Back to Archived Journals » Advances and Applications in Bioinformatics and Chemistry » Volume 8
Parallel computing in genomic research: advances and applications
Authors Ocaña K, de Oliveira D
Received 1 July 2015
Accepted for publication 21 September 2015
Published 13 November 2015 Volume 2015:8 Pages 23—35
DOI https://doi.org/10.2147/AABC.S64482
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Juan Fernandez-Recio
Kary Ocaña,1 Daniel de Oliveira2
1National Laboratory of Scientific Computing, Petrópolis, Rio de Janeiro, 2Institute of Computing, Fluminense Federal University, Niterói, Brazil
Abstract: Today's genomic experiments have to process the so-called "biological big data" that is now reaching the size of Terabytes and Petabytes. To process this huge amount of data, scientists may require weeks or months if they use their own workstations. Parallelism techniques and high-performance computing (HPC) environments can be applied for reducing the total processing time and to ease the management, treatment, and analyses of this data. However, running bioinformatics experiments in HPC environments such as clouds, grids, clusters, and graphics processing unit requires the expertise from scientists to integrate computational, biological, and mathematical techniques and technologies. Several solutions have already been proposed to allow scientists for processing their genomic experiments using HPC capabilities and parallelism techniques. This article brings a systematic review of literature that surveys the most recently published research involving genomics and parallel computing. Our objective is to gather the main characteristics, benefits, and challenges that can be considered by scientists when running their genomic experiments to benefit from parallelism techniques and HPC capabilities.
Keywords: high-performance computing, genomic research, cloud computing, grid computing, cluster computing, parallel computing
Introduction
Bioinformatics is a multidisciplinary field that is in constant evolution due to technological advances in correlated sciences (eg, computer science, biology, mathematical, chemistry, and medicine).1 Thus, it requires skills from these domains of sciences for modeling, gathering, storing, manipulating, analyzing, and interpreting biological information, ie, “biological big data”.2 Since biological big data is generated by several different bioinformatics/biological/biomedical experiments, it can be presented as structured or unstructured data. Due to the complexity in the nature of the biological big data, a shift to discovery-driven data science is under way, especially in the genomic field.2,3
Genomic research is the most representative domain in bioinformatics, as it is the initial step of several types of experiments and it is also required in several other bioinformatics fields. It compares genomic features – DNA sequences, genes, regulatory sequences, or other genomic structural components – of different organisms. In general, comparative genomics starts with the alignment of genomic orthologues sequences (ie, sequences that share a common ancestry) for checking the level of similarity (conservation) among sequences (or genomes). Then evolutionary inferences can be performed over these results to infer, for example, the phylogenetic relationships or population genetics.4 Up-to-date, comparative genomics needs to process biological big data, then it expanded their experiments at large-scale scenarios (increasing both, the amount and complexity of data or tasks). For this reason, it makes extensive use of novel techniques, technologies, and specialized computing infrastructure to make possible the managing and parallel processing for comparing several available genomes (maybe hundreds or thousands of whole genomes).
Due to the increase of the number of experiments (that are also becoming more complex) involving genomic research as well as the advances of DNA sequencing technologies (ie, the next-generation sequencing [NGS]5 methods), the amount and complexity of biological data is being increased. It directly affects the performance of the computational execution of bioinformatics experiments. Due to the aforementioned huge volume of produced data, it is almost impossible to process all data in an ordinary desktop machine in standalone executions.
Scientists need to use high-performance computing (HPC) environments together with parallelism techniques to process all the produced data in a feasible time. Several large-scale bioinformatics projects already benefit from parallelism techniques in HPC infrastructures as clusters, grids, graphics processing units, and clouds.6–8 In this scenario, bioinformatics provides interesting opportunities for research in HPC applications for the next years. Some vast, rich, and complex bioinformatics areas related to genomics can also benefit from HPC infrastructures and parallel techniques, such as the NGS, proteomics, transcriptomics, metagenomics, and structural bioinformatics.1
However, integrating biological and bioinformatics experiments with parallel techniques and HPC environments is far from simple. One strategy could focus on redesigning bioinformatics applications (eg, FASTA, BLAST, HMMER, ClustalW, and RAxML) to their parallel versions (using MPI or MPJ,9 for instance). A second strategy can be related to the development of pipelines for bioinformatics, which are mainly conceptualized to automate the process. These pipelines can also be represented as scientific workflows, managed with scientific workflow systems.10 Their executions can benefit from the use of HPC environments (eg, clusters, grids, or clouds). For instance, cloud computing is an interesting strategy emerging as a solution applied in several bioinformatics areas. Tavaxy,11 Pegasus,12 Swift/T,13 and SciCumulus14 are some examples of scientific workflow systems that are able to manage bioinformatics experiments in cloud infrastructures.
The amount of published scientific articles evidences that the use of parallel computing in genomic research has emerged as a viable and interesting solution that is being already adopted by many projects. Several technologies, techniques, systems, platforms, applications, infrastructures, and standard protocols have been already proposed. Considering the huge interest on this area, we present in this article a systematic review of literature (SRL) for parallel computing applied to the genomic field.
The main objective of this review is to present and discuss about the main solutions that were implemented to achieve improvements in the execution times in bioinformatics analyses based on distributed and parallel approaches. It may serve as a guideline for other data analysis projects in bioinformatics and computer science using infrastructures and concepts integrating HPC, big data and in large-scale bioinformatics overview. The authors believe that this article will be useful to the scientific community in future work to compare different approaches that provide parallel computing capabilities for genomic experiments.
Systematic review of literature
The SRL is an interesting way for designing systematic reviews, as we are focusing at identifying, evaluating, and comparing available published articles associated to a particular topic area of interest for answering a specific scientific question.15 SRL follows a protocol that allows replicating studies for other researchers. As proposed by Kitchenham et al,15 a SRL has three main phases: i) planning, ii) conduction, and iii) analysis of results. In the planning phase, we must have a clear goal of our research since here is set the protocol that will be followed in the conduction phase.
We follow the PRISMA statement16 to develop the systematic review based on qualitative and quantitative synthesis to analyze research articles of interest. PRISMA stands for preferred reporting items for systematic reviews and metaanalyses and was efficiently used for structuring our SRL, and define the methodological strategy followed in our research, particularly to sustain the evaluation and critical appraisal about the publications elected to be included in this article.
In the context of this article, we prepared two research questions that should be answering for concluding our research:
- RQ1: What approaches provide HPC capabilities for genomic analysis?
- RQ2: Which parallel techniques coupled to those approaches provide HPC capabilities?
Therefore, our search strategy consisted of identifying approaches in published articles that cover main concepts (or terms) related to genomic researches, HPC, and parallel and distributed techniques.
Here, we define the search string (as presented in Figure 1) used for conducting our search strategy in existing electronic databases (summarized in Table 1) for the scientific literature search.
 | Figure 1 Defined search string with genomics and HPC-related key terms. |
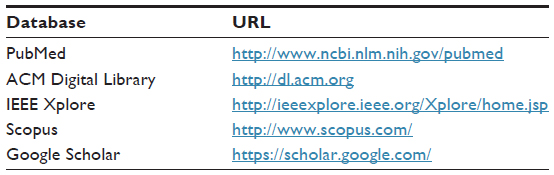 | Table 1 Electronic scientific databases selected as sources |
We used the logical operator “AND” to connect the key terms (ie, genomic research and HPC) and the “OR” operator to connect the possible variations derived from any key terms. Then, the search string defined in Figure 1 was used for querying a set of existing electronic databases as presented in Table 1. Five electronic databases were selected based on the following criteria: i) the publication of articles is regularly updated, ii) all articles are available for download and analysis, and iii) all articles are revised using a peer-review process.
Many of the returned articles were considered as irrelevant for the goals proposed in this research. So we defined two additional criteria (inclusion/exclusion) to include articles in our research. The criterion for inclusion refers to the study presented in the article, which must involve both genomic and parallel computing terms. For instance, two articles can present the same research, but only the latest published article would be considered. For excluding articles (at the exclusion criterion), we consider the following topics: i) articles must be available for reading on the Internet, ii) articles must be presented in electronic format, and iii) articles should be written in English. Then, we searched the literature by applying these inclusion/exclusion criteria and by techniques of hand-searching key journals, to identify the existing approaches to qualitative meta-synthesis.
Once our protocol was defined, we conducted the SRL between April and May 2015. Initially, the record or the number of citations returned by searching the key terms defined in Figure 1 was 8,090. This number of records was reduced to 7,900 by eliminating records that were duplicated in searching or, for example, those that were not written in English. We reduced drastically the records (from 7,900 to 329 and finally to 303) as we detected that many articles following the key terms “genomic” or “genome” were related to in vivo or in vitro experiment and not to in silico (bioinformatics) experiments (this was performed by a in house script). As we are interested in determining the correlation between the genomic and parallel computing terms, only records for in silico (bioinformatics-genomics) and HPC-related key terms were included in our analyzes. The final and refined reduction (303–30 articles) was done according to the exclusion criterion: 1) about the availability of articles (ie, free full-text articles were included) and 2) the exclusion of articles that present very similar researches (often belonging to the same research group), in which only the article published in the most important journal (eg, with the highest impact factor) was included.
Finally, 30 articles were found in our SLR, as detailed in Table 2. Interestingly, we observed that those articles were published in the last 8 years (since 2008), and that they were defined in three distinct approaches of qualitative synthesis (Table 2, column “Type of study”): research articles, methodology articles, and software articles.
 | Table 2 Main information about publications related to genomics and parallel computing |
In the next section, we discuss the most relevant articles returned by our aforementioned methodology in order to compare existing approaches that combine genomic and parallel computing researches.
Analysis of the selected articles
This section details the results (articles) of the conducted SRL method used to evaluate research obtained by querying the electronic databases presented in Table 1. Table 2 summarizes information about the articles returned by the key terms: genomics, HPC, and parallel and distributed computing and following authors include a discussion about these selected articles.
Table 2 was organized following the classification of articles returned by our SRL protocol based on both columns: “Execution main finding” and “Bioinformatics field”. For “main finding”, the articles were sorted by three classification types of the HPC approaches used in the referred article’s methodology: SE for standalone/serial execution, PT for parallel techniques, and HPC for high-performance computing techniques. On the other hand, “Bioinformatics Field” returned the classification of articles following the main bioinformatics areas explored in the articles: comparative genomics, phylogenomics, transcriptomics, homology modeling, proteomics, and evolutionary genomics.
Other columns of Table 2 are the following: publication source, type of study, year, and research country that respectively indicate the name of the journal, the type of research/study used in articles, the year of publication and the country of the research. The column type of study return three classification for the published articles: MA for methodology article; SA: software article; RA: research article. The HPC infrastructure column present the environment used for the experiment execution in the article (ie, network PC, clusters, grids, and cloud) or the workflow management system (if used) (eg, Hadoop, Galaxy, and SciCumulus).
Bernardes et al17 proposed an improved method for the construction of profiles hidden Markov models (pHMMs) for detecting remote (or distant) homologous sequences using structural alignments. Remote homologous is a type of sequence that presents very low level of similarity or identity in regions (mainly 20% of conservation in multiple sequence alignment, also called the “midnight zone”) and traditional methods of detection such as BLAST or FASTA are not able to detect this very low level of conservation within sequences (ie, in genes or genomes). One alternative for increasing the specificity and sensibility of detecting true positives of remote homologous sequences is using three-dimensional (3D) methodologies of alignment and searching. Then the authors compared the performance of structural and sequence pHMM programs at detecting remote homologous sequences. Bernardes et al’s method was executed in parallel in an in-house cluster.
AMPHORA18 is a pipeline for phylogenomic analyses designed to automate sequential executions. Several of the most popular bioinformatics tools are available to be used with AMPHORA, for example, multiple sequence alignment (MSA) tools such as ClustalW or Muscle; orthologs searching tools such as BLAST or HMMER and phylogenomic construction trees with RAxML. In the article, authors show that AMPHORA is scalable and efficient in HPC environments by constructing a phylogenomic tree composed by 578 bacterial species and by assigning phylotypes to 18,607 markers of metagenomic data collected from the Sargasso Sea. However, as reported by the authors, the execution presented in the article was performed in desktop machines with multiple processors instead of using HPC environments such as grids or clusters.
Ahmed et al19 present a genomic analysis focused on the comparison of several assembly genome approaches in HPC scenarios. Nowadays, performing assembly genomes executions in a feasible time is an open, yet important, challenge for bioinformaticians. The reason is that the assembly of large size genomes is considered as a very computing intensive process, consuming up to weeks or months (eg, in eukaryotic complex genomes) of total processing time. Due to that, several sequential assemblers that perform execution in a feasible time (ie, diminishing the total execution time) have been proposed to assist in the process of the genome assembly. However, a few algorithms efficiently parallelize the assembly process to speed up the required processing time, then very little has been done to investigate how to use parallel algorithms and metrics of parallel computing paradigm of assembly genomes to ascertain their scalability and efficiency.
The Java-based approach names Hadoop-BAM20 aims at manipulating the several formats of files used in most of the several bioinformatics experiments (ie, NGS). Hadoop-BAM coupled to the traditional Hadoop framework the well-known and popularly used applications Picard and SAMtool. The file formats that are supported Hadoop-BAM are BAM, SAM, FASTQ, FASTA, QSEQ, BCF, and VCF. A disadvantage of using Hadoop-BAM is that the command line tools, which should be friendly and understandable to users, are limited in scope and hard-to-use by scientists with no expertise in the use of Hadoop. In addition, depending on the version used of Hadoop, the performance of Hadoop-BAM can be affected since Hadoop presents some limitations, especially when the analysis has very short maps and reduces invocations.
Blom et al21 propose EDGAR, an approach for executing comparative analysis of prokaryotic genomes. EDGAR is designed to analyze the produced information (ie, similarities or differences) obtained from genomic comparisons. EDGAR’s implementation is based on a three-layer architecture, the core of EDGAR is implemented in Perl, all data is stored in a SQLite database, and the user interface is implemented using JavaScript. As input datasets, EDGAR needs related genomes in multi-fasta format files to be consumed. In addition, EDGAR needs the National Center for Biotechnology Information protein table and BLAST database to execute the genomic comparisons. EDGAR is also able to generate phylogenetic trees. Since this task is computing intensive, EDGAR is designed to be executed in computing clusters. In the article, authors state that EDGAR was evaluated by performing an all-against-all comparison against ten genomes of Xanthomonas in a local cluster using the Sun Grid Engine. Although this approach is useful for several purposes, it is limited from the scalability perspective. Since EDGAR is designed to execute only on computing clusters, it cannot benefit from other infrastructures such as grids or clouds, unless important adaptations are performed.
Armadillo22 is an open-source workflow system designed for modeling and executing phylogenomic analyses. It allows for scientists to develop their own application, that authors named as modules (considered in other articles as tasks or activities45) and adding them to the structure of a workflow, thus creating new and complex genomic analyses. Differently from general-purpose workflow systems such as Pegasus and Swift/T, Armadillo is focused on providing facilities for bioinformaticians such as allowing them to interconnect a set of existing bioinformatics applications with others in a dataflow, thus easing the effective use of Armadillo for scientists with no computational expertise. The bioinformatics applications that are already provided by Armadillo are MSA such as ProbCons; searching homologous sequences using BLAST; testing evolutionary model search with ProtTest; building phylogenetic tree using the neighbor-joining algorithm with PHYLIP or the maximum likelihood (ML) algorithm with PhyML; and other evolutionary inferences with PAML. Nevertheless, no information is presented about how Armadillo was coupled to HPC infrastructures (cloud, grid, or cluster) to parallelize these executions.
Severin et al23 propose eHive, a distributed system, to support comparative genomic analyses modeled as scientific workflows. The eHive system is composed of three different workflows that can be executed by the scientists: i) a workflow that executes the pairwise whole genome alignments, ii) a workflow that executes the multiple whole genome alignments, and iii) a workflow that executes the gene trees with protein homology inference. The eHive relies on a MySQL database to store all data consumed and produced by the dataflows. The modeled workflows can be parallelized, and since they consume several fasta files as input, the content of each file can be also processed in parallel. Authors showed that eHive is more efficient than the existing job scheduling systems, such as Portable Batch System,46 that are based on central job queues, which may become a bottleneck in some cases. Besides this performance advantage, another important advantage of eHive is that scientists are able to modify the structure of the workflow during the execution course of the analysis. Scientists are able to create new jobs during the execution (by providing more data), but they can also change programs that are part of the pipeline, add/remove control rules, etc. The main drawback of eHive is the use of the MySQL database since MySQL presents severe overheads when it has several concurrent accesses. The eHive was evaluated using the Sun Grid Engine47 and Portable Batch System.
Tavaxy11 is a system for modeling and executing bioinformatics workflows based on the integration of the Taverna and Galaxy workflow systems. Tavaxy supports execution in a single (sequential) environment or in clouds. It offers a set of new features that simplify and enhance the development of sequence analysis applications, covering several areas of bioinformatics as NGS, assembly, sequence analysis, metagenomics, proteomics, or comparative genomics. The focus of Tavaxy is facilitating the efficient execution of bioinformatics analysis tasks on HPC infrastructures and cloud computing systems. Tavaxy can be downloaded or directly used as a service in clouds (http://www.tavaxy.org).
Bioconductor24 is a software project that integrates more than 1,024 software packages, 887 annotation packages, and 241 experimental data packages, covering the main areas in bioinformatics experiments. Inside the classification of “software packages”, the “research field” shows the areas of bioinformatics that are covered by Bioconductor. They are biomedical information, cell biology, chemioinformatics, functional genomics, genetics, lipidomics, metabolomics, metagenomics, pharmacogenetics, pharmacogenomics, proteomics, and system biology. Summarizing, the packages in Bioconductor follows several organization and scientists need to decide which area or packages can be adapted better to their own experiment. Bioconductor aims for supporting scientists at analyzing and for the better comprehension of high-throughput data in genomics and molecular biology, but other areas of bioinformatics are also covered such as phylogeny, proteomics, NGS, transcriptomics, RNA-differential analyses, and several statistics analysis for bioinformatics. The project aims to enable interdisciplinary research, collaboration, and rapid development of scientific software. Bioconductor is based on the statistical programming language R and the several interoperable packages contributed by a large, diverse community of scientists. Packages cover a range of bioinformatics and statistical applications. Bioconductor packages can be downloaded at http://www.bioconductor.org/ or it is also available as an AMI (Amazon Machine Image) and a series of Docker images.
A fundamental problem in bioinformatics is genome assembly due its computing intensive execution requirements. As NGS technologies produce huge quantity of volumes of fragmented genome reads, large amounts of memory is required to assemble the complete genome efficiently. Kleftogiannis et al25 compare current memory-efficient techniques for genome assembly with respect to quality, memory consumption, and execution time. Then by combining existing methodologies, they propose two general assembly strategies that can improve short-read assembly approaches and results in reduction of the memory footprint. They are the following: i) Diginorm-MSP-Assembly and ii) Zeromemory assembly. Finally, they implement the genome assembly experiment in Amazon Elastic Compute Cloud (Amazon EC2) cloud infrastructure and discuss about the several characteristics in performance of each of the assembly application used in the methodology of the article and the benefits of using clouds for parallelizing the execution.
Proteomics-related experiments are considered as high computational complexity tasks and the implementation details of the parallelized algorithms of these methods as well as their computational performance have not been provided. Enumeration of all amino acid compositions is an important step and computationally expensive task in several proteomics workflows, including peptide mass fingerprinting, mass defect labeling, mass defect filtering, and de novo peptide sequencing. Nefedov and Sadygov26 present a parallel method for enumerating all amino acid compositions up to a given length and discuss about the computational times for their proposed method, which was executed on a HPC cluster computer. As the authors reported, this is the first detailed description of a computational method for a complete and unbiased enumeration of all theoretically possible peptides. They demonstrated that the parallelization of this type of tasks can be improved at using HPC infrastructures and may be significantly improved and extended to other several proteomics studies. Ongoing works are related and explore the accuracy of protein identification in real mass spectrometry data. The software is available for download at https://ispace.utmb.edu/users/rgsadygo/Proteomics/ParallelMethod.
Yabi27 is a workflow system that is focused on deploying scientific analyses modeled as workflows in several HPC resources in a transparent form. The idea behind Yabi is to allow for scientists to focus on science instead of managing a complex HPC environment. Yabi allows for scientists to model their workflow using a huge set of applications (including their own code) and then save the modeled workflow for a posteriori reuse. Although Yabi was designed for general-purpose usage (ie, it can be applied in a variety of domains), it is mostly used by the genomic community since its Web-based environment and drag-and-drop tools are almost mandatory in bioinformatics experiments. Yabi is able to execute genomic analyses in compute clusters, grids, and clouds, and it was evaluated using several bioinformatics/biomedical experiments as cases of study, such as analyses from genomics, transcriptomics, and proteomics (ie, using their respective related programs Repeatmasker, genscan, MzXML2Search, Peptide Prophet, and Mascot).
The Crossbow28 tool was designed for identifying single nucleotide polymorphisms in whole-genome sequencing (WGS) data, based on the real need of predicting the occurrence of diseases in patients. Crossbow is specialized in alignment and variant-calling activities, and it is composed of the applications Bowtie (ie, aligner) and SOAPsnp (ie, genotyper), which are invoked in a coherent flow designed to perform several different analyses. Crossbow is based on Hadoop,48,49 which means it is able to execute genomic analyses in both clusters and clouds. However, as Crossbow presents limitations for large-scale WGS projects related to data management issues and scalability issues, Rainbow was proposed.29 Rainbow is an open-source, scalable, and cloud-based system that allows for the automation of large-scale WGS experiments. The main advantages of Rainbow is that it is able to handle BAM and FASTQ file types; to split large sequence files and to log performance metrics related to processing and monitoring data using multiple virtual machines in Amazon EC2 cloud, thus allowing for Rainbow to improve the performance based on past collected results.
As genomic data analysis in evolutionary biology is becoming so computing intensive, several techniques for scaling computations through parallelization of calculations and advanced programming techniques were discussed. BioNode30 shows how a bioinformatics workflow can be effectively modeled and executed into virtual machines in a virtual cluster in different cloud environments. BioNode is based on Debian Linux and can run both on personal computers in a local network and in the cloud. Approximately 200 bioinformatics programs closely related to biological evolutionary experiments are included. Examples of representative software included in BioNode are PAML, Muscle, MAFFT, MrBayes, and BLAST. In addition, BioNode configuration allows for those scripts to parallelize these aforementioned bioinformatics software. BioNode supports designing and open-sourcing virtual machine images for the community. BioNode can be deployed on several operating systems (Windows, OSX, Linux), architectures, and in the cloud.
Dong et al31 propose a prediction and analysis tool named ProteinSPA, which employs a specific protein structure prediction workflow designed to be executed in grid environments that integrates several bioinformatics tools in parallel. The parallelism is needed since protein structure prediction is considered as a very computing intensive task. The ProteinSPA tool is mainly based on mpiBLAST, which allows for parallel execution. It can be deployed both on clusters and on grids.
Bionimbus32 is an open source and cloud-based system used by a variety of genomic experiments. Bionimbus is based on OpenStack, and it aims at creating virtual machines in the cloud on demand, depending on the need of the experiment. Bionimbus presents the portal called Tukey that acts as a single entry point for various resources available in Bionimbus. The authors used an acute myeloid leukemia-sequencing project as case study for testing Bionimbus. Bionimbus provides several applications for quality control, alignment, variant calling, and annotation and also an infrastructure that supports large-scale executions. For example, each simple input data generates BAM files with sizes ranged between 5 and 10 GB and the alignment step requires eight central processing units for approximately 12 hours. Bionimbus also offers a community cloud that contains a set of several public biological datasets, including the 1,000 genomes biological database.
Singh et al33 present a computational infrastructure for grids which accelerates the execution bioinformatics experiments that are computing intensive. The infrastructure is based on a hybrid computing model that provides two different types of parallelism: one that is based on volunteer computing infrastructures (eg, peer-to-peer network) and another that uses graphical processing units for fast sequence alignment. The case of study presented in this article evaluates all-against-all genomic comparisons between a set of microbial organisms, ie, each gene from a genome is compared to all genes from the other genomes. Then, the phenotype–genotype explorer PheGee33 was used to analyze results, ie, linking the candidate genes supposed to be responsible for a given phenotype. The Smith–Waterman algorithm13 was the chosen methodology to perform the pairwise alignment of the gene sequences since it shows better sensitivity values for low-scoring alignment14 than faster traditional algorithms such as BLAST.15
iTree34 is a pipeline for automating phylogenomic analysis executions. It was designed to be executed in parallel in grid environments using multi-threaded programming. iTree addresses aims at easing the installation and setting up of the environment, the choice of the reference dataset, and other features related to the experimental processing of some bioinformatics algorithms and applications as MSA and phylogenetic tree building. Nevertheless, iTtree does not provide information about large-scale executions in clouds or in clusters.
El-Kalioby et al35 propose a software package named elasticHPC that aims at easing the daily duties of scientists that need HPC capabilities to run their experiments. The main idea behind elasticHPC is to provide a variety of resources in the cloud and in each resource, and then a set of applications would be already deployed. For example, we may find a virtual machine in the cloud where sequence analysis tools such as BLAST are already installed and ready for use. Then, as clouds provide the pay-as-you-go model for the execution, scientists will pay only for the time required for executing their experiments. This approach is very similar to the CloudBioLinux, but the main difference is that elasticHPC allows for horizontal and vertical scaling of the environment, thus benefiting from the elasticity characteristic of clouds.
Reid et al36 propose the workflow Mercury for comparative genomic analysis. Mercury is composed of the following main activities: 1) generation of sequence reads and base call confidence values from sequencing raw data; 2) processing and mapping reads against a reference genome with BWA, thus producing a BAM file; 3) merging individual BAM files for variant calling; 4) identifying variants with Atlas-SNP and Atlas-indel for producing variant files (VCF); and 5) annotation of biological and functional information contained into the variant lists and then formatting for publication. Mercury can be efficiently deployed in local machines or in cloud environments (eg, Amazon EC2) using the DNAnexus platform. The main idea is that scientists are able to instantiate as many virtual machines as they need to process the workflow in parallel.
Minevich et al37 propose CloudMap, a pipeline that aims at simplifying the analysis of mutant genome sequences, allowing scientists to identify genetic differences (or sequence variations) among individuals. CloudMap is composed of “template” workflows and implemented using the Galaxy workflow management system. Then, CloudMap can be executed in the scientists’ desktops or in the cloud, specifically in the Amazon EC2 cloud. Authors demonstrated the effectiveness of CloudMap for WGS analysis of Caenorhabditis elegans and Arabidopsis genomes. The advantage of CloudMap basically is associated with its implementation in the traditional workflow systems as Galaxy. Then, it benefits from the advantages provided by this workflow system, for example, the ability to create virtual machines in the cloud providing parallelism and distribution of executions. However, Galaxy presents a limited scalability in comparison with other workflow systems such as Swift/T or Pegasus.
Wall et al proposed the pipeline Roundup6 that is modeled and implemented on top of the Hadoop framework48 and designed to be deployed in Amazon EC2 clouds. Roundup improves the parallelism of the comparative genomic algorithm called reciprocal smallest distance. Roundup orchestrates the execution of programs and packages that aim at comparing whole genomes and reconstructing the evolutionary relationships. Roundup uses BLAST for all-in-all comparisons, ClustalW for constructing MSA, PAML for the ML estimation of the of evolutionary distance and Python scripts that intermediate several processes, for example, format conversion, etc. The main idea behind this article is to show how cloud computing can be more interesting from the economic perspective than local computing infrastructures such as clusters or grids. The authors showed that although clouds present several disadvantages as pointed by Armbrust et al,7 they represent an interesting alternative to providing parallel capabilities for comparative genomic experiments. The use of Hadoop by the authors is the main advantage and disadvantage of the approach at the same time. The advantage is that scientists did not require designing solutions for scheduling, fault-tolerance, etc. However, as stated by Ding et al,50 Hadoop presents severe overheads, mainly when the experiment presents short tasks.
Krampis et al38 propose the use of virtual machines on cloud infrastructures as an alternative to in-house architectures, ie, small clusters. The proposed approach CloudBioLinux38 offers an analysis framework for executing genomic experiments in cloud computing platforms. The idea behind CloudBioLinux is not to propose an experiment for genomic analysis. Instead, it provides the necessary infrastructure for scientists to run their experiments. The virtual machine image created for CloudBioLinux contains a set of bioinformatics applications (more than 135) for constructing MSA, clustering, assembly, display and editing, and phylogenetic analyzes. CloudBioLinux was initially designed to run in the Amazon EC2, but authors have already tested it on a private Eucalyptus cloud installed at their research center. Scientists are allowed for accessing a huge variety of computational resources to execute their analysis sequentially or in parallel.
Finally, we presented a set of bioinformatics scientific workflows proposed by our research group build on top of the scientific workflow management system SciCumulus14 and deployed on the Amazon EC2 cloud. The main goal covered by these workflows is to allow scientists to design/execute their bioinformatics experiments in clouds, also analyzing the provenance data (at runtime) by querying the provenance database of SciCumulus. The scientific workflows are SciHmm, SciPhy, SciPhylomics, SciEvol, SciDock, and SciSamma, which will be presented in more details as follows.
SciHmm39 is a scientific workflow for comparative genomics build on top of SciCumulus scientific workflow engine and deployed on Amazon EC2 cloud. It aims at identifying homologous sequences by constructing/applying pHMMs using the HMMER package. Therefore, it is possible to obtain some interesting parameters and bioinformatics information that can be further used in a posteriori phylogenetic/evolutionary experiments, as the best MSA method (based on the quality of the MSA or trees obtained and the computational time required) and the e-value. SciHmm is composed of five main activities: i) MSA construction (using MAFFT), ii) pHMM build (using HMMER-hmmbuild), iii) pHMM search (using HMMER-hmmsearch), iv) cross-validation procedure that uses a leave-one-multifasta-out algorithm (using Perl scripts), and v) Receiver-Operating Characteristic curves generation (using Perl scripts).
SciPhy40 is a scientific workflow for phylogenetic analyses. It aims at constructing ML phylogenetic trees using the RAxML program. SciPhy is composed of four activities: i) MSA construction (using MAFFT), ii) MSA format conversion (using ReadSeq), iii) search for the best evolutionary model (using ModelGenerator), and iv) phylogenetic trees build (using RAxML).
SciPhylomics41 is a scientific workflow for phylogenomic analyses. It aims at constructing ML phylogenomic trees using the RAxML program. SciPhylomics is composed of nine activities: the first four activities belong to SciPhy and the following are specific from SciPhylomics. Thus, we considered SciPhy as the SciPhylomics’ sub-workflow. After the execution of the SciPhy sub-workflow and with the phylogenetic trees in hand, the following activities are executed: v) the data quality analysis that filters results based on the given quality criteria informed by scientists, vi) the MSA concatenation that generates superalignments (using Perl scripts), vii) the evolutionary model election (using Perl scripts), viii) the phylogenomic trees construction (using RAxML), and ix) the phylogenomic tree election (using Perl scripts). At the end of the execution, one or more phylogenetic and phylogenomic trees are generated.
SciEvol42 is a scientific workflow for molecular evolutionary analyses build on top of SciCumulus and deployed on Amazon Web Services. It aims at detecting positive Darwinian selection on genomic data, ie, determining the selective pressure (positive, negative, or neutral) is being exerted in biological sequences. SciEvol is composed of eleven activities: i) fasta file preprocessing for stop codons removal (using Perl scripts); ii) MSA construction (using MAFFT); iii) MSA format conversion to the PHYLIP format (using ReadSeq); iv) phylogenetic tree construction (using RAxML); v–x) evolutionary analysis execution that executes six codon substitution models (using codeml for M0, M1, M2, M3, M7, M8), and xi) evolutionary data analysis (using Perl scripts) that applies the likelihood ratio test on nested models (M0 vs M3; M1 vs M2; M7 vs M8) and reports statistical results.
SciDock43 is a scientific workflow for molecular docking-based virtual screening analyses build on top of SciCumulus and deployed on Amazon Web Services. It executes/manages molecular docking data-intensive experiments for discovering novel drug targets using AutoDock and AutoDock Vina, as they are the most popular and frequently used tools for docking. SciDock is composed by eight activities: i) ligand transformation from SDF to Sybyl Mol2 (using Babel); ii) ligand preparation (using MGLTools); iii) receptor preparation (using MGLTools); iv) AutoGrid’s parameter preparation (using MGLTools); v) receptor’s coordinates map generation (using AutoGrid), vi) docking filter (Perl scripts), vii) docking parameter preparation (using MGLTools), and viii) docking execution (using AutoDock or AutoDock Vina).
SciSamma44 is a scientific workflow for structural approach and molecular modeling analyses (ie, homology modeling analyses) build on top of SciCumulus and deployed on Amazon Web Services. It aims at predicting 3D models from a biological sequence in order to discover new drugs. SciSamma is composed of eight activities: i) homologous detection (using BLAST), ii) template election (using Perl Script) that extracts important information from the protein data bank file, iii) alignment construction (using MAFFT) that aligns the target sequence with the template, iv) model building (using MODELLER) that build the target sequence’ 3D-model based on the template structure/alignment, v) best model selection (using Perl script) that elects the best protein data bank model with the lowest value of the DOPE assessment score, vi) model refining (using MODELLER), vii) model prediction (using MODELLER), and vii) model evaluation (using MODELLER).
Analyzing the presented articles and researches, we can state that the association of genomic research and parallel computing is a fertile field. Different genomic applications of different genomic fields are applied in different HPC environments. To summarize the presented approaches, Table 2 shows the main characteristics of each of the aforementioned articles.
Discussion
This article focuses on presenting the characteristics of the existing approaches that focuses on comparative genomic techniques that are supported by parallel computing and HPC environments. The increase in genomic research projects is a direct result of advances of DNA sequencing technologies (eg, NGS). Likewise, the amount and complexity of biological data is continuously increasing, fostering the use of HPC and their parallel capabilities that are now mandatory to process this data in a feasible time. Bioinformatics fields such as genomics, proteomics, transcriptomics, metagenomics, or structural bioinformatics can be supported by HPC experts using well-known technologies and infrastructures already applied in other domains of science such as engineering and astronomy.
Having outlined the range of research articles identified belonging to the areas of genomics and HPC parallel and distributed techniques, we now focus on analyzing how we can classify, characterize, and compare one research to another since they come from many different science areas. The first point is to turn articles that join the multidisciplinary sciences, electing those articles that reflect the connection between these sciences based on the knowledge and expertise of the reviewers who analyze the articles. Second, it is needed to analytically understand about the details of the research, for instance, how the genomic research was covered? ie, as a research article or a methodological research? or if the experiment focus on in vivo, in vitro, or in silico genomic experiments since we are interested only by the in silico (bioinformatics) one. What is the bioinformatics methodology implemented in the article? and what about the HPC environment used as computational infrastructure?
In terms of quality assessment, it might be important to consider the research context in which these various articles were developed. We extracted all the articles that involve two main key terms: genomics (for bioinformatics) and parallel/distributed computing in HPC environments (for computational science) but other are emerged concepts can be explored as we did in the present study.
Conclusion
A broad range of well-known bioinformatics applications are discussed in the surveyed publications covered in this article (as summarized in Table 2) following the two proposed questions (RQ1 and RQ2). We present the relevant publications that show the use and benefit of using parallel computing techniques coupled with genomic applications with the goal of improving the performance in large-scale comparative genomic executions. Current parallel computing techniques and technologies including clusters, grids, and compute clouds are used in several different scenarios of genomics research.
This article enables readers to access a set of articles involving complex bioinformatics applications and experiments with larger/richer datasets executed benefits from powerful parallel computing approaches. By associating both bioinformatics and parallel computing fields, scientists are able to conduct relevant advances in several application sciences by deciphering the biological information contained in genomes, better understanding about complex genetic diseases, designing customized and personal-directed drug therapies, and understanding the evolutionary history of genes and genomes. The authors believe this article will be useful to the scientific community for developing or future works to evaluate and compare different genomic approaches that benefit from parallel computing. We believe that following the classifying approaches presented in this article, specialists may consider which approaches meet their needs. New solutions for parallel computing in genomics are available, many others are under development, which makes the field very fertile and hard to be understood and classified.
Acknowledgments
The authors would like to thank National Council of Technological and Scientific Development (CNPq) (grant 478878/2013-3) and FAPERJ (grant E-26/111.370/2013) for partially sponsoring this research.
Author contributions
All authors contributed toward data analysis, drafting and revising the paper and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
Pevsner J. Bioinformatics and Functional Genomics. Hoboken, NJ: John Wiley & Sons, Inc.; 2009. | |
Dai L, Gao X, Guo Y, Xiao J, Zhang Z. Bioinformatics clouds for big data manipulation. Biol Direct. 2012;7(1):43. | |
Marx V. Biology: the big challenges of big data. Nature. 2013; 498(7453):255–260. | |
Miller W, Makova KD, Nekrutenko A, Hardison RC. Comparative genomics. Annu Rev Genomics Hum Genet. 2004;5(1):15–56. | |
Koboldt DC, Steinberg KM, Larson DE, Wilson RK, Mardis ER. The next-generation sequencing revolution and its impact on genomics. Cell. 2013;155(1):27–38. | |
Wall DP, Kudtarkar P, Fusaro VA, Pivovarov R, Patil P, Tonellato PJ. Cloud computing for comparative genomics. BMC Bioinformatics. 2010;11(1):259. | |
Armbrust M, Fox A, Griffith R, et al. A view of cloud computing. Commun ACM. 2010;53(4):50–58. | |
Buyya R, Broberg J, Goscinski AM. Cloud Computing: Principles and Paradigms. 1st ed. Wiley, New Jersey, NJ; 2011. | |
Carpenter B, Getov V, Judd G, Skjellum A, Fox G. MPJ: MPI-like message passing for Java. Concurr Comput. 2000;12(11):1019–1038. | |
Ailamaki A, Ioannidis YE, and Livny M. Scientific workflow management by database management. In: Proceedings of the Tenth International Conference on Scientific and Statistical Database Management, Capri, Italy, 1998. Washington, DC: IEEE Computer Society; 1998: 190–199. | |
Abouelhoda M, Issa S, Ghanem M. Tavaxy: integrating Taverna and Galaxy workflows with cloud computing support. BMC Bioinformatics. 2012;13:77. | |
Lee K, Paton NW, Sakellariou R, Deelman E, Fernandes AAA, Mehta G. Adaptive workflow processing and execution in Pegasus. In: Proceedings of the 3rd International Conference on Grid and Pervasive Computing, Kunming, China, 2008. Washington, DC: IEEE Computer Society; 1998:99–106. | |
Wozniak JM, Armstrong TG, Wilde M, Katz DS, Lusk E, Foster IT. Swift/T: large-scale application composition via distributed-memory dataflow processing. In: Proceedings of the 13th IEEE/ACM International Symposium on Cluster, Cloud and Grid Computing (CCGrid). Washington, DC: IEEE Computer Society; 2013:95–102. | |
Oliveira D, Ogasawara E, Baião F, Mattoso M. SciCumulus: a lightweight cloud middleware to explore many task computing paradigm in scientific workflows. In: Proceedings of the 3rd International Conference on Cloud Computing. Washington, DC: IEEE Computer Society; 2010:378–385. | |
Kitchenham B, Brereton P, Turner M, et al. The impact of limited search procedures for systematic literature reviews: a participant-observer case study. In: Proceedings of the 2009 3rd International Symposium on Empirical Software Engineering and Measurement, Lake Buena Vista, FL, USA, 15–16 October 2009. Washington, DC: IEEE Computer Society; 2009:336–345. | |
Urrútia G, Bonfill X. Declaración PRISMA: una propuesta para mejorar la publicación de revisiones sistemáticas y metaanálisis. Med Clín. 2010;135(11):507–511. | |
Bernardes JS, Dávila AM, Costa VS, Zaverucha G. Improving model construction of profile HMMs for remote homology detection through structural alignment. BMC Bioinformatics. 2007;8(1):435. | |
Wu M, Scott AJ. Phylogenomic analysis of bacterial and archaeal sequences with AMPHORA2. Bioinformatics. 2012;28(7):1033–1034. | |
Ahmed M, Ahmad I, Khan SU. A comparative analysis of parallel computing approaches for genome assembly. Interdiscip Sci. 2011;3(1):57–63. | |
Niemenmaa M, Kallio A, Schumacher A, Klemela P, Korpelainen E, Heljanko K. Hadoop-BAM: directly manipulating next generation sequencing data in the cloud. Bioinformatics. 2012;28(6):876–877. | |
Blom J, Albaum SP, Doppmeier D, et al. EDGAR: a software framework for the comparative analysis of prokaryotic genomes. BMC Bioinformatics. 2009;10(1):154. | |
Lord E, Leclercq M, Boc A, Diallo AB, Makarenkov V. Armadillo 1.1: an original workflow platform for designing and conducting phylogenetic analysis and simulations. PLoS One. 2012;7(1):e29903. | |
Severin J, Beal K, Vilella AJ, et al. eHive: an artificial intelligence workflow system for genomic analysis. BMC Bioinformatics. 2010; 11(1):240. | |
Huber W, Carey VJ, Gentleman R, et al. Orchestrating high-throughput genomic analysis with Bioconductor. Nat Methods. 2015; 12(2):115–121. | |
Kleftogiannis D, Kalnis P, Bajic VB. Comparing memory-efficient genome assemblers on stand-alone and cloud infrastructures. PLoS One. 2013;8(9):e75505. | |
Nefedov AV, Sadygov RG. A parallel method for enumerating amino acid compositions and masses of all theoretical peptides. BMC Bioinformatics. 2011;12(1):432. | |
Hunter AA, Macgregor AB, Szabo TO, Wellington CA, Bellgard MI. Yabi: an online research environment for grid, high performance and cloud computing. Source Code Biol Med. 2012;7(1):1. | |
Gurtowski J, Schatz MC, Langmead B. Genotyping in the cloud with crossbow. In Baxevanis AD, Petsko GA, Stein LD, Stormo GD, editors. Current Protocols in Bioinformatics. Hoboken, NJ: John Wiley & Sons, Inc.; 2012. | |
Zhao S, Prenger K, Smith L, et al. Rainbow: a tool for large-scale whole-genome sequencing data analysis using cloud computing. BMC Genomics. 2013;14:425. | |
Prins P, Belhachemi D, Möller S, Smant G. Scalable computing for evolutionary genomics. Methods Mol Biol. 2012;856:529–545. | |
Dong S, Liu P, Cao Y, Du Z. Grid computing methodology for protein structure prediction and analysis. In: Chen G, Pan Y, Guo M, Lu J, editors. Parallel and Distributed Processing and Applications – ISPA 2005 Workshops. Berlin: Springer; 2005:257–266. | |
Heath AP, Greenway M, Powell R, et al. Bionimbus: a cloud for managing, analyzing and sharing large genomics datasets. J Am Med Inform Assoc. 2014;21(6):969–975. | |
Singh A, Chen C, Liu W, Mitchell W, Schmidt B. A hybrid computational grid architecture for comparative genomics. IEEE Trans Inf Technol Biomed. 2008;12(2):218–225. | |
Moustafa A, Bhattacharya D, Allen AE. iTree: a high-throughput phylogenomic pipeline. In: 5th Cairo International Biomedical Engineering Conference (CIBEC), Cairo, Egypt, 16 December 2010. Washington, DC: IEEE Computer Society; 2010:103–107. | |
El-Kalioby M, Abouelhoda M, Krüger J, et al. Personalized cloud-based bioinformatics services for research and education: use cases and the elasticHPC package. BMC Bioinformatics. 2012;13(Suppl 17):S22. | |
Reid JG, Carroll A, Veeraraghavan N, et al. Launching genomics into the cloud: deployment of Mercury, a next generation sequence analysis pipeline. BMC Bioinformatics. 2014;15(1):30. | |
Minevich G, Park DS, Blankenberg D, Poole RJ, Hobert O. CloudMap: a cloud-based pipeline for analysis of mutant genome sequences. Genetics. 2012;192(4):1249–1269. | |
Krampis K, Booth T, Chapman B, et al. Cloud BioLinux: pre-configured and on-demand bioinformatics computing for the genomics community. BMC Bioinformatics. 2012;13(1):42. | |
Ocaña KACS, de Oliveira D, Dias J, Ogasawara E, Mattoso M. Designing a parallel cloud based comparative genomics workflow to improve phylogenetic analyses. Future Gener Comput Syst. 2013;29(8):2205–2219. | |
Ocaña KACS, de Oliveira D, Ogasawara E, Dávila AMR, Lima AAB, Mattoso M. SciPhy: a cloud-based workflow for phylogenetic analysis of drug targets in protozoan genomes. In: de Souza ON, Telles GP, Palakal M, editors. Advances in Bioinformatics and Computational Biology. Berlin: Springer; 2011:66–70. | |
Oliveira D, Ocaña KACS, Ogasawara E, et al. Performance evaluation of parallel strategies in public clouds: a study with phylogenomic workflows. Future Gener Comput Syst. 2013;29(7):1816–1825. | |
Ocaña KACS, de Oliveira D, Horta F, Dias J, Ogasawara E, Mattoso M. Exploring molecular evolution reconstruction using a parallel cloud based scientific workflow. In: de Souto MC, Kann MG, editors. Advances in Bioinformatics and Computational Biology. Berlin: Springer; 2012:179–191. | |
Ocaña K, Benza S, Oliveira D, Dias J, Mattoso M. Exploring large scale receptor-ligand pairs in molecular docking workflows in HPC clouds. In: IEEE 28th International Parallel and Distributed Processing Symposium Workshops. 13th IEEE International Workshop on High Performance Computational Biology (HiComb 2014), Phoenix, AZ, USA, 19–23 May 2014. Washington, DC: IEEE Computer Society; 2014:536–545. | |
Ocaña KACS, Oliveira D, Silva V, Benza S, Mattoso MLQ. Exploiting the parallel execution of homology workflow alternatives in HPC compute clouds. In: Toumani F, Pernici, B, Grigori, D, et al, editors. Service-Oriented Computing – ICSOC 2014 Workshops. Berlin: Springer; 2014:336–350. | |
Deelman E, Gannon D, Shields M, Taylor I. Workflows and e-Science: an overview of workflow system features and capabilities. Future Gener Comput Syst. 2009;25(5):528–540. | |
Bayucan A, Henderson RL, Jones JP. Portable Batch System Administration Guide. Mountain View, CA: Veridian Systems; 2000. | |
Gentzsch W. Sun Grid Engine: towards creating a compute power grid. In: Proceedings of First IEEE/ACM International Symposium on Cluster Computing and the Grid, Brisbane, QLD, 15–18 May 2001. Washington, DC: IEEE Computer Society; 2001:35–36. | |
Apache Software Foundation. Hadoop. Forest Hill, MD: Apache Software Foundation; 2009. | |
Dean J, Ghemawat S. MapReduce: simplified data processing on large clusters. In: Proceedings of the 6th Conference on Symposium on Opearting Systems Design and Implementation, volume 6. Berkeley, CA: USENIX Association; 2004:10–10. | |
Ding M, Zheng L, Lu Y, Li L, Guo S, Guo M. More convenient more overhead: the performance evaluation of Hadoop streaming. In: Proceedings of the 2011 ACM Symposium on Research in Applied Computation. New York, NY: ACM; 2011:307–313. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.