Back to Journals » Cancer Management and Research » Volume 12
PARP Inhibitors in Endometrial Cancer: Current Status and Perspectives
Authors Musacchio L ![]() , Caruso G
, Caruso G ![]() , Pisano C, Cecere SC, Di Napoli M, Attademo L, Tambaro R, Russo D
, Pisano C, Cecere SC, Di Napoli M, Attademo L, Tambaro R, Russo D ![]() , Califano D
, Califano D ![]() , Palaia I
, Palaia I ![]() , Muzii L, Benedetti Panici P, Pignata S
, Muzii L, Benedetti Panici P, Pignata S
Received 25 April 2020
Accepted for publication 8 July 2020
Published 22 July 2020 Volume 2020:12 Pages 6123—6135
DOI https://doi.org/10.2147/CMAR.S221001
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Xueqiong Zhu
Lucia Musacchio,1 Giuseppe Caruso,1 Carmela Pisano,2 Sabrina Chiara Cecere,2 Marilena Di Napoli,2 Laura Attademo,2 Rosa Tambaro,2 Daniela Russo,3 Daniela Califano,3 Innocenza Palaia,1 Ludovico Muzii,1 Pierluigi Benedetti Panici,1 Sandro Pignata2
1Department of Maternal and Child Health and Urological Sciences, University “Sapienza”, Policlinico Umberto I, Rome, Italy; 2Department of Urology and Gynecology, Istituto Nazionale Tumori IRCCS “Fondazione G. Pascale”, Naples, Italy; 3Functional Genomic Unit, Istituto Nazionale Tumori IRCCS “Fondazione G. Pascale”, Naples, Italy
Correspondence: Sandro Pignata
Department of Urology and Gynecology, Istituto Nazionale Tumori IRCCS “Fondazione G. Pascale”, Via Semmola, Naples 80131, Italy
Tel +39 0815903409
Fax +39 0815903861
Email [email protected]
Abstract: Advanced, recurrent and metastatic endometrial cancer (EC) has a dismal prognosis due to poor response rates to conventional treatments. In the era of precision medicine, the improved understanding of cancer genetics and molecular biology has led to the development of targeted therapies, such as poly (ADP-ribose) polymerase (PARP) inhibitors. This class of drugs that inhibit PARP enzymes has been investigated in many different types of tumors and its use in the treatment of gynecological malignancies has rapidly increased over the past few years. Data from several clinical trials showed that PARP inhibitors have a beneficial role in cancers with a defect in the homologous DNA recombination system, regardless of the BRCA mutational status. Since EC frequently shows mutations in PTEN and TP53 genes, indirectly involved in the homologous DNA recombination pathway, several in vivo and in vitro studies investigated the efficacy of PARP inhibitors in EC, showing promising results. This review will discuss the use of PARP inhibitors in endometrial cancer, summarizing data from preclinical studies and providing an overview of the ongoing trials, with a special focus on the development of combined treatment strategies with PARP inhibitors and immune checkpoint inhibitors.
Keywords: endometrial cancer, PARP inhibitors, PTEN mutation, P53 mutation, homologous recombination deficiency, immune checkpoint inhibitors
Introduction
Endometrial cancer (EC) is the most common gynecological malignancy in developed countries.1 While patients with early-stage and low-risk disease present an excellent prognosis with five-year survival rates of over 95%, women with advanced, recurrent and metastatic EC have extremely poor outcomes, owing to the low response rate to standard systemic chemotherapy.2
EC is a heterogeneous disease consisting of various histological subtypes with different pathogenesis, prognosis and sensitivity to therapeutic agents.3,4 Given the improved knowledge of cancer genetics and biology, in 2013, The Cancer Genome Atlas (TGCA) proposed a new endometrial cancer molecular classification, based on the following four groups: POLE-ultramutated, microsatellite instability-hypermutated (MSI-H), copy-number low, and copy-number high.3
POLE-ultramutated tumors, representing 6.4% of low-grade and 17.4% of high-grade endometrioid tumors, are characterized by a high mutation rate and are associated with a good prognosis. In this group, PTEN, PIK3R1, PIK3CA, and RAS genes are frequently mutated.5 MSI-hypermutated (MSI-H) group represents 28.6% of low-grade and 54.3% of high-grade endometrioid EC (EEC).6,7 These tumors show MSI and a high mutation rate owing to defects in the mismatch repair (MMR) system, with MLH1, MSH2, MSH6, and PMS2 representing the most involved genes. Moreover, mutations of the PTEN gene and the PTEN-PIK3CA pathway are frequent in this subgroup.8
Copy-number low tumors represent 60% of low-grade and only 8.7% of high-grade EC. This group shows microsatellite stability (MSS) and a low mutation rate. Mutations of the PTEN gene and the PIK3CA pathway occur in 77% and 53% of cases, respectively.3,6 In addition, this subgroup presents high progesterone receptor (PgR) expression levels.9
Copy-number high (serous-like) subgroup represents mainly serous (97.7%) and mixed-histology (75%) tumors. It presents a low mutation rate and is associated with a poor prognosis. The TP53 gene is commonly mutated (92%), whereas mutations in KRAS and PTEN genes are less frequent.6,7
Basically, the first three of these subgroups consist mainly of endometrioid EC and are mostly associated with a mutation in the PTEN gene, whereas the copy-number high subgroup includes almost exclusively serous tumors and presents a pathogenetic variant of P53.3
Homologous Recombination System and BRCA Mutational Status in EC
An important area to explore is the correlation between the BRCA mutational status and the risk of developing endometrial cancer, which remains still unclear. Only a few studies have been conducted so far, suggesting that the risk is different between BRCA1 and BRCA2 mutation carriers. In 1999, it has been reported that the BRCA2 mutation is a risk factor for several cancers but not for uterine cancer.10 Conversely, in 2002, a large cohort study assessed the risk of developing other cancers in BRCA1-mutated patients and found that women with BRCA1 mutations had an increased rate of uterine cancer as compared to general population cancer rates (uterine body, RR = 2.65).11 Moreover, in 2004, a study of 27 patients evaluated whether BRCA1/2 mutations are related to a greater risk of developing uterine papillary serous carcinoma (UPSC). Four patients (20%) had a germline BRCA1 mutation, thus suggesting that UPSC may represent a manifestation of the BRCA-mutation syndrome.12 Nevertheless, a previous study published in 2000 by Goshen et al reported no somatic BRCA mutations among 56 UPSC patients.13
Recently, a multicenter, prospective study aimed to assess the risk of uterine cancer in 1083 BRCA-mutated patients after undergoing risk-reducing salpingo-oophorectomy (RRSO) without hysterectomy. Although the overall risk of uterine cancer was not significantly increased (8 cases reported versus 4.3 expected), five cases of UPSC were reported, four of which occurred in BRCA1-mutated patients, thus exceeding the expected rate of UPSC among BRCA1-mutated women.14
In February 2020, during the SGO Winter Meeting, Fehniger et al presented the results from a study conducted to evaluate the prevalence of BRCA1/2 somatic mutations among 89 women treated for advanced and recurrent EC. Five patients had a mutation in BRCA1, 9 patients in BRCA2, and 1 in both genes. In addition, among the 15 BRCA-mutated patients, 7 had MSI-H and 9 high-TMB (tumor mutational burden) tumors.15 These findings mean that most of the somatic mutations could have been passenger mutations in hypermutated tumors. Moreover, these results could provide a rationale for the use of PARP inhibitors (PARPi) in this subset of EC patient.
Interestingly, a strong similarity between copy-number high, serous-like EC, high-grade serous ovarian cancer (HGSOC), and basal-like breast cancer has been suggested. Both HGSOC and basal-like breast cancer, showing a deficiency in the homologous recombination (HR) system caused by mutations in BRCA1/2 genes, are part of the same hereditary BRCA-related breast and ovarian cancer syndrome (HBOC syndrome).16,17
Potential PARPi Biomarkers in EC
The HR system is essential for the DNA double-strand breaks (DSBs) repair. Although the Homologous Recombination Deficiency (HRD) has been commonly associated with mutations in the BReast CAncer (BRCA) 1/2 genes, it is now clear that other genetic and epigenetic alterations may play a crucial role, involving the following genes: ARID1A, ATM, ATR, BAP1, BARD1, BLM, BRIP1, CHEK1/2, FANCA/C/D2/E/F/G/L, MRE11A, NBN, PALB2, RAD50, RAD51, RAD51B, and WRN (Figure 1).18–21 The exact role of HRD in all tumor lineages has not yet been assessed. Although several molecular studies have been conducted for ovarian, breast and prostate cancer, few and discordant data are reported in literature as regards the relationship between HRD and EC molecular subgroups. De Jonge et al showed that HRD was significantly associated with non-endometrioid histologies and P53-mutated patients, whereas no correlation was found between HRD and the loss of PTEN expression. Moreover, authors confirmed that mutant P53 is more frequent in somatic copy number alteration (SCNA)-high, serous-like EC group than other subclasses according to TGCA.22
 |
Figure 1 Overview of the HR pathway in PTEN-deficient EC cells. When DSBs occur, ATM, ATR and CHK1/2 kinases phosphorylate BRCA1, that is stabilized by BARD1. BRCA2, whose correct conformation is maintained by DSS1 and PALB2, carries RAD51 to the site of DNA damage, where it forms nucleoprotein filaments and stabilizes DNA double strands. The EGFR activates PI3K/AKT pathway, that in turn inhibits p53. Conversely, PTEN inhibits the PI3K pathway and thus activates p53, that induces cell apoptosis and cycle arrest and increases the expression of the RAD51 and the MRN complex (RAD50, MRE11, NBS1), involved in the DSBs repair. Moreover, PTEN upregulates RAD51. Therefore, mutant PTEN inhibits p53 and RAD51 expression, thus impairing the HR system. Similarly, mutant p53 downregulates the MRN complex and RAD51 levels. |
In 2019, Heeke et al reviewed the molecular profiles of 52,426 tumors through the Next Generation Sequencing (NGS) technique, including 5540 endometrial cancers, in order to identify pathogenetic mutations in HR pathway genes. The results showed that the highest frequency of HRD was achieved in EC (34.4%), with the most frequent mutations involving the gene ARID1A (27%), followed by ATM (4.61%), ATRX (3.13%) and BRCA2 (3.05%).23
Nevertheless, Shen et al demonstrated that also the PTEN protein contributes in maintaining the genomic integrity through the upregulation of RAD51 expression levels. The loss of RAD51 function causes a deficiency in the HR system (HRD), thus preventing DNA DSBs from being repaired.24 Indeed, it has been reported that RAD51 levels are markedly reduced in PTEN-mutated cells compared to wild-type PTEN.
Since somatic PTEN mutations are extremely frequent in POLE-ultramutated, MSI-hypermutated, and copy-number low EC, it can be assumed that a deficiency in the HR system could play a crucial role in these subgroups.
Recently, novel targeted therapies have been explored with the aim to maximize the clinical benefits, while reducing the adverse events. One of the most promising drug classes are the poly (ADP-ribose) polymerase (PARP) inhibitors (PARPi), that have already been shown to improve the Progression-Free Survival (PFS) in ovarian cancer patients. Currently, BRCA1/2 are clinically validated biomarkers for the use of PARPi in ovarian cancer, prostate cancer, breast cancer, and pancreatic cancer, and several authors suggested their potential role in other malignancies.
The molecular subgroup analyses of several ovarian cancer clinical trials suggested that PARPi are more active in BRCA-mutated patients, followed by HR-deficient (HRD) and HR-proficient subgroups.25–28 Therefore, several studies have been conducted to evaluate which tumors could benefit from PARi, including EC.
Furthermore, since it has been observed that the DNA damage promotes the immune priming through the upregulation of programmed cell death protein-1 (PD-1) and programmed death-ligand 1 (PD-L1) expression, the combination of PARPi with anti-PD-1/PD-L1 antibodies appears as a promising strategy in EC patients.29
Based on results from preclinical data demonstrating the sensitivity of EC cell lines to PARPi, recently, several Phase I/II clinical trials have been developed. In this review, we discuss the scientific rationale behind the use of PARPi in EC, focusing on the most promising ongoing trials.
Background on DNA Damage and Its Mechanism Repair
DNA is highly susceptible to various genotoxic insults, both endogenous (eg, reactive oxygen radicals, ROS) and exogenous (environmental agents, such as ultraviolet light, ionizing radiation, and chemicals), that can cause several types of DNA damage, including oxidation, alkylation, hydrolysis, depyrimidination, depurination, deamination, single-strand breaks (SSBs), and double-strand breaks (DSBs).30 Preserving genomic integrity is essential for adequate cell replication, growth and survival. In the presence of DNA damage, the cell can either repair the damage or induce cell apoptosis if the damage exceeds the repair capacity, thus preventing mutagenesis and the progression to cancer. Collectively, all the mechanisms activated in response to the DNA damage, including DNA repair mechanisms, damage tolerance and cell-cycle checkpoints, form a strictly controlled complex network of cellular pathways, known as the DNA Damage Response (DDR).31
Six main DNA repair mechanisms have been recognised to repair DNA damage. DSBs, which are more cytotoxic, are repaired through two distinct mechanisms: homologous recombination (HR) and non-homologous end-joining (NHEJ). Conversely, SSBs are fixed via base excision repair (BER), nucleotide excision repair (NER), mismatch repair (MMR), and translesion synthesis (TS), using the undamaged complementary strand, which acts as a useful template.
Homologous recombination is a high-fidelity system, that repairs DNA damage through the guide of the homologous chromatid, although it acts exclusively in S and G2 cell cycle phases. Crucial proteins involved in HR pathway include BRCA1, BRCA2, RAD51, and PALB2 genes.32 However, the exact molecular details are still controversial. HR is the most important process for repairing DSBs, being a conservative process that tends to restore the original DNA sequence at the sites of damage. When HR is deficient, DSBs repair is performed through NHEJ, which in contrast to HR: (a) binds directly the ends of a DSB together; (b) acts very rapidly throughout the entire cell cycle; (c) represents an error-prone process.33
PARP Family
Poly (ADP-ribose) polymerase (PARP) represents a family of 17 structurally and functionally different proteins. The role of PARP1 and PARP2 is to detect and orchestrates DNA SSBs repair via BER pathway.34 PARP is composed of four domains: a DNA-binding domain, a caspase-cleaved domain, an auto-modification domain, and a catalytic domain. Briefly, in the presence of DNA SSBs, the DNA-binding domain will bind the DNA and induce a conformational shift, which activates the catalytic domain and acts as a signal for the recruitment of other DNA-repairing enzymes, including DNA ligase III, DNA polymerase beta and scaffolding proteins.16 The auto-modification domain is responsible for releasing PARP from the DNA after catalysis. However, some findings have recently suggested that the main role of PARP1/2 is to stabilize and regulate the DNA stalled replication forks.
In the past few years, PARPi have shown to be a promising targeted therapy in gynecologic oncology. They block PARP enzymes both inhibiting their catalytic activity and, as more recently suggested, especially by trapping PARP-DNA complexes at the damaged sites and thus preventing DNA repair, replication, and transcription.35
In HR-deficient cancer cells, in which the major DNA DSBs repair mechanism is dysfunctional, the use of PARPi, blocking DNA SSBs repair via BER and trapping PARP enzymes, determines a huge DNA damage with the accumulation of DSBs which is beyond repair capacity. This therapeutic strategy exploits the phenomenon of synthetic lethality.36
Methods
For this review of the literature, a non-systematic search was performed in PubMed using the following keywords to retrieve preclinical data: “endometrial cancer”, ‘PARP inhibitors’, ‘p53’, ‘PTEN’, “olaparib”, “rucaparib”, “niraparib”, “talazoparib”, and “veliparib”. In addition, we queried Clinicaltrials.gov for ongoing trials using the same keywords. Literature search strategy was performed up to March 2020, without temporal limits. Only English written publications were selected. Titles and abstracts of search results were screened to determine eligibility in the manuscript.
PARPi in EC
In vitro Studies
Considering that the PTEN gene is involved in the regulation of the DNA HR system, some authors evaluated whether endometrioid endometrial cancer (EEC) cell lines lacking PTEN expression were sensitive to PARP inhibition.
In 2010, Dedes et al examined 8 EEC cells (6 PTEN-deficient and 2 wild-type PTEN) and studied the correlation between the PTEN status and the sensitivity to the PARP inhibitor KU0058948. The PTEN deficiency in EC cells provided a significantly greater sensitivity to PARPi than wild-type PTEN.37 These data were confirmed by Dinkic et al, who evaluated the PARP expression in four different EC cell lines and showed that the addition of olaparib, a PARP inhibitor, to carboplatin/paclitaxel doublet sensitized EC cells to paclitaxel-induced apoptosis, thus suggesting that the combination of cytotoxic agents with PARPi can represent a promising therapeutic option.38
However, in 2014, Miyasaka et al published discordant results, showing that the expression of PTEN was not correlated with a high sensitivity to PARPi. The response to olaparib was evaluated in 16 EC cell lines: 12 PTEN-deficient and 4 wild-type PTEN. One of the 4 wild-type PTEN cells and only 3 of the 12 PTEN-mutated cells were sensitive. Furthermore, they assessed whether PTEN-deficiency was related to a downregulation of RAD51, showing that RAD51 expression levels were not associated with the PTEN status.39
In 2017, Philip et al evaluated four EC cell lines, two PTEN-deficient and two wild-type, and found that PTEN-deficient cell lines were more sensitive to the PARPi olaparib and talazoparib than wild-type PTEN cell lines. In addition, since PTEN-mutated cells showed an over-activation of the PI3K/mTOR pathway, the use of PI3K inhibitors reduced the RAD51 foci formation in PTEN-mutated cells and further improved the sensitivity of these cells to PARPi.40
Since the PTEN protein inhibits the activation of the phosphoinositide 3-kinase (PI3K)/AKT pathway,41,42 which is a cell pro-survival signalling cascade in EC, Bian et al conducted a study to assess the sensitivity of four PTEN-deficient EC cell lines to the combination regimen of olaparib and buparlisib, a P13K-inhibitor, and showed that this combination treatment strategy synergistically inhibited the growth of all the PTEN-deficient ECC cell lines examined.43
In vivo Studies
In 2013, Janzen et al evaluated the efficacy of olaparib at low and high estrogen concentrations in a PTEN-deficient EC mouse model, showing a 6-fold decrease in tumor size when olaparib was administered in a low estrogen milieu. In contrast, the same treatment was ineffective in mice exposed to high estrogen levels. CYP3A41 is an estrogen-regulated enzyme involved in olaparib metabolism,44,45 whose concentration has been reported to be higher in the livers of mice treated with estrogens. This may explain why the concentration of olaparib was lower in estradiol-supplemented mice. Moreover, RAD51 expression and recruitment to DNA damage sites were significant in PTEN-deficient epithelia exposed to high estrogen levels. In the context of a low estrogen milieu, the RAD51 protein levels and so its effects appear to be reduced in PTEN-deficient epithelia.46 Therefore, these findings suggested that the expression of RAD51 could be related to the concentration of estrogen in PTEN-deficient cells and that PARP inhibition in combination with hormonal therapy may increase antitumor efficacy.
In 2011, Forster et al presented the case of a 58-year-old woman with recurrent EEC, showing brain metastases. In light of her sensitivity to previous platinum-based treatments, she was treated with olaparib as part of a Phase I study. The molecular analysis of the tumor biopsy was negative for somatic BRCA1/2 mutations but revealed the absence of PTEN expression in tumor cells. Magnetic Resonance Imaging (MRI) performed 10 weeks after starting the treatment with olaparib showed a reduction in the size of brain metastases with a decrease in perilesional edema. Moreover, lung, liver, bone, and peritoneum disease appeared stable on Computed Tomography (CT) scan. After 8 months, the radiological evaluation showed a disease progression and the patient discontinued olaparib.47
Recently, Gockley et al reported the case of a 43-year-old woman with low-grade EEC treated with a PARP inhibitor. At the time of pelvic relapse, the patient received 4 cycles of carboplatin and gemcitabine. Given her germline and somatic BRCA2 mutation and her sensitivity to platinum-based chemotherapy, she started olaparib 300 mg orally twice daily as maintenance treatment. The pelvic MRI performed 10 months after olaparib initiation showed a partial response (PR) with a decrease in size of the adnexal mass. Therefore, she continued olaparib with clinical follow-ups every 2 months for over 15 months with stable disease documented on PET/CT scan and MRI at the time of published data.48
PARPi Plus Immune Checkpoint Inhibitors
In the era of precision medicine, PARPi and immune checkpoint inhibitors (ICIs), have revolutionized the outcome of cancer patients, improving both PFS and overall survival (OS). Literature data showed that ICIs improved clinical outcomes in cancers with an increased mutational load,49,50 which is 10–100 times higher in MMR-deficient tumors compared to MMR-proficient.51,52 Therefore, it has been suggested that ICIs can represent a promising therapeutic strategy for EC patients, especially in MMR-deficient and MSI-H groups. In 2017, the Food and Drug Administration (FDA) approved pembrolizumab, an anti PD-1 antibody, for patients with unresectable or metastatic MSI-H or MMR-deficient solid tumors, including EC, presenting a progressive disease after a prior treatment and no satisfactory alternative treatments.
Interestingly, a recent study including 60 patients with advanced urothelial carcinomas suggested that, besides the mutations in MMR proteins, defects in DNA Damage Response (DDR) pathways may also promote the anti-PD-1/PD-L1 antitumor activity, as they were associated with an increase in the response rate of around 60%.53 The DDR deficiency, which is predominantly related to the HR system, seems to act as a stimulator of interferon genes (STING), promoting the expression of IFN and other inflammatory cytokines and, therefore, increasing the immune infiltrate.54 For example, in breast cancer, the DDR deficiency is associated with a significantly higher level of CD8 T-cell infiltration, while BRCA1/2-mutated ovarian cancers present increased PD-L1 expression levels and T-cell infiltration. Moreover, several in vivo studies showed that PARP inhibition increased PD-L1 expression. In 2017, Jiao et al presented interesting data about the correlation between PARPi and PD-1/PD-L1 expression in breast cancer cell lines, xenograft tumors, and syngeneic tumors treated with olaparib and talazoparib. It was found that PARP inhibition upregulated the PD-L1 expression in breast cancer cell lines and animal models and that combining PARPi with anti-PD-L1 agents significantly increased the therapeutic efficacy in vivo, compared with each single agent alone.55
A few data on the combination of PARPi with ICIs in ovarian cancer patients have been collected. In Phase I/II study TOPACIO, the niraparib/pembrolizumab combination demonstrated an overall response rate (ORR) of 25% in platinum-resistant ovarian cancer setting, regardless of the DDR status.56 Furthermore, in MEDIOLA study, the olaparib/durvalumab combination demonstrated an ORR of 63% in relapsed platinum-sensitive, BRCA1/2-mutated ovarian cancer, which is higher than the one reported for PARPi monotherapy in the same setting.57
Based on available preclinical, clinical and translational data, several clinical trials are ongoing to evaluate the strategic role of the combination between PARPi and ICIs in endometrial cancer.
Ongoing Clinical Trials
Currently, there are several Phase I and II clinical trials evaluating the role of PARPi (olaparib, niraparib, rucaparib, and talazoparib) in metastatic, advanced, and recurrent EC, alone or in combination with other drugs (Table 1).
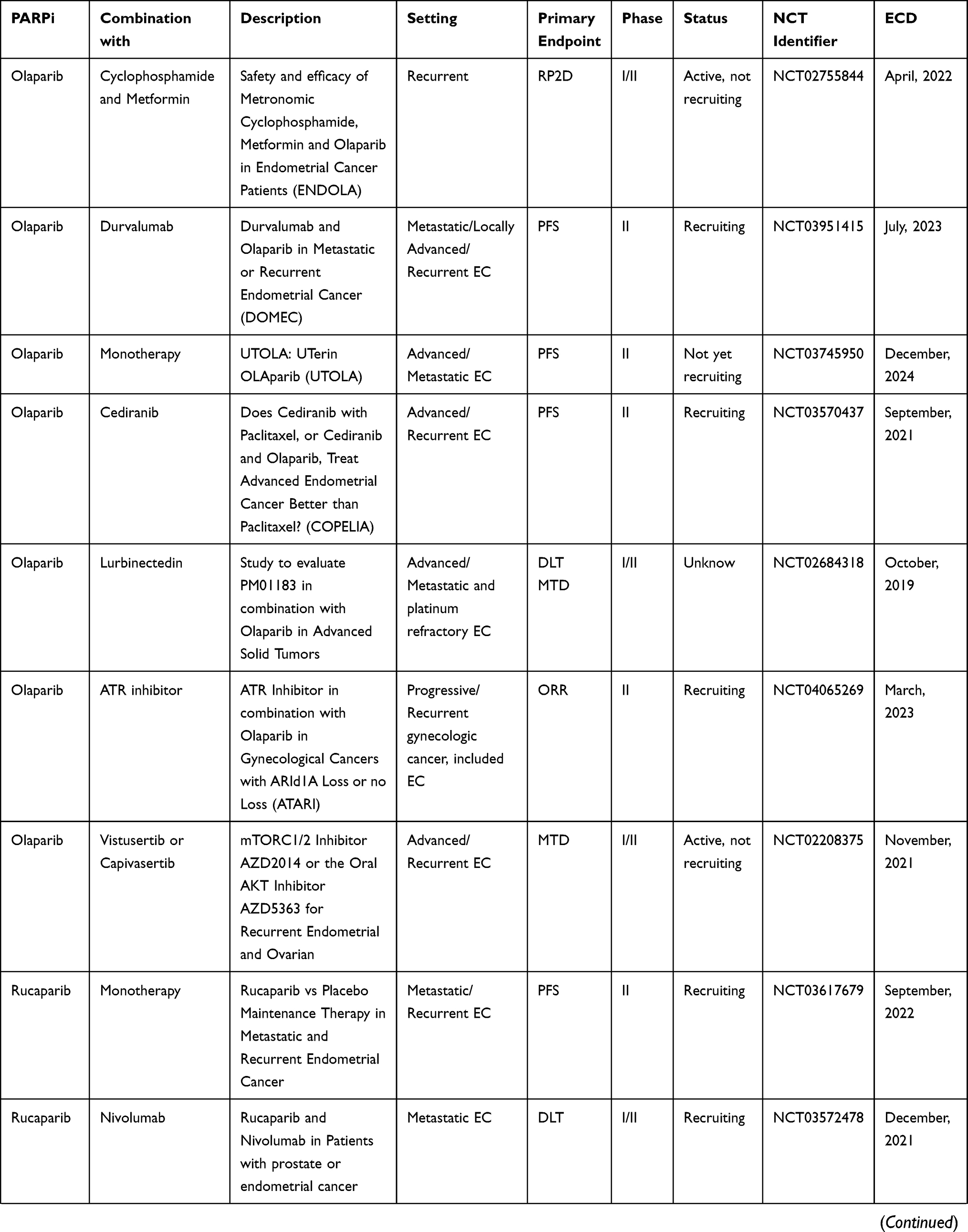 |  |  |
Table 1 Ongoing Clinical Trials |
Olaparib is under evaluation in many Phase I and II clinical trials. NCT02208375 is a Phase Ib/II to evaluate the maximum tolerated dose (MTD) of olaparib and vistusertib (mTOR inhibitor) or olaparib and capivasertib (AKT kinase inhibitor) when given together in treating patients with recurrent endometrial cancer.58 ENDOLA trial is a Phase I/II study evaluating the safety and efficacy of olaparib in combination with metronomic cyclophosphamide plus metformin in recurrent/metastatic EC. The rationale behind this combined therapy is that metronomic cyclophosphamide may increase the anti-proliferative effect of olaparib and exert anti-angiogenic effects, while metformin can also increase the anti-proliferative effect of olaparib without further toxicity. The primary endpoint is the recommended Phase II trial (RP2D) dose of olaparib in combination with metformin and metronomic cyclophosphamide.59
DOMEC (NCT03951415) is a prospective, multicenter, Phase II study that aims to assess the efficacy of olaparib in combination with durvalumab (anti PD-L1) in advanced, recurrent and metastatic EC. Patients with prior chemotherapy failure, unwilling to undergo chemotherapy, or chemo-naive not suitable for chemotherapy are enrolled in this trial and they receive olaparib tablets 300 mg twice daily and durvalumab 1500 mg intravenously (IV) every 28 days. The primary endpoint is the PFS.60
UTOLA (NCT03745950) is a multicenter, double-blind, randomized Phase II trial assessing the efficacy of olaparib as maintenance after platinum-based chemotherapy in advanced and recurrent EC patients. Patients randomized in the experimental arm will receive olaparib 300 mg orally twice daily as maintenance until progression disease according to RECIST 1.1 or unacceptable toxicity.61
Moreover, olaparib is also being tested in combination with cediranib, an anti-VEGF antibody, in a Phase II, randomized, three arms, open-label clinical trial.
In COPELIA study (NCT03570437), patients with recurrent and advanced EC will be randomized in three arms: in cohort A, patients receive paclitaxel 80 mg/mq administered in days 1, 8 and 15 of a 28-day cycle up to 6 cycles; in cohort B, cediranib 20 mg orally daily for 28 days is added to the treatment with paclitaxel for 6 cycles; in cohort C, cediranib 20 mg orally is administered daily in combination with olaparib 300 mg orally twice daily for 28 days. Patients enrolled in cohort B and C with at least stable disease will be able to continue cediranib alone (cohort B) or cediranib/olaparib (cohort C) daily until disease progression. The primary endpoint is the PFS.62
NCT02684318 is a Phase I/II study to evaluate the efficacy and tolerability of PM01183 (Lurbinectedin) in combination with olaparib in patients with advanced or metastatic solid tumors, including EC. The primary objective of Phase Ib is to establish the safety [dose limiting toxicity (DLT), MTD, and RP2D] of orally administered olaparib in combination with PM01183, whereas the primary objective of Phase II is to assess the efficacy of PM01183 in combination with olaparib in terms of tumor response rate according to RECIST 1.1 criteria.63
Furthermore, rucaparib is being tested in several Phase I/II clinical trials. NCT03572478 is a Phase I/II study to assess the safety (Phase I: DLT) and efficacy (Phase II: time to disease progression) of the combination of an immune checkpoint inhibitor (nivolumab) with a PARP inhibitor (rucaparib) in patients with metastatic or recurrent EC.64 NCT03552471 is a Phase I study to determine the recommended Phase II dose for the combination of mirvetuximab soravtansine with rucaparib camsylate (rucaparib) in recurrent EC patients. Secondary objectives of this study are to determine the safety and tolerability of combining these drugs in the study population, to explore the objective antitumor activity (complete or partial response) according to RECIST criteria, to measure the PFS, and to evaluate the pharmacokinetics of mirvetuximab soravtansine and rucaparib in combination.65
NCT03617679 is a Phase II, randomized, double-blind, clinical trial evaluating the efficacy of rucaparib as maintenance treatment in patients with metastatic and recurrent EC after the first-line chemotherapy. Patients within the experimental arm will receive rucaparib 600 mg orally twice daily until disease progression or other indications for discontinuation. The PFS is used as a primary endpoint.66
The efficacy of rucaparib is also tested in association with other drugs, such as bevacizumab and atezolizumab. ENDOBARR (NCT03694262) is an open-label, non randomized, Phase II clinical trial investigating the efficacy and safety of rucaparib in combination with atezolizumab and bevacizumab in recurrent, progressive EC patients. The rationale behind the combined use of PARPi and bevacizumab (anti-VEGF) can be explained through the results of some studies showing that the hypoxia induced by the antiangiogenic therapy causes a deficit in the HR pathway. Therefore, HR-deficient hypoxic tumor cells are sensitized to the action of PARPi.67 In the ENDOBARR trial patients will receive rucaparib 600 mg orally twice daily plus bevacizumab 15 mg/kg IV on day 1 of every 21-day cycle plus atezolizumab 1200 mg IV on day 1 of every cycle. The primary endpoint is the ORR.68
NCT03476798 is a Phase II clinical trial that aims to determine the PFS in recurrent EC patients who receive rucaparib 600 mg orally twice daily plus bevacizumab 15mg/kg IV on day 1 of each 21-day cycle.69
NCT03586661 is a Phase I clinical trial to determine the MTD and RP2D of the combination of niraparib and copanlisib in patients with recurrent EC. Patients in the experimental arm will receive niraparib PO daily on days 1–28 and copanlisib IV on days 1,8 and 15. Cycles repeat every 28 days in the absence of disease progression or unacceptable toxicity.70 NCT03016338 and NCT04080284 are two Phase II clinical trials investigating the efficacy of nivolumab, alone or in combination with other drugs. The main goal of NCT03016338 is to assess whether combining niraparib with TSR-042 (dostarlimab, an anti PD-1) increases the clinical outcomes in recurrent EC. Patients in the experimental arm will receive niraparib 200 or 300 mg daily for a 21-day cycle and dostarlimab 500 mg IV on day 1 of every cycle followed by 1000 mg IV every 6 weeks for a maximum of 2 years. The primary endpoint is the clinical benefit rate.71 On the other hand, NCT04080284 is a clinical trial that analyzes the efficacy of niraparib as maintenance in patients with advanced or platinum-sensitive recurrent uterine serous carcinoma. The primary endpoint is the PFS.72
Finally, talazoparib is an emerging PARP inhibitor under evaluation in several Phase I and II clinical trials. NCT03968406 is a Phase I to determine the safety, tolerability, and MTD of talazoparib combining talazoparib and fractionated radiotherapy in patients with refractory or recurrent EC.73 NCT02912572 is a Phase II, two-group, two-stage, open-label study of avelumab (an anti-PD-L1) in patients with MSS, MSI-H and POLE-ultramutated recurrent or persistent EC and of avelumab/talazoparib in patients with MSS recurrent or persistent EC. Talazoparib will be administered in cohort C MSS patients in combination with avelumab. The PFS is used as a primary endpoint.74
Discussion
The advent of PARPi and their use in the treatment of ovarian cancer has meaningfully revolutionized the clinical outcome of these patients, improving both PFS and OS.75 Based on the results from three Phase III clinical trials,25,26,76 the FDA has approved, respectively, olaparib, niraparib and rucaparib in different setting. Specifically, olaparib has been approved as maintenance monotherapy after platinum-based chemotherapy in BRCA-mutated advanced ovarian cancer.77 Moreover, recently, olaparib has been approved in combination with bevacizumab for first-line maintenance treatment of patients with advanced epithelial ovarian, fallopian tube, or primary peritoneal cancer who are in complete or partial response to first-line platinum-based chemotherapy and whose cancer is associated with HR-deficiency positive status, defined by either a deleterious or suspected deleterious BRCA mutation, and/or genomic instability.78 Niraparib has been approved for the maintenance treatment of patients with advanced epithelial ovarian cancer with or without BRCA-mutation who are in a complete or partial response to first-line platinum-based chemotherapy,28 whereas rucaparib has been approved for patients with high-grade, advanced ovarian cancer, with or without BRCA-mutation, who received at least two prior lines of platinum-based chemotherapy.26 Since then, several studies have been carried out to better understand genetic alterations and to evaluate the role of PARPi in other gynecological cancers, including EC.36
Although no conclusive literature data have been yet published, clinical evidence suggests that the use of PARPi could be extremely effective in tumors with HRD. Several clinical trials showed that the deficiency of the tumor suppressor PTEN gene is associated with HR defects.79–81 Therefore, the concept of synthetic lethality appears applicable to PTEN-deficient cells treated with PARPi, as well as for BRCA-deficient cells.82
However, discordant data are reported in literature. Dedes et al showed that PTEN-deficient EEC cell lines are sensitive to single-agent olaparib,37 whereas other authors suggested that olaparib should be combined with the PI3K inhibitor BKM120/buparlisib in order to be effective.33 Moreover, Janzen et al showed that the sensitivity to olaparib treatment was higher when EC cells were exposed to a low estrogen milieu.46 Therefore, these conflicting results suggested that the response of PTEN-deficient cells to olaparib could be context-specific.
- Although few data have been published on the relationship between EC and the BRCA-mutational status, some authors described germline alterations in other HR-related genes (eg, ATM, BARD1, BRIP1, CHEK2, NBN, RAD51C).83 It has been suggested that a mutation in the PTEN gene causes a downregulation of RAD51, a key protein implicated in the HR repair system.84,85
Moreover, both copy-number high, serous-like EC and HGSOC present a mutation in the TP53 gene in more than 90% of cases. Although authors found that p53 plays multiple roles in the regulation of the HR pathway, the exact mechanisms are still unclear. P53 is a tumor suppressor protein that prevents oncogenic transformation and maintains the genomic stability, by activating or repressing several target genes involved in various signalling pathway.86 There is increasing evidence suggesting a key role of p53 mutations on DNA damage response. Previous literature data detected an interaction between the mutant forms of p53 and the protein MRE11, which is part of the MRN complex (MRE11/RAD50/NBS1) involved in the initial sensing of DNA DSBs and subsequent recruitment of ATM.87 Moreover, authors showed that mutant p53 determines the loss of RAD51 function and, subsequently, a deficiency in the HR system.84,85 Finally, several studies showed that certain p53 mutants acquire a “gain-of-function” (GOF) phenotype leading to oncogenesis and drug resistance. Indeed, mutant p53 can activate the expression of those genes that are normally suppressed by wild-type p53, such as MDR1, VEGFR, and EGFR, which are involved in angiogenesis and cellular proliferation.88–90 In light of these observations, it can be explained the potential role of PARPi both in POLE-ultramutated, MSI-H, and copy-number low EC groups, which are associated with mutations of the PTEN gene, and in the copy-number high group, which correlates with mutations of the TP53 gene, as both PTEN and p53 proteins could play a key role in the HR pathway. Moreover, a study conducted on breast cancer and animal models showed that PARPi upregulated the PD-L1 expression.55
In conclusion, despite the lack of clinical data, the preliminary results of the studies conducted on ovarian cancer suggest that also in EC the combination of PARPi with anti-PD-1/PD-L1 therapy could significantly increase the therapeutic efficacy compared to monotherapy. Results from ongoing clinical trials will provide more data to establish the real clinical benefit of these drugs.
Author Contributions
All authors contributed to data analysis, drafting or revising the article, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–424. doi:10.3322/caac.21492
2. Colombo N, Preti E, Landoni F, et al. Endometrial cancer: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2013;24(Suppl 6):vi33–8. doi:10.1093/annonc/mdt353
3. Kandoth C, Schultz N, Cherniack AD, et al. Integrated genomic characterization of endometrial carcinoma. Nature. 2013;497(7447):67–73. doi:10.1038/nature12113
4. Reid-Nicholson M, Iyengar P, Hummer AJ, Linkov I, Asher M, Soslow RA. Immunophenotypic diversity of endometrial adenocarcinomas: implications for differential diagnosis. Mod Pathol. 2006;19(8):1091–1100. doi:10.1038/modpathol.3800620
5. Murali R, Soslow RA, Weigelt B. Classification of endometrial carcinoma: more than two types. Lancet Oncol. 2014;15(7):e268–78. doi:10.1016/S1470-2045(13)70591-6
6. Gargiulo P, Della Pepa C, Berardi S, et al. Tumor genotype and immune microenvironment in POLE-ultramutated and MSI-hypermutated endometrial cancers: new candidates for checkpoint blockade immunotherapy? Cancer Treat Rev. 2016;48:61–68. doi:10.1016/j.ctrv.2016.06.008
7. Le Gallo M, Bell DW. The emerging genomic landscape of endometrial cancer. Clin Chem. 2014;60(1):98–110. doi:10.1373/clinchem.2013.205740
8. Oda K, Stokoe D, Taketani Y, McCormick F. High frequency of coexistent mutations of PIK3CA and PTEN genes in endometrial carcinoma. Cancer Res. 2005;65(23):10669–10673. doi:10.1158/0008-5472.CAN-05-2620
9. Wilczyński M, Danielska J, Wilczyński J. An update of the classical Bokhman’s dualistic model of endometrial cancer. Prz Menopauzalny. 2016;15(2):63–68. doi:10.5114/pm.2016.61186
10. Breast Cancer Linkage Consortium. Cancer risks in BRCA2 mutation carriers. J Natl Cancer Inst. 1999;91(15):1310–1316. doi:10.1093/jnci/91.15.1310
11. Thompson D, Easton DF. Cancer incidence in BRCA1 mutation carriers. J Natl Cancer Inst. 2002;94(18):1358–1365. doi:10.1093/jnci/94.18.1358
12. Lavie O, Hornreich G, Ben-Arie A, et al. BRCA germline mutations in Jewish women with uterine serous papillary carcinoma. Gynecol Oncol. 2004;92(2):521–524. doi:10.1016/j.ygyno.2003.11.009
13. Goshen R, Chu W, Elit L, et al. Is uterine papillary serous adenocarcinoma a manifestation of the hereditary breast-ovarian cancer syndrome? Gynecol Oncol. 2000;79(3):477–481. doi:10.1006/gyno.2000.6003
14. Shu CA, Pike MC, Jotwani AR, et al. Uterine cancer after risk-reducing salpingo-oophorectomy without hysterectomy in women with BRCA mutations. JAMA Oncol. 2016;2(11):1434–1440. doi:10.1001/jamaoncol.2016.1820
15. Fehniger J, Levine DA, Pothuri B BRCA1/2 somatic mutations in patients with advanced or recurrent endometrial cancer.
16. Konstantinopoulos PA, Ceccaldi R, Shapiro GI, D’Andrea AD. Homologous recombination deficiency: exploiting the fundamental vulnerability of ovarian cancer. Cancer Discov. 2015;5(11):1137–1154. doi:10.1158/2159-8290.CD-15-0714
17. Lord CJ, Ashworth A. BRCAness revisited. Nat Rev Cancer. 2016;16(2):110–120. doi:10.1038/nrc.2015.21
18. Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474(7353):609–615. doi:10.1038/nature10166
19. Pennington KP, Walsh T, Harrell MI, et al. Germline and somatic mutations in homologous recombination genes predict platinum response and survival in ovarian, fallopian tube, and peritoneal carcinomas. Clin Cancer Res. 2014;20(3):764–775. doi:10.1158/1078-0432.CCR-13-2287
20. Ledermann JA, Drew Y, Kristeleit RS. Homologous recombination deficiency and ovarian cancer. Eur J Cancer. 2016;60:49–58. doi:10.1016/j.ejca.2016.03.005
21. McCabe N, Turner NC, Lord CJ, et al. Deficiency in the repair of DNA damage by homologous recombination and sensitivity to poly(ADP-ribose) polymerase inhibition. Cancer Res. 2006;66(16):8109–8115. doi:10.1158/0008-5472.CAN-06-0140
22. de Jonge MM, Auguste A, van Wijk LM, et al. Frequent homologous recombination deficiency in high-grade endometrial carcinomas. Clin Cancer Res. 2019;25(3):1087–1097. doi:10.1158/1078-0432.CCR-18-1443
23. Heeke AL, Pishvaian MJ, Lynce F, et al. Prevalence of homologous recombination-related gene mutations across multiple cancer types. JCO Precis Oncol. 2018;2018. doi:10.1200/PO.17.00286
24. Shen WH, Balajee AS, Wang J, et al. Essential role for nuclear PTEN in maintaining chromosomal integrity. Cell. 2007;128(1):157–170. doi:10.1016/j.cell.2006.11.042
25. Mirza MR, Monk BJ, Herrstedt J, et al. Niraparib maintenance therapy in platinum-sensitive, recurrent ovarian cancer. N Engl J Med. 2016;375(22):2154–2164. doi:10.1056/NEJMoa1611310
26. Coleman RL, Oza AM, Lorusso D, et al. Rucaparib maintenance treatment for recurrent ovarian carcinoma after response to platinum therapy (ARIEL3): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. 2017;390(10106):1949–1961. doi:10.1016/S0140-6736(17)32440-6
27. Coleman RL, Fleming GF, Brady MF, et al. Veliparib with first-line chemotherapy and as maintenance therapy in ovarian cancer. N Engl J Med. 2019;381(25):2403–2415. doi:10.1056/NEJMoa1909707
28. González-Martín A, Pothuri B, Vergote I, et al. Niraparib in patients with newly diagnosed advanced ovarian cancer. N Engl J Med. 2019;381(25):2391–2402. doi:10.1056/NEJMoa1910962
29. Stewart RA, Pilié PG, Yap TA. Development of PARP and immune-checkpoint inhibitor combinations. Cancer Res. 2018;78(24):6717–6725. doi:10.1158/0008-5472.CAN-18-2652
30. Chatterjee N, Walker GC. Mechanisms of DNA damage, repair, and mutagenesis. Environ Mol Mutagen. 2017;58(5):235–263.
31. Giglia-Mari G, Zotter A, Vermeulen W. DNA damage response. Cold Spring Harb Perspect Biol. 2011;3(1):a000745. doi:10.1101/cshperspect.a000745
32. Li X, Heyer WD. Homologous recombination in DNA repair and DNA damage tolerance. Cell Res. 2008;18(1):99–113. doi:10.1038/cr.2008.1
33. Davis AJ, Chen DJ. DNA double strand break repair via non-homologous end-joining. Transl Cancer Res. 2013;2(3):130–143. doi:10.3978/j.issn.2218-676X.2013.04.02
34. Herceg Z, Wang ZQ. Functions of poly(ADP-ribose) polymerase (PARP) in DNA repair, genomic integrity and cell death. Mutat Res. 2001;477(1–2):97–110. doi:10.1016/S0027-5107(01)00111-7
35. Murai J, Huang SY, Das BB, et al. Trapping of PARP1 and PARP2 by clinical PARP inhibitors. Cancer Res. 2012;72(21):5588–5599. doi:10.1158/0008-5472.CAN-12-2753
36. Tomao F, Santangelo G, Musacchio L, et al. Targeting cervical cancer: is there a role for poly (ADP-ribose) polymerase inhibition? J Cell Physiol. 2020;235(6):5050–5058. doi:10.1002/jcp.29440
37. Dedes KJ, Wetterskog D, Mendes-Pereira AM, et al. PTEN deficiency in endometrioid endometrial adenocarcinomas predicts sensitivity to PARP inhibitors. Sci Transl Med. 2010;2(53):53ra75. doi:10.1126/scitranslmed.3001538
38. Dinkic C, Jahn F, Zygmunt M, et al. PARP inhibition sensitizes endometrial cancer cells to paclitaxel-induced apoptosis. Oncol Lett. 2017;13(4):2847–2851. doi:10.3892/ol.2017.5795
39. Miyasaka A, Oda K, Ikeda Y, et al. Anti-tumor activity of olaparib, a poly (ADP-ribose) polymerase (PARP) inhibitor, in cultured endometrial carcinoma cells. BMC Cancer. 2014;14(1):179. doi:10.1186/1471-2407-14-179
40. Philip CA, Laskov I, Beauchamp MC, et al. Inhibition of PI3K-AKT-mTOR pathway sensitizes endometrial cancer cell lines to PARP inhibitors. BMC Cancer. 2017;17(1):638. doi:10.1186/s12885-017-3639-0
41. Liu P, Cheng H, Roberts TM, Zhao JJ. Targeting the phosphoinositide 3-kinase pathway in cancer. Nat Rev Drug Discov. 2009;8(8):627–644. doi:10.1038/nrd2926
42. Chalhoub N, Baker SJ. PTEN and the PI3-kinase pathway in cancer. Annu Rev Pathol. 2009;4(1):127–150. doi:10.1146/annurev.pathol.4.110807.092311
43. Bian X, Gao J, Luo F, et al. PTEN deficiency sensitizes endometrioid endometrial cancer to compound PARP-PI3K inhibition but not PARP inhibition as monotherapy. Oncogene. 2018;37(3):341–351. doi:10.1038/onc.2017.326
44. Samol J, Ranson M, Scott E, et al. Safety and tolerability of the poly(ADP-ribose) polymerase (PARP) inhibitor, olaparib (AZD2281) in combination with topotecan for the treatment of patients with advanced solid tumors: a Phase I study. Invest New Drugs. 2012;30(4):1493–1500. doi:10.1007/s10637-011-9682-9
45. Dean E, Middleton MR, Pwint T, et al. Phase I study to assess the safety and tolerability of olaparib in combination with bevacizumab in patients with advanced solid tumours. Br J Cancer. 2012;106(3):468–474. doi:10.1038/bjc.2011.555
46. Janzen DM, Paik DY, Rosales MA, et al. Low levels of circulating estrogen sensitize PTEN-null endometrial tumors to PARP inhibition in vivo. Mol Cancer Ther. 2013;12(12):2917–2928. doi:10.1158/1535-7163.MCT-13-0572
47. Forster MD, Dedes KJ, Sandhu S, et al. Treatment with olaparib in a patient with PTEN-deficient endometrioid endometrial cancer. Nat Rev Clin Oncol. 2011;8(5):302–306. doi:10.1038/nrclinonc.2011.42
48. Gockley AA, Kolin DL, Awtrey CS, Lindeman NI, Matulonis UA, Konstantinopoulos PA. Durable response in a woman with recurrent low-grade endometrioid endometrial cancer and a germline BRCA2 mutation treated with a PARP inhibitor. Gynecol Oncol. 2018;150(2):219–226. doi:10.1016/j.ygyno.2018.05.028
49. Goodman AM, Kato S, Bazhenova L, et al. Tumor mutational burden as an independent predictor of response to immunotherapy in diverse cancers. Mol Cancer Ther. 2017;16(11):2598–2608. doi:10.1158/1535-7163.MCT-17-0386
50. Gandara DR, Paul SM, Kowanetz M, et al. Blood-based tumor mutational burden as a predictor of clinical benefit in non-small-cell lung cancer patients treated with atezolizumab. Nat Med. 2018;24(9):1441–1448. doi:10.1038/s41591-018-0134-3
51. Le DT, Uram JN, Wang H, et al. PD-1 blockade in tumors with mismatch-repair deficiency. N Engl J Med. 2015;372(26):
52. Chalmers ZR, Connelly CF, Fabrizio D, et al. Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med. 2017;9(1):34. doi:10.1186/s13073-017-0424-2
53. Teo MY, Seier K, Ostrovnaya I, et al. Alterations in DNA damage response and repair genes as potential marker of clinical benefit from PD-1/PD-L1 blockade in advanced urothelial cancers. J Clin Oncol. 2018;36(17):1685–1694. doi:10.1200/JCO.2017.75.7740
54. Chen Q, Sun L, Chen ZJ. Regulation and function of the cGAS-STING pathway of cytosolic DNA sensing. Nat Immunol. 2016;17(10):1142–1149. doi:10.1038/ni.3558
55. Jiao S, Xia W, Yamaguchi H, et al. PARP inhibitor upregulates PD-L1 expression and enhances cancer-associated immunosuppression. Clin Cancer Res. 2017;23(14):3711–3720. doi:10.1158/1078-0432.CCR-16-3215
56. Konstantinopoulos PA, Waggoner SE, Vidal GA, et al. TOPACIO/keynote-162 (NCT02657889): a phase 1/2 study of niraparib + pembrolizumab in patients (pts) with advanced triple-negative breast cancer or recurrent ovarian cancer (ROC)—results from ROC cohort. J Clin Oncol. 2018;36(15_suppl):106. doi:10.1200/JCO.2018.36.15_suppl.106
57. Drew Y, de Jonge M, Hong SH, et al. An open-label, Phase II basket study of olaparib and durvalumab (MEDIOLA): results in germline BRCA-mutated (gBRCAm) platinum-sensitive relapsed (PSR) ovarian cancer (OC). Gynecol Oncol. 2018;149(Supplement 1):246–247. doi:10.1016/j.ygyno.2018.04.555
58. Clinicaltrials.gov. Available from: https://clinicaltrials.gov/ct2/show/NCT02208375.
59. Clinicaltrials.gov. Available from: https://clinicaltrials.gov/ct2/show/NCT02755844.
60. Clinicaltrials.gov. Available from: https://clinicaltrials.gov/ct2/show/NCT03951415.
61. Clinicaltrials.gov. Available from: https://clinicaltrials.gov/ct2/show/NCT03745950.
62. Clinicaltrials.gov. Available from: https://clinicaltrials.gov/ct2/show/NCT03570437.
63. Clinicaltrials.gov. Available from: https://clinicaltrials.gov/ct2/show/NCT02684318.
64. Clinicaltrials.gov. Available from: https://clinicaltrials.gov/ct2/show/NCT03572478.
65. Clinicaltrials.gov. Available from: https://clinicaltrials.gov/ct2/show/NCT03552471.
66. Clinicaltrials.gov. Available from: https://clinicaltrials.gov/ct2/show/NCT03617679.
67. Chan N, Pires IM, Bencokova Z, et al. Contextual synthetic lethality of cancer cell kill based on the tumor microenvironment. Cancer Res. 2010;70(20):8045–8054. doi:10.1158/0008-5472.CAN-10-2352
68. Clinicaltrials.gov. Available from: https://clinicaltrials.gov/ct2/show/NCT03694262.
69. Clinicaltrials.gov. Available from: https://clinicaltrials.gov/ct2/show/NCT03476798.
70. Clinicaltrials.gov. Available from: https://clinicaltrials.gov/ct2/show/NCT03586661.
71. Clinicaltrials.gov. Available from: https://clinicaltrials.gov/ct2/show/NCT03016338.
72. Clinicaltrials.gov. Available from: https://clinicaltrials.gov/ct2/show/NCT04080284.
73. Clinicaltrials.gov. Available from: https://clinicaltrials.gov/ct2/show/NCT03968406.
74. Clinicaltrials.gov. Available from: https://clinicaltrials.gov/ct2/show/NCT02912572.
75. Tomao F, Musacchio L, Di Mauro F, et al. Is BRCA mutational status a predictor of platinum-based chemotherapy related hematologic toxicity in high-grade serous ovarian cancer patients? Gynecol Oncol. 2019;154(1):138–143. doi:10.1016/j.ygyno.2019.04.009
76. Moore K, Colombo N, Scambia G, et al. Maintenance olaparib in patients with newly diagnosed advanced ovarian cancer. N Engl J Med. 2018;379(26):2495–2505. doi:10.1056/NEJMoa1810858
77. Pujade-Lauraine E, Ledermann JA, Selle F, et al. Olaparib tablets as maintenance therapy in patients with platinum-sensitive, relapsed ovarian cancer and a BRCA1/2 mutation (SOLO2/ENGOT-Ov21): a double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Oncol. 2017;18(9):1274–1284. doi:10.1016/S1470-2045(17)30469-2
78. Ray-Coquard I, Pautier P, Pignata S, et al. Olaparib plus bevacizumab as first-line maintenance in ovarian cancer. N Engl J Med. 2019;381(25):2416–2428. doi:10.1056/NEJMoa1911361
79. McEllin B, Camacho CV, Mukherjee B, et al. PTEN loss compromises homologous recombination repair in astrocytes: implications for glioblastoma therapy with temozolomide or poly(ADP-ribose) polymerase inhibitors. Cancer Res. 2010;70(13):5457–5464. doi:10.1158/0008-5472.CAN-09-4295
80. Fraser M, Zhao H, Luoto KR, et al. PTEN deletion in prostate cancer cells does not associate with loss of RAD51 function: implications for radiotherapy and chemotherapy. Clin Cancer Res. 2012;18(4):1015–1027. doi:10.1158/1078-0432.CCR-11-2189
81. Kechagioglou P, Papi RM, Provatopoulou X, et al. Tumor suppressor PTEN in breast cancer: heterozygosity, mutations and protein expression. Anticancer Res. 2014;34(3):1387–1400.
82. Mendes-Pereira AM, Martin SA, Brough R, et al. Synthetic lethal targeting of PTEN mutant cells with PARP inhibitors. EMBO Mol Med. 2009;1(6–7):315–322. doi:10.1002/emmm.200900041
83. Ring KL, Bruegl AS, Allen BA, et al. Germline multi-gene hereditary cancer panel testing in an unselected endometrial cancer cohort. Mod Pathol. 2016;29(11):1381–1389. doi:10.1038/modpathol.2016.135
84. Susse S, Janz C, Janus F, Deppert W, Wiesmuller L. Role of heteroduplex joints in the functional interactions between human Rad51 and wild-type p53. Oncogene. 2000;19(39):4500–4512. doi:10.1038/sj.onc.1203809
85. Buchhop S, Gibson MK, Wang XW, Wagner P, Stürzbecher HW, Harris CC. Interaction of p53 with the human Rad51 protein. Nucleic Acids Res. 1997;25(19):3868–3874. doi:10.1093/nar/25.19.3868
86. Menon V, Povirk L. Involvement of p53 in the repair of DNA double strand breaks: multifaceted roles of p53 in homologous recombination repair (HRR) and non-homologous end joining (NHEJ). Subcell Biochem. 2014;85:321–336.
87. Xu Y. DNA damage: a trigger of innate immunity but a requirement for adaptive immune homeostasis. Nat Rev Immunol. 2006;6(4):261–270. doi:10.1038/nri1804
88. Thottassery JV, Zambetti GP, Arimori K, Schuetz EG, Schuetz JD. p53-dependent regulation of MDR1 gene expression causes selective resistance to chemotherapeutic agents. Proc Natl Acad Sci U S A. 1997;94(20):11037–11042. doi:10.1073/pnas.94.20.11037
89. Sampath J, Sun D, Kidd VJ, et al. Mutant p53 cooperates with ETS and selectively up-regulates human MDR1 not MRP1. J Biol Chem. 2001;276(42):39359–39367. doi:10.1074/jbc.M103429200
90. Roemer K. Mutant p53: gain-of-function oncoproteins and wild-type p53 inactivators. Biol Chem. 1999;380(7–8):879–887. doi:10.1515/BC.1999.108
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.