Back to Journals » OncoTargets and Therapy » Volume 12
Paradoxical effects of cellular senescence-inhibited gene involved in hepatocellular carcinoma migration and proliferation by ERK pathway and mesenchymal-like markers
Authors Cheng Q, Tong TJ, Li Z ![]() , Hu S, Chen D, Wang SQ, Zhu JY
, Hu S, Chen D, Wang SQ, Zhu JY
Received 23 September 2018
Accepted for publication 3 January 2019
Published 15 March 2019 Volume 2019:12 Pages 2035—2046
DOI https://doi.org/10.2147/OTT.S188449
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Sanjeev K. Srivastava
Qian Cheng,1 Tan-jun Tong,2 Zhao Li,1 Shi-hua Hu,1 Ding-bao Chen,1 Si-qi Wang,1 Ji-ye Zhu1
1Peking University Institute of Organ Transplantation, Peking University Center of Liver Cancer Diagnosis and Treatment, Beijing Key Surgical Basic Research Laboratory of Liver Cirrhosis and Liver Cancer, Department of Hepatobiliary Surgery, Peking University People’s Hospital, Beijing, 100044, China; 2Peking University Research Center on Aging, Department of Biochemistry and Molecular Biology, Peking University Health Science Center, Beijing, 100191, China
Background: Cellular senescence-inhibited gene (CSIG) strongly prolongs the progression of replicative senescence. However, roles and mechanisms of CSIG in tumor progression have not been studied widely.
Methods: Roles of CSIG in migration and proliferation of SMMC7721 and Huh7 cells were analyzed by transwell or cell viability assays, respectively. Tumorigenicity assays were used to study whether CSIG knockdown could affect SMMC7721 proliferation in vivo. Next, Western blotting and RT-PCR were preformed to evaluate the effects of CSIG on P-ERK cascade and epithelial mesenchymal transformation markers. Then, the location and expression of CSIG protein was detected by immunofluorescence and Western blotting, respectively. Finally, the Cancer Genome Atlas dataset was used to analyze CSIG mRNA levels in hepatocellular carcinoma (HCC) and adjacent non-tumor tissues.
Results: In this study, we found that CSIG overexpression promoted SMMC7721 cell migration, and CSIG knockdown suppressed tumorigenicity of SMMC7721 cells. In contrast to expectation, CSIG up-regulation could significantly inhibit Huh7 cell growth and migration. CSIG could promote P-ERK activation and levels of mesenchymal-like markers in SMMC7721 cells, whereas CSIG suppressed P-ERK activation and levels of mesenchymal-like markers in Huh7 cells. CSIG protein was located in nucleoli as well as nucleoplasm of SMMC7721 cells, whereas CSIG protein was mainly expressed in the nucleoli rather than nucleoplasm of Huh7 cells. Finally, due to individual differences, raised or down-regulated trends of CSIG in HCC as compared with adjacent non-tumor tissues are different among various patient populations.
Conclusion: In summary, these results indicate that CSIG might play different roles in SMMC7721 and Huh7 cells through regulating P-ERK pathway and mesenchymal-like markers. The differential distribution of CSIG might be an important factor that causes its different functions in SMMC7721 and Huh7 cells. CSIG might play different roles in various patient populations.
Keywords: cellular senescence-inhibited gene, hepatocellular carcinoma, migration, proliferation, P-extracellular regulated protein kinases, mesenchymal-like markers
Introduction
Hepatocellular carcinoma (HCC) ranks as the sixth most common malignancy and more than 700,000 new patients are diagnosed per year.1–3 HCC is the third most common cause of cancer-death worldwide, and the 5-year survival rate for HCC is very low.4,5 The poor prognosis of HCC is due to metastasis and recurrence after partial hepatectomy and liver transplantation.6–8 Previous studies report that undetected intrahepatic lesions lead to 60%–70% of recurrences, while 30%–40% come from de novo HCC lesions.9 To provide new prognostic indicators and novel therapeutic strategies for improved clinical management, the underlying molecular mechanisms of HCC proliferation and metastasis need to be further investigated.
Cellular senescence-inhibited gene (CSIG) is composed of nine exons and it is located on chromosome 16p13.3 (Genebank accession no AY154473).10–12 CSIG protein is a nucleolar protein with a ribosomal L1 domain in its N terminus and a lysine-rich domain in its C terminus, so it is also known as RSL1D1.10,11 CSIG translocates to the nucleoplasm in response to nucleolar stress, caused by different factors including low doses of actinomycin D, doxorubicin, and knockdown of TIF-IA.13
CSIG protein is involved in many biological processes including cell senescence, rRNA processing, apoptosis, etc.11–14 CSIG can significantly delay cell senescence by interacting with PTEN mRNA and inhibiting its translation.11 CSIG binds to the NOC1 mRNA 5′-UTR and inhibits NOCL1 mRNA stabilization, finally inducing rRNA processing.12 Our previous study demonstrated that CSIG facilitates proliferation of several HCC cells through interacting with c-myc protein and promoting its stability.15 However, roles and mechanisms of CSIG in HCC metastasis and prognosis remain unknown.
Carcinoma cells that have activated an epithelial-to-mesenchymal transition (EMT) program often exhibit enhanced migratory and invasive abilities.16–18 In particular, as occurs in multiple tissues, epithelial carcinoma cells are able to obtain mesenchymal-like characteristics by activation of EMT program.16,19 Activation of the EMT program elicits changes in a number of fundamental aspects of cellular physiology that include: alterations in the cytoskeletal organization, associated changes in cell morphology, dissolution of epithelial cell–cell junctions as well as an ability to degrade and reorganize the extracellular matrix, enabling cell invasion and migration.20,21
In this study, we investigated effects of CSIG on migration and proliferation of SMMC7721 and Huh7 cells. To clarify mechanisms of CSIG involved in HCC progression, we further studied whether P-ERK pathway and EMT markers were regulated by CSIG. The present study may provide new insight into the mechanism of therapeutic intervention for HCC.
Materials and methods
Cell culture
Cells involved in this study included the immortalized human hepatic cell line L02, and HCC cells (SMMC7721, Huh7, MHCC97L, MHCC97H, HepG2, and Bel7402). The usage and methods of cell lines in cell-based experiments were approved by the ethical and institutional review board of Peking University People’s Hospital. The L02 cells were purchased from China Center for Type Culture Collection (CCTCC, Wu Han, China). HepG2 and Huh7 cells were obtained from American Type Culture Collection (ATCC, Manassas, VA, USA). The MHCC97L and MHCC97H cells were obtained from the Liver Cancer Institute of Fudan University (Shanghai, China). SMMC7721 was obtained from Department of Experimental Hematology, Beijing Institute of Radiation Medicine (Beijing, China), and verified by short tandem repeat profiling. Bel7402 cells were obtained from the Department of Biology, Peking University Health Science Center (Beijing, China) and verified by short tandem repeat profiling. All cells were maintained in DMEM or RPMI 1640 supplemented with 10% FBS in a humidified incubator at 37°C with 5% CO2.
Transient transfection
For CSIG overexpression experiments, PIRES-flag-CSIG and PIRES-flag plasmids transfections were performed by using Lipofectamine 2000 reagents, and analyzed 48 hours or 72 hours after transfection. For siRNA knockdown assays, siRNA targeting CSIG sequences were 5′-AGAAGGAACAGACCCCAGA-3′ and negative control siRNA sequences were 5′-UUCUCCGAACGUGUCACGU-3′. Cells were transfected with siRNA duplexes for 48 hours or 72 hours by using Lipofectamine RNAiMAX (Invitrogen, Thermo Fisher Scientific, Waltham, MA, USA).
Virus packaging for constructing CSIG knockdown cells
The CSIG-specific shRNA expression vectors and the scrambled ineffective shRNA cassette in the pMCV-puro-miR30 plasmid were purchased from Generay Biotech (Shanghai, China). The human CSIG-specific shRNA target sequences were 5′-AGAAGGAACAGACCCCAGAGC-3′. Virus packaging was performed in Phoenix Retroviral Expression System after transfection of pMCV-puro-miR30-shCSIG and pMCV-puro-miR30-shNC plasmids.22 Forty-eight hours after transfection, supernatants were collected. SMMC7721 cells were infected with viral supernatant and stable cell lines were obtained through selection with puromycin.
qRT-PCR
Total RNAs from HCC cells were extracted with RNA Extraction Kit (Aidlab, Beijing, China), and reverse transcription was performed with PrimeScript RT Master Mix (Takara, Dalian, China). Gene-specific primers were: CSIG forward and reverse: 5′-CGTATTGGTCACGTTGGAATGC-3′, 5′-CCACTTCTCTGGCAATTTTTCTG-3′;15 E-cadherin forward and reverse: 5′-TTCCTCCCAATACATCTCCC-3′, 5′-TAACCGTTAGTAGTCACCCACC-3′; Vimentin forward and reverse: 5′-AGTCCACTGAGTACCGGAGAC-3′, 5′-CATTTCACGCATCTGGCGTTC-3′; N-cadherin forward and reverse: 5′-AGCTCCATTCCGACTTAGACA-3′, 5′-CAGCCTGAGCACGAAGAGTG-3′; MMP9 forward and reverse: 5′-TGTACCGCTATGGTTACACTCG-3′, 5′-GGCAGGGACAGTTGCTTCT-3′; GAPDH forward and reverse: 5′-CCTCCGGGAAACTGTGGCGTGATGG-3′,5′-AGACGGCAGGTCAGGTCCACCACTG-3′.23 Primer sequences of Vimentin, N-cadherin, and MMP9 were from PrimerBank, which is a public resource for PCR primers. RT-PCR was carried out using the SYBR Green Realtime PCR Master Mix on an ABI Prism 7000 system (Applied Biosystems, Thermo Fisher Scientific). Relative mRNA levels were expressed as 2−ΔΔCt value.
Western blotting
Western blotting was carried out as previously described.13,15 Anti-CSIG antibody was from Tanjun Tong’s laboratory (reference 11) or purchased from Sigma-Aldrich Co (St Louis, MO, USA).11 Anti-phospho-ERK, anti-ERK, anti-MMP9, anti-E-cadherin, anti-Vimentin, anti-N-cadherin, and anti-GAPDH antibodies were purchased from Cell Signaling Technology (Danvers, MA, USA).
Cell viability assays
Firstly, SMMC7721-shCSIG and SMMC7721-scramble cells were seeded in 96-well plates, and the viable cells were measured with MTT after serval days. Then, cells were treated with dimethyl sulfoxide and absorbance was measured at a wavelength of 490 nm to estimate the viability of cells.
Similarly, Huh7 cells were plated in 96-well plates following transfection with PIRES-flag-CSIG or PIRES-flag plasmids. After transfection for 1 day, 2 days, 3 days, and 4 days, cells were incubated with Cell Counting Kit-8 every day. Subsequently, absorbance at a wavelength of 450 nm was measured.
Colony formation assays
An amount of 1,000 HCC cells were seeded in 60 mm dishes. Several days later, cells were fixed in 4% formaldehyde for 20 minutes and washed twice with PBS, then stained with 0.1% crystal violet for 15 minutes, and washed with PBS twice.
Tumorigenicity assays in nude mice
The animal experiments were approved by the Institutional Animal Care and Use Committee at Peking University Health Science Center, and all animal studies and procedures were performed in accordance with the UK Animals (Scientific Procedures) Act of 1986 and the associated animal care guidelines. Female nude mice (6–8 weeks old) were purchased from Weitonglihua Company (Beijing, China) and raised under specified pathogen-free conditions. SMMC7721-shCSIG and SMMC7721-scramble cells (3×106) were subcutaneously injected into the right side and the left side of mice, respectively. Approximately 7 weeks after injection, mice were sacrificed and tumors were weighed.
Cell migration assays
Cell migration assays were assessed using transwell chambers (8 μm transwell inserts, Falcon, Thermo Fisher Scientific). HCC cells in serum-free DMEM were seeded in the upper chamber, and medium containing 10% FBS was added in the lower chamber as the chemoattractant. After 24 or 48 hours of incubation, the penetrated cells on the filters were fixed and stained with 0.1% crystal violet. Migranted cells were counted in five random fields (×10 magnifications) and imaged using SPOT imaging software.
Immunofluorescence
Cells seeded on coverslips were fixed with 4% paraformaldehyde for 15 minutes, then washed with PBS and permeabilized with 0.5% Triton X-100 for 10 minutes. Samples were blocked with 2% BSA for 1 hour at room temperature and then incubated with CSIG antibody (Sigma-Aldrich Co.). Next, the samples were washed with PBS and incubated with Alexa Fluor® 488-conjugated secondary antibody for 1 hour at room temperature. Samples were washed again with PBS and stained with DAPI. Immunofluorescence images were captured with a fluorescent microscope.
Datasets
The Cancer Genome Atlas (TCGA, https://tcga-data.nci.nih.gov/tcga/) dataset was used to evaluate mRNA levels of CSIG in HCC and adjacent non-tumor tissues.
Statistical analyses
The data were presented as mean ± SD from at least three independent experiments. Differences between variables were assessed by Student t-tests, and statistical significance was described as *P<0.05, **P<0.01, and ***P<0.001.
Results
Knockdown of CSIG suppressed SMMC7721 cell proliferation in vitro and in vivo, CSIG overexpression could promote SMMC7721 cell migration
As our recent studies showed that CSIG overexpression could promote proliferative and tumorigenic abilities of SMMC7721 cells, we determined whether CSIG knockdown could inhibit SMMC7721 cell growth and tumorigenicity in this research. Firstly, we established CSIG knockdown cell line SMMC7721-shCSIG and its corresponding negative control cell SMMC7721-scramble by retrovirus mediation. The down-regulation of CSIG protein was confirmed by Western blotting (Figure 1A). Next, we determined cancer cell proliferation by viability and colony formation assays. Consequently, the proliferative ability of SMMC7721-shCSIG was decreased as compared with control group (Figure 1B and C). To further detect the effect of CSIG knockdown on HCC cell growth in vivo, SMMC7721-shCSIG and SMMC7721-scramble cells were subcutaneously injected into nude mice. After approximately 6 weeks, xenografts were photographed and weighed. Consequently, tumor weights in SMNC7721-shCSIG group were strongly decreased, compared to SMMC7721-control group (Figure 1D).
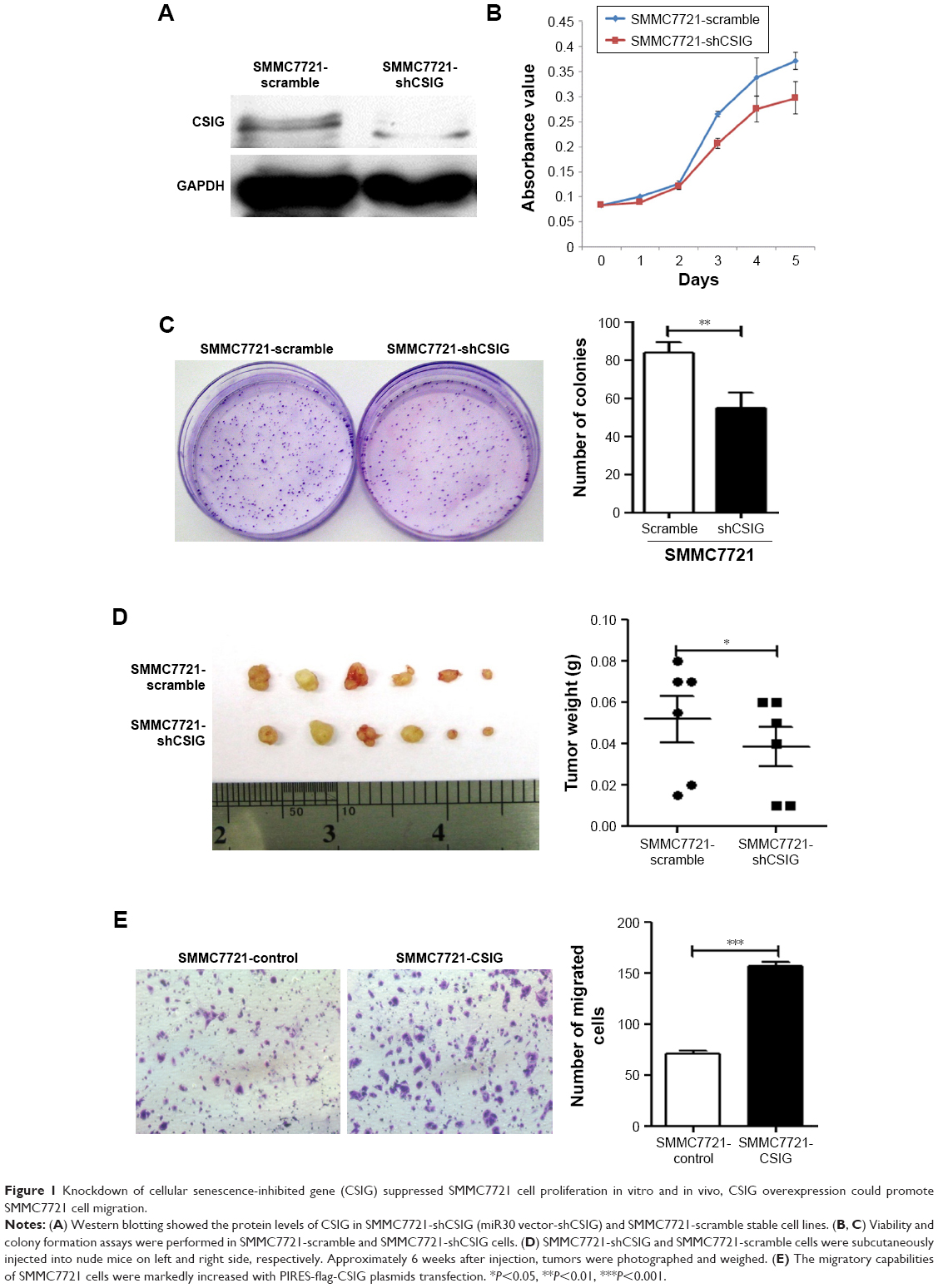 | Figure 1 Knockdown of cellular senescence-inhibited gene (CSIG) suppressed SMMC7721 cell proliferation in vitro and in vivo, CSIG overexpression could promote SMMC7721 cell migration. |
HCC recurrences are mainly due to intrahepatic and extrahepatic metastasis.9 Thus, cell migration assays were performed to determine the role of CSIG in HCC migration. The migratory ability of SMMC7721 cells transfected with flag-PIIRES-CSIG plasmids was strongly increased as compared with control cells treated with flag-PIIRES vectors (Figure 1E).
CSIG suppressed proliferative and migratory capabilities of Huh7 cells
We analyzed the effect of up-regulating CSIG on growth and migration of Huh7. RT-PCR and Western blotting assays showed that CSIG mRNA and protein levels were overexpressed in Huh7 cells transfected with PIRES-flag-plasmids, respectively (Figure 2A). In contrast to expectation, overexpressing CSIG could significantly inhibit Huh7 cells’ growth (Figure 2B). The migrating ability of Huh7 cells transfected with flag-PIIRES-CSIG plasmids was strongly decreased as compared with control cells (Figure 2C and D). Therefore, CSIG played a completely opposite role between SMMC7721 and Huh7 cell progression.
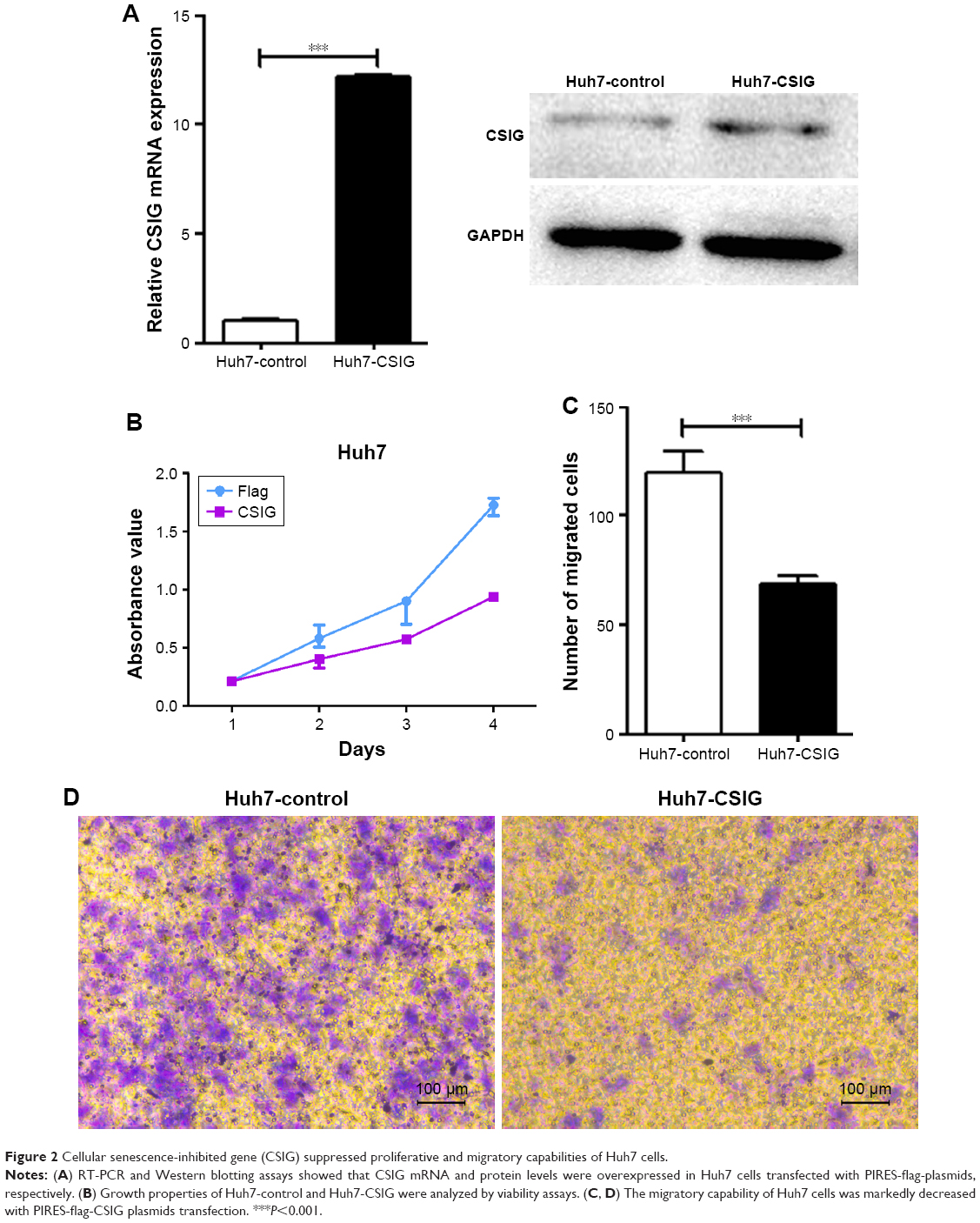 | Figure 2 Cellular senescence-inhibited gene (CSIG) suppressed proliferative and migratory capabilities of Huh7 cells. |
CSIG could promote P-ERK and levels of mesenchymal-like markers in SMMC7721 cells
To elucidate the molecular mechanisms by which CSIG affects proliferation and migration of HCC cells, we measured the expression levels of several signaling pathways and proteins in HCC cells. P-ERK cascade plays important roles in the proliferation and migration of HCC cells.24–26 Our data showed that CSIG overexpression could activate P-ERK in SMMC7721 cells, inversely, CSIG knockdown could inhibit P-ERK pathway in SMMC7721 cells (Figure 3A and B).
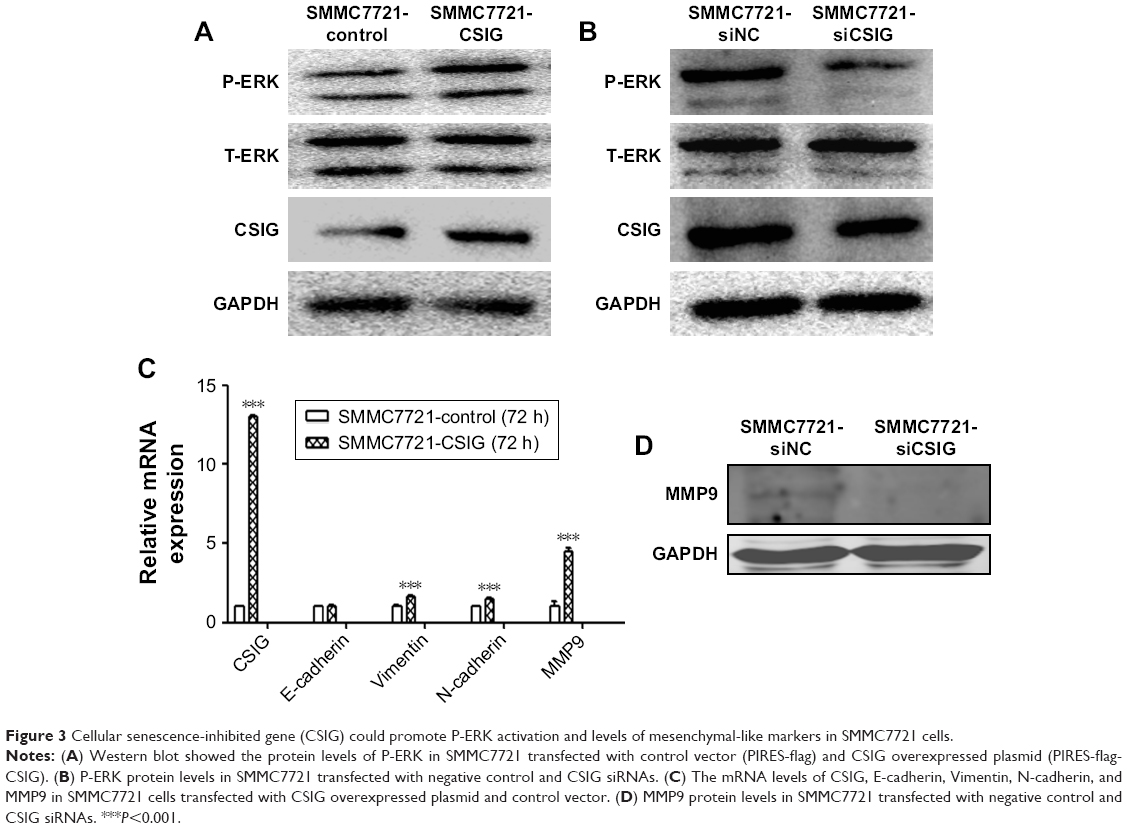 | Figure 3 Cellular senescence-inhibited gene (CSIG) could promote P-ERK activation and levels of mesenchymal-like markers in SMMC7721 cells. |
EMT plays a key role in HCC metastasis.27 Thus, we determined if CSIG could regulate the EMT in SMMC7721 cells. The mRNA level of epithelial cell marker E-cadherin was not changed significantly in SMMC7721-CSIG cells as compared with control group. However, mRNA levels of mesenchymal cell markers (Vimentin, N-cadherin, MMP9) were significantly increased in CSIG-overexpressed SMMC7721 cells (Figure 3C). Among these mesenchymal-like markers, the fold change of MMP9 was the most obvious (Figure 3C). Further findings showed that MMP9 protein was decreased in SMMC7721 cells transfected with CSIG siRNA (Figure 3D). Therefore, the capability of CSIG to promote SMMC7721 migration mainly depends on MMP9 enzyme.
Taken together, CSIG obviously activated P-ERK cascade and mesenchymal cell markers, thereby increasing proliferation and migration capacities of SMMC7721 cells.
CSIG could repress P-ERK activation and levels of mesenchymal-like markers in Huh7 cells
In view of different roles of CSIG in Huh7 cells as compared with SMMC7721 cells, P-ERK pathway in Huh7 cells transfected with PIRES-flag-CSIG plasmids was detected by Western blotting. Our data showed that CSIG overexpression could inhibit P-ERK activation in Huh7 cells (Figure 4A). The mRNA and protein levels of EMT markers were detected by RT-PCR and Western blotting assays, respectively. Consequently, mRNA levels of Vimentin, N-cadherin, and MMP9 were down-regulated in CSIG-overexpressed Huh7 cells; E-cadherin mRNA was not changed in CSIG-overexpressed Huh7 cells as compared with control group (Figure 4B). Moreover, CSIG overexpression could inhibit protein levels of Vimentin and MMP9 and did not affect E-cadherin and N-cadherin protein significantly in Huh7 cells (Figure 4C and D).
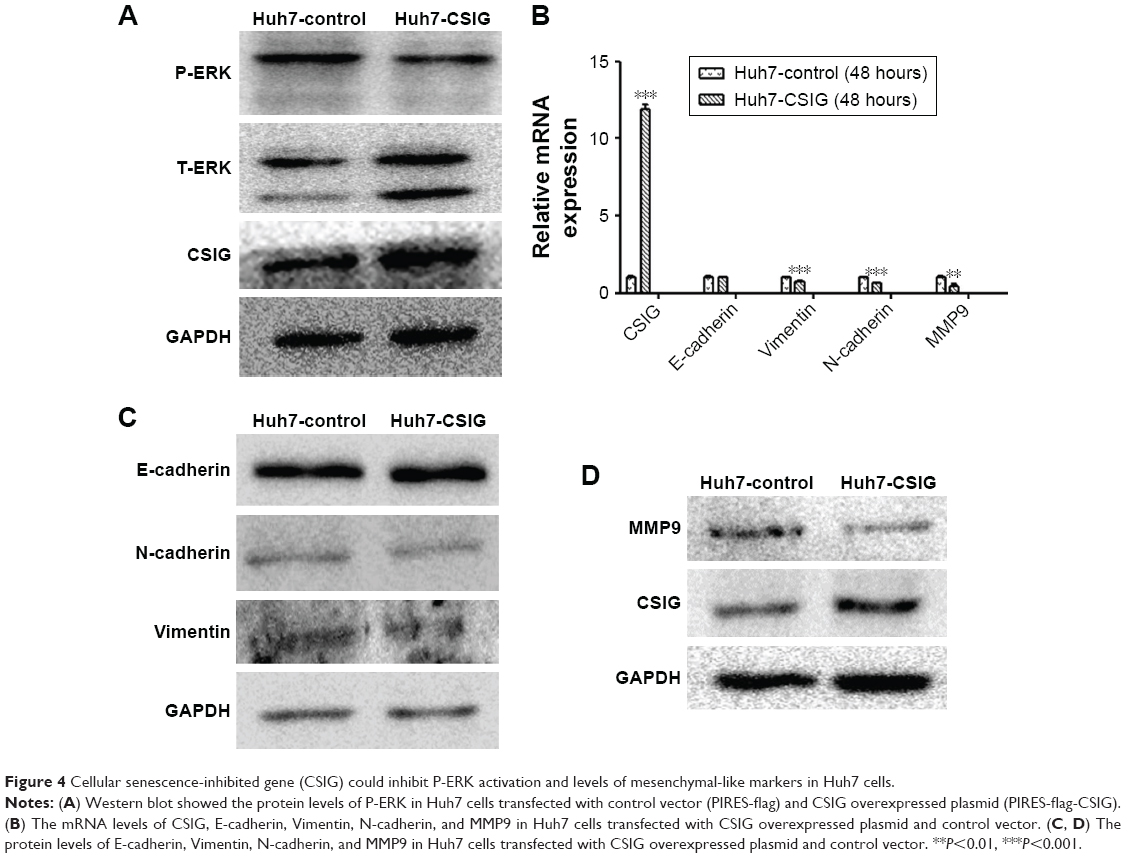 | Figure 4 Cellular senescence-inhibited gene (CSIG) could inhibit P-ERK activation and levels of mesenchymal-like markers in Huh7 cells. |
Taken together, CSIG obviously inhibited P-ERK cascade as well as protein levels of Vimentin and MMP9, thereby suppressing proliferation and migration capacities of Huh7 cells.
The localization and expression of CSIG in HCC cells
In view of different roles of CSIG in Huh7 cells as compared with SMMC7721 cells, localization of CSIG was examined by immunofluorescence analysis. CSIG protein was mainly located in the nucleus including nucleoli as well as nucleoplasm, and it was also detected in the cytoplasm of SMMC7721 cells (Figure 5A). Similar results were obtained in MHCC97H cells (Figure 5B). Whereas CSIG protein was mainly expressed in the nucleoli rather than nucleoplasm of Huh7 cells (Figure 5C).
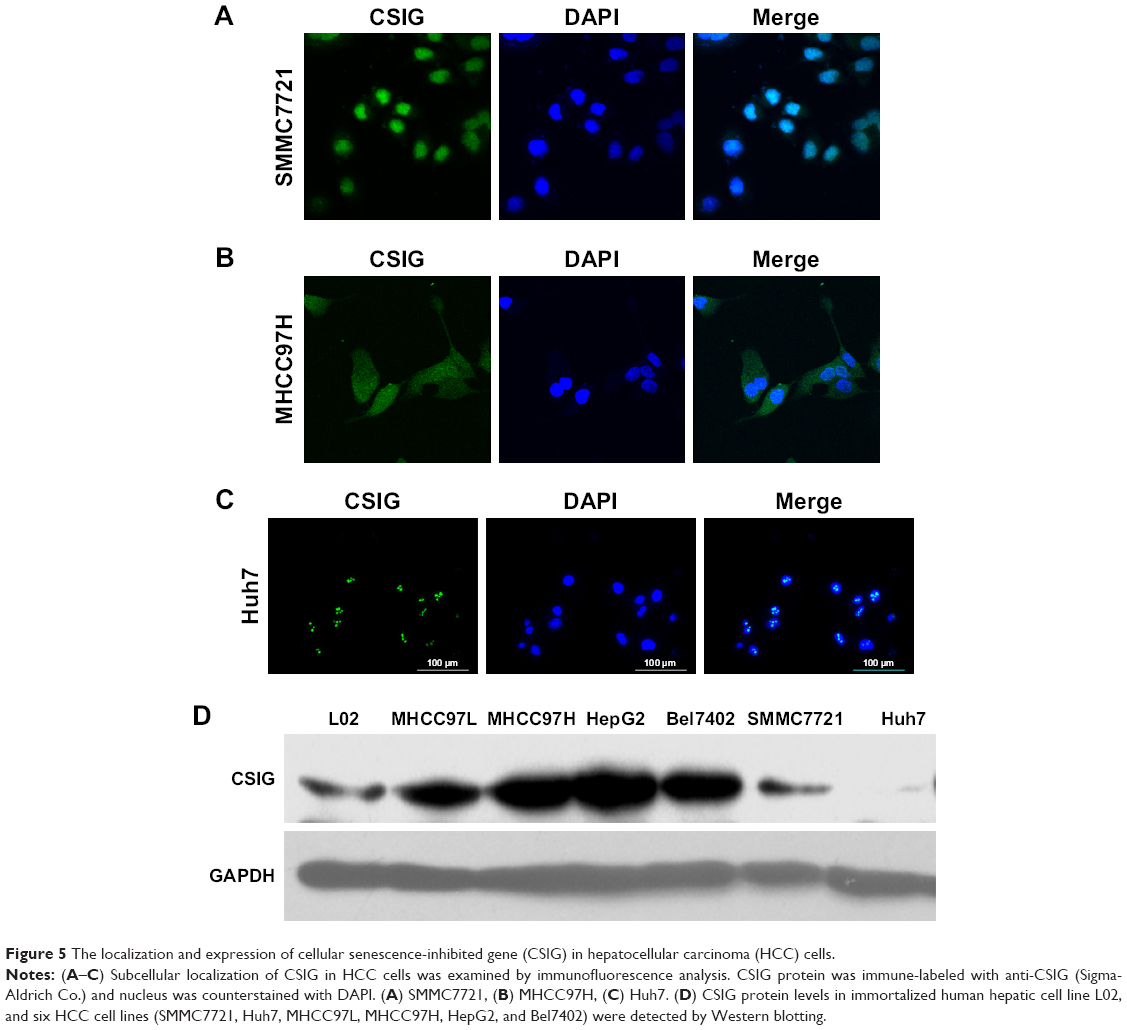 | Figure 5 The localization and expression of cellular senescence-inhibited gene (CSIG) in hepatocellular carcinoma (HCC) cells. |
CSIG protein levels were higher in the four HCC cell lines (MHCC97L, MHCC97H, HepG2, Bel7402) as compared with that of normal liver cell L02, and CSIG protein in SMMC7721 cells was approximately equal to that in L02 cells (Figure 5D). In contrast to expectation, CSIG protein was lower in Huh7 cells than that in L02 cells (Figure 5D).
CSIG levels are expressed differently in different tissues
TCGA database was used to analyze whether CSIG expression varies between HCC and corresponding non-tumor tissues. In some patients, mRNA levels of CSIG in HCC tissue were higher than that in the paracancerous tissue (n=8, high CSIG-expression); however, in other patients, mRNA levels of CSIG in HCC tissue were lower than that in the paracancerous tissue (n=10, low CSIG-expression) (Figure 6A and B). Consequently, due to individual differences, raised or down-regulated trends of CSIG in HCC as compared with adjacent non-tumor tissues are different among various patient populations. The ratio of recurrent cases in low CSIG-expression group was approximately 40% (4/10), while the ratio of recurrent cases was approximately 75% (6/8) in high CSIG-expression group (Figure 6C). The ratio of TNM stage 1 cases in low CSIG-expression group was approximately 66.7% (6/9), while the ratio of TNM stage 1 cases was approximately 25% (2/8) in high CSIG-expression group (Figure 6D). The patients in the low CSIG-expression group achieved better overall survival and disease-free survival than the rates observed in the high CSIG-expression group (Figure 6E and F).
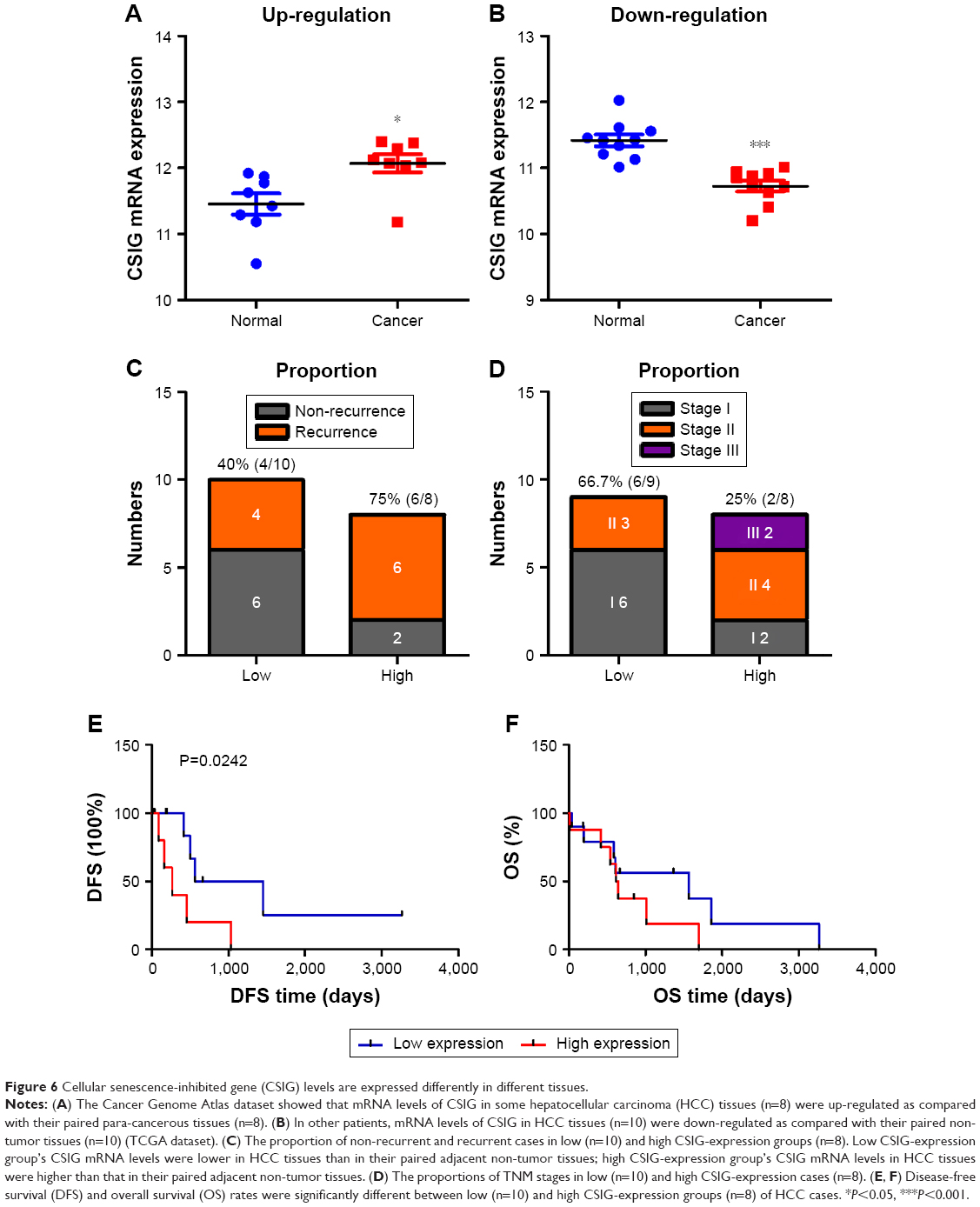 | Figure 6 Cellular senescence-inhibited gene (CSIG) levels are expressed differently in different tissues. |
Discussion
Increasing evidence suggests that CSIG plays important roles in many physiological and pathophysiological processes including cell senescence, nucleolar stress response, and UV-induced apoptosis.11,13,14 However, the role of CSIG in cancer development and progression has not been studied widely. In this study, we clarified whether CSIG could affect HCC migration, proliferation, and the EMT process.
Our previous study showed that CSIG overexpression could promote proliferative and tumorigenic abilities of SMMC7721 and HepG2 cells.15 In this study, we found that CSIG knockdown significantly suppressed growth and tumorigenic capacities of SMMC7721 cells, which was consistent with our previous findings.15 In contrast to expectation, CSIG overexpression significantly suppressed Huh7 cell growth. Importantly, we first demonstrated that migratory abilities of SMMC7721 cells were strongly increased with CSIG overexpressed plasmids transfection. Conversely, CSIG could inhibit Huh7 cell migration. In general, the role of CSIG in Huh7 cell progression was completely opposite to that in SMMC7721 and HepG2 cells. Furthermore, functions of CSIG in regulating migration in many tumors have not been reported. First, we found that CSIG served as a novel regulator for liver cancer metastasis.
In view of the different roles of CSIG in Huh7 cells as compared with SMMC7721 cells, we measured the expression levels of several signaling pathways in these two cell lines. Our data showed that CSIG could activate P-ERK in SMMC7721 cells, conversely, CSIG repressed P-ERK activation in Huh7 cells. P-ERK cascade plays important roles in the proliferation and migration of HCC cells. Therefore, as CSIG has different roles in regulating P-ERK pathway in different cells, the role of CSIG in Huh7 cell growth was opposite to that in SMMC7721 cells.24–26 In addition, P-ERK could activate many downstream genes including c-myc, MMP9, and CCND1, etc.24–26 Our previous research showed that CSIG could promote c-myc mRNA and protein levels in three HCC cell lines (SMMC7721, HepG2, and MHCC97H).15 Therefore, we could speculate that CSIG might promote the expression of c-myc mRNA by activating the P-ERK cascade.
EMT plays a key role in HCC metastasis.27 Our findings demonstrated that CSIG facilitated mesenchymal-like markers including Vimentin, N-cadherin, and MMP9 in SMMC7721 cells and inhibited mesenchymal-like markers including Vimentin and MMP9 in Huh7 cells. As CSIG plays different roles in regulating mesenchymal-like markers in different cells, the role of CSIG in Huh7 cell growth was opposite to that in SMMC7721 cells.
CSIG protein is a nucleolar protein and regulates ribosome RNA processing.10,11 Protein complex shuttles between the nucleolus and nucleoplasm are important for many non-ribosomal processes such as regulation of stress responses, cell growth and death, mitosis, and the cell cycle.28,29 In this study, we found that CSIG protein was located in nucleoli as well as nucleoplasm in SMMC7721 and MHCC97H cells, whereas it was only located in the nucleoli of Huh7 cells. So we could speculate that the differential distribution of CSIG is an important factor that causes its different functions in SMMC7721 (or MHCC97H) and Huh7 cells.
CSIG protein is involved in many biological process through directly interacting with its target mRNA or protein.11,12 For instance, CSIG can significantly delay cell senescence by interacting with PTEN mRNA and inhibiting its translation.11 CSIG binds to the NOC1 mRNA 5′-UTR, and inhibits NOCL1 mRNA stabilization in HEK293 cells.12 Further research will be needed to find proteins or mRNAs which directly interact with CSIG protein, so that the molecular mechanism of CSIG regulating P-ERK pathway and EMT process can be clarified in-depth.
CSIG protein was lower in Huh7 cells than that in normal liver cell L02, however, CSIG protein levels were higher in the four HCC cell lines (MHCC97L, MHCC97H, HepG2, and Bel7402) than in L02 cells. CSIG protein levels in SMMC7721 cells were approximately equal to that in L02 cells. These results might explain that CSIG plays completely opposite roles in Huh7 cells as compared to other cells. In addition, TCGA database was used to analyze whether CSIG expression varies between HCC and its corresponding non-tumor tissues. In some patients, mRNA levels of CSIG in the HCC tissue was higher than that in the paracancerous tissue (n=8); however, in other patients, mRNA levels of CSIG in the HCC tissue was lower than that in the paracancerous tissue (n=10). Consequently, due to individual differences, raised or down-regulated trends of CSIG in HCC as compared with adjacent non-tumor tissues are different among various patient populations. Therefore, CSIG might play different roles in various patient populations.
Conclusion
In summary, our study demonstrated that CSIG protein was located in nucleoli as well as nucleoplasm of SMMC7721 and MHCC97H cells, whereas it was only located in the nucleoli of Huh7 cells. As summarized in Figure 7, CSIG promoted SMMC7721 progression by activating P-ERK signal cascade and metastasis-related protein mesenchymal-like markers, conversely, CSIG suppressed Huh7 progression by activating P-ERK signal cascade and mesenchymal-like markers. The differential distribution of CSIG was found to be an important factor that causes its different functions in SMMC7721 and Huh7 cells. CSIG might play different roles in various patient populations. This study might provide good evidence for HCC tumor heterogeneity and theoretical clues for future precision medicine.
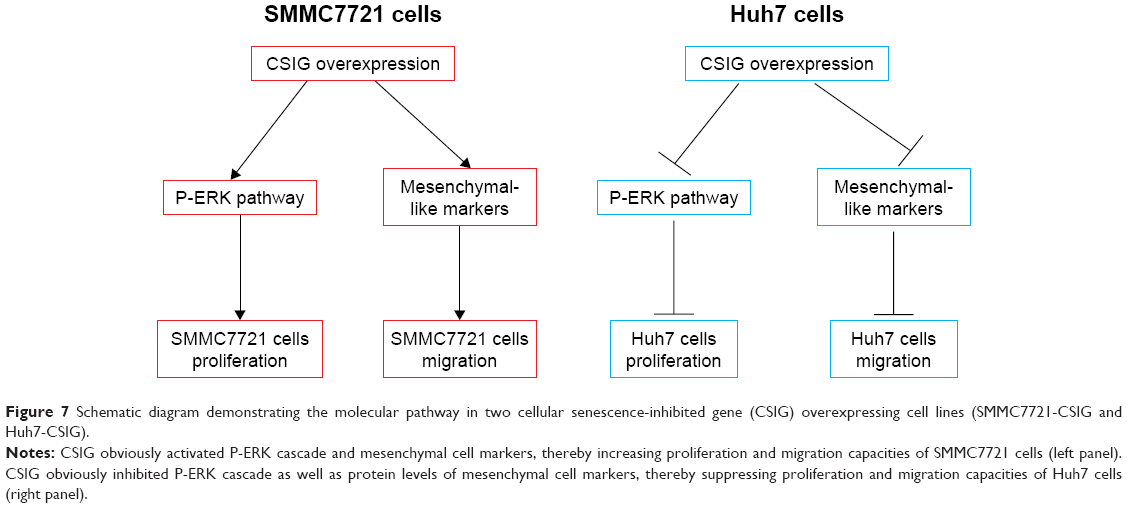 | Figure 7 Schematic diagram demonstrating the molecular pathway in two cellular senescence-inhibited gene (CSIG) overexpressing cell lines (SMMC7721-CSIG and Huh7-CSIG). |
Acknowledgment
This work was supported by National Natural Science Foundation of China (81570590, 81502581).
Disclosure
The authors report no conflicts of interest in this work.
References
Lo Re O, Fusilli C, Rappa F, et al. Induction of cancer cell stemness by depletion of macrohistone H2A1 in hepatocellular carcinoma. Hepatology. 2018;67(2):636–650. | ||
Slotta JE, Kollmar O, Ellenrieder V, Ghadimi BM, Homayounfar K. Hepatocellular carcinoma: surgeon’s view on latest findings and future perspectives. World J Hepatol. 2015;7(9):1168–1183. | ||
Wu YH, Liu W, Zhang L, et al. Effects of microRNA-24 targeting c-myc on apoptosis, proliferation and cytokine expressions in chondrocytes of rats with osteoarthritis via MAPK signaling pathway. J Cell Biochem. 2018;119(10):7944–7958. | ||
Singal AG, El-Serag HB. Hepatocellular carcinoma from epidemiology to prevention: translating knowledge into practice. Clin Gastroenterol Hepatol. 2015;13(12):2140–2151. | ||
Lanton T, Shriki A, Nechemia-Arbely Y, et al. Interleukin 6-dependent genomic instability heralds accelerated carcinogenesis following liver regeneration on a background of chronic hepatitis. Hepatology. 2017;65(5):1600–1611. | ||
Han D, Li J, Wang H, et al. Circular RNA circMTO1 acts as the sponge of microRNA-9 to suppress hepatocellular carcinoma progression. Hepatology. 2017;66(4):1151–1164. | ||
Tung-Ping Poon R, Fan ST, Wong J. Risk factors, prevention, and management of postoperative recurrence after resection of hepatocellular carcinoma. Ann Surg. 2000;232(1):10–24. | ||
Liu Y, Zhang Y, Wang S, et al. Prospero-related homeobox 1 drives angiogenesis of hepatocellular carcinoma through selectively activating interleukin-8 expression. Hepatology. 2017;66(6):1894–1909. | ||
Llovet JM, Schwartz M, Mazzaferro V. Resection and liver transplantation for hepatocellular carcinoma. Semin Liver Dis. 2005;25(02):181–200. | ||
Guo S, Zhang Z, Tong T. Cloning and characterization of cellular senescence-associated genes in human fibroblasts by suppression subtractive hybridization. Exp Cell Res. 2004;298(2):465–472. | ||
Ma L, Chang N, Guo S, et al. CSIG inhibits PTEN translation in replicative senescence. Mol Cell Biol. 2008;28(20):6290–6301. | ||
Yuan F, Zhang Y, Ma L, et al. Enhanced NOLC1 promotes cell senescence and represses hepatocellular carcinoma cell proliferation by disturbing the organization of nucleolus. Aging Cell. 2017;16(4):726–737. | ||
Xie N, Ma L, Zhu F, et al. Regulation of the MDM2-p53 pathway by the nucleolar protein CSIG in response to nucleolar stress. Sci Rep. 2016;6(1):36171. | ||
Li N, Zhao G, Chen T, et al. Nucleolar protein CSIG is required for p33ING1 function in UV-induced apoptosis. Cell Death Dis. 2012;3(3):e283. | ||
Cheng Q, Yuan F, Lu F, et al. CSIG promotes hepatocellular carcinoma proliferation by activating c-MYC expression. Oncotarget. 2015;6(7):4733–4744. | ||
Nieto MA, Huang RY-J, Jackson RA, Thiery JP. EMT: 2016. Cell. 2016;166(1):21–45. | ||
Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci Unit States Am. 2003;100(7):3983–3988. | ||
Pattabiraman DR, Weinberg RA. Tackling the cancer stem cells – what challenges do they pose? Nat Rev Drug Discov. 2014;13(7):497–512. | ||
Bierie B, Pierce SE, Kroeger C, et al. Integrin-β4 identifies cancer stem cell-enriched populations of partially mesenchymal carcinoma cells. Proc Natl Acad Sci Unit States Am. 2017;114(12):E2337–E2346. | ||
Shibue T, Weinberg RA. EMT, CSCs, and drug resistance: the mechanistic link and clinical implications. Nat Rev Clin Oncol. 2017;14(10):611–629. | ||
Taube JH, Herschkowitz JI, Komurov K, et al. Core epithelial-to-mesenchymal transition interactome gene-expression signature is associated with claudin-low and metaplastic breast cancer subtypes. Proc Natl Acad Sci Unit States Am. 2010;107(35):15449–15454. | ||
Wang P, Han L, Shen H, et al. Protein kinase D1 is essential for Ras-induced senescence and tumor suppression by regulating senescence-associated inflammation. Proc Natl Acad Sci Unit States Am. 2014;111(21):7683–7688. | ||
Zhang G, Cheng Y, Zhang Q, et al. ATX-LPA axis facilitates estrogen–induced endometrial cancer cell proliferation via MAPK/ERK signaling pathway. Mol Med Rep. 2018;17(3):4245–4252. | ||
Li D, Liu X, Zhou J, et al. Long noncoding RNA HULC modulates the phosphorylation of YB-1 through serving as a scaffold of extracellular signal-regulated kinase and YB-1 to enhance hepatocarcinogenesis. Hepatology. 2017;65(5):1612–1627. | ||
Jt L, Zhao WD, He W, Wei W. Hedgehog signaling pathway mediates invasion and metastasis of hepatocellular carcinoma via ERK pathway. Acta Pharmacol Sin. 2012;33(5):691–700. | ||
Jia YL, Shi L, Zhou JN, et al. Epimorphin promotes human hepatocellular carcinoma invasion and metastasis through activation of focal adhesion kinase/extracellular signal-regulated kinase/matrix metalloproteinase-9 axis. Hepatology. 2011;54(5):1808–1818. | ||
Giannelli G, Koudelkova P, Dituri F, Mikulits W. Role of epithelial to mesenchymal transition in hepatocellular carcinoma. J Hepatol. 2016;65(4):798–808. | ||
Boulon S, Westman BJ, Hutten S, Boisvert FM, Lamond AI. The nucleolus under stress. Mol Cell. 2010;40(2):216–227. | ||
Pederson T, Tsai RY. In search of nonribosomal nucleolar protein function and regulation. J Cell Biol. 2009;184(6):771–776. |
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.