Back to Journals » Drug Design, Development and Therapy » Volume 10
Pasireotide: a novel treatment for patients with acromegaly
Authors Cuevas-Ramos D, Fleseriu M
Received 28 August 2015
Accepted for publication 28 November 2015
Published 11 January 2016 Volume 2016:10 Pages 227—239
DOI https://doi.org/10.2147/DDDT.S77999
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Wei Duan
Daniel Cuevas-Ramos,1 Maria Fleseriu2,3
1Department of Endocrinology and Metabolism, Neuroendocrinology Clinic, Instituto Nacional de Ciencias Médicas y Nutrición Salvador Zubirán, Mexico City, Mexico; 2Department of Medicine (Endocrinology), 3Department of Neurological Surgery, Northwest Pituitary Center, Oregon Health & Science University, Portland, OR, USA
Abstract: Morbidity and mortality rates in patients with active acromegaly are higher than the general population. Adequate biochemical control restores mortality to normal rates. Now, medical therapy has an increasingly important role in the treatment of patients with acromegaly. Somatostatin receptor ligands (SRLs) are considered the standard medical therapy, either after surgery or as a first-line therapy when surgery is deemed ineffective or is contraindicated. Overall, octreotide and lanreotide are first-generation SRLs and are effective in ~20%–70% of patients. Pegvisomant, a growth hormone receptor antagonist, controls insulin-like growth factor 1 in 65%–90% of cases. Consequently, a subset of patients (nonresponders) requires other treatment options. Drug combination therapy offers the potential for more efficacious disease control. However, the development of new medical therapies remains essential. Here, emphasis is placed on new medical therapies to control acromegaly. There is a focus on pasireotide long-acting release (LAR) (Signifor LAR®), which was approved in 2014 by the US Food and Drug Administration and the European Medicine Agency for the treatment of acromegaly. Pasireotide LAR is a long-acting somatostatin multireceptor ligand. In a Phase III clinical trial in patients with acromegaly (naïve to medical therapy or uncontrolled on a maximum dose of first-generation SRLs), 40 and 60 mg of intramuscular pasireotide LAR achieved better biochemical disease control than octreotide LAR, and tumor shrinkage was noted in both pasireotide groups. Pasireotide LAR tolerability was similar to other SRLs, except for a greater frequency and degree of hyperglycemia and diabetes mellitus. Baseline glucose may predict hyperglycemia occurrence after treatment, and careful monitoring of glycemic status and appropriate treatment is required. A precise definition of patients with acromegaly who will derive the greatest therapeutic benefit from pasireotide LAR remains to be established. Lastly, novel therapies and new potential delivery modalities (oral octreotide) are summarized.
Keywords: pasireotide, somatostatin analogs, emerging treatments, growth hormone, insulin-like growth factor 1, somatostatin receptor ligand
An introduction to disease management
Acromegaly is most commonly caused by a somatotroph pituitary adenoma with autonomous overproduction of growth hormone (GH) (Table 1).1–7 Its worldwide prevalence is ~40–70 cases per million and incidence is approximately three to ten cases per million.8–10
 | Table 1 Etiology of acromegaly |
Somatotrophs comprise ~45% of anterior pituitary cells, and GH comprises a single-chain polypeptide of 191 amino acids.11 The excess GH causes increased transcription, synthesis, and release of insulin-like growth factor 1 (IGF-1) from the liver.11–13 Clinical manifestations include acral enlargement, diaphoresis, and facial malformations.
If untreated, serious consequences such as diabetes mellitus, hypertension, sleep apnea, pulmonary hypertension, and heart failure can increase patients’ mortality by up to 2.5–3.5 times in comparison with the general population.14,15 Disease control by reducing GH to <2.5 μg/L (as determined by radioimmunoassay) or <1 μg/dL (as determined by sensitive immunometric assays) and normalizing IGF-1 (adjusted for age and sex) may lessen the increased mortality risk.14,16,17 In addition to normalizing GH and IGF-1, treatment aims to prevent tumor growth or, ideally, induce tumor shrinkage. Currently, therapeutic modalities for acromegaly include neurosurgical intervention, medical therapies, and radiotherapy.11,18–23
Medical therapy, such as somatostatin receptor ligands (SRLs) and dopamine agonists or the GH-receptor antagonist pegvisomant target pituitary adenoma GH secretion or block peripheral GH action, respectively, and are mostly used to treat persistent or recurrent acromegaly after noncurative neurosurgery.8,9,24–26 These medical therapies can be also used as a primary therapy for patients in whom surgery is contraindicated or as a short-term therapy before the intervention.27–29 However, even with these therapies, a substantial proportion of patients (30%–60%) require further treatment for ongoing disease.21,25 The multireceptor-targeted SRL pasireotide (Signifor®) was initially approved in 2012 by the US Food and Drug Administration (FDA) for use in patients who were considered uncontrolled after surgical treatment of a corticotroph pituitary adenoma (Cushing’s disease).30 Somatotroph pituitary adenomas predominantly express somatostatin receptor (SSTR) types 2 and 5. The efficacy of pasireotide in reducing GH and IGF-1 levels and in shrinking tumor in patients with acromegaly has been evaluated in Phase III clinical trials. Injectable pasireotide long-acting release (LAR) (Signifor LAR) was approved in 2014 by the US FDA31 for the treatment of acromegaly in patients who have experienced an inadequate response to surgery or for those in whom surgery is not an option. This review outlines the role of pasireotide LAR for the medical management of patients with acromegaly.
Comorbidities
Acromegaly is characterized by a high incidence of cardiovascular disease and represents the main cause of morbidity and mortality. Hypertension is present in about one-third of all patients; however, some authors have reported a prevalence of 60%.32 Cardiomyopathy is a consequence of chronic GH exposure, causing biventricular concentric hypertrophy with thickened ventricle walls but normal sized chambers.33 Functional alterations are common such as cardiac insufficiency with decreased ejection fraction, valve abnormalities, and high incidence of arrhythmias. By the time of diagnosis, arrhythmias, hypertension, and valvular heart disease are present in up to 60% of patients.11,34 Patients with acromegaly have swelling of the nasopharyngeal tissue, sleep apnea, and lethargy.
Excess of GH induces insulin resistance by counteracting the ability of insulin to suppress gluconeogenesis, decreasing peripheral glucose utilization, and reducing insulin receptor numbers and binding affinity.33 Excess of GH also counteracts insulin action on lipid metabolism. Altered glucose metabolism is the most frequent metabolic complication in patients with acromegaly. The prevalence of impaired glucose tolerance can be up to 46%, and diabetes mellitus is present in 19%–56% of cases.33
Despite initial reports, cancer incidence per se is not increased in patients with acromegaly.35,36 Moreover, recent epidemiological studies in the cohorts of patients with acromegaly found that cancer death rates were similar to those in the general population.16,37 Patients with acromegaly have an increased risk of colonic polyps that may transform to colorectal cancer, and colonoscopy screening, while controversial, is recommended.38,39 In addition to increased GH and IGF-1 levels, hyperinsulinemia may also play a significant role in the development of hyperplasic polyps, adenomatous polyps, and adenocarcinoma.40
Acromegalic arthropathy affects up to 70% of patients and is the most significant cause of functional disability. Symptomatic carpal tunnel syndrome is also common in patients with acromegaly.11–13
Overview of current and emerging therapeutic strategies
Somatostatin physiology
Somatostatin (SST) is a cyclical peptide of 14 (SST-14) or 28 (SST-28) amino acids with a short half-life (<3 minutes) (Figure 1). SST-producing cells can be found throughout the central nervous system (CNS) and peripheral nervous systems and in the endocrine pancreas and gut.41 In the CNS, after SST is released by the hypothalamus, GH secretion is inhibited via action on SSTRs.
 | Figure 1 Somatostatin and pasireotide structure. |
SSTRs are five 7-domain G-protein-coupled receptors named SSTR1 to SSTR5. The adult human pituitary gland mainly expresses SSTR1, SSTR2, SSTR3, and SSTR5. In normal human pituitary, SSTR5 is highly expressed, followed by SSTR2, SSTR1, and SSTR3. SSTR4 is expressed in extremely low levels.42 SSTR2 and SSTR5 are expressed in 95% and 85% of GH-secreting pituitary adenomas, respectively, whereas SSTR1 and SSTR3 are expressed in ~40%.43,44
At the level of the pituitary, the main effect of SST is the inhibition of both hormone secretion and cell growth;42,45,46 a potent antiproliferative mechanism and antisecretory action are also evident.47 SSTRs are an important pharmacological treatment target. SRLs were developed to increase SST half-life and action, decrease multiple and simultaneous actions in different organs, and also reduce post-SST infusion rebound in GH, insulin, and glucagon. SRLs are classified as first- generation (predominantly acting on SSTR; octreotide and lanreotide) and second-generation or multiligand acting on SSTR5 and SSTR2 (pasireotide).
Pharmacodynamic characteristics
First-generation SRLs
SST analogs, or SRLs, such as octreotide or lanreotide, are considered the mainstay of medical therapeutic options for the treatment of acromegaly. Octreotide and lanreotide display high-affinity binding to SSTR2, with half-lives of 2 hours and <1 hour, respectively. Both drugs show a small volume of distribution and a low clearance that results in a longer duration of exposure and long-lasting biological activity compared with SST. Moreover, rebound hormonal hypersecretion is not present; thus, these SRLs are optimal for clinical use.48
Mixing octreotide with microspheres of carboxymethylcellulose sodium as biodegradable glucose polymers and mannitol and water as diluent resulted in an octreotide LAR formulation, which remains therapeutic for 24–42 days.48
Lanreotide is available in two LAR/slow-release formulations. Slow-release lanreotide was created after mixing lanreotide with microspheres of lactide/glycolide copolymers, which allows for administration of every 7–28 days.11 A second formulation is lanreotide autogel, which is a viscous aqueous formulation supplied in ready-to-use prefilled syringes and is administered every 28–56 days (Table 2).
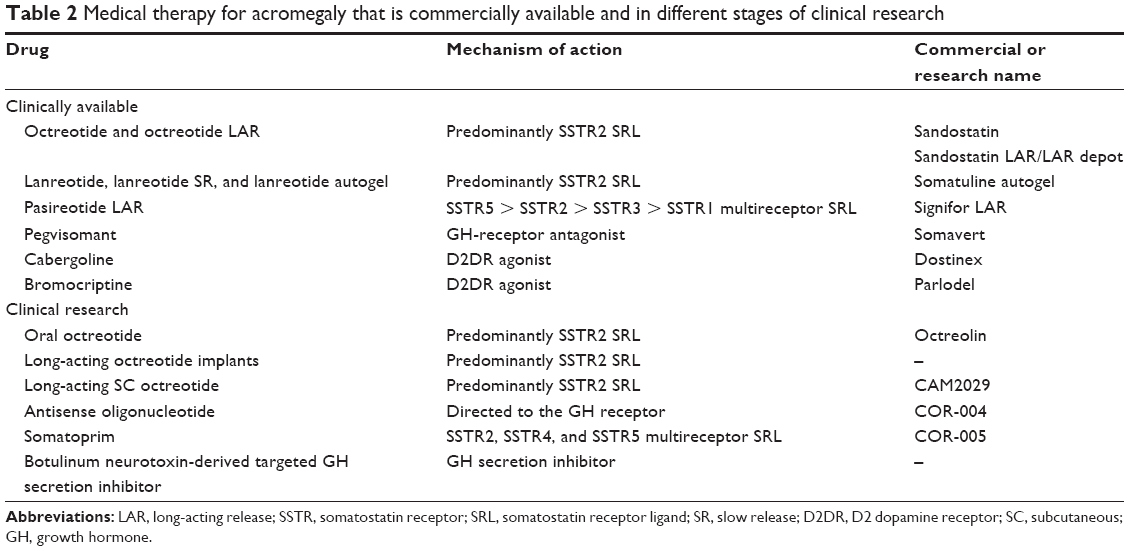 | Table 2 Medical therapy for acromegaly that is commercially available and in different stages of clinical research |
Octreotide LAR (Sandostatin® LAR Depot) and lanreotide autogel (Somatuline® Depot) are US FDA approved as a long-term maintenance therapy in patients with acromegaly who have had an inadequate response to surgery and/or radiotherapy or for whom surgery and/or radiotherapy is not an option.11,20,23,49
Second-generation SRLs
Pasireotide (SOM230) is a novel multireceptor-targeted SST created by the incorporation of four synthetic and two essential amino acids of SST in a novel cyclohexapeptide structure (Figure 1). Pasireotide has a high affinity for SSTR5 followed by SSTR2, SSTR3, and SSTR1 (Table 2 and Figure 2). When compared with octreotide, pasireotide displays a 40, 30, and five times higher binding affinity to SSTR5, SSTR1, and SSTR3, respectively, and a 2.5 times lower binding affinity to SSTR2.50 Pasireotide has 106-fold higher affinity for SSTR5 in comparison with lanreotide.50 Pasireotide LAR was developed using biodegradable polymers using a method similar to that of octreotide LAR.20,23
 | Figure 2 Pasireotide effects. |
Since somatotroph tumors express higher levels of SSTR2 and SSTR5, the SSTR-targeting profile of pasireotide adds a potential benefit compared with more selective SSTR2 ligands such as lanreotide or octreotide.
Pharmacology, mechanism of action, and pharmacokinetics of pasireotide LAR
Pharmacodynamics and mechanism of action in acromegaly
Many G-protein-coupled receptors, including SSTR1–SSTR5, regulate responsiveness to continued agonist effect with different degrees of receptor internalization and degradation. Therefore, continued exposure to the peptide causes progressive reduction of the inhibitory effects. However, the desensitization of SSTRs in patients with acromegaly who respond well to SRLs shows a persistent inhibitory effect of GH secretion without escape after prolonged treatment periods.51 Pasireotide modulates SSTR trafficking differently than octreotide, resulting in quicker recycling of SSTRs, particularly SSTR2, to the plasma membrane after endocytosis.52 Pasireotide stimulates phosphorylation of serine residues 341 and 343 of SSTR2. This mechanism results in accelerated recycling that may counteract the desensitization of SSTR2.53
In contrast to receptor desensitization, the expression of SSTRs is highly variable in pituitary adenomas. SSTR2 mRNA expression correlates positively with in vivo GH suppression induced by SRLs.54–56 In patients treated with octreotide or lanreotide, which mainly acts on the SSTR2 receptor, the expression of SSTR2 is a key determinant for responsiveness and efficient inhibition of GH release. However, the loss of SSTR2 is not the only determinant. Some partially SRL-sensitive GH-secreting adenomas have a loss of SSTR2 expression. In such tumors, SSTR5 mRNA expression is higher than SSTR2, and this would seem to explain the incomplete sensitivity of the SRLs.54,57 Also, a SSTR2- and SSTR5-bispecific compound was shown to exhibit better GH suppression when compared with a selective agonist to each receptor.58 Therefore, some GH-secreting adenomas show better response to SSTR2-specific ligands, while in others, SSTR5-specific ligands are more potent.
Pharmacokinetics
Pasireotide LAR administered via intramuscular injection is widely distributed, primarily through plasma,31,59 and exhibits 88% protein binding.
After three 28-day (3 months) intramuscular injections of pasireotide LAR (20, 40, or 60 mg/month) in patients with acromegaly, steady-state circulating levels were reached.60 After the first injection, independent of the dose, pasireotide LAR quickly increased (within the first 2–10 hours), and pasireotide plasma levels then plateaued or decreased. A dose–response serum increment was also observed.60 Furthermore, a higher response rate was noted with an increased pasireotide LAR dose.61 Pasireotide exhibits high metabolic stability; however, it may inhibit cytochrome P450 enzyme;61 therefore, some concomitant drugs need to be avoided if possible, as some are contraindicated, or a dose adjustment may be required (Table 3). Pasireotide is cleared through liver metabolism, and ~60% of the drug is eliminated in the feces or urine after 10 days following a single subcutaneous (SC) dose.31,59 Therefore, pasireotide-circulating levels may increase in patients with moderate-to-severe hepatic impairment compared with normal liver function.62 In patients with kidney failure, an increment of circulating pasireotide is also expected.59 The pharmacokinetic parameters of pasireotide were shown not to be influenced by age, body weight, sex, or race.59
 | Table 3 Potential drug interactions with pasireotide LAR |
Clinical efficacy, safety, and tolerability of pasireotide LAR
Clinical efficacy
Approval of pasireotide LAR for the treatment of adults with acromegaly was based on data from two large, international, multicenter, double-blinded randomized Phase III studies, C2305 and C2402. These studies confirmed higher rates of composite biochemical control (defined as mean GH level <2.5 μg/L and normal IGF-1 levels) than the first-generation SRLs.
A proof-of-concept trial compared a single dose of pasireotide (100 or 250 μg) with 100 μg octreotide. GH circulation levels were significantly reduced with all three interventions, with a marked dose-dependent reduction in the two pasireotide groups (100 or 250 μg).63 Interestingly, those cases with presumably SSTR2 predominant expression responded to both octreotide and pasireotide treatments. In others, pasireotide was more effective in reducing GH circulating levels that are likely due to the higher abundance of SSTR5. However, in some cases, there was a better response to octreotide, presumably due to predominant SSTR2 and low SSTR5 expression. A subsequent Phase II, multicenter, open-label, randomized, cross-over trial of pasireotide was conducted.64 A total of 60 patients with active acromegaly were initially given 100 μg octreotide over the course of 1 month to evaluate the response to standard treatment. Patients were then randomized to 200, 400, or 600 μg/day of pasireotide. After 1 month, patients taking pasireotide showed a full response rate with normalization of both GH and IGF-1 levels in 14%, 12%, and 30% with 200, 400, and 600 μg, respectively. After 3 months, 38% of patients achieved normal IGF-1 levels, and in 49%, GH levels were <2.5 μg/L. Tumor volume decreased by >20% in 20 patients (39%).64
Phase III trials with pasireotide LAR
C2305 in patients naïve to medical therapy (efficacy: biochemical control and tumor shrinkage)
The C2305 study was a multicenter, randomized, double-blinded study conducted in 358 patients with active acromegaly naïve to medical treatment, who had persistent disease despite neurosurgical intervention or were ineligible for surgery. Patients were randomized to receive pasireotide LAR 40 mg/28 days (n=176) with the option to uptitrate to 60 mg or to receive octreotide LAR 20 mg/28 days (n=182) for 12 months with the option to uptitrate to 30 mg. Key exclusion criteria included previous therapy with SRLs, dopamine agonists or GH-receptor antagonist (pegvisomant), pituitary irradiation within the past 10 years, significant cardiovascular morbidity, and glycated hemoglobin (HbA1c) >8%.65 At baseline, 42% of patients had undergone at least one pituitary surgery with a median time since surgery of 7 months. GH levels were 22 and 19 μg/L in the pasireotide and octreotide groups, respectively, and IGF-1 levels were 3.1 times the upper limit of normal (ULN) in both groups. Mean tumor volume was 2,421 versus 2,259 mm3, respectively. After 12 months, biochemical control (defined earlier) was met in 31% versus 19% in patients with pasireotide LAR and octreotide LAR, respectively (P=0.007). Also, biochemical control was achieved early with pasireotide LAR (by month 3) in 30%. Dose uptitration was recommended in all uncontrolled patients but was implemented in just 51% of patients with pasireotide and 67% of patients with octreotide. Lack of uptitration has been shown to be one of the causes of reduced response in either SRLs66 or pegvisomant-treated patients.67 Health-related quality of life questionnaires showed similar reduction of ring size and less symptom severity score in both the treatment groups.65 Interestingly, IGF-1 levels were significantly lower with pasireotide LAR than octreotide LAR (38.6% vs 23.6%, P=0.002, respectively), despite a similar GH suppression with pasireotide and octreotide. The exact mechanism of improved IGF-1 suppression is currently unknown. Biochemical control was higher in patients who had surgery in both groups; however, remarkably, ~60% of patients in this study did not undergo surgery.
A significant tumor reduction was achieved in 80% and 77% of patients with pasireotide LAR and octreotide LAR, respectively. Mean tumor volume reduction was 40% in both groups. Tumor reduction or no change in tumor volume was seen in 98% of patients with pasireotide LAR at month 12. Contrary to other studies, no differences in tumor shrinkage were observed between postsurgery or de novo patients.65
C2305: extension study
Results of a 12-month, double-blinded, multicenter extension phase study demonstrated that pasireotide LAR provided sustained biochemical control in patients with acromegaly.68 After month 12, patients who achieved biochemical control continued their study treatment. At month 26, 49% and 46% of patients in each group, respectively, achieved biochemical control.68 Eighty-one (45%) patients who had not achieved disease control after 12 months switched from octreotide LAR to pasireotide LAR, and 38 (22%) patients switched from pasireotide LAR to octreotide LAR at month 13. After a further year of treatment, biochemical control was successful in an additional 17% (n=14) of patients who switched to pasireotide LAR and 0% of patients who switched to octreotide LAR. Also, tumor volume decreased to 25% in the pasireotide group and 18% in the octreotide group. In each group, 54% and 42% of patients, respectively, achieved >20% tumor volume reduction.69,70
C2402 (PAOLA study): patients inadequately controlled
The pasireotide versus continued treatment with octreotide or lanreotide in patients with inadequately controlled acromegaly (PAOLA) study was a randomized study evaluating the efficacy and safety of double-blind pasireotide LAR (40 and 60 mg) versus continued open-label pretrial octreotide or lanreotide.61 Patients under combination therapy with GH-receptor antagonist (14%) or dopamine agonist (32%) were eligible for inclusion if such agents were discontinued at least 8 weeks before receiving the study drug. Patients with a positive history of pituitary irradiation within the past 10 years, significant cardiovascular morbidity, or HbA1c >8% were excluded. Inadequate control was defined by a five-point 2-hour mean GH levels of >2.5 μg/L and IGF-1 levels >1.3 times sex- and age-adjusted ULN. Biochemical control at 6 months with pasireotide LAR 40 or 60 mg versus continued pretrial octreotide or lanreotide therapy was met for both pasireotide LAR dose groups. Specifically, 15% and 20% of patients treated with pasireotide LAR 40 and 60 mg, respectively, achieved full GH and IGF-1 biochemical control at 6 months compared with 0% in the pretrial therapy control arm. Biochemical control was achieved by month 3 in 15% and 18% of 40 and 60 mg pasireotide LAR, respectively; 81% and 70% of patients treated with pasireotide LAR 40 and 60 mg had either a reduction or no change in tumor volume from baseline (as assessed by magnetic resonance imaging) at month 6, respectively.61 A reduction of 25% or more in tumor volume by week 24 was achieved in more patients receiving pasireotide than active control (18% for 40 mg, 11% for 60 mg of pasireotide LAR vs 1.5% with active control). Clinically, Acromegaly Quality of Life Questionnaire scores of patients significantly improved; greater improvements were recorded with pasireotide LAR treatment.61,71
During the 28-week extension of PAOLA study, biochemical control was maintained with pasireotide LAR and was generally well tolerated in patients inadequately controlled by first-generation SST analogs. This was consistent with data from the core study, where ~20% of patients in the active control group achieved biochemical control after switching to pasireotide LAR in the extension study.72
Safety and tolerability of pasireotide LAR
Most adverse events associated with pasireotide LAR are mild to moderate in severity.61,68
Pasireotide-induced hyperglycemia
Hyperglycemia was more common with pasireotide LAR (57%) than octreotide LAR (22%) in the C2305 Phase III trial with the higher incidence of diabetes mellitus (26% vs 4%, respectively).50 Eight (4.5%) patients in the pasireotide LAR group discontinued treatment due to serious drug-adverse event, the most common of which was elevated blood glucose.65 After 26 months of treatment in the C2305 study (core and extension), hyperglycemia-related events with pasireotide LAR 40 mg/month were 63% compared with 25% with octreotide LAR 20 mg/month (68).68 Similarly, in the PAOLA study (C2402 trial), pasireotide-induced hyperglycemia was more common (~30%) versus active control (14%) as well as diabetes mellitus (~25% vs 9%, respectively).61 Five patients discontinued pasireotide LAR treatment because of serious adverse events, all of which were related to pasireotide-induced hyperglycemia or diabetes. HbA1c levels, which increased in the first 3 months of pasireotide LAR treatment, were stable by month 26. Mean glucose and HbA1c levels slightly increased in the octreotide LAR group.61,68
The exact mechanism of pasireotide-induced hyperglycemia in patients with acromegaly is unknown. Data from a dose–response study in healthy volunteers suggested that pasireotide-associated hyperglycemia was due to insulin suppression. Also, mild inhibition of glucagon was reported.73 Further clinical research using a hyperglycemic clamp test in healthy volunteers showed that hyperglycemia associated with pasireotide treatment was not due to changes in hepatic/peripheral insulin sensitivity. Decreased gastrointestinal incretin hormones with the subsequent reduction on insulin secretion would seem to explain the increment in glucose levels. During an oral glucose tolerance test, glucagon-like peptide 1 and glucose-dependent insulinotropic polypeptide were significantly decreased compared with baseline.74
Dipeptidyl peptidase-4 inhibitors (vildagliptin), glucagon-like peptide 1 analogs (liraglutide), and insulin secretagogues (nateglinide) have been also tested to treat pasireotide-related hyperglycemia in 90 healthy male volunteers. Comparison between vildagliptin 50 mg bid, nateglinide 60 mg tid, or liraglutide 0.6 mg SC qd for 7 days showed that glucose increment after pasireotide treatment was lower in the groups treated with metformin (60%), nateglinide (49%), vildagliptin (38%), and liraglutide (19%), suggesting a benefit of antihyperglycemic drugs to mitigate pasireotide-related hyperglycemia.75 Clinical trials comparing antihyperglycemic drugs or insulin are warranted to establish optimal treatment in patients with pasireotide-induced hyperglycemia.
Although the treatment of pasireotide-induced hyperglycemia in healthy volunteers has been focused on increasing incretin levels or using incretin analogs, optimal therapy in patients with acromegaly remains to be elucidated.65 Metformin-based antidiabetic therapy appears to effectively control pasireotide-associated hyperglycemia in selected patients with acromegaly. During the 12-month study Phase III clinical trial (C2305), 24 subjects received metformin alone, 19 subjects received metformin plus another oral antidiabetic therapy, and ten subjects were treated with insulin alone or in addition to an oral agent.76 Results showed lower mean levels of HbA1c at month 12 in groups with metformin (6.6%) or metformin plus another oral antidiabetic agent (6.7%) than insulin with or without other therapy (7.2%).
After pasireotide is stopped, hyperglycemia appears to be reversible since patients (n=38) who switched from pasireotide to octreotide after 13 months of treatment showed improvement of their glucose levels (from 127 to 104 mg/dL) and HbA1c (from 6.7% to 6.1%). In contrast, those patients who switched from octreotide to pasireotide at month 13 (n=81) showed an increment in mean glucose levels (from 104 to 141 mg/dL) and HbA1c (6.1%–7% at month 3 and stabilizing to 6.7% at month 12).55 In the C2402 (PAOLA) study, glycemic control improved when an early treatment adjustment was established in the first 2 weeks of pasireotide, regardless of diabetes status.77
Pasireotide: other adverse events
Mild gastrointestinal disturbances were reported in the Phase II randomized, open-label, cross-over, multicenter pasireotide (SOM230) study.64 Regardless of causality, the most common adverse events related with pasireotide in clinical Phase III trials were nausea (25%), diarrhea (22%), abdominal pain (12%), and flatulence (10%). Headache and nasopharyngitis were also more common with pasireotide LAR. Interestingly, cholelithiasis was more common with octreotide LAR (36%) than pasireotide LAR (26%).61,65 As expected, the frequency of adverse events was higher in patients naïve to medical treatment with SRLs (40%–45%) versus patients previously treated with SRLs (5%–20%).61,65 All pituitary hormones should be monitored (thyroid, adrenal, and gonadal) in patients taking pasireotide LAR, due to potential hormonal inhibition. In clinical trials, pasireotide LAR has been rarely associated with adrenal insufficiency.61,65 Pasireotide may induce bradycardia and QT interval prolongation. Bradycardia-related adverse events in the Phase III clinical trials were 3%–8% for pasireotide LAR and 0% for active controls.61,65 One patient receiving pasireotide LAR 40 mg required dose interruption due to liver injury.61,65
Potential drug–drug interactions related to cytochrome P450-3A4 metabolism may cause QT prolongation or cardiac arrhythmias, which should be assessed in all patients (Table 3).
Place in therapeutic armamentarium
As mentioned previously, pasireotide LAR was approved in 2014 by the US FDA31 and the European Medicines Agency in Europe,59 as an injectable suspension, for intramuscular use, for the treatment of patients with acromegaly who have had an inadequate response to surgery and/or for whom surgery is not an option. Regulatory approval of pasireotide LAR for this indication in several other countries is awaited.
Surgical removal of the GH-secreting pituitary adenoma is the treatment of choice for acromegaly if the tumor is considered resectable, and the surgery is performed by an experienced surgeon.49 Current treatment guidelines recommend normal serum IGF-1 for age- and sex-matched and serum GH <1.0 μg/L as treatment targets.49 If active disease persists following surgery, patients have a tumor that cannot be resected, or in those cases whereby a patient chooses not to undergo surgery, medical management is recommended (Table 2).
Occasionally, medical therapy is used as a primary treatment before surgery.78
Radiation of an adenoma as a third-line therapy might be necessary in selected cases, such as with aggressive tumors.
Pharmacotherapy includes SRLs, pegvisomant, and dopamine agonists. When uncontrolled acromegaly persists, a pharmaceutical combination may improve the efficacy and minimize the potential adverse effects with increased doses of individual agents.49,79
In pre- and clinical trials, pasireotide LAR was superior for biochemical disease control in patients with active acromegaly naïve to medial therapy or uncontrolled on a maximum dose of first-generation SRLs treatment. Biochemical control was rapid in the first 3 months, and control was sustained up to 26 months. Tumor volume was also decreased.
In a subset of patients with acromegaly, remission can be achieved with pasireotide LAR treatment if patients were previously uncontrolled on SRLs.61,80 Tumors that are large, aggressive, have less SSTR2 expression, or are sparsely granulated are more resistant to first-generation SRLs.55,56 Additional studies are needed to define which patients will likely benefit the most from pasireotide LAR treatment. Similarly, the use of pasireotide in combination with other drug therapies requires further investigation.
Pasireotide LAR should be administered via deep intramuscular injection with a recommended initial dose of 40 mg every 28 days. If after 3 months the patients’ disease remains active, the dose may be increased to 60 mg/28 days. Adverse reactions should be carefully monitored. Patients with mild liver or kidney failure do not require dose adjustment; however, pasireotide LAR should be avoided in cases with severe hepatic or renal failure. Glycemic status with fasting plasma glucose and HbA1c levels should be assessed prior to starting treatment and then every 1–3 months.
In the C2402 (PAOLA) study, 47% of all patients treated with pasireotide LAR (40 or 60 mg) did not receive antidiabetic medication at any time during the study. It has been suggested that the effects of pasireotide LAR on glucose homeostasis may be attenuated by increased insulin sensitivity due to the compensatory increase in IGF-binding protein-2.81
Metformin, if not contraindicated, should be initiated early.77 Also, other antidiabetic agents or insulin should be considered to increase glycemic control. Guidelines for the treatment of hyperglycemia and diabetes should be followed when patients with acromegaly already have or develop diabetes during the follow-up.82 If hyperglycemia persists despite adequate treatment, pasireotide dose should be reduced or discontinued. Liver function tests and a gallbladder ultrasound should be performed periodically with pasireotide LAR treatment. An electrocardiogram may be indicated in patients with cardiac disease or with risk factors for bradycardia or arrhythmias. Drug–drug interactions should be carefully monitored (Table 3).83
Other emerging medical therapies for acromegaly
Oral octreotide
Oral octreotide appears to be an attractive option for patients with acromegaly that is biochemically controlled with injectable SRLs. A newly developed transient permeability enhancer to improve the absorption of pharmacologically active drug has been shown to increase the intestinal absorption of an oral formulation of octreotide and may represent an alternative to the parenteral formulation in patients with acromegaly. Oral octreotide is absorbed within 1 hour of administration. Equivalent pharmacokinetic and pharmacodynamics in a Phase II study were demonstrated using 20 mg of oral octreotide or 100 μg of SC octreotide.84 Furthermore, a single dose of 20 mg of oral octreotide resulted in the significant suppression of both basal GH- and arginine-GH-releasing hormone stimulated levels. A large, multicenter, Phase III study in 151 patients with acromegaly during 13 months showed biochemical normalization in 62% of cases after switching from injectable SRLs (from 88.7% at the baseline visit while on injectable SRLs).85 The effect was durable, and 85% of subjects initially controlled on oral octreotide maintained the response for ~1 year. GH levels were reduced compared with baseline and acromegaly-related symptoms improved.85 Approximately half of the patients did require >40 mg doses to maintain response. Good baseline control with injectable SRLs (IGF-1 ≤1× ULN/GH <2.5 ng/mL) predicted responsiveness to oral octreotide.85 The dose of injectable SRLs was not a good predictor of responsiveness. Over 70% of patients on low- or mid-dose injectable SRLs responded to capsules, but also half of the patients on high-dose injections were responders after switching to oral capsules. As expected, the safety profile was similar to that of other SRLs. Most adverse events occurred at the beginning of treatment. Gastrointestinal adverse events were transient and mostly resolved within 2 weeks.85
Long-acting subcutaneous octreotide and octreotide implants
CAM2029 is a long-acting octreotide formulation that is based on a proprietary fluid crystal (FC) delivery system, for the treatment of acromegaly. Octreotide is delivered such that rapid onset and long-acting octreotide release is provided. CAM2029 has been evaluated in Phase I trials, specifically assessing pharmacokinetics and pharmacodynamics. In healthy volunteers, octreotide FC provided greater bioavailability with a more rapid onset and similar duration of effect compared with octreotide LAR. The FC formulation offered enhanced convenience as it is supplied in a prefilled syringe with a thin needle.86 A Phase III study of octreotide FC in patients with acromegaly is planned.
Octreotide implants for long-acting continuous release have been studied in Phase II and Phase III clinical trials. In comparison with monthly octreotide LAR, biochemical control and safety parameters were maintained >24 weeks with an octreotide implant. Diarrhea and headache were more frequently reported with the octreotide implant, whereas cholecystitis and hypertension were more commonly reported with octreotide LAR.87
Antisense oligonucleotide
ATL1103 is a second-generation antisense oligomer drug targeted to block the GH receptor, thereby lowering IGF-1 levels. ATL1103 is a 20mer with a phosphorothioate backbone and 2′-O-methoxyethyl modifications of the five nucleotides at either end intended to increase its plasma half-life and affinity for the target RNA to allow posthybridization RNaseH degradation of the GHr RNA strand. Animal studies showed a rapid distribution from plasma to peripheral tissues (Cmax in 2–4 hours), with a tissue clearance half-life of 2–4 weeks.
ATL1103 (COR-004) was evaluated in a Phase II randomized, open-label, multicenter, parallel group study in patients with active acromegaly. Patients were randomized to receive either ATL1103 200 mg once or twice weekly for 13 weeks (three doses in the first week). The primary endpoint was to evaluate the safety and to understand the single-dose and multiple-dose pharmacokinetic profiles. Thirty-five patients were recruited (average age 50.4 years [26–80 years]; eleven males), and all completed the treatment. There was a significant fall in serum IGF-1 of 26% by week 14 (1 week past the last dose) with 200 mg ATL1103 twice weekly (577±198 vs 411±174 ng/mL [mean ± standard deviation], P<0.0001). Once-weekly dosing did not result in a significant fall in IGF-1. The fall in IGF-1 was associated with a mean reduction in ring size and serum GH. ATL1103 was well tolerated with mild-to-moderate injection site reactions, the most common drug-related adverse event. ATL1103 offers a novel therapeutic approach for patients with active acromegaly.88
Somatoprim
Somatoprim (DG3173-COR-005) is a multireceptor SRL that binds SSTR2, SSTR4, and SSTR5. Somatoprim is unique in comparison to other SRLs. In particular, in vivo studies showed an additional 40% response in patients with pituitary adenomas that were resistant to octreotide and less of a hyperglycemic effect since insulin-suppressing activity is less potent.89 A Phase II study showed lower GH secretion with reduced effects on insulin secretion and glucose levels as compared with octreotide in two single ascending dose in patients with acromegaly.89
Targeted secretion inhibitor
A botulinum neurotoxin-derived targeted secretion inhibitor was designed to target pituitary somatotroph cells and suppress GH secretion.90 The recombinant protein was created by modifying the GH-releasing hormone domain and the endopeptidase domain of botulinum toxin. The N-ethylmaleimide-sensitive fusion attachment protein receptor (SNARE) was depleted in vitro, blocking the GH exocytosis after GH-releasing hormone binding to its receptor. Administration in vivo in rats produced a dose-dependent inhibition of GH synthesis, storage, and secretion with IGF-1 reduction.90 In human samples of pituitary adenoma from 25 patients with acromegaly and 47 patients with nonfunctioning pituitary adenoma, SNARE proteins were variably expressed.91 However, the targeted secretion inhibitor did not inhibit GH secretion in somatotroph cell cultures.91 Novel targeted secretion inhibitors may have future potential with therapeutic benefit, but further research is needed.
Conclusion
Treating patients with acromegaly can be extremely challenging, and inadequate disease control may lead to serious consequences. Pegvisomant and combination therapies have been used to manage patients uncontrolled on first-generation SRLs. Novel medical therapies provide different or enhanced mechanisms to control elevated circulating GH and IGF-1 levels. Pasireotide LAR seems a promising medical therapy for patients with acromegaly that cannot be controlled with available SRLs. In recent Phase III trials, pasireotide LAR has been shown to be more efficacious than first-generation SRLs. However, hyperglycemia developed in approximately half of the patients. Interestingly, the rates of hyperglycemia were similar in responders versus nonresponders, but baseline fasting plasma glucose level could predict hyperglycemia. Proactive monitoring and early intervention in the management of pasireotide-induced hyperglycemia are recommended. Other novel pharmaceutical therapies for patients with acromegaly, as well as new potential delivery modalities, including the first oral octreotide therapy are on the horizon and show early promise. Overall, vigilant and judicious care is advised in treating individual patients with acromegaly.
Acknowledgment
The authors thank Shirley McCartney, PhD, Oregon Health & Science University, for editorial assistance with the manuscript.
Disclosure
The authors did not receive any support for writing of this manuscript. Dr Fleseriu is the principal investigator on research grants to Oregon Health & Science University from Chiasma, Cortendo, Ipsen, Novartis, and Pfizer and an ad hoc scientific consultant to Cortendo, Chiasma, and Novartis. Dr Cuevas-Ramos has no conflicts of interest in this work.
References
Asa SL, Bilbao JM, Kovacs K, et al. Hypothalamic neuronal hamartoma associated with pituitary growth hormone cell adenoma and acromegaly. Acta Neuropathol. 1980;52:231–234. | ||
Asa SL, Scheithauer BW, Bilbao JM, et al. A case for hypothalamic acromegaly: a clinicopathological study of six patients with hypothalamic gangliocytomas producing growth hormone-releasing factor. J Clin Endocrinol Metab. 1984;58:796–803. | ||
Beckers A, Lodish MB, Trivellin G, et al. X-linked acrogigantism syndrome: clinical profile and therapeutic responses. Endocr Relat Cancer. 2015;22:353–367. | ||
Beuschlein F, Strasburger CJ, Siegerstetter V, et al. Acromegaly caused by secretion of growth hormone by a non-Hodgkin’s lymphoma. N Engl J Med. 2000;342:1871–1876. | ||
Melmed S, Ezrin C, Kovacs K, et al. Acromegaly due to secretion of growth hormone by an ectopic pancreatic islet-cell tumor. N Engl J Med. 1985;312:9–17. | ||
Shimon I, Melmed S. Growth hormone- and growth-hormone-releasing hormone-producing tumors. Cancer Treatment and Research. Los Angeles, CA: Springer Science + Business Media; 1997:1–24. | ||
Trivellin G, Daly AF, Faucz FR, et al. Gigantism and acromegaly due to Xq26 microduplications and GPR101 mutation. N Engl J Med. 2014;371:2363–2374. | ||
Daly AF, Rixhon M, Adam C, et al. High prevalence of pituitary adenomas: a cross-sectional study in the province of Liège, Belgium. J Clin Endocrinol Metab. 2006;91:4769–4775. | ||
Fernandez A, Karavitaki N, Wass JAH. Prevalence of pituitary adenomas: a community-based, cross-sectional study in Banbury (Oxfordshire, UK). Clin Endocrinol (Oxf). 2010;72:377–382. | ||
Hoskuldsdottir GT, Fjalldal SB, Sigurjonsdottir HA. The incidence and prevalence of acromegaly, a nationwide study from 1955 through 2013. Pituitary. 2015;18:803–807. | ||
Melmed S. Acromegaly. N Engl J Med. 2006;355:2558–2573. | ||
Melmed S. Acromegaly pathogenesis and treatment. J Clin Invest. 2009;119:3189–3202. | ||
Melmed S. Pathogenesis of pituitary tumors. Nat Rev Endocrinol. 2011;7:257–266. | ||
Holdaway IM, Bolland MJ, Gamble GD. A meta-analysis of the effect of lowering serum levels of GH and IGF-I on mortality in acromegaly. Eur J Endocrinol. 2008;159:89–95. | ||
Reid TJ, Post KD, Bruce JN, et al. Features at diagnosis of 324 patients with acromegaly did not change from 1981 to 2006: acromegaly remains under-recognized and under-diagnosed. Clin Endocrinol (Oxf). 2010;72:203–208. | ||
Holdaway IM, Rajasoorya RC, Gamble GD. Factors influencing mortality in acromegaly. J Clin Endocrinol Metab. 2004;89:667–674. | ||
McCabe J, Ayuk J, Sherlock M. Treatment factors that influence mortality in acromegaly. Neuroendocrinology. Epub 2015 Jan 30. | ||
Ayuk J, Clayton RN, Holder G, et al. Growth hormone and pituitary radiotherapy, but not serum insulin-like growth factor-I concentrations, predict excess mortality in patients with acromegaly. J Clin Endocrinol Metab. 2004;89:1613–1617. | ||
Biermasz NR. Pituitary gland: mortality in acromegaly reduced with multimodal therapy. Nat Rev Endocrinol. 2014;10:708–710. | ||
Fleseriu M. Advances in the pharmacotherapy of patients with acromegaly. Discov Med. 2014;17:329–338. | ||
Giustina A, Chanson P, Kleinberg D, et al. Expert consensus document: a consensus on the medical treatment of acromegaly. Nat Rev Endocrinol. 2014;10:243–248. | ||
Jane JA, Starke RM, Elzoghby MA, et al. Endoscopic transsphenoidal surgery for acromegaly: remission using modern criteria, complications, and predictors of outcome. J Clin Endocrinol Metab. 2011; 96:2732–2740. | ||
Melmed S, Kleinberg D, Bonert V, et al. Acromegaly: assessing the disorder and navigating therapeutic options for treatment. Endocr Pract. 2014;20:7–17. | ||
Albarel F, Castinetti F, Morange I, et al. Outcome of multimodal therapy in operated acromegalic patients, a study in 115 patients. Clin Endocrinol. 2013;78:263–270. | ||
Cuevas-Ramos D, Carmichael JD, Cooper O, et al. A structural and functional acromegaly classification. J Clin Endocrinol Metab. 2015;100:122–131. | ||
Mercado M, Espinosa E, Ramirez C. Current status and future directions of the pharmacological therapy of Acromegaly. Minerva Endocrinol. Epub 2015 Oct 20. | ||
Colao A, Auriemma RS, Pivonello R. The effects of somatostatin analogue therapy on pituitary tumor volume in patients with acromegaly. Pituitary. Epub 2015 Aug 20. | ||
Espinosa E, Ramirez C, Mercado M. The multimodal treatment of acromegaly: current status and future perspectives. Endocr Metab Immune Disord Drug Targets. 2014;14:169–181. | ||
Kleinberg DL. Primary therapy for acromegaly with somatostatin analogs and a discussion of novel peptide analogs. Rev Endocr Metab Disord. 2005;6:29–37. | ||
US Food and Drug Administration. FDA Approves Signifor, a New Orphan Drug for Cushing’s Disease. 2012. Available from: http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm332351.htm. Accessed July 17, 2015. | ||
Novartis Pharmaceuticals. Signifor LAR Prescribing Information. 2014. Available from: http://www.pharma.us.novartis.com/product/pi/pdf/signifor_lar.pdf. Accessed March 14, 2015. | ||
Bondanelli M, Ambrosio MR, degli Uberti EC. Pathogenesis and prevalence of hypertension in acromegaly. Pituitary. 2001;4: 239–249. | ||
Colao A, Ferone D, Marzullo P, et al. Systemic complications of acromegaly: epidemiology, pathogenesis, and management. Endocr Rev. 2004;25:102–152. | ||
Gurnell M, Powlson AS. Cardiovascular disease and sleep disordered breathing in acromegaly. Neuroendocrinology. Epub 2015 Jul 28. | ||
Melmed S. Acromegaly and cancer: not a problem? J Clin Endocrinol Metab. 2001;86:2929–2934. | ||
Orme SM, McNally RJQ, Cartwright RA, et al. Mortality and cancer incidence in acromegaly: a retrospective cohort study 1. J Clin Endocrinol Metab. 1998;83:2730–2734. | ||
Mestron A, Webb S, Astorga R, et al. Epidemiology, clinical characteristics, outcome, morbidity and mortality in acromegaly based on the Spanish Acromegaly Registry (Registro Espanol de Acromegalia, REA). Eur J Endocrinol. 2004;151:439–446. | ||
Lois K, Bukowczan J, Perros P, et al. The role of colonoscopic screening in acromegaly revisited: review of current literature and practice guidelines. Pituitary. 2014;18:568–574. | ||
Renehan AG, Brennan BM. Acromegaly, growth hormone and cancer risk. Best Pract Res Clin Endocrinol Metab. 2008;22:639–657. | ||
Colao A, Pivonello R, Auriemma RS, et al. The association of fasting insulin concentrations and colonic neoplasms in acromegaly: a colonoscopy-based study in 210 patients. J Clin Endocrinol Metab. 2007;92:3854–3860. | ||
Lamberts SW, van der Lely AJ, de Herder WW, et al. Octreotide. N Engl J Med. 1996;334:246–254. | ||
Ben-Shlomo A, Melmed S. Pituitary somatostatin receptor signaling. Trends Endocrinol Metab. 2010;21:123–133. | ||
Reubi J, Waser B, Schaer J-C, et al. Somatostatin receptor sst1–sst5 expression in normal and neoplastic human tissues using receptor autoradiography with subtype-selective ligands. Eur J Nucl Med. 2001;28:836–846. | ||
Taboada GF, Luque RM, Bastos W, et al. Quantitative analysis of somatostatin receptor subtype (SSTR1-5) gene expression levels in somatotropinomas and non-functioning pituitary adenomas. Eur J Endocrinol. 2007;156:65–74. | ||
Sellers LA, Alderton F, Carruthers AM, et al. Receptor isoforms mediate opposing proliferative effects through gbeta gamma-activated p38 or Akt pathways. Mol Cell Biol. 2000;20:5974–5985. | ||
Theodoropoulou M, Stalla GK, Spengler D. ZAC1 target genes and pituitary tumorigenesis. Mol Cell Endocrinol. 2010;326:60–65. | ||
Cuevas-Ramos D, Fleseriu M. Treatment of Cushing’s disease: a mechanistic update. J Endocrinol. 2014;223:R19–R39. | ||
Ben-Shlomo A, Melmed S. Somatostatin agonists for treatment of acromegaly. Mol Cell Endocrinol. 2008;286:192–198. | ||
Katznelson L, Laws ER, Melmed S, et al. Acromegaly: an endocrine society clinical practice guideline. J Clin Endocrinol Metab. 2014;99:3933–3951. | ||
Bruns C, Lewis I, Briner U, et al. SOM230: a novel somatostatin peptidomimetic with broad somatotropin release inhibiting factor (SRIF) receptor binding and a unique antisecretory profile. Eur J Endocrinol. 2002;146:707–716. | ||
Hofland LJ, Lamberts SWJ. The pathophysiological consequences of somatostatin receptor internalization and resistance. Endocr Rev. 2003;24:28–47. | ||
Lesche S, Lehmann D, Nagel F, et al. Differential effects of octreotide and pasireotide on somatostatin receptor internalization and trafficking in vitro. J Clin Endocrinol Metab. 2009;94:654–661. | ||
Nagel F, Doll C, Pöll F, et al. Structural determinants of agonist-selective signaling at the sst 2A somatostatin receptor. Mol Endocrinol. 2011;25:859–866. | ||
Brzana J, Yedinak CG, Gultekin SH, et al. Growth hormone granulation pattern and somatostatin receptor subtype 2A correlate with postoperative somatostatin receptor ligand response in acromegaly: a large single center experience. Pituitary. 2012;16:490–498. | ||
Gadelha MR, Kasuki L, Korbonits M. Novel pathway for somatostatin analogs in patients with acromegaly. Trends Endocrinol Metab. 2013;24:238–246. | ||
Wildemberg LE, Neto LV, Costa DF, et al. Low somatostatin receptor subtype 2, but not dopamine receptor subtype 2 expression predicts the lack of biochemical response of somatotropinomas to treatment with somatostatin analogs. J Endocrinol Invest. 2013;36:38–43. | ||
Nielsen S, Mellemkjær S, Rasmussen LM, et al. Expression of somatostatin receptors on human pituitary adenomas in vivo and ex vivo. J Endocrinol Invest. 2001;24:430–437. | ||
Ren SG, Taylor J, Dong J, et al. Functional association of somatostatin receptor subtypes 2 and 5 in inhibiting human growth hormone secretion. J Clin Endocrinol Metab. 2003;88:4239–4245. | ||
European Medicines Agency. Summary of Product Characteristics: Signifor Powder and Solvent for Suspension for Injection. 2014. Available from: www.ema.europa.eu/ema. Accessed May 14, 2015. | ||
Petersenn S, Bollerslev J, Arafat AM, et al. Pharmacokinetics, pharmacodynamics, and safety of pasireotide LAR in patients with acromegaly: a randomized, multicenter, open-label, phase I study. J Clin Pharmacol. 2014;54:1308–1317. | ||
Gadelha MR, Bronstein MD, Brue T, et al. Pasireotide versus continued treatment with octreotide or lanreotide in patients with inadequately controlled acromegaly (PAOLA): a randomised, phase 3 trial. Lancet Diabetes Endocrinol. 2014;2:875–884. | ||
Horsmans Y, Hu K, Ruffin M, et al. Effect of hepatic impairment on the pharmacokinetics of pasireotide (SOM230): results from a multicenter phase I study. J Clin Pharmacol. 2012;52:552–558. | ||
van der Hoek J, de Herder WW, Feelders RA, et al. A single-dose comparison of the acute effects between the new somatostatin analog SOM230 and octreotide in acromegalic patients. J Clin Endocrinol Metab. 2004;89:638–645. | ||
Petersenn S, Schopohl J, Barkan A, et al. Pasireotide (SOM230) demonstrates efficacy and safety in patients with acromegaly: a randomized, multicenter, phase II trial. J Clin Endocrinol Metab. 2010;95:2781–2789. | ||
Colao A, Bronstein MD, Freda P, et al. Pasireotide versus octreotide in acromegaly: a head-to-head superiority study. J Clin Endocrinol Metab. 2014;99(3):791–799. | ||
Fleseriu M. Clinical efficacy and safety results for dose escalation of somatostatin receptor ligands in patients with acromegaly: a literature review. Pituitary. 2010;14:184–193. | ||
van der Lely AJ, Biller BMK, Brue T, et al. Long-term safety of pegvisomant in patients with acromegaly: comprehensive review of 1288 subjects in ACROSTUDY. J Clin Endocrinol Metab. 2012;97: 1589–1597. | ||
Sheppard M, Bronstein MD, Freda P, et al. Pasireotide LAR maintains inhibition of GH and IGF-1 in patients with acromegaly for up to 25 months: results from the blinded extension phase of a randomized, double-blind, multicenter, phase III study. Pituitary. 2014;18:385–394. | ||
Fleseriu M, Sheppard M, Bronstein MD, et al. Switching patients with acromegaly from octreotide LAR to pasireotide LAR improves biochemical control: crossover extension to a randomized, double-blind, multicenter, phase III study. In: The Endocrine Society’s 94th Annual Meeting and Expo; June 23–26, 2012, Presentation Number: SUN-741; Houston, TX, 2012. | ||
Freda PU, Fleseriu M, AJvd L, et al. Improvement of biochemical control in patients with acromegaly switched from octreotide LAR to pasireotide LAR: crossover extension to a double-blind, multicenter, randomized, phase III study. In: The Endocrine Society’s 95th Annual Meeting and Expo; June 15–18, 2013, Poster Board SUN-95; San Francisco, CA, 2013. | ||
Fleseriu M, Gadelha M, Bronstein MD, et al. OR7-7: pasireotide LAR provides superior efficacy over octreotide LAR and lanreotide ATG in patients with inadequately controlled acromegaly: a phase III, multicenter, randomized study (PAOLA). Growth Horm IGF Res. 2014;24:S20–S21. | ||
Gadelha MR, Bronstein MD, Brue T, et al. Biochemical control is maintained with pasireotide LAR in patients with acromegaly: results from the extension of a randomized phase III study (PAOLA). In: The Endocrine Society’s 97th Annual Meeting and Expo; Mar 5–8, 2015, Presentation Number: PP09-01; San Diego, CA, 2015. | ||
Shenouda M, Maldonado M, Wang Y, et al. An open-label dose-escalation study of once-daily and twice-daily pasireotide in healthy volunteers. Am J Ther. 2014;21:164–173. | ||
Henry RR, Ciaraldi TP, Armstrong D, et al. Hyperglycemia associated with pasireotide: results from a mechanistic study in healthy volunteers. J Clin Endocrinol Metab. 2013;98:3446–3453. | ||
Breitschaft A, Hu K, Hermosillo Reséndiz K, et al. Management of hyperglycemia associated with pasireotide (SOM230): healthy volunteer study. Diabetes Res Clin Pract. 2014;103:458–465. | ||
Colao AM, Gu F, Gadelha MR, et al. Metformin-based oral antidiabetic therapy proved effective in hyperglycaemia associated with pasireotide in patients with acromegaly. Endocr Abstr. 2015;37:E800. | ||
Gadelha MR, Brue T, Fleseriu M, et al. Proactive monitoring and early intervention in the management of pasireotide-induced hyperglycemia: lessons from the phase III, 24-week paola study. In: The Endocrine Society’s 97th Annual Meeting and Expo; Mar 5–8, 2015, Presentation Number: LBT-079; San Diego, CA, 2015. | ||
Fleseriu M, Hoffman AR, Katznelson L, et al. American Association of Clinical Endocrinologists and American College of Endocrinology Disease State Clinical Review: management of acromegaly patients: what is the role of pre-operative medical therapy? Endocr Pract. 2015;21:668–673. | ||
Fleseriu M. The role of combination medical therapy in acromegaly. Curr Opin Endocrinol Diabetes Obes. 2013;20(4):321–329. | ||
Ben-Shlomo A. Pharmacotherapy for acromegaly. Endocrinol Metab Clin North Am. 2015;44:35–41. | ||
Schmid H, Brue T, Colao A, et al. Effect of pasireotide on GH, IGF1, IGFBP2, IGFBP3, HbA1C and glucose in patients with inadequately controlled acromegaly: exploratory results from a multicentre, randomized, 24-week study (PAOLA). Endocr Abstr. 2014;35:905. | ||
Colao A, De Block C, Gaztambide MS, et al. Managing hyperglycemia in patients with Cushing’s disease treated with pasireotide: medical expert recommendations. Pituitary. 2013;17:180–186. | ||
McKeage K. Pasireotide in acromegaly: a review. Drugs. 2015;75:1039–1048. | ||
Tuvia S, Atsmon J, Teichman SL, et al. Oral octreotide absorption in human subjects: comparable pharmacokinetics to parenteral octreotide and effective growth hormone suppression. J Clin Endocrinol Metab. 2012;97:2362–2369. | ||
Melmed S, Popovic V, Bidlingmaier M, et al. Safety and efficacy of oral octreotide in acromegaly: results of a multicenter phase III trial. J Clin Endocrinol Metab. 2015;100:1699–1708. | ||
Roberts J, Linden M, Cervin C, et al. Octreotide fluid crystal provides sustained octreotide bioavailability and similar IGF1 suppression to that of octreotide LAR (Sandostatin LAR): randomized, open-label, Phase I, repeat-dose study in healthy volunteers. Endocr Abstr. 2014;35:914. | ||
Chieffo C, Cook D, Xiang Q, et al. Efficacy and safety of an octreotide implant in the treatment of patients with acromegaly. J Clin Endocrinol Metab. 2013;98:4047–4054. | ||
Störmann S, Schopohl J. Emerging drugs for acromegaly. Expert Opin Emerg Drugs. 2014;19:79–97. | ||
Plockinger U, Hoffmann U, Geese M, et al. DG3173 (somatoprim), a unique somatostatin receptor subtypes 2-, 4- and 5-selective analogue, effectively reduces GH secretion in human GH-secreting pituitary adenomas even in Octreotide non-responsive tumours. Eur J Endocrinol. 2011;166:223–234. | ||
Somm E, Bonnet N, Martinez A, et al. A botulinum toxin-derived targeted secretion inhibitor downregulates the GH/IGF1 axis. J Clin Invest. 2012;122:3295–3306. | ||
Garcia EA, Trivellin G, Aflorei ED, et al. Characterization of SNARE proteins in human pituitary adenomas: targeted secretion inhibitors as a new strategy for the treatment of acromegaly? J Clin Endocrinol Metab. 2013;98:E1918–E1926. | ||
Cuevas-Ramos D, Fleseriu M. Somatostatin receptor ligands and resistance to treatment in pituitary adenomas. J Mol Endocrinol. 2014;52:R223–R240. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.