–13
The underlying mechanisms driving the development of airway remodeling are largely unclear and likely to be extremely complex and multifaceted. While for many years airway remodeling was thought to result from the presence of chronic inflammation within the asthmatic airway, this has more recently been questioned. Structural changes in the airways of preschool wheezers do not correlate with inflammatory cell counts in bronchoalveolar lavage fluid.11 It is possible that different features of airway remodeling differ in the underlying mechanisms driving them. The following section will discuss the potential mechanisms responsible for the development and progression of airway remodeling in asthma.
Potential Mechanisms Driving Airway Remodeling
Airway inflammation has long been thought to drive the development of asthmatic airway remodeling. Asthma is largely driven by TH2 inflammation associated with interleukin-4 (IL4), interleukin-5 (IL5), and interleukin-13 (IL13), and TH2 inflammation remains a crucial target in asthma therapy development. However, a cluster analysis of asthmatic patients has suggested that patients with fixed airflow obstruction and evident airway remodeling have predominantly TH17 rather than TH2 driven inflammation.14
Further evidence of a link between inflammation and airway remodeling comes from in vivo and in silico models of asthma. A theoretical model of airway remodeling demonstrates that inflammation is sufficient to promote thickening of the airway wall towards the lumen, although increased thickening occurs when biomechanical contractile forces and inflammation are modeled simultaneously,15 suggesting interplay of multiple pathways. Additionally, numerous mouse models have highlighted a potential link between inflammation and remodeling. For example, Interleukin-33 (IL33) can exacerbate allergen-induced inflammation and remodeling in a mouse model,16 and M2 macrophages, which IL33 promotes polarization towards,17,18 has been associated with allergen-induced remodeling in mice.19
From the studies described above it is clear that the mechanistic link between airway inflammation and airway remodeling is still ambiguous. The fact that remodeling occurs very early in the disease course, including in young children with wheeze prior to a diagnosis of asthma,11–13 suggests that chronic inflammation may not be the sole driver of airway remodeling.
An alternative possibility is that the mechanical environment of the asthmatic airway drives remodeling changes. This was initially suggested in 2011 when Grainge et al20 demonstrated remodeling changes in response to bronchoconstriction in the absence of additional inflammation. Mechanistically, contraction of ASM cells and airways causes activation of the pro-remodeling cytokine TGFβ and downstream remodeling changes.21–23 Moreover, pharmacological inhibition of transient receptor potential vanilloid-1 (TRPV1), which can modulate ASM tone, reduces airway remodeling in vivo.24,25 Mathematical modeling has also suggested that airway contraction contributes to remodeling.15
In addition to contractile mechanical forces promoting airway remodeling it is also possible that non-contractile biomechanical forces contribute.26 ECM proteins within the asthmatic airway wall can promote proliferation of ASM cells27 and drive remodeling changes in vivo.28 Additionally, altered mechanics due to a stiffer airway wall may drive remodeling changes. Increased matrix stiffness promotes epithelial–mesenchymal transition,29 collagen production by fibroblasts,30 and ASM cell proliferation,31 all of which may contribute to airway remodelling. Recently, a link between matrix crosslinking, which stiffens ECM, and the development of asthmatic airway remodeling has been described whereby the matrix crosslinking enzyme lysyl oxidase-like-2 (LOXL2) has been implicated.32 Crucially, LOXL2 levels were increased in asthmatic ASM cells and pharmacological inhibition of LOXL2 in vivo reduced allergen-induced airway remodeling.32
Integrins
Integrins are heterodimeric transmembrane receptors that facilitate cell–cell and cell–matrix interactions. They provide a direct link between the environment outside of the cell and the cytoskeleton within the cell, and involved in the transmission of biomechanical signals. The family is composed of 24 mammalian members, made up by a variety of combinations of alpha (α) and a beta (β) subunit; there are eight distinct β subunits and 18 distinct α subunits.33 The α subunit is responsible for the ligand binding properties of integrins, while the downstream intracellular signaling events are co-ordinated by the β subunit. Some integrins can bind to only one type of ligand, while other integrins are able to recognize several ECM proteins.
Integrins can mediate bi-directional signals through the cell membrane; inside-out signalling regulates extracellular binding activity of integrins and thereby switching into active conformation. On the other hand, binding of ECM proteins on integrins activate signals that are transmitted into the cells known as outside-in signaling.33 These signaling events modulate roles in cell attachment, survival, proliferation, leukocyte trafficking, cell differentiation, cytoskeleton organization, cell migration, gene expression, tumorigenicity, and intracellular pH.
Integrins combine with multiple proteins to form integrin adhesion complexes (IAC), also known as the integrin adhesome, to activate downstream signaling pathways. To date the literature suggests such complexes involve at least 232 distinct integrin-associated proteins (IAP),34 including talin, paxillin, kindlins, filamin, vinculin, integrin-linked kinase (ILK), focal adhesion kinase (FAK), Src family protein tyrosine kinases (SFK), and GTPases of the Rho family. Such complexes can be split into four compartments: the ECM, the integrin, IAPs, and the actin cytoskeleton.34 The wide-ranging and diverse functions of just 24 distinct integrins are largely dependent on the complexity and diversity of IACs.
Several integrins are expressed within the lung and have roles in lung development, including branching morphogenesis, epithelial cell polarization, and differentiation.35,36 Expression of integrins varies across lung cell types and at varying times of development. Within the airway epithelium eight integrins are expressed, namely α2β1, α3β1, α5β1, α6β4, α9β1, αvβ5, αvβ6, and αvβ8.36–38 In some cases, integrin subunit expression in the epithelium is dramatically increased during inflammation or repair, most notably for the epithelially-restricted integrin αvβ6.37,39–41 Within the lung mesenchymal cells expression of α5β1, αvβ3, α2β1, α4β1, α5β1, αvβ5, and α7β5 have all been reported.22,42,43 Lung inflammatory cells also express integrin receptors; macrophages express β2 integrins, α4β1 and α5β1,44,45 and T lymphocytes are known to express α4β1, α5β1, αEβ7 and β2 integrins.42 Eosinophils, which have an important role in the pathophysiology of asthma, have a distinctive combination of eight integrins, α4β1, α6β1, αLβ2, αDβ2, αMβ2, αXβ2, and α4β7.46,47
The known function of integrins and integrin adhesomes make them attractive candidates for understanding how mechanical cues, including contractile forces and matrix stiffening, might influence airway remodeling processes. Furthermore, integrins are well-known for regulating leukocyte and inflammatory cell trafficking, which could also have important implications for asthma development and progression and for airway remodeling. The following section will discuss the role of integrin superfamily members in mediating specific airway remodeling processes in a variety of lung cells important to asthma pathogenesis. We have summarised how specific integrin heterodimers might be involved in asthmatic airway remodeling process in Table 1 and Figure 1.
Epithelial Changes in Airway Remodeling
The epithelial layer serves as a physical barrier to the exterior environment. As a result, it is the lungs’ first line of defence against foreign bodies inhaled during breathing. In addition, the healthy airway epithelium modulates immune responses and promotes the expulsion of inhaled particles through mucous production and cilia movement. The asthmatic airway epithelium undergoes dramatic phenotypic changes resulting in loss of epithelial integrity through epithelial shedding and increased mucous production via mucous gland hyperplasia.
Loss of airway epithelium is a well-documented phenomenon in asthma48–51 and is linked with airway hyper-reactivity.48,50 Loss of epithelial integrity occurs early in the disease course,49 and is thought to result from cellular apoptosis, senescence, and ineffective repair mechanisms.52,53 The asthmatic airway epithelium expresses markers of cellular injury/repair including increased epidermal growth factor receptor (EGFR),54,55 transforming growth factor β (TGFβ),56,57 and decreased E-cadherin.58 Furthermore, apoptosis and proliferative pathways are altered.59
Senescence of the epithelium occurs in asthma53 and may promote asthma development by compromising epithelial integrity and barrier function. Moreover, epithelial cell senescence drives thymic stromal lymphopoietin (TSLP)-induced airway remodeling.53 Crucially, airway epithelial senescence can be driven by a deficiency in integrin β4 expression in a P53 dependent manner,60 and the asthmatic human bronchial airway epithelium has reduced integrin β4 expression.61 In the ovalbumin mouse model of asthma, integrin β4 expression is reduced on the airway epithelium and is associated with structural disruption of the epithelial layer.62 Together, these studies in human asthmatic patients and animal models of asthma suggest a crucial role for β4 integrins in maintaining epithelial integrity in the airway.
In addition to loss of epithelial integrity, the asthmatic airway produces excessive quantities of mucous. MUC5AC and MUC5B are polymeric mucins that are significantly increased in the asthmatic airway and MUC5AC levels correlate with clinical measures of asthma including fractional exhaled nitric oxide (FeNO), sputum eosinophils, and airway hyper-responsiveness.63 A key driver of increased mucous production is goblet cell hyperplasia, which is evident in mild through to severe asthma.64,65 Additionally, mucous over-production can be driven by paracrine interactions with underlying airway smooth muscle cells.66 Overall, mucous gland hyperplasia and excessive mucus production can lead to mucous plugging of the airway, reduced airway lumen area, and airflow obstruction.67 Integrins have been implicated in mucous overproduction and goblet cell hyperplasia. β1 integrins have recently been shown to regulate cellular and secreted MUC5AC and MUC5B production in lung epithelial cells.68,69 Conversely, interactions between Mfge8 and integrin β3 subunits protect against allergen induced airway remodeling changes, including goblet cell hyperplasia.70
Increased Airway Smooth Muscle Mass (ASM)
Thickening of the airway smooth muscle (ASM) layer is a common and prominent feature of asthmatic airway remodeling. In the healthy airway, ASM cells are thought to play an important role in modulating respiratory airway tone. During disease processes, however, they have an important role in inflammatory and remodeling processes, releasing chemokines, pro-inflammatory and/or pro-fibrotic cytokines, and ECM proteins,22,26,71–73 which contributes to asthma pathogenesis.
In the asthmatic airway increased ASM mass appears to be driven by both increased myocyte size (hypertrophy) and increased myocyte number (hyperplasia), which are in turn associated with disease duration and severity.74 Some studies have suggested that the increase is due to hyperplasia rather than hypertrophy75 and others have suggested that hyperplasia only occurs in cases of fatal asthma.76 The causes of increased ASM mass in asthma are likely to be multifaceted. Interactions between ASM cells and airway epithelial cells can promote increased ASM cell proliferation and production of inflammatory cytokines and chemokines,77 suggesting a role for paracrine signaling between the two cell types. Furthermore, interactions between ASM cells and CD4+ T lymphocytes, known to be crucial to the pathogenesis of asthma, can increase ASM cell proliferation.78 Numerous ASM cell mitogens have been implicated in asthma, including Platelet derived growth factor (PDGF),79 TGFβ80 epidermal growth factor (EGF),78 heparin-binding EGF,81 and vascular endothelial growth factor (VEGF).82 In certain cases, such as PDGF,83 these mitogens can also promote ASM cell migration, which may contribute to the thickening of the ASM layer and expansion of the airway wall. Regardless of the underlying mechanism, during an asthma exacerbation, the thickened ASM bundle contributes to the airway-constricting capacity of the muscle84 and is thought to contribute to fixed airflow obstruction in severe asthma.
Several integrins have been linked with the contractile function of ASM cells. The fibronectin binding α5β1 integrins are involved in ASM cell contraction; functional blockade of α5β1 interrupts the function of focal adhesions, reduces interleukin-13 (IL13)-induced contraction of tracheal rings and inhibits airway hyper-responsiveness in vivo.85 Crucially, pharmacological inhibition of α5β1 had no effect on baseline tone of the smooth muscle rings and only reduced contraction in response to asthma-relevant contractile agonists, making it a potentially attractive approach for therapeutic targeting in asthma as the homeostatic functions of ASM could be preserved.85 A similar role has recently been identified for α2β1 integrins in regulating IL13-induced contraction, in this case through interrupting ASM cell tethering to collagen I and laminin-111.86
Contraction of ASM cells occurs via force transmission through polymerization and reorganization of the actin cytoskeleton. The cytoplasmic tail of β integrins binds to actin filaments through “linker” proteins such as vinculin, talin, and α-actinin, whereas the extracellular component of integrins interacts with the extracellular matrix to tether the cell.87 Force transmission between the cell and the extracellular matrix is therefore delivered by the actin–integrin–matrix complex. Actin filament polymerization and myosin activation are two concurrent biochemical mechanisms that are critical for smooth muscle contraction homeostasis, however, inhibiting actin polymerization limits smooth muscle force generation with minimal impact on myosin light chain phosphorylation.88–90 Crucially, actin-regulatory proteins are involved in regulating proliferation of smooth muscle cells,91 demonstrating how force transmission through integrins may influence cell proliferation and remodeling. Finally, TGFβ, which can be activated by ASM cells via integrins in response to reorganization of the actin cytoskeleton,22 augments ASM cell contraction in a RhoA-independent manner.92 This suggests a perpetual feedback loop whereby bronchoconstriction causes integrin-mediated TGFβ activation to promote airway remodeling, which in turn increases the contractility of the ASM cells and contributes to fixed airflow obstruction by increasing the baseline tone of the ASM layer.
In addition to promoting cell contractility through interactions with actin, integrin superfamily members are also involved in negative regulation of ASM contraction. Ligation of α8β1 integrins on ASM cells by milk fat globule-EGF factor-8 (Mfge8) proteins prevents IL13-induced ASM contraction.93 α9β1 integrins are also capable of negatively regulating ASM contraction. Loss of, or inhibition of, α9β1 integrins in mice increases airway contraction.94 These studies all highlight the importance of ASM cell interactions with matrix proteins through cell surface integrins to regulate ASM contraction and airway narrowing. As discussed previously, uncontrolled bronchoconstriction can promote airway remodeling via integrin-mediated activation of the pro-remodeling cytokine TGFβ.21–23 Taken together, it is clear that integrins have a potentially crucial role in regulating both pathological ASM contraction and downstream pro-remodeling effects, representing a direct link between uncontrolled asthma symptoms and the development of airway remodeling through a mechanobiological mechanism.
In addition to effects on ASM contraction, integrins expressed by ASM cells may also promote migration and proliferation of ASM cells, which is thought to contribute to thickening of the ASM layer and airway lumen narrowing in airway remodeling.95 Global blockade of RGD-binding integrins with a synthetic RGDS peptide attenuates allergen-induced ASM hyperplasia and hypercontractility, suggesting a crucial role for this subset of integrins in ASM remodeling.96 β1 integrins are highly expressed in ASM cells plus other mesenchymal cells in the lung, including myofibroblasts, and have recently been shown to localize key adaptor proteins at the leading edge of migrating ASM cells.97 Additionally, β1 integrins have been implicated in pro-proliferative responses of ASM cells to increasing matrix stiffness.31 α2β1, α4β1, and α5β1 have all been shown to regulate ASM cell proliferation.98 The matrix protein fibulin-5 has been implicated in this process through binding to β1 integrins to promote ASM cell proliferation via the mechanosensing YAP/TAZ pathway.99 Furthermore, laminin binding to α7β1 integrins promotes ASM cell survival and differentiation to a contractile phenotype.100 All together these studies support an important role of β1 integrins in regulating increased ASM mass in asthmatic airway remodeling.
Subepithelial Fibrosis
Subepithelial fibrosis in the asthmatic airway occurs in the lamina reticularis, just below the basement membrane, where ECM proteins such as interstitial collagens, fibronectin, tenascin, and proteoglycan accumulate.101 Subepithelial fibrosis is linked to asthma severity; collagen expression in the airway wall is higher in patients with moderate or severe asthma compared with those with mild disease,57 and the degree of subepithelial fibrosis is inversely correlated with FEV1.102 Increased deposition and decreased degradation of extracellular matrix (ECM) proteins is one of the major hallmarks of fibrosis regardless of organ or tissue type, and is primarily controlled by fibroblasts and myofibroblasts. Within the asthmatic airway, the number of myofibroblasts present correlates with the amount of collagens and tenascin detected in the subepithelial region.102 Furthermore, fibrocytes, which can differentiate into myofibroblasts, are increased in asthma and may contribute to subepithelial fibrosis.103
Information relating to a direct role for integrins in regulating matrix deposition in asthma is limited. In vitro studies have shown that treatment of ASM cells with the pro-remodeling cytokine TGFβ leads to increased fibronectin deposition via an α5β1 mediated mechanism involving ERK signaling.104 Additionally, in murine models it has been reported that interleukin-32 (IL32) reduces allergen-induced fibrosis via suppression of the integrin-FAK-paxillin signaling axis.105
Transforming growth factor-β (TGFβ) is thought to be a key driver of subepithelial fibrosis in asthma. TGFB1 mRNA is increased in bronchial biopsies from asthmatic individuals and levels correlate with the degree of subepithelial fibrosis.106 Furthermore, all three isoforms of TGFβ are increased in the asthmatic airway.56,57,107–109 TGFβ causes transdifferentiation of airway fibroblasts into highly synthetic, matrix producing myofibroblasts110,111 and increases production of matrix proteins by fibroblasts/myofibroblasts.112,113 Crucial evidence from murine animal models shows that inhibition of both TGFβ1 and 2 with isoform-specific function blocking antibodies reduced allergen-induced subepithelial collagen deposition,114 and intra-tracheal instillation of TGFβ1 is sufficient to cause subepithelial fibrosis.115 Finally, there is recent evidence suggesting that human bronchial fibroblast responses to TGFβ are altered in asthma, with pro-fibrotic responses being increased while anti-fibrotic responses are decreased.116 Together, these studies highlight a crucial role for TGFβ in regulating subepithelial fibrosis in asthma.
αvβ8 integrins are capable of activating TGFβ via recruitment of matrix metalloproteinases, which proteolytically cleave the latent TGFβ complex on the cell surface.117 Proteolytic cleavage of TGFβ has been previously reported,71,118 however, αvβ8 is the only integrin described thus far that mediates TGFβ activation via proteolysis. Importantly, expression of αvβ8 integrins is increased in asthma119 and expression of MMP-9 and MMP-8 in the airway inversely correlate with FEV1.120,121 Other cell types are capable of activating TGFβ via integrins including myofibroblasts, lung epithelial cells, and ASM cells.22,122–124 This raises the possibility that integrin-mediated TGFβ contributes to matrix deposition in the asthmatic airway, although this remains to be definitively proved.
Integrins have been implicated in inflammatory cell-mediated mechanisms of airway remodeling. Inhibiting RGD-binding integrins peptide on eosinophils using an RGDS blocks their ability to bind to ASM cells and interrupts the eosinophil-induced increased in ECM gene expression.125,126 Despite these studies it has been shown that a blockade of RGD-binding integrins, again using a synthetic RGDS peptide, reduces markers of ASM remodeling in vivo but has no effect on airway fibrosis, suggesting that this subset of integrins does not mediate subepithelial fibrosis.96 Finally a limited study has shown that collagen deposition in the asthmatic airway is inversely correlated with expression of α2 subunits on blood and CD4+ cells.
Angiogenesis
Angiogenesis refers to the process of forming new blood vessels. Increased vascularity of the asthmatic airway is a common observation and is evident in newly diagnosed asthma patients.127–129 The implications of increased vascularity on the pathogenesis of asthma and airway remodeling are still somewhat unclear. Correlations between increased vascularity and decreased lung function in asthma are inconsistent, with some reports finding a correlation128 and others reporting no link.127 Furthermore, animal models have shown that reducing angiogenesis experimentally using the inhibitor of angiogenesis, tumstatin, does not improve lung function.130 Although a link between increased vascularity and decreased lung function in asthma is still unclear, angiogenesis within the asthmatic airway wall enhances inflammatory cell recruitment and can cause edema, which may contribute to asthma pathogenesis.129
VEGF is one of the most potent activators of endothelial cell growth and promotes vascular permeability. VEGF levels in bronchial biopsies, serum, and bronchoalveolar lavage fluid are increased in asthma,131–133 and VEGF expression within airway cells correlates with the number of vessels.134 ASM cells isolated from asthmatics can drive angiogenesis via increased VEGF secretion.135 Crucially, pharmacological inhibition of VEGF signaling has shown promise in experimental models of asthma by reducing expression of growth factors, improving epithelial barrier function,136,137 and reducing markers of airway remodeling.138
Integrins have long been implicated in angiogenic processes, with the earliest descriptions demonstrating links between αvβ3 and αvβ5 and angiogenesis.139,140 Single nucleotide polymorphisms (SNPs) within the ITGB3 gene are associated with asthma pathogenesis.141 Additionally, pharmacological inhibition of αvβ3 prevents blood vessel maturation.142 However, genetic knockout of either β3 or β5 subunits does not alter vascular development.143,144
Genetic knockdown of integrin subunits has highlighted some potentially important roles in angiogenesis during development, which may also be important in disease. For example, genetic loss of integrin α5, which binds to fibronectin, leads to vascular defects and mice that are embryonic lethal, similar to the fibronectin knockout animals.145 This suggests a crucial role of α5 integrins and fibronectin in early angiogenesis. However a separate study found that inhibiting α5β1 with a small molecule inhibitor alpha5beta1 Integrin blockade inhibits lymphangiogenesis in airway inflammation and interrupts lymphatic vessel development without affecting blood vessel development.146 Finally, an important role for endothelial cell α2β1 integrin in promoting lumen formation in new capillaries has been described.147
Integrins in Airway Remodeling: Inflammation
Chronic airway inflammation is a hallmark of asthma and, as has been discussed previously in this article, has the potential to influence pro-remodeling pathways. Several integrins including α2β1, α5β1, αvβ3, and αvβ1 have been linked with increased cytokine release when ASM cells are cultured on collagen and fibronectin, suggesting that an altered mechanical environment may influence the inflammatory environment within the airway wall.148
Eosinophils are thought to be important to the pathogenesis of asthma and they express numerous integrins. Integrins have a key role in mediating migration of eosinophils from the blood into the lung, where they accumulate in asthma.149 Integrins, particularly β2 integrins such as αmβ2 and α4 integrins, have been implicated in eosinophil degranulation and inflammatory mediator release.150–152 In addition, α4 integrin binding to its ligand fibronectin via Fas antigen signaling increases the eosinophil survival, which may contribute to airway eosinophilia in asthma.153
Airway neutrophilia is associated with increased asthma severity and asthma that is refractory to corticosteroids, the backbone of asthma treatment.154 There is a paucity of research focused directly upon a potential role for integrins in driving airway neutrophilia in asthma; however, integrins, particularly β2 integrins, are well known to regulate neutrophil recruitment to sites of inflammation.155,156 Furthermore, neutrophils and their products have been implicated in lung fibrogenesis in other chronic lung diseases such as interstitial lung disease (ILD). For example αMβ2 integrins can regulate neutrophil extracellular trap (NET) formation in ILD,157 and secretory leukocyte protease inhibitor (SLPI), which inhibits neutrophil elastase, has differential effects on collagen expression in mouse lung tissue.158 Previous work has shown that integrin expression by sputum neutrophils in asthmatic patients is aberrant compared with healthy controls,159 however, whether such changes in integrin expression affect the overall activity of neutrophils in asthma and the impact this has on airway remodeling is yet to be elucidated.
Exposure to allergens causes an increase in TH2 cell infiltration and TH2 cytokine expression in asthmatic patients. TH2 cells co-ordinate allergy-induced asthmatic inflammatory responses through Th2 cytokines (IL-4 and IL-5), causing eosinophil infiltration and hyper-responsiveness of the airways.160 Airway epithelial cells, by acting as antigen presentation cells (APCs), can cause T-cell activation and proliferation, and silencing β4 integrins in asthmatic airway epithelial cells impairs their antigen presentation capacity and decreases T-cell proliferation.161 This is one possible integrin-dependent mechanism that may contribute to TH2 inflammation bias in asthmatic airways.
Therapeutic Targeting of Integrins to Impact Airway Remodeling
To date no drug has been developed that specifically targets the development and progression of airway remodeling. Corticosteroids, which are the mainstay of asthma treatment and primarily target airway inflammation, can reduce several markers of airway remodeling, including ASM proliferation,162 TGFβ expression in fibroblasts,163 and VEGF expression by epithelial cells,164 and can reconstitute epithelial structure.165 Despite these effects, airway remodeling persists in asthmatic patients despite long-term treatment with inhaled or oral corticosteroids, suggesting there is no overwhelming impact of corticosteroids on airway remodeling in asthmatic patients.
In recent years several new biological therapies have been developed and approved, particularly for the treatment of severe asthma, some of which have shown some effects on airway remodeling. Mepolizumab, a clinically approved anti-IL5 monoclonal antibody, has been shown to reduce airway wall thickness in CT scans166 and reduce matrix protein deposition in bronchial biopsies.167 Benralizumab is another monoclonal antibody that targets IL5 signaling, which computational modeling has suggested reduces ASM mass and the number of tissue myofibroblasts present in the airway wall.168 Omalizumab targets IgE for the treatment of allergic asthma and has been shown to reduce airway wall thickness when measured by computed tomography.169 Research into the effects of other new monoclonal antibody therapies such as dupilumab (anti-Il4 receptor) and reslizumab (anti-Il5) are yet to be published, however, the former studies suggest that inhibiting TH2 inflammation may reduce asthmatic airway remodeling in severe asthma patients. Whether such treatments can sufficiently reduce airway remodeling to lead to long-term positive effects on fixed airflow obstruction or slow the decline in lung function seen in asthmatics, which is thought to be driven by airway remodeling, is likely to be the focus of ongoing studies into the utility of biological therapies. Another key question that remains to be answered is whether therapeutic treatment of airway remodeling will be sufficient or whether prophylactic treatment much earlier in the disease course will be required for the biggest clinical benefit.
Research Dilemmas in Airway Remodeling
As discussed above, airway remodeling is a complex and diverse collection of structural changes involving many tissues and cell types. Despite the introduction of various new therapies for asthma in recent years including various biological treatments targeting airway inflammation, there has yet to be an effective treatment for airway remodeling. This is potentially a result of the many specific challenges associated with researching the underlying mechanisms driving airway remodeling, which were highlighted in detail in an American Thoracic Society statement in 2017,170 and which will be discussed briefly here.
Lack of Appropriate Animal Models
A major hindrance to research investigating airway remodeling and asthma pathogenesis more widely is the lack of an appropriate animal model. Mice are the most commonly used species for in vivo models of asthma and airway remodeling, however, rats, guinea pigs, and larger species including pigs, sheep, and horses are also used.171
A significant drawback to animal models of asthma is that asthma is a human disease that does not spontaneously occur within the animal kingdom, with the exception of eosinophilic bronchitis in cats and heaves in horses, both of which are obstructive airway diseases with some similarities to asthma. Animal models are therefore largely dependent upon sensitizing animals experimentally to an allergen and then delivering that allergen to the airways to elicit an allergic inflammatory response.171 Such models are advantageous when studying how allergy and/or inflammation drive features of asthma; however, as discussed above, the relative roles of these processes in driving airway remodeling is still largely unclear and so using such models to drive airway remodeling in animals may not accurately reflect the pathogenesis driving remodeling in man.
Size and anatomical differences between human lungs and the species used for models of asthma and airway remodeling also have the potential to negatively impact the utility of findings from such models. For example the human lung has a vastly greater number of branching airways compared with mouse lungs, the effect of which on the development of remodeling is unclear with our insufficient understanding of the mechanisms driving remodeling.170 Recent methodological advances in assessing airway remodeling in airways of various sizes in murine models of asthma172 may aid our understanding of the heterogeneic nature of remodeling, albeit within the confines of a rodent disease model discussed above.
Lack of Uniformity in Core Experimental and Technological Design
Aside from species differences, the ability to compare results across studies is further complicated by methodologies used to assess airway remodeling. Airway remodeling is often quantified across large- and medium-sized airways by measuring airway wall thickness; however, bronchioles and other smaller bronchi, because of their diverse components and structures, may have different impacts on the evolution of airway remodeling. Even at the cellular level, distinct morphological, synthetic, and epigenetic differences between lung compartments exist, as has been described for fibroblasts isolated from airways compared with distal lung regions.173,174 Existing whole-organ/whole-body imaging modalities do not have enough resolution to distinguish particular cell types and can only assess various degrees of wall thickness.170
Quantifying airway remodeling in human airways largely depends upon measuring indices within airway biopsy samples, or imaging modalities such as high-resolution computed tomography (HR-CT), both of which can predict fixed airflow obstruction in asthmatic patients.7,175 These techniques present challenges when attempting to study the longitudinal development and slow progression of airway remodeling in asthma patients due to either their invasive nature (biopsy) or high radiation exposure and cost (HR-CT). Several studies have suggested potential biomarkers of airway remodeling including TGFβ and periostin,176 galectin-3,177 hyaluronan,178 however, they have yet to be widely validated, which restricts their utility in clinical research. It is clear that both mechanistic studies of airway remodeling and clinical trials testing potential interventions that target airway remodeling remain incredibly difficult due to a lack of consensus on which AR index to use, cost effectiveness, safety, ability to make repeated measurements, plus sensitivity and specificity of measurement.
Concluding Remarks
As our understanding of the underlying mechanisms driving airway remodeling in asthma improves, so does our knowledge of how cell surface integrins play a critical role in the development and progression of airway remodeling. There is still much that we do not fully understand including the relative importance of mechanical and inflammatory cues to the development of airway remodeling. However, what is clear from research in recent years is that integrins may be involved in multiple aspects of airway remodeling across all lung cells types (see Figure 1). In the years to come, therapeutic targeting of airway remodeling may improve morbidity and lung function in patients with severe, uncontrolled asthma. With the advent of biological therapies in recent years we have begun to observe some positive effects on features of airway remodeling in the most severe asthmatics. Questions remain, however, about whether these effects are sufficient to produce long-term and long lasting impacts on airway remodeling that would improve fixed airflow obstruction and slow the decline in lung function that is observed in asthma. While the effects of some biologics on airway remodeling are encouraging we believe targeting airway remodeling specifically, rather than as a bi-product of targeting inflammatory pathways, will lead to the biggest clinical improvement in airway remodeling in the years to come. Such targeting could include approaches to target integrin mediated pathways since we have hopefully demonstrated in this review that integrins are integral to many pathways involved in airway remodeling pathogenesis. Targeting integrins directly to impact airway remodeling could be a useful adjunct to existing therapies that target airway inflammation to enable both fundamental features of asthma to be treated simultaneously.
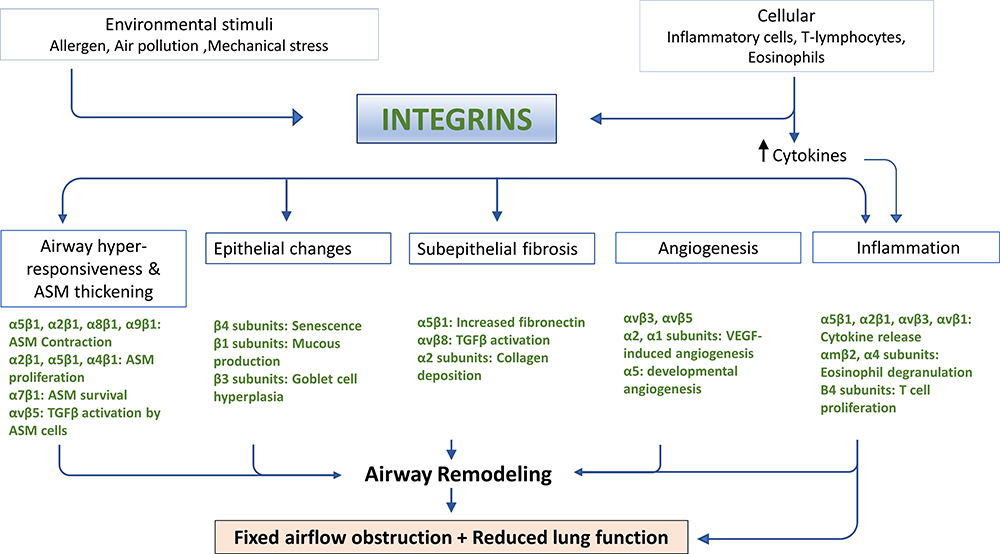 |
Figure 1 Schematic diagram giving an overview of how different integrin heterodimers expressed by a variety of lung cell types may contribute to the development and/or progression of airway remodeling in asthma. Both environmental and cellular stimuli converge upon integrin signaling pathways in a variety of cell types to contribute to airway hyper-responsiveness and ASM thickening, mucous over-production, subepithelial fibrosis, new blood vessel formation, and airway inflammation. |
Disclosure
Amanda Tatler reports grants from Medical research foundation, Asthma UK, and Biogen during the conduct of the study and personal fees from Pliant therapeutics outside the submitted work. The authors report no other potential conflicts of interest for this work.
References
1. Vos T, Lim SS, Abbafati C, et al. Global burden of 369 diseases and injuries in 204 countries and territories, 1990–2019: a systematic analysis for the Global Burden of Disease Study 2019. Lancet. 2020;396:1204–1222. doi:10.1016/S0140-6736(20)30925-9
2. Hough KP, Curtiss ML, Blain TJ, et al. Airway remodeling in asthma. Front Med. 2020;7:191. doi:10.3389/fmed.2020.00191
3. O’Byrne PM, Pedersen S, Lamm CJ, et al. Severe exacerbations and decline in lung function in asthma. Am J Respir Crit Care Med. 2009;179:19–24. doi:10.1164/rccm.200807-1126OC
4. Sorkness RL, Bleecker ER, Busse WW, et al. Lung function in adults with stable but severe asthma: air trapping and incomplete reversal of obstruction with bronchodilation. J Appl Physiol. 2008;104:394–403. doi:10.1152/japplphysiol.00329.2007
5. Ortega H, Yancey SW, Keene ON, et al. Asthma exacerbations associated with lung function decline in patients with severe eosinophilic asthma. J Allergy Clin Immunol Pract. 2018;6:980–986 e981. doi:10.1016/j.jaip.2017.12.019
6. Krings JG, Goss CW, Lew D, et al. Quantitative CT metrics are associated with longitudinal lung function decline and future asthma exacerbations: results from SARP-3. J Allergy Clin Immunol. 2021;148:752–762. doi:10.1016/j.jaci.2021.01.029
7. Kasahara K, Shiba K, Ozawa T, Okuda K, Adachi M. Correlation between the bronchial subepithelial layer and whole airway wall thickness in patients with asthma. Thorax. 2002;57:242–246. doi:10.1136/thorax.57.3.242
8. Kozlik P, Zuk J, Bartyzel S, et al. The relationship of airway structural changes to blood and bronchoalveolar lavage biomarkers, and lung function abnormalities in asthma. Clin Exp Allergy. 2020;50:15–28. doi:10.1111/cea.13501
9. Niimi A, Matsumoto H, Amitani R, et al. Airway wall thickness in asthma assessed by computed tomography. Relation to clinical indices. Am J Respir Crit Care Med. 2000;162:1518–1523. doi:10.1164/ajrccm.162.4.9909044
10. Aysola RS, Hoffman EA, Gierada D, et al. Airway remodeling measured by multidetector CT is increased in severe asthma and correlates with pathology. Chest. 2008;134:1183–1191. doi:10.1378/chest.07-2779
11. Lezmi G, Gosset P, Deschildre A, et al. Airway remodeling in preschool children with severe recurrent wheeze. Am J Respir Crit Care Med. 2015;192:164–171. doi:10.1164/rccm.201411-1958OC
12. Saglani S, Payne DN, Zhu J, et al. Early detection of airway wall remodeling and eosinophilic inflammation in preschool wheezers. Am J Respir Crit Care Med. 2007;176:858–864. doi:10.1164/rccm.200702-212OC
13. O’Reilly R, Ullmann N, Irving S, et al. Increased airway smooth muscle in preschool wheezers who have asthma at school age. J Allergy Clin Immunol. 2013;131:
14. Ye WJ, Xu W-G, Guo X-J, et al. Differences in airway remodeling and airway inflammation among moderate-severe asthma clinical phenotypes. J Thorac Dis. 2017;9:2904–2914. doi:10.21037/jtd.2017.08.01
15. Hill MR, Philp CJ, Billington CK, et al. A theoretical model of inflammation- and mechanotransduction-driven asthmatic airway remodelling. Biomech Model Mechanobiol. 2018;17:1451–1470. doi:10.1007/s10237-018-1037-4
16. Sjoberg LC, Nilsson AZ, Lei Y, et al. Interleukin 33 exacerbates antigen driven airway hyperresponsiveness, inflammation and remodeling in a mouse model of asthma. Sci Rep. 2017;7:4219. doi:10.1038/s41598-017-03674-0
17. John AE, Wilson MR, Habgood A, et al. Loss of epithelial G q and G 11 signaling inhibits TGFβ production but promotes IL-33–mediated macrophage polarization and emphysema. Sci Signal. 2016;9:ra104. doi:10.1126/scisignal.aad5568
18. Kurowska-Stolarska M, Stolarski B, Kewin P, et al. IL-33 amplifies the polarization of alternatively activated macrophages that contribute to airway inflammation. J Immunol. 2009;183:6469–6477. doi:10.4049/jimmunol.0901575
19. Wang Q, Hong L, Chen M, et al. Targeting M2 macrophages alleviates airway inflammation and remodeling in asthmatic mice via miR-378a-3p/GRB2 pathway. Front Mol Biosci. 2021;8:717969. doi:10.3389/fmolb.2021.717969
20. Grainge CL, Lau LCK, Ward JA, et al. Effect of bronchoconstriction on airway remodeling in asthma. N Engl J Med. 2011;364:2006–2015. doi:10.1056/NEJMoa1014350
21. Oenema TA, Smit M, Smedinga L, et al. Muscarinic receptor stimulation augments TGF-beta1-induced contractile protein expression by airway smooth muscle cells. Am J Physiol. 2012;303:L589–597. doi:10.1152/ajplung.00400.2011
22. Tatler AL, John AE, Jolly L, et al. Integrin alphavbeta5-mediated TGF-beta activation by airway smooth muscle cells in asthma. J Immunol. 2011;187:6094–6107. doi:10.4049/jimmunol.1003507
23. Oenema TA, Maarsingh H, Smit M, et al. Bronchoconstriction induces TGF-beta release and airway remodelling in guinea pig lung slices. PLoS One. 2013;8:e65580. doi:10.1371/journal.pone.0065580
24. Yocum GT, Chen J, Choi CH, et al. Role of transient receptor potential vanilloid 1 in the modulation of airway smooth muscle tone and calcium handling. Am J Physiol. 2017;312:L812–L821. doi:10.1152/ajplung.00064.2017
25. Choi JY, Lee HY, Hur J, et al. TRPV1 blocking alleviates airway inflammation and remodeling in a chronic asthma murine model. Allergy Asthma Immunol Res. 2018;10:216–224. doi:10.4168/aair.2018.10.3.216
26. Noble PB, Pascoe CD, Lan B, et al. Airway smooth muscle in asthma: linking contraction and mechanotransduction to disease pathogenesis and remodelling. Pulm Pharmacol Ther. 2014;29:96–107. doi:10.1016/j.pupt.2014.07.005
27. Johnson PR, Burgess JK, Underwood PA, et al. Extracellular matrix proteins modulate asthmatic airway smooth muscle cell proliferation via an autocrine mechanism. J Allergy Clin Immunol. 2004;113:690–696. doi:10.1016/j.jaci.2003.12.312
28. Zhang C, Wang W, Liu C, Lu J, Sun K. Role of NF-kappaB/GATA3 in the inhibition of lysyl oxidase by IL-1beta in human amnion fibroblasts. Immunol Cell Biol. 2017;95:943–952. doi:10.1038/icb.2017.73
29. Brown AC, Fiore VF, Sulchek TA, Barker TH. Physical and chemical microenvironmental cues orthogonally control the degree and duration of fibrosis-associated epithelial-to-mesenchymal transitions. J Pathol. 2013;229:25–35. doi:10.1002/path.4114
30. Humphrey JD, Dufresne ER, Schwartz MA. Mechanotransduction and extracellular matrix homeostasis. Nat Rev Mol Cell Biol. 2014;15:802–812. doi:10.1038/nrm3896
31. Shkumatov A, Thompson M, Choi KM, et al. Matrix stiffness-modulated proliferation and secretory function of the airway smooth muscle cells. Am J Physiol. 2015;308:L1125–1135. doi:10.1152/ajplung.00154.2014
32. Jopeth Ramis RM, Pappalardo F, Cairns J, et al. LOXL2 mediates airway smooth muscle cell matrix stiffness and drives asthmatic airway remodelling. BioRxivs. 2020. doi:10.1101/2020.11.16.384792
33. Cox D, Brennan M, Moran N. Integrins as therapeutic targets: lessons and opportunities. Nat Rev Drug Discov. 2010;9:804–820. doi:10.1038/nrd3266
34. Green HJ, Brown NH. Integrin intracellular machinery in action. Exp Cell Res. 2019;378:226–231. doi:10.1016/j.yexcr.2019.03.011
35. Coraux C, Delplanque A, Hinnrasky J, et al. Distribution of integrins during human fetal lung development. J Histochem Cytochem. 1998;46:803–810. doi:10.1177/002215549804600703
36. Wu JE, Santoro SA. Differential expression of integrin alpha subunits supports distinct roles during lung branching morphogenesis. Dev Dyn. 1996;206:169–181. doi:10.1002/(SICI)1097-0177(199606)206:2<169::AID-AJA6>3.0.CO;2-G
37. Pilewski JM, Latoche JD, Arcasoy SM, Albelda SM. Expression of integrin cell adhesion receptors during human airway epithelial repair in vivo. Am J Physiol. 1997;273:L256–263. doi:10.1152/ajplung.1997.273.1.L256
38. Mette SA, Pilewski J, Buck CA, Albelda SM. Distribution of integrin cell adhesion receptors on normal bronchial epithelial cells and lung cancer cells in vitro and in vivo. Am J Respir Cell Mol Biol. 1993;8:562–572. doi:10.1165/ajrcmb/8.5.562
39. Tatler AL, Habgood A, Porte J, et al. Reduced Ets domain-containing protein Elk1 promotes pulmonary fibrosis via increased integrin alphavbeta6 expression. J Biol Chem. 2016;291:9540–9553. doi:10.1074/jbc.M115.692368
40. Tatler AL, Goodwin AT, Gbolahan O, et al. Amplification of TGFbeta induced ITGB6 gene transcription may promote pulmonary fibrosis. PLoS One. 2016;11:e0158047. doi:10.1371/journal.pone.0158047
41. Sheppard D, Cohen DS, Wang A, Busk M. Transforming growth factor beta differentially regulates expression of integrin subunits in Guinea pig airway epithelial cells. J Biol Chem. 1992;267:17409–17414. doi:10.1016/S0021-9258(18)41941-2
42. Teoh CM, Tan S, Tran T, et al. Integrins as therapeutic targets for respiratory diseases. Curr Mol Med. 2016;15:714–734. doi:10.2174/1566524015666150921105339
43. Roman J, Little CW, McDonald JA. Potential role of RGD-binding integrins in mammalian lung branching morphogenesis. Development. 1991;112:551–558. doi:10.1242/dev.112.2.551
44. Albert RK, Embree LJ, McFeely JE, Hickstein DD. Expression and function of beta 2 integrins on alveolar macrophages from human and nonhuman primates. Am J Respir Cell Mol Biol. 1992;7:182–189. doi:10.1165/ajrcmb/7.2.182
45. McNally AK, Anderson JM. Beta1 and beta2 integrins mediate adhesion during macrophage fusion and multinucleated foreign body giant cell formation. Am J Pathol. 2002;160:621–630. doi:10.1016/s0002-9440(10)64882-1
46. Barthel SR, Johansson MW, McNamee DM, Mosher DF. Roles of integrin activation in eosinophil function and the eosinophilic inflammation of asthma. J Leukoc Biol. 2008;83:1–12. doi:10.1189/jlb.0607344
47. Barthel SR, Annis DS, Mosher DF, Johansson MW. Differential engagement of modules 1 and 4 of vascular cell adhesion molecule-1 (CD106) by integrins alpha4beta1 (CD49d/29) and alphaMbeta2 (CD11b/18) of eosinophils. J Biol Chem. 2006;281:32175–32187. doi:10.1074/jbc.M600943200
48. Jeffery PK, Wardlaw AJ, Nelson FC, Collins JV, Kay AB. Bronchial biopsies in asthma. An ultrastructural, quantitative study and correlation with hyperreactivity. Am Rev Respir Dis. 1989;140:1745–1753. doi:10.1164/ajrccm/140.6.1745
49. Zhou C, Yin G, Liu J, Liu X, Zhao S. Epithelial apoptosis and loss in airways of children with asthma. J Asthma. 2011;48:358–365. doi:10.3109/02770903.2011.565848
50. Laitinen LA, Heino M, Laitinen A, Kava T, Haahtela T. Damage of the airway epithelium and bronchial reactivity in patients with asthma. Am Rev Respir Dis. 1985;131:599–606. doi:10.1164/arrd.1985.131.4.599
51. Faul JL, Tormey VJ, Leonard C, et al. Lung immunopathology in cases of sudden asthma death. Eur Respir J. 1997;10:301–307. doi:10.1183/09031936.97.10020301
52. Heijink IH, Kuchibhotla VN, Roffel MP, et al. Epithelial cell dysfunction, a major driver of asthma development. Allergy. 2020;75:1902–1917. doi:10.1111/all.14421
53. Wu J, Dong F, Wang R-A, et al. Central role of cellular senescence in TSLP-induced airway remodeling in asthma. PLoS One. 2013;8:e77795. doi:10.1371/journal.pone.0077795
54. Hirota N, Risse P-A, Novali M, et al. Histamine may induce airway remodeling through release of epidermal growth factor receptor ligands from bronchial epithelial cells. FASEB J. 2012;26:1704–1716. doi:10.1096/fj.11-197061
55. Puddicombe SM, Polosa R, Richter A, et al. Involvement of the epidermal growth factor receptor in epithelial repair in asthma. FASEB J. 2000;14:1362–1374. doi:10.1096/fasebj.14.10.1362
56. Brown SD, Baxter KM, Stephenson ST, et al. Airway TGF-beta1 and oxidant stress in children with severe asthma: association with airflow limitation. J Allergy Clin Immunol. 2012;129:
57. Chakir J, Shannon J, Molet S, et al. Airway remodeling-associated mediators in moderate to severe asthma: effect of steroids on TGF-beta, IL-11, IL-17, and type I and type III collagen expression. J Allergy Clin Immunol. 2003;111:1293–1298. doi:10.1067/mai.2003.1557
58. Trautmann A, Krüger K, Akdis M, et al. Apoptosis and loss of adhesion of bronchial epithelial cells in asthma. Int Arch Allergy Immunol. 2005;138:142–150. doi:10.1159/000088436
59. Cohen L, E X, Tarsi J, et al. Epithelial cell proliferation contributes to airway remodeling in severe asthma. Am J Respir Crit Care Med. 2007;176:138–145. doi:10.1164/rccm.200607-1062OC
60. Yuan L, Du X, Tang S, et al. ITGB 4 deficiency induces senescence of airway epithelial cells through p53 activation. FEBS J. 2019;286:1191–1203. doi:10.1111/febs.14749
61. Liu C, Xiang Y, Liu H, et al. Integrin beta4 was downregulated on the airway epithelia of asthma patients. Acta Biochim Biophys Sin. 2010;42:538–547. doi:10.1093/abbs/gmq058
62. Liu C, Liu H-J, Xiang Y, et al. Wound repair and anti-oxidative capacity is regulated by ITGB4 in airway epithelial cells. Mol Cell Biochem. 2010;341:259–269. doi:10.1007/s11010-010-0457-y
63. Tajiri T, Matsumoto H, Jinnai M, et al. Pathophysiological relevance of sputum MUC5AC and MUC5B levels in patients with mild asthma. Allergol Int. 2021. doi:10.1016/j.alit.2021.09.003
64. Ordonez CL, Khashayar R, Wong H, et al. Mild and moderate asthma is associated with airway goblet cell hyperplasia and abnormalities in mucin gene expression. Am J Respir Crit Care Med. 2001;163:517–523. doi:10.1164/ajrccm.163.2.2004039
65. Aikawa T, Shimura S, Sasaki H, Ebina M, Takishima T. Marked goblet cell hyperplasia with mucus accumulation in the airways of patients who died of severe acute asthma attack. Chest. 1992;101:916–921. doi:10.1378/chest.101.4.916
66. Faiz A, Weckmann M, Tasena H, et al. Profiling of healthy and asthmatic airway smooth muscle cells following interleukin-1beta treatment: a novel role for CCL20 in chronic mucus hypersecretion. Eur Respir J. 2018;52:1800310. doi:10.1183/13993003.00310-2018
67. Yoshida Y, Takaku Y, Nakamoto Y, et al. Changes in airway diameter and mucus plugs in patients with asthma exacerbation. PLoS One. 2020;15:e0229238. doi:10.1371/journal.pone.0229238
68. Iwashita J, Murata J. Integrin beta1 subunit regulates cellular and secreted MUC5AC and MUC5B production in NCI-H292 human lung epithelial cells. Biochem Biophys Rep. 2021;28:101124. doi:10.1016/j.bbrep.2021.101124
69. Iwashita J, Yamamoto T, Sasaki Y, Abe T. MUC5AC production is downregulated in NCI-H292 lung cancer cells cultured on type-IV collagen. Mol Cell Biochem. 2010;337:65–75. doi:10.1007/s11010-009-0286-z
70. Zhi Y, Huang H, Liang L. MFG-E8/integrin beta3 signaling contributes to airway inflammation response and airway remodeling in an ovalbumin-induced murine model of asthma. J Cell Biochem. 2018;119:8887–8896. doi:10.1002/jcb.27142
71. Tatler AL, Porte J, Knox A, Jenkins G, Pang L. Tryptase activates TGFbeta in human airway smooth muscle cells via direct proteolysis. Biochem Biophys Res Commun. 2008;370:239–242. doi:10.1016/j.bbrc.2008.03.064
72. John AE, Zhu YM, Brightling CE, Pang L, Knox AJ. Human airway smooth muscle cells from asthmatic individuals have CXCL8 hypersecretion due to increased NF-kappaB p65, C/ EBPbeta, and RNA polymerase II binding to the CXCL8 promoter. J Immunol. 2009;183:4682–4692. doi:10.4049/jimmunol.0803832
73. Clifford RL, Patel JK, John AE, et al. CXCL8 histone H3 acetylation is dysfunctional in airway smooth muscle in asthma: regulation by BET. Am J Physiol. 2015;308:L962–972. doi:10.1152/ajplung.00021.2015
74. Benayoun L, Druilhe A, Dombret MC, Aubier M, Pretolani M. Airway structural alterations selectively associated with severe asthma. Am J Respir Crit Care Med. 2003;167:1360–1368. doi:10.1164/rccm.200209-1030OC
75. Woodruff PG, Dolganov GM, Ferrando RE, et al. Hyperplasia of smooth muscle in mild to moderate asthma without changes in cell size or gene expression. Am J Respir Crit Care Med. 2004;169:1001–1006. doi:10.1164/rccm.200311-1529OC
76. James AL, Elliot JG, Jones RL, et al. Airway smooth muscle hypertrophy and hyperplasia in asthma. Am J Respir Crit Care Med. 2012;185:1058–1064. doi:10.1164/rccm.201110-1849OC
77. O’Sullivan MJ, Jang JH, Panariti A, et al. Airway epithelial cells drive airway smooth muscle cell phenotype switching to the proliferative and pro-inflammatory phenotype. Front Physiol. 2021;12:687654. doi:10.3389/fphys.2021.687654
78. Al Heialy S, Risse P-A, Zeroual MA, et al. T cell-induced airway smooth muscle cell proliferation via the epidermal growth factor receptor. Am J Respir Cell Mol Biol. 2013;49:563–570. doi:10.1165/rcmb.2012-0356OC
79. Hirota JA, Ask K, Farkas L, et al. In vivo role of platelet-derived growth factor–BB in airway smooth muscle proliferation in mouse lung. Am J Respir Cell Mol Biol. 2011;45:566–572. doi:10.1165/rcmb.2010-0277OC
80. Pan Y, Liu L, Li S, et al. Activation of AMPK inhibits TGF-beta1-induced airway smooth muscle cells proliferation and its potential mechanisms. Sci Rep. 2018;8:3624. doi:10.1038/s41598-018-21812-0
81. Wang Q, Li H, Yao Y, et al. HB-EGF-promoted airway smooth muscle cells and their progenitor migration contribute to airway smooth muscle remodeling in asthmatic mouse. J Immunol. 2016;196:2361–2367. doi:10.4049/jimmunol.1402126
82. Kim SH, Pei Q-M, Jiang P, et al. Effect of active vitamin D3 on VEGF-induced ADAM33 expression and proliferation in human airway smooth muscle cells: implications for asthma treatment. Respir Res. 2017;18:7. doi:10.1186/s12931-016-0490-9
83. Parameswaran K, Cox G, Radford K, et al. Cysteinyl leukotrienes promote human airway smooth muscle migration. Am J Respir Crit Care Med. 2002;166:738–742. doi:10.1164/rccm.200204-291OC
84. Ijpma G, Panariti A, Lauzon AM, Martin JG. Directional preference of airway smooth muscle mass increase in human asthmatic airways. Am J Physiol. 2017;312:L845–L854. doi:10.1152/ajplung.00353.2016
85. Sundaram A, Chen C, Khalifeh-Soltani A, et al. Targeting integrin alpha5beta1 ameliorates severe airway hyperresponsiveness in experimental asthma. J Clin Invest. 2017;127:365–374. doi:10.1172/JCI88555
86. Liu S, Ngo U, Tang X-Z, et al. Integrin alpha2beta1 regulates collagen I tethering to modulate hyperresponsiveness in reactive airway disease models. J Clin Invest. 2021;131. doi:10.1172/JCI138140
87. Gunst SJ, Tang DD. The contractile apparatus and mechanical properties of airway smooth muscle. Eur Respir J. 2000;15:600–616. doi:10.1034/j.1399-3003.2000.15.29.x
88. Wang Y, Liao G, Wang R, Tang DD. Acetylation of Abelson interactor 1 at K416 regulates actin cytoskeleton and smooth muscle contraction. FASEB J. 2021;35:e21811. doi:10.1096/fj.202100415R
89. Wang T, Cleary RA, Wang R, Tang DD. Role of the adapter protein Abi1 in actin-associated signaling and smooth muscle contraction. J Biol Chem. 2013;288:20713–20722. doi:10.1074/jbc.M112.439877
90. Wang R, Cleary RA, Wang T, Li J, Tang DD. The association of cortactin with profilin-1 is critical for smooth muscle contraction. J Biol Chem. 2014;289:14157–14169. doi:10.1074/jbc.M114.548099
91. Jia L, Wang R, Tang DD. Abl regulates smooth muscle cell proliferation by modulating actin dynamics and ERK1/2 activation. Am J Physiol. 2012;302:C1026–1034. doi:10.1152/ajpcell.00373.2011
92. Ojiaku CA, Cao G, Zhu W, et al. TGF-beta1 evokes human airway smooth muscle cell shortening and hyperresponsiveness via Smad3. Am J Respir Cell Mol Biol. 2018;58:575–584. doi:10.1165/rcmb.2017-0247OC
93. Khalifeh-Soltani A, Gupta D, Ha A, Podolsky MJ. The Mfge8-alpha8beta1-PTEN pathway regulates airway smooth muscle contraction in allergic inflammation. FASEB J. 2018;fj201800109R. doi:10.1096/fj.201800109R
94. Chen C, Kudo M, Rutaganira F, et al. Integrin alpha9beta1 in airway smooth muscle suppresses exaggerated airway narrowing. J Clin Invest. 2012;122:2916–2927. doi:10.1172/JCI60387
95. Kaminska M, Foley S, Maghni K, et al. Airway remodeling in subjects with severe asthma with or without chronic persistent airflow obstruction. J Allergy Clin Immunol. 2009;124:
96. Dekkers BG, Bos IS, Gosens R, Halayko AJ, Zaagsma J, Meurs H. The integrin-blocking peptide RGDS inhibits airway smooth muscle remodeling in a Guinea pig model of allergic asthma. Am J Respir Crit Care Med. 2010;181:556–565. doi:10.1164/rccm.200907-1065OC
97. Wang R, Liao G, Wang Y, Tang DD. Distinctive roles of Abi1 in regulating actin-associated proteins during human smooth muscle cell migration. Sci Rep. 2020;10:10667. doi:10.1038/s41598-020-67781-1
98. Nguyen TT, Ward JP, Hirst SJ. beta1-Integrins mediate enhancement of airway smooth muscle proliferation by collagen and fibronectin. Am J Respir Crit Care Med. 2005;171:217–223. doi:10.1164/rccm.200408-1046OC
99. Fu J, Zheng M, Zhang X, et al. Fibulin-5 promotes airway smooth muscle cell proliferation and migration via modulating Hippo-YAP/TAZ pathway. Biochem Biophys Res Commun. 2017;493:985–991. doi:10.1016/j.bbrc.2017.09.105
100. Tran T, Teoh CM, Tam JKC, et al. Laminin drives survival signals to promote a contractile smooth muscle phenotype and airway hyperreactivity. FASEB J. 2013;27:3991–4003. doi:10.1096/fj.12-221341
101. Roche WR, Beasley R, Williams JH, Holgate ST. Subepithelial fibrosis in the bronchi of asthmatics. Lancet. 1989;1:520–524. doi:10.1016/S0140-6736(89)90067-6
102. Hoshino M, Nakamura Y, Sim J, Shimojo J, Isogai S. Bronchial subepithelial fibrosis and expression of matrix metalloproteinase-9 in asthmatic airway inflammation. J Allergy Clin Immunol. 1998;102:783–788. doi:10.1016/s0091-6749(98)70018-1
103. Wang CH, Huang C-D, Lin H-C, et al. Increased circulating fibrocytes in asthma with chronic airflow obstruction. Am J Respir Crit Care Med. 2008;178:583–591. doi:10.1164/rccm.200710-1557OC
104. Moir LM, Burgess JK, Black JL. Transforming growth factor beta 1 increases fibronectin deposition through integrin receptor alpha 5 beta 1 on human airway smooth muscle. J Allergy Clin Immunol. 2008;121:1034–1039 e1034. doi:10.1016/j.jaci.2007.12.1159
105. Hong GH, Park S-Y, Kwon H-S, et al. IL-32gamma attenuates airway fibrosis by modulating the integrin-FAK signaling pathway in fibroblasts. Respir Res. 2018;19:188. doi:10.1186/s12931-018-0863-3
106. Vignola AM, Chanez P, Chiappara G, et al. Transforming growth factor-beta expression in mucosal biopsies in asthma and chronic bronchitis. Am J Respir Crit Care Med. 1997;156:591–599. doi:10.1164/ajrccm.156.2.9609066
107. Redington AE, Madden J, Frew A, et al. Transforming growth factor-beta 1 in asthma. Measurement in bronchoalveolar lavage fluid. Am J Respir Crit Care Med. 1997;156:642–647. doi:10.1164/ajrccm.156.2.9605065
108. Torrego A, Hew M, Oates T, Sukkar M, Fan Chung K. Expression and activation of TGF-beta isoforms in acute allergen-induced remodelling in asthma. Thorax. 2007;62:307–313. doi:10.1136/thx.2006.063487
109. Batra V, Musani AI, Hastie AT, et al. Bronchoalveolar lavage fluid concentrations of transforming growth factor (TGF)-beta1, TGF-beta2, interleukin (IL)-4 and IL-13 after segmental allergen challenge and their effects on alpha-smooth muscle actin and collagen III synthesis by primary human lung fibroblasts. Clin Exp Allergy. 2004;34:437–444. doi:10.1111/j.1365-2222.2004.01885.x
110. Walker EJ, Heydet D, Veldre T, Ghildyal R. Transcriptomic changes during TGF-beta-mediated differentiation of airway fibroblasts to myofibroblasts. Sci Rep. 2019;9:20377. doi:10.1038/s41598-019-56955-1
111. Guo W, Shan B, Klingsberg RC, Qin X, Lasky JA. Abrogation of TGF-beta1-induced fibroblast-myofibroblast differentiation by histone deacetylase inhibition. Am J Physiol. 2009;297:L864–870. doi:10.1152/ajplung.00128.2009
112. Sidhu SS, Yuan S, Innes AL, et al. Roles of epithelial cell-derived periostin in TGF-beta activation, collagen production, and collagen gel elasticity in asthma. Proc Natl Acad Sci U S A. 2010;107:14170–14175. doi:10.1073/pnas.1009426107
113. Frangogiannis N. Transforming growth factor-beta in tissue fibrosis. J Exp Med. 2020;217:e20190103. doi:10.1084/jem.20190103
114. Bottoms SE, Howell JE, Reinhardt AK, Evans IC, McAnulty RJ. Tgf-Beta isoform specific regulation of airway inflammation and remodelling in a murine model of asthma. PLoS One. 2010;5:e9674. doi:10.1371/journal.pone.0009674
115. Kenyon NJ, Ward RW, McGrew G, Last JA. TGF-beta1 causes airway fibrosis and increased collagen I and III mRNA in mice. Thorax. 2003;58:772–777. doi:10.1136/thorax.58.9.772
116. Wnuk D, Paw M, Ryczek K, et al. Enhanced asthma-related fibroblast to myofibroblast transition is the result of profibrotic TGF-beta/Smad2/3 pathway intensification and antifibrotic TGF-beta/Smad1/5/(8)9 pathway impairment. Sci Rep. 2020;10:16492. doi:10.1038/s41598-020-73473-7
117. Mu D, Cambier S, Fjellbirkeland L, et al. The integrin alpha(v)beta8 mediates epithelial homeostasis through MT1-MMP-dependent activation of TGF-beta1. J Cell Biol. 2002;157:493–507. doi:10.1083/jcb.200109100
118. Tatler AL, Jenkins G. TGF-beta activation and lung fibrosis. Proc Am Thorac Soc. 2012;9:130–136. doi:10.1513/pats.201201-003AW
119. Ling KM, Sutanto EN, Iosifidis T, et al. Reduced transforming growth factor beta1 (TGF-beta1) in the repair of airway epithelial cells of children with asthma. Respirology. 2016;21:1219–1226. doi:10.1111/resp.12810
120. Prikk K, Maisi P, Pirilä E, et al. Airway obstruction correlates with collagenase-2 (MMP-8) expression and activation in bronchial asthma. Lab Investig. 2002;82:1535–1545. doi:10.1097/01.lab.0000035023.53893.b6
121. Suzuki R, Kato T, Miyazaki Y, et al. Matrix metalloproteinases and tissue inhibitors of matrix metalloproteinases in sputum from patients with bronchial asthma. J Asthma. 2001;38:477–484. doi:10.1081/jas-100105868
122. Wipff PJ, Rifkin DB, Meister JJ, Hinz B. Myofibroblast contraction activates latent TGF- 1 from the extracellular matrix. J Cell Biol. 2007;179:1311–1323. doi:10.1083/jcb.200704042
123. Xu MY, Porte J, Knox AJ, et al. Lysophosphatidic acid induces {alpha}v{beta}6 integrin-mediated TGF-{beta} activation via the LPA2 receptor and the small G protein G{alpha}q. Am J Pathol. 2009;174:1264–1279. doi:10.2353/ajpath.2009.080160
124. Jenkins RG, Su X, Su G, et al. Ligation of protease-activated receptor 1 enhances alpha(v)beta6 integrin-dependent TGF-beta activation and promotes acute lung injury. J Clin Invest. 2006;116:1606–1614. doi:10.1172/JCI27183
125. Januskevicius A, Gosens R, Sakalauskas R, et al. Suppression of eosinophil integrins prevents remodeling of airway smooth muscle in asthma. Front Physiol. 2016;7:680. doi:10.3389/fphys.2016.00680
126. Janulaityte I, Januskevicius A, Kalinauskaite-Zukauske V, Bajoriuniene I, Malakauskas K. In vivo allergen-activated eosinophils promote collagen I and fibronectin gene expression in airway smooth muscle cells via TGF-beta1 signaling pathway in asthma. Int J Mol Sci. 2020;21:1837. doi:10.3390/ijms21051837
127. Tanaka H, Yamada G, Saikai T, et al. Increased airway vascularity in newly diagnosed asthma using a high-magnification bronchovideoscope. Am J Respir Crit Care Med. 2003;168:1495–1499. doi:10.1164/rccm.200306-727OC
128. Orsida BE, Li X, Hickey B, et al. Vascularity in asthmatic airways: relation to inhaled steroid dose. Thorax. 1999;54:289–295. doi:10.1136/thx.54.4.289
129. Salvato G. Quantitative and morphological analysis of the vascular bed in bronchial biopsy specimens from asthmatic and non-asthmatic subjects. Thorax. 2001;56:902–906. doi:10.1136/thorax.56.12.902
130. Van der Velden J, Harkness LM, Barker DM, et al. The effects of tumstatin on vascularity, airway inflammation and lung function in an experimental sheep model of chronic asthma. Sci Rep. 2016;6:26309. doi:10.1038/srep26309
131. Lee HY, Min KH, Lee SM, Lee JE, Rhee CK. Clinical significance of serum vascular endothelial growth factor in young male asthma patients. Korean J Intern Med. 2017;32:295–301. doi:10.3904/kjim.2014.242
132. Lee SY, Kwon S, Kim KH, et al. Expression of vascular endothelial growth factor and hypoxia-inducible factor in the airway of asthmatic patients. Ann Allergy Asthma Immunol. 2006;97:794–799. doi:10.1016/S1081-1206(10)60971-4
133. Feltis BN, Wignarajah D, Zheng L, et al. Increased vascular endothelial growth factor and receptors: relationship to angiogenesis in asthma. Am J Respir Crit Care Med. 2006;173:1201–1207. doi:10.1164/rccm.200507-1105OC
134. Chetta A, Zanini A, Foresi A, et al. Vascular endothelial growth factor up-regulation and bronchial wall remodelling in asthma. Clin Exp Allergy. 2005;35:1437–1442. doi:10.1111/j.1365-2222.2005.02360.x
135. Simcock DE, Kanabar V, Clarke GW, et al. Induction of angiogenesis by airway smooth muscle from patients with asthma. Am J Respir Crit Care Med. 2008;178:460–468. doi:10.1164/rccm.200707-1046OC
136. Zhang R, Dong H, Zhao H, et al. 1,25-Dihydroxyvitamin D3 targeting VEGF pathway alleviates house dust mite (HDM)-induced airway epithelial barrier dysfunction. Cell Immunol. 2017;312:15–24. doi:10.1016/j.cellimm.2016.11.004
137. Turkeli A, Yilmaz Ö, Karaman M, et al. Anti-VEGF treatment suppresses remodeling factors and restores epithelial barrier function through the E-cadherin/beta-catenin signaling axis in experimental asthma models. Exp Ther Med. 2021;22:689. doi:10.3892/etm.2021.10121
138. Yuksel H, Yilmaz O, Karaman M, et al. Role of vascular endothelial growth factor antagonism on airway remodeling in asthma. Ann Allergy Asthma Immunol. 2013;110:150–155. doi:10.1016/j.anai.2012.12.015
139. Brooks PC, Clark RA, Cheresh DA. Requirement of vascular integrin alpha v beta 3 for angiogenesis. Science. 1994;264:569–571. doi:10.1126/science.7512751
140. Friedlander M, Brooks PC, Shaffer RW, et al. Definition of two angiogenic pathways by distinct α v integrins. Science. 1995;270:1500–1502. doi:10.1126/science.270.5241.1500
141. Thompson EE, Pan L, Ostrovnaya I, et al. Integrin beta 3 genotype influences asthma and allergy phenotypes in the first 6 years of life. J Allergy Clin Immunol. 2007;119:1423–1429. doi:10.1016/j.jaci.2007.03.029
142. Drake CJ, Cheresh DA, Little CD. An antagonist of integrin alpha v beta 3 prevents maturation of blood vessels during embryonic neovascularization. J Cell Sci. 1995;108(Pt 7):2655–2661. doi:10.1242/jcs.108.7.2655
143. Hodivala-Dilke KM, McHugh KP, Tsakiris DA, et al. Beta3-integrin-deficient mice are a model for Glanzmann thrombasthenia showing placental defects and reduced survival. J Clin Invest. 1999;103:229–238. doi:10.1172/JCI5487
144. Huang X, Griffiths M, Wu J, Farese RV, Sheppard D. Normal development, wound healing, and adenovirus susceptibility in beta5-deficient mice. Mol Cell Biol. 2000;20:755–759. doi:10.1128/MCB.20.3.755-759.2000
145. Yang JT, Rayburn H, Hynes RO. Embryonic mesodermal defects in alpha 5 integrin-deficient mice. Development. 1993;119:1093–1105. doi:10.1242/dev.119.4.1093
146. Okazaki T, Ni A, Ayeni OA, et al. alpha5beta1 Integrin blockade inhibits lymphangiogenesis in airway inflammation. Am J Pathol. 2009;174:2378–2387. doi:10.2353/ajpath.2009.080942
147. Davis GE, Camarillo CW. An alpha 2 beta 1 integrin-dependent pinocytic mechanism involving intracellular vacuole formation and coalescence regulates capillary lumen and tube formation in three-dimensional collagen matrix. Exp Cell Res. 1996;224:39–51. doi:10.1006/excr.1996.0109
148. Peng Q, Lai D, Nguyen TT-B, et al. Multiple β 1 integrins mediate enhancement of human airway smooth muscle cytokine secretion by fibronectin and type I collagen. J Immunol. 2005;174:2258–2264. doi:10.4049/jimmunol.174.4.2258
149. Weller PF, Rand TH, Goelz SE, Chi-Rosso G, Lobb RR. Human eosinophil adherence to vascular endothelium mediated by binding to vascular cell adhesion molecule 1 and endothelial leukocyte adhesion molecule 1. Proc Natl Acad Sci USA. 1991;88:7430–7433. doi:10.1073/pnas.88.16.7430
150. Nagata M, Sedgwick JB, Kita H, Busse WW. Granulocyte macrophage colony-stimulating factor augments ICAM-1 and VCAM-1 activation of eosinophil function. Am J Respir Cell Mol Biol. 1998;19:158–166. doi:10.1165/ajrcmb.19.1.3001
151. Kato M, Kita H, Tokuyama K, Morikawa A. Cross-linking of the beta2 integrin, CD11b/CD18, on human eosinophils induces protein tyrosine phosphorylation and cellular degranulation. Int Arch Allergy Immunol. 1998;117(Suppl 1):68–71. doi:10.1159/000053576
152. Nagata M, Sedgwick JB, Bates ME, Kita H, Busse WW. Eosinophil adhesion to vascular cell adhesion molecule-1 activates superoxide anion generation. J Immunol. 1995;155:2194–2202.
153. Higashimoto I, Chihara J, Kakazu T, et al. Regulation of eosinophil cell death by adhesion to fibronectin. Int Arch Allergy Immunol. 1996;111(Suppl 1):66–69. doi:10.1159/000237420
154. Ray A, Kolls JK. Neutrophilic inflammation in asthma and association with disease severity. Trends Immunol. 2017;38:942–954. doi:10.1016/j.it.2017.07.003
155. Sekheri M, Othman A, Filep JG. beta2 integrin regulation of neutrophil functional plasticity and fate in the resolution of inflammation. Front Immunol. 2021;12:660760. doi:10.3389/fimmu.2021.660760
156. Fan Z, McArdle S, Marki A, et al. Neutrophil recruitment limited by high-affinity bent beta2 integrin binding ligand in cis. Nat Commun. 2016;7:12658. doi:10.1038/ncomms12658
157. Khawaja AA, Chong DLW, Sahota J, et al. Identification of a novel HIF-1alpha-alphaMbeta2 integrin-NET axis in fibrotic interstitial lung disease. Front Immunol. 2020;11:2190. doi:10.3389/fimmu.2020.02190
158. Habgood AN, Tatler AL, Porte J, et al. Secretory leukocyte protease inhibitor gene deletion alters bleomycin-induced lung injury, but not development of pulmonary fibrosis. Lab Investig. 2016;96:623–631. doi:10.1038/labinvest.2016.40
159. Maestrelli P, De Fina O, Bertin T, et al. Integrin expression on neutrophils and mononuclear cells in blood and induced sputum in stable asthma. Allergy. 1999;54:1303–1308. doi:10.1034/j.1398-9995.1999.00337.x
160. Holgate ST, Davies DE. Rethinking the pathogenesis of asthma. Immunity. 2009;31:362–367. doi:10.1016/j.immuni.2009.08.013
161. Liu C, Qin X, Liu H, Xiang Y. Downregulation of integrin beta4 decreases the ability of airway epithelial cells to present antigens. PLoS One. 2012;7:e32060. doi:10.1371/journal.pone.0032060
162. Fernandes D, Guida E, Koutsoubos V, et al. Glucocorticoids inhibit proliferation, cyclin D1 expression, and retinoblastoma protein phosphorylation, but not activity of the extracellular-regulated kinases in human cultured airway smooth muscle. Am J Respir Cell Mol Biol. 1999;21:77–88. doi:10.1165/ajrcmb.21.1.3396
163. Shull S, Meisler N, Absher M, Phan S, Cutroneo K. Glucocorticoid-induced down regulation of transforming growth factor-beta 1 in adult rat lung fibroblasts. Lung. 1995;173:71–78. doi:10.1007/BF02981467
164. Bandi N, Kompella UB. Budesonide reduces vascular endothelial growth factor secretion and expression in airway (Calu-1) and alveolar (A549) epithelial cells. Eur J Pharmacol. 2001;425:109–116. doi:10.1016/s0014-2999(01)01192-x
165. Laitinen LA, Laitinen A, Haahtela T. A comparative study of the effects of an inhaled corticosteroid, budesonide, and a beta 2-agonist, terbutaline, on airway inflammation in newly diagnosed asthma: a randomized, double-blind, parallel-group controlled trial. J Allergy Clin Immunol. 1992;90:32–42. doi:10.1016/s0091-6749(06)80008-4
166. Haldar P, Brightling CE, Hargadon B, et al. Mepolizumab and exacerbations of refractory eosinophilic asthma. N Engl J Med. 2009;360:973–984. doi:10.1056/NEJMoa0808991
167. Flood-Page P, Menzies-Gow A, Phipps S, et al. Anti-IL-5 treatment reduces deposition of ECM proteins in the bronchial subepithelial basement membrane of mild atopic asthmatics. J Clin Invest. 2003;112:1029–1036. doi:10.1172/JCI17974
168. Chachi L, Diver S, Kaul H, et al. Computational modelling prediction and clinical validation of impact of benralizumab on airway smooth muscle mass in asthma. Eur Respir J. 2019;54:1900930. doi:10.1183/13993003.00930-2019
169. Tajiri T, Niimi A, Matsumoto H, et al. Comprehensive efficacy of omalizumab for severe refractory asthma: a time-series observational study. Ann Allergy Asthma Immunol. 2014;113:470–475 e472. doi:10.1016/j.anai.2014.06.004
170. Prakash YS, Halayko AJ, Gosens R, et al. An Official American Thoracic Society Research Statement: current challenges facing research and therapeutic advances in airway remodeling. Am J Respir Crit Care Med. 2017;195:e4–e19. doi:10.1164/rccm.201611-2248ST
171. Kianmeher M, Ghorani V, Boskabady MH. Animal model of asthma, various methods and measured parameters: a methodological review. Iran J Allergy Asthma Immunol. 2016;15:445–465.
172. Tatler AL, Philp CJ, Hill MR, et al. Differential remodelling in small and large murine airways revealed by novel whole lung airway analysis. BioRxivs. 2022. doi:10.1101/2022.01.15.476324
173. Kotaru C, Schoonover KJ, Trudeau JB, et al. Regional fibroblast heterogeneity in the lung: implications for remodeling. Am J Respir Crit Care Med. 2006;173:1208–1215. doi:10.1164/rccm.200508-1218OC
174. Clifford RL, Yang CX, Fishbane N, et al. TWIST1 DNA methylation is a cell marker of airway and parenchymal lung fibroblasts that are differentially methylated in asthma. Clin Epigenetics. 2020;12:145. doi:10.1186/s13148-020-00931-4
175. Gupta S, Siddiqui S, Haldar P, et al. Qualitative analysis of high-resolution CT scans in severe asthma. Chest. 2009;136:1521–1528. doi:10.1378/chest.09-0174
176. Cianchetti S, Cardini C, Puxeddu I, et al. Distinct profile of inflammatory and remodelling biomarkers in sputum of severe asthmatic patients with or without persistent airway obstruction. World Allergy Organ J. 2019;12:100078. doi:10.1016/j.waojou.2019.100078
177. Riccio AM, Mauri P, De Ferrari L, et al. Galectin-3: an early predictive biomarker of modulation of airway remodeling in patients with severe asthma treated with omalizumab for 36 months. Clin Transl Allergy. 2017;7:6. doi:10.1186/s13601-017-0143-1
178. Ayars AG, Altman LC, Potter-Perigo S, et al. Sputum hyaluronan and versican in severe eosinophilic asthma. Int Arch Allergy Immunol. 2013;161:65–73. doi:10.1159/000343031
179. Kitamura H, Cambier S, Somanath S, et al. Mouse and human lung fibroblasts regulate dendritic cell trafficking, airway inflammation, and fibrosis through integrin alphavbeta8-mediated activation of TGF-beta. J Clin Invest. 2011;121:2863–2875. doi:10.1172/JCI45589
180. Macias-Perez I, Borza C, Chen X, et al. Loss of integrin alpha1beta1 ameliorates Kras-induced lung cancer. Cancer Res. 2008;68:6127–6135. doi:10.1158/0008-5472.CAN-08-1395
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.