Back to Journals » Drug Design, Development and Therapy » Volume 9
Paracellular permeation-enhancing effect of AT1002 C-terminal amidation in nasal delivery
Authors Song K, Kim S, Shim C, Chung S, Kim D ![]() , Rhee S, Choi GJ, Kim C, Kim K
, Rhee S, Choi GJ, Kim C, Kim K
Received 16 December 2014
Accepted for publication 17 February 2015
Published 27 March 2015 Volume 2015:9 Pages 1815—1823
DOI https://doi.org/10.2147/DDDT.S79383
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Professor Shu-Feng Zhou
Keon-Hyoung Song,1 Sang-Bum Kim,2 Chang-Koo Shim,2 Suk-Jae Chung,2 Dae-Duk Kim,2 Sang-Ki Rhee,1 Guang J Choi,1 Chul-Hyun Kim,3 Kiyoung Kim4
1Department of Pharmaceutical Engineering, Soonchunhyang University, Asan, Republic of Korea; 2College of Pharmacy and Research Institute of Pharmaceutical Sciences, Seoul National University, Seoul, Republic of Korea; 3Department of Sports Medicine, 4Department of Medical Biotechnology, Soonchunhyang University, Asan, Republic of Korea
Background: The identification of permeation enhancers has gained interest in the development of drug delivery systems. A six-mer peptide, H-FCIGRL-OH (AT1002), is a tight junction modulator with promising permeation-enhancing activity. AT1002 enhances the transport of molecular weight markers or agents with low bioavailability with no cytotoxicity. However, AT1002 is not stable in neutral pH or after incubation under physiological conditions, which is necessary to fully uncover its permeation-enhancing effect. Thus, we increased the stability or mitigated the instability of AT1002 by modifying its terminal amino acids and evaluated its subsequent biological activity.
Methods: C-terminal-amidated (FCIGRL-NH2, Pep1) and N-terminal-acetylated (Ac-FCIGRL, Pep2) peptides were analyzed by liquid chromatography–mass spectrometry. We further assessed cytotoxicity on cell monolayers, as well as the permeation-enhancing activity following nasal administration of the paracellular marker mannitol.
Results: Pep1 was nontoxic to cell monolayers and showed a relatively low decrease in peak area compared to AT1002. In addition, administration of mannitol with Pep1 resulted in significant increases in the area under the plasma concentration–time curve and peak plasma concentration at 3.63-fold and 2.68-fold, respectively, compared to mannitol alone. In contrast, no increase in mannitol concentration was shown with mannitol/AT1002 or mannitol/Pep2 compared to the control. Thus, Pep1 increased the stability or possibly reduced the instability of AT1002, which resulted in an increased permeation-enhancing effect of AT1002.
Conclusion: These results suggest the potential usefulness of C-terminal-amidated AT1002 in enhancing nasal drug delivery, which may lead to the development of a practical drug delivery technology for drugs with low bioavailability.
Keywords: N-terminal acetylation, stability, permeation enhancer
Introduction
The identification of permeation enhancers has gained interest in the development of drug delivery systems to improve the bioavailability of therapeutic agents that need to reach the systemic circulation. Tight junctions between adjacent epithelial cells are major absorption barriers for therapeutic agents.1 Studies of permeation enhancers have focused on modulating the tight junction barrier; ideal candidates should have potent, reversible, and nontoxic enhancing properties at the cellular membrane or cytoskeleton.2
The six-mer peptide H-FCIGRL-OH (AT1002) is a promising permeation enhancer that modulates tight junctions. AT1002 was identified from modifications of the zonula occludens toxin (Zot, 399 amino acids, 44.8 kDa), which binds to a specific receptor on the luminal membrane surface, and reversibly opens tight junctions between epithelial cells in a nontoxic manner.3–11 The Zot fragment of AT1002 (amino acid residues 288–293) enhances the transport of molecular weight markers or agents with low bioavailability such as mannitol, PEG4000, inulin, cyclosporin A, calcitonin, ritonavir, and saquinavir, with no cytotoxicity after intranasal, intraduodenal, and intratracheal administration.12–17 For example, the area under the curve (AUC0–t) and maximum concentration (Cmax) of molecular weight markers were significantly increased, respectively, by 2.35-fold and 3.57-fold for PEG-4000, and by 2.92-fold and 3.15-fold for inulin (P<0.05) following intranasal administration with 10 mg/kg AT1002.16
Continuing studies have demonstrated that the permeation-enhancing effect of AT1002 is synergistically increased by the bioadhesive polymer carrageenan. The AUC0–t and Cmax of mannitol, inulin, calcitonin, saquinavir, and ritonavir were significantly increased by 2.17-fold/3.14-fold, 6.60-fold/6.89-fold, 4.08-fold/6.06-fold, 3.51-fold/5.77-fold, and 2.48-fold/2.55-fold, respectively (P<0.01), after intranasal administration with AT1002 (5 mg/kg) in the presence of carrageenan (1%, weight per volume [w/v]).12–14 By contrast, 5 mg/kg AT1002 alone did not significantly increase the AUC0–t and Cmax of inulin, calcitonin, saquinavir, or ritonavir (1.10-fold/1.26-fold, 1.43-fold/1.49-fold, 1.02-fold/1.02-fold, and 1.06-fold/1.15-fold, respectively).12,14 It was assumed that the synergistic permeation-enhancing effect resulted from the bioadhesive activity of carrageenan, which might increase the membrane adhesion of AT1002 and drugs. In addition, the synergistic effect between AT1002 and carrageenan was due to reduced instability of AT1002 by the viscosity of carrageenan, which protected the functional state of the peptide under stressful environmental conditions. The relationship between the effects and stability of AT1002 might be supported by results that show that AT1002 is unstable in neutral to basic pH conditions and with increasing incubation time.13
An unstable peptide or protein, due to degradation or aggregation, may have reduced activity and solubility. Chemical degradation and protein aggregation may be induced by a variety of physical factors such as temperature and ionic strength, or with time;18 degradation products such as C-terminal fragments can change significantly with varying pH.19 In addition, the stability of proteins can be modified by soluble and nontoxic excipients, such as dextrose, or by changing their structural characteristics towards those of thermophilic proteins by introducing negatively charged residues at the N-cap or positively charged residues at the C-cap of the helix.18 The latter approach is effective because the helical structure of short peptides and proteins is stabilized by capping interactions between side chains and unfulfilled peptide groups at the N- and C-termini.20,21 The hypothesis of this study is that the stability and permeation-enhancing activity of AT1002 can be further enhanced by modifying the amino acids of AT1002. In the experiments described, peptide terminal modification, C-terminal amidation, and N-terminal acetylation were applied to improve the stability of AT1002 in the nasal delivery of mannitol as a paracellular marker.
Materials and methods
Materials
Peptides (>95%) of FCIGRL (H-FCIGRL-OH, NH2-FCIGRL-COOH, AT1002), FCIGRL-NH2 (H-FCIGRL-NH2, NH2-FCIGRL-CONH2, Pep1), Ac-FCIGRL (Ac-FCIGRL-OH, COCH3-NH-FCIGRL-COOH, Pep2), and Ac-FCIGRL-NH2 (Ac-FCIGRL-OH, COCH3-NH-FCIGRL-CONH2, Pep3) were purchased from Peptron (Daejeon, Republic of Korea), and stored at −70°C before use. Citric acid, dextrose, hydrochloric acid, sodium phosphate monobasic, sodium phosphate dibasic, sodium tetraborate decahydrate, trifluoroacetic acid (TFA), Dulbecco’s Modified Eagle’s Medium, nonessential amino acid solution, penicillin–streptomycin, and Hank’s balanced salt solution (HBSS) were purchased from Sigma-Aldrich Co. (St Louis, MO, USA). Methanol and acetonitrile were high-performance liquid chromatography (HPLC)-grade (Thermo Fisher Scientific, Waltham, MA, USA). All of the solutions and buffers were prepared with HPLC-grade water purified with a Zeneer Power I (Human Corp., Seoul, Republic of Korea) and cellulose nitrate membrane filters (47 mm, 0.2 μm, Whatman, Little Chalfont, UK). [3H]-mannitol (1 mCi/mL) was purchased from American Radiolabeled Chemicals (St Louis, MO, USA). Universal scintillation counting cocktail was purchased from ICN (Cost Mesa, CA, USA). Rompun for injection was purchased from Bayer Korea (Seoul, Republic of Korea). Zoletil 50 was purchased from Virbac (Carros, France). Polyethylene tubing (PE-50) was obtained from BD (Franklin Lakes, NJ, USA). All of the surgical supplies were purchased from Professional Hospital Furnishers (Punjab, Pakistan). The Caco-2 cell line was obtained from the American Type Culture Collection (Manassas, VA, USA). Trypsin–EDTA (Life Technologies Inc., Gaithersburg, MD, USA) and fetal bovine serum (HyClone Laboratories, Logan, UT, USA) were used as purchased. Transwells were purchased from Corning Inc. (Corning, NY, USA). All of the other reagents were of analytical grade or better.
Peptide stability study
Stock solutions (1 mg/mL) were prepared by dissolving each peptide in 70% methanol. Aliquots of the stock solution containing specified amounts of each peptide were immediately placed in polypropylene tubes and evaporated using a centrifugal evaporator (CVE-3000; Eyela, Tokyo, Japan). Reconstitution solution (1 mL) was added to each tube followed by vortexing for 5 seconds to yield a peptide concentration of 10 µg/mL.
The reconstitution solution was prepared with various buffer solutions and dextrose solution using a citrate–phosphate buffer containing 0.02 M citric acid and 0.02 M sodium phosphate dibasic at a pH of 2.5, 4.0, and 5.0; phosphate buffer containing 0.01 M sodium phosphate monobasic/0.02 M sodium phosphate dibasic at pH 7.4; and borate buffer containing 0.01 M hydrochloric acid/0.02 M sodium tetraborate decahydrate at pH 9.0. Dextrose solutions were prepared at concentrations of 0.5%, 2%, and 5% (w/v). The effect of incubation time on the peak area of AT1002 was assessed at every hour from 0 hour to 5 hours. The capped reaction vial tube was then placed into a tray at a constant temperature within the autosampler. Samples were periodically removed from each tube, and directly analyzed by liquid chromatography–mass spectrometry (LC-MS).
LC-MS conditions
The HPLC system (Shimadzu Corp., Kyoto, Japan) consisted of a binary pump (LC-20AD), autosampler (SIL-20A), and degasser. Aliquots (2 µL) of the reconstituted solutions were injected onto the LC column. Separation of each peptide was performed on a C18 reversed-phase column (2.1×150 mm, 3.5 µm particle size; Vydac, Hesperia, CA, USA) using gradient elution at a flow rate of 0.3 mL/minute for 15 minutes. The mobile phases were composed of acetonitrile containing 0.1% TFA (eluent A), and water containing 0.1% TFA (eluent B). Eluent A was increased from 20% to 34% for 7 minutes for the gradient program for AT1002. The outlet from the HPLC column was directly connected to the mass analyzer. Mass spectra were recorded on an LCMS-2020 mass spectrometer system (Shimadzu Corp.) equipped with an electrospray ionization (ESI) source. Nitrogen (>99.999%) produced from a GeniSys NitroGenerator (Texol Gasgen, Glasgow, Scotland) was used as the nebulizing and drying gas. The MS instrument was operated in positive-ion, selected-ion monitoring (SIM) mode with LabSolutions software (Shimadzu Corp.) under the following MS tuning conditions: desolvation line temperature of 250°C; heat block at 200°C; nebulizing gas of 1.5 L/minute; drying gas of 20 L/minute. The area of the mass peak of AT1002 in SIM chromatograms was calculated and compared to that of each control.
Peptide cytotoxicity study
Cytotoxicity was determined using a lactate dehydrogenase (LDH) bioassay (Cytotoxicity Detection KitPLUS; Hoffman-La Roche Ltd., Basel, Switzerland) as previously described.17 Briefly, Caco-2 cells were grown as monolayers at 37°C in an atmosphere of 5% CO2 and 90% relative humanity. The integrity of the cell monolayers was evaluated by measuring the transepithelial electrical resistance with an epithelial voltohmmeter (EVOM; World Precision Instruments, Sarasota, FL, USA). Caco-2 cell monolayers were incubated with AT1002, Pep1, and Pep2 in phosphate-buffered saline (PBS) for 1 hour, respectively. LDH activity was assessed as a function of time by the addition of LDH kit reagents to an aliquot of the solution in the Transwell plate at the end of each incubation period according to the manufacturer’s manual. PBS and Triton X-100 (5%) were used as the negative and positive control, respectively, and the absorbance was measured at 490 nm.
Animals
Male Sprague Dawley (SD) rats weighing 280–290 g were purchased from Orient Bio (Seongnam, Republic of Korea). Rats were housed individually in cages and allowed to acclimate for at least 2 days after arrival. Rats were fed standard rat chow and water ad libitum and maintained on a 12-hour light/dark cycle. Prior to the study, rats were fasted overnight with water ad libitum. The protocol for animal studies was approved by the Soonchunhyang University and Seoul National University Institutional Animal Care and Use Committee.
Intranasal administration of formulations with each peptide as a permeation enhancer
The formulation for [3H]-mannitol (20 μCi/kg) was obtained by mixing [3H]-mannitol with 5% dextrose solution (mannitol alone). After the addition of an appropriate amount of each peptide (2.5 mg/kg) to mannitol solution alone, peptide treatment solutions were prepared as mannitol/AT1002, mannitol/Pep1, and mannitol/Pep2. The formulation with Pep3 for nasal administration could not be prepared because Pep3 was not completely soluble in 5% dextrose solution at the concentration for nasal administration (2.5 mg/kg) to rats. It was assumed that terminally modified Pep3 increased the hydrophobic nature of the phenylalanine and leucine of AT1002. All of the dosing solutions were prepared immediately prior to intranasal administration to the femoral vein-cannulated rats. Male SD rats were anesthetized with an intramuscular injection of Zoletil 50 (20 mg/kg) and Rompun (2.3 mg/kg), and the femoral vein was cannulated using PE-50. The following formulations were prepared for intranasal administration to rats (n=3–4 per group): 1) mannitol alone; 2) mannitol/AT1002; 3) mannitol/Pep1; and 4) mannitol/Pep2. The rats were manually restrained in a supine position, and the head was tilted back slightly while the formulations were instilled into the nostrils. The dosing solutions of 35 μL per nostril were administered.24 Blood samples (250 μL) were drawn via the femoral cannula into heparinized syringes at 10, 20, 40, 60, 120, 180, 240, and 360 minutes for the mannitol study, and were immediately centrifuged (15,000× g for 10 minutes) to obtain the plasma (100 μL). Scintillation cocktail for the samples of radiolabeled mannitol was added, and samples were analyzed for radioactivity by Beckman Coulter LS6500 multipurpose scintillation counter (Brea, CA, USA).
Data analysis
The area of the peak for each peptide from LC-MS was converted into the relative peak area (%) compared to that of each peptide in buffer (pH 2.5) after 0 hours’ incubation when comparing the stability of each peptide. For intranasal administration, the amount of absorbed radiolabeled mannitol was converted into a concentration using the specific activities of serially diluted solutions of the radiolabeled stock solution. The pharmacokinetic parameters were calculated using the noncompartmental analysis WinNonlin® pharmacokinetic software package (Pharsight, Mountain View, CA, USA). The AUC0–t was calculated using the linear trapezoidal method; the Cmax of each peptide and time to reach the peak (Tmax) following intranasal administration was determined from the observed data. All of the data are expressed as the mean and standard error of the mean (mean ± SEM). The statistical significance of differences between treatments and/or controls was evaluated using Student’s t-test and one-way analysis of variance (ANOVA) followed by Dunnett’s post-hoc test where appropriate (SPSS for Windows; v12.0; SPSS, Chicago, IL, USA), and the level of significance was set at P<0.05 or P<0.01.
Results and discussion
LC-MS chromatograms of AT1002 terminally modified peptides
Figure 1 shows that each single-charged ion with a protonated molecule [M + H]+ appeared at m/z 708.4, 707.4, 750.4, and 749.4 in the full-scan spectra under positive ESI, which corresponded precisely to the molecular mass of AT1002 and the terminally modified AT1002 peptides (Pep1, Pep2, Pep3). A peptide including one cysteine can be easily oxidized to form dimerized disulfide bonds, which can result in peptide instability due to aggregation or polymerization.18,22,23 However, we did not observe dimerization of AT1002, Pep1, Pep2, or Pep3 in each full-scan spectra due to the instability of AT1002, as shown in our previous report,13 and due to its degradation in solution. We considered that peptide dimerization was not due to the hydrophobic characteristics of phenylalanine and isoleucine flanking each cysteine in AT1002, Pep1, Pep2, and Pep3.
 | Figure 1 Full-scan spectra and SIM chromatograms at each m/z of FCIGRL-NH2 (Pep1), Ac-FCIGRL (Pep2), Ac-FCIGRL-NH2 (Pep3), and FCIGRL (AT1002). |
The LC conditions were slightly modified for analysis of all peptides compared to our previous report.13 Under the described conditions, AT1002, Pep1, Pep2, and Pep3 had peak retention times of 4.9, 3.5, 8.8, and 7.7 minutes, respectively, in the SIM mode, and no interference was observed by the aqueous solutions.
Effect of pH and dextrose on stability of AT1002 terminally modified peptides
The peak areas of peptides in buffer solutions over a range of pH values (2.5–9.0) and dextrose solutions at concentrations of 0.5%, 2%, and 5% (w/v) were monitored by LC-MS. Stability was expressed as the relative peak area of each peptide, and each point was reported as a peak area ratio compared to the initial peak area of the same peptide at pH 2.5 with time 0 incubation as the control (100%±2.32% for AT1002, 100%±4.22% for Pep1, 100%±4.11% for Pep2, and 100%±4.81% for Pep3; n=3 per group). This pH was chosen because it showed the highest peak area in all peptides.
Figure 2 shows that the stability of all of the peptides was affected by pH. The peak ratios of all of the peptides decreased with increasing pH, indicating that the peptides were unstable under neutral and basic conditions. The decreasing peak ratios of AT1002 corresponded to the instability of a common peptide at neutral and basic conditions, in accordance with our previously reported results.13 Although the peak ratios of Pep2 and Pep3 exhibited decreased stability with increasing pH (to pH 7.4) compared to AT1002, there were no significant differences in the peak ratios of AT1002 at each pH point. There was increased stability of Pep1, the C-terminal-amidated peptide of AT1002, by increasing pH in parallel to AT1002, but the slope of the decrease was relatively low and the peak area ratios of Pep1 were 82.5%±3.08% and 70.2%±2.13% at pH 5.0 and pH 7.4, respectively, which was significantly higher (P<0.01) than those of AT1002 (70.5%±2.90% and 60.8%±2.50%) at pH 5.0 and 7.4, respectively, indicating that Pep1 was less sensitive to pH changes compared to AT1002.
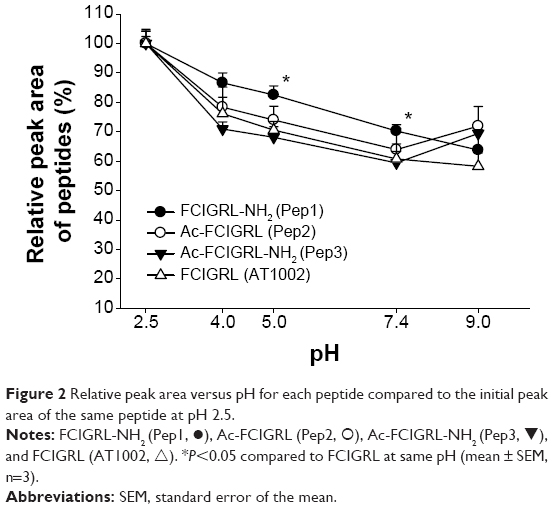 | Figure 2 Relative peak area versus pH for each peptide compared to the initial peak area of the same peptide at pH 2.5. |
The effects of dextrose on the stability of AT1002 terminally modified peptides are shown in Figure 3. Dextrose is a stabilizing excipient of proteins,19 which is used as an additive for nasal administration solutions during in vivo studies.24 Our previous report indicated that dextrose has stabilizing effects on AT1002.13 Dextrose increased the peak areas of Pep1, Pep2, and Pep3 in the same manner as AT1002, indicating that it is effective at increasing stability or at least reducing the instability of the terminally modified peptides. The relative peak areas of all of the peptides increased with increasing dextrose concentration. For example, the peak area ratios of AT1002 were 68.8%±4.85%, 79.9%±6.36%, 94.3%±7.02%, and 99.5%±2.80%, and those of Pep1 were 75.0%±4.67%, 82.9%±5.63%, 91.2%±4.84%, and 97.2%±3.12%, at dextrose concentrations of 0.0%, 0.5%, 2.0%, and 5.0% (w/v), respectively. No significant differences in relative peak areas of terminally modified peptides compared to those of AT1002 at the same concentration of dextrose were found.
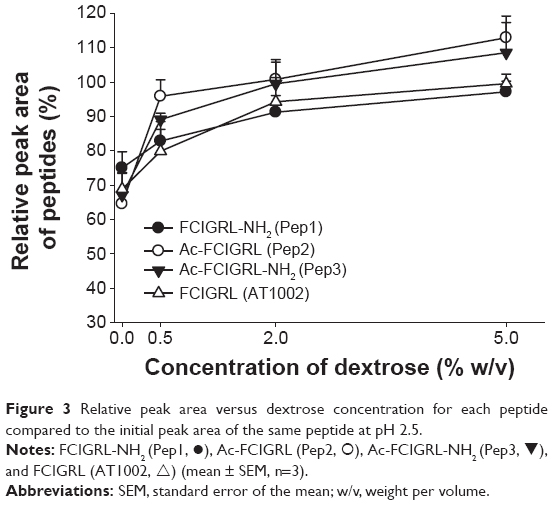 | Figure 3 Relative peak area versus dextrose concentration for each peptide compared to the initial peak area of the same peptide at pH 2.5. |
The reduced instability of terminally modified peptides by dextrose was confirmed in an incubation study of each peptide in a 5.0% dextrose solution for 5 hours (Figure 4). Pep1 showed a relatively low decrease in peak area compared to AT1002 at each incubation time point. The peak area ratios of Pep1 with 3, 4, and 5 hours’ incubation time were 96.3%±6.77%, 94.7%±5.70%, and 95.7%±5.10%, which were significantly higher than those of AT1002 with 3 hours (76.1%±1.95%), 4 hours (71.7%±3.57%), and 5 hours (68.7%±4.61%) of incubation time, respectively (P<0.05), compared to the initial peak area of the same peptide at time 0 incubation time. In addition, when comparing only the change in the peptides themselves over incubation time, Pep1 showed no significant difference in peak areas, indicating that the peptide remained stable for at least 5 hours. Consequently, the stabilizing effect of dextrose was shown to be more effective in peptides modified with C-terminal amidation.
 | Figure 4 Temporal profiles of the relative peak area of each peptide compared to the initial peak area of the same peptide at 0 hours of incubation time. |
Cytotoxicity of AT1002 terminally modified peptides
An LDH assay was used to examine the toxic effects of terminally modified AT1002 peptides on epithelial cells. An increase in LDH released from the cytosol of cells resulted from an increase in dead or plasma membrane–damaged cells. Caco-2 cell monolayers were incubated with AT1002, Pep1, and Pep2 in PBS for 1 hour. The viability of cells was not significantly different for each peptide compared to the PBS negative control (Figure 5). These results suggest that the terminally modified peptides of AT1002 did not produce a cytotoxic response to Caco-2 cells during the incubation time. AT1002 was also not cytotoxic, which correlates with our previous results.17
 | Figure 5 The LDH assay for FCIGRL (AT1002), FCIGRL-NH2 (Pep1), Ac-FCIGRL (Pep2), PBS (negative control), and Triton X-100 (positive control) at 1 hour incubation time in Caco-2 cell monolayers. |
Intranasal administration of mannitol with AT1002 terminally modified peptides
The formulations of [3H]-mannitol in 5% dextrose solution with or without AT1002, Pep1, and Pep2 (2.5 mg/kg) were administered intranasally to femoral vein-cannulated SD male rats.
The pharmacokinetic profiles were characterized for each formulation, and the parameters were calculated using noncompartmental analysis. The mean (± SEM) plasma concentration versus time profiles following the intranasal administration of mannitol formulations are shown in Figure 6. Significantly higher permeation of mannitol was found in the formulations of mannitol/Pep1 compared to the control formulation of mannitol alone after 40 minutes, indicating a significant enhancement in the intranasal absorption of mannitol with Pep1 (P<0.01). The administration of mannitol with Pep1 produced a significant increase (3.63-fold) in AUC0–360 minutes (22.08±3.54 min ng/mL) and an increase (2.68-fold) in Cmax (82.06±12.10 pg/mL) over the control formulation of mannitol alone (AUC0–360 minutes =6.08±1.33 min ng/mL, Cmax =30.58±6.02 pg/mL; P<0.01; Table 1). However, there were no significant differences in mannitol concentration at each time point between the formulations of mannitol/AT1002, mannitol/Pep2, and mannitol alone. The enhancement of mannitol by Pep1 was in accordance with no statistically significant difference over all of the incubation times in the peak areas of Pep1 compared to AT1002 or Pep2 (Figure 4), suggesting that C-terminal amidation of AT1002 increased the stability or at least reduces the instability of AT1002, which exposed the inherent permeation-enhancing effect of AT1002.
 | Figure 6 Average plasma concentration of [3H]-mannitol versus time in femoral vein-cannulated SD rats following the intranasal administration of each peptide formulation. |
 | Table 1 Pharmacokinetic parameters in femoral vein-cannulated SD rats after intranasal administration of [3H]-mannitol (20 μCi/kg) with AT1002 and terminally modified AT1002 (2.5 mg/kg) |
In addition, the enhancement of mannitol by Pep1 was more pronounced at lower doses of Pep1 and with the absence of carrageenan, which promoted the effect of AT1002. The permeation-enhancing effect of Pep1 further increased the AUC0–360 minutes and Cmax of mannitol (20 μCi/kg) despite the lower dose of Pep1 (2.5 mg/kg). These data were in contrast to those from our previous study, which showed that AT1002 (5 mg/kg) increased the absorption of mannitol (40 μCi/kg) with a 2.17-fold increase in AUC0–360 minutes and a 3.14-fold increase in Cmax compared to the control of mannitol/carrageenan, and compared to the study of mannitol without carrageenan, which showed that the permeation of mannitol did not change or increase with AT1002 (5 mg/kg).13 Enhanced mannitol permeation with Pep1 was comparable to that seen with the delta G fragment of Zot (amino acid residues 265–399) when the differences in the administration dose and route were disregarded. Delta G enhanced the transport of mannitol (40 μCi/kg) by 2.57-fold of AUC0–360 minutes and 2.00-fold of Cmax, respectively, after intraduodenal administration to rats.25 Therefore, these data indicate that the C-terminal amidation of AT1002 increased the intranasal permeation of mannitol.
Conclusion
Terminal modification of AT1002 affected the stability and effectiveness of paracellular permeation enhancement. Pep1 is a C-terminal-amidated peptide of AT1002 that is nontoxic to cell monolayers and showed a relatively low decrease in peak area compared to AT1002 in an incubation study. Furthermore, Pep1 produced a statistically significant increase in AUC0–360 minutes (by 3.63-fold) and Cmax (by 2.68-fold) compared to the control in the nasal administration of mannitol, whereas no significant difference in the mannitol concentration was observed for the formulations of mannitol/AT1002 or mannitol/Pep2 compared to the control of mannitol alone. Thus, Pep1 increased the stability or at least reduced the instability of AT1002, which exposed the inherent permeation-enhancing effect of AT1002. These results suggest the potential usefulness of C-terminal-amidated AT1002 in enhancing nasal drug delivery, and may lead to the development of a practical drug delivery technology for drugs with low bioavailability.
Acknowledgment
This research was supported by the Soonchunhyang University Research Fund.
Disclosure
The authors report no conflicts of interest in this work.
References
Davis SS, Illum L. Absorption enhancers for nasal drug delivery. Clin Pharmacokinet. 2003;42(13):1107–1128. | ||
Saaber D, Wollenhaupt S, Baumann K, Reichl S. Recent progress in tight junction modulation for improving bioavailability. Expert Opin Drug Discov. 2014;9(4):367–381. | ||
Cox DS, Raje S, Gao H, Salama NN, Eddington ND. Enhanced permeability of molecular weight markers and poorly bioavailable compounds across Caco-2 cell monolayers using the absorption enhancer, zonula occludens toxin. Pharm Res. 2002;19(11):1680–1688. | ||
Cox DS, Gao H, Raje S, Scott KR, Eddington ND. Enhancing the permeation of marker compounds and enaminone anticonvulsants across Caco-2 monolayers by modulating tight junctions using zonula occludens toxin. Eur J Pharm Biopharm. 2001;52(2):145–150. | ||
Lu R, Wang W, Uzzau S, Vigorito R, Zielke HR, Fasano A. Affinity purification and partial characterization of the zonulin/zonula occludens toxin (Zot) receptor from human brain. J Neurochem. 2000;74(1):320–326. | ||
Fasano A. Innovative strategies for the oral delivery of drugs and peptides. Trends Biotechnol. 1998;16(4):152–157. | ||
Fasano A. Modulation of intestinal permeability: an innovative method of oral drug delivery for the treatment of inherited and acquired human diseases. Mol Genet Metab. 1998;64(1):12–18. | ||
Fasano A. Novel approaches for oral delivery of macromolecules. J Pharm Sci. 1998;87(11):1351–1356. | ||
Fasano A, Uzzau S, Fiore C, Margaretten K. The enterotoxic effect of zonula occludens toxin on rabbit small intestine involves the paracellular pathway. Gastroenterology. 1997;112(3):839–846. | ||
Fasano A, Uzzau S. Modulation of intestinal tight junctions by Zonula occludens toxin permits enteral administration of insulin and other macromolecules in an animal model. J Clin Invest. 1997;99(6):1158–1164. | ||
Fasano A, Fiorentini C, Donelli G, et al. Zonula occludens toxin modulates tight junctions through protein kinase C-dependent actin reorganization, in vitro. J Clin Invest. 1995;96(2):710–720. | ||
Song KH, Eddington ND. The impact of AT1002 on the delivery of ritonavir in the presence of bioadhesive polymer, carrageenan. Arch Pharm Res. 2012;35(5):937–943. | ||
Song KH, Eddington ND. The influence of stabilizer and bioadhesive polymer on the permeation-enhancing effect of AT1002 in the nasal delivery of a paracellular marker. Arch Pharm Res. 2012;35(2):359–366. | ||
Song KH, Eddington ND. The influence of AT1002 on the nasal absorption of molecular weight markers and therapeutic agents when co-administered with bioadhesive polymers and an AT1002 antagonist, AT1001. J Pharm Pharmacol. 2012;64(1):30–39. | ||
Gopalakrishnan S, Pandey N, Tamiz AP, et al. Mechanism of action of ZOT-derived peptide AT-1002, a tight junction regulator and absorption enhancer. Int J Pharm. 2009;365(1–2):121–130. | ||
Song KH, Fasano A, Eddington ND. Enhanced nasal absorption of hydrophilic markers after dosing with AT1002, a tight junction modulator. Eur J Pharm Biopharm. 2008;6(1):231–237. | ||
Song KH, Fasano A, Eddington ND. Effect of the six-mer synthetic peptide (AT1002) fragment of zonula occludens toxin on the intestinal absorption of cyclosporine A. Int J Pharm. 2008;351(1–2):8–14. | ||
Wang W. Instability, stabilization, and formulation of liquid protein pharmaceuticals. Int J Pharm. 1999;185(2):129–188. | ||
Schrier JA, Kenley RA, Williams R, et al. Degradation pathways for recombinant human macrophage colony-stimulating factor in aqueous solution. Pharm Res. 1993;10(7):933–944. | ||
Forood B, Feliciano EJ, Nambiar KP. Stabilization of alpha-helical structures in short peptides via end capping. Proc Natl Acad Sci U S A. 1993;90(3):838–842. | ||
Lyu PC, Wemmer DE, Zhou HX, Pinker RJ, Kallenbach NR. Capping interactions in isolated alpha helices: position-dependent substitution effects and structure of a serine-capped peptide helix. Biochemistry. 1993;32(2):421–425. | ||
Shahrokh Z, Eberlein G, Buckley D, et al. Major degradation products of basic fibroblast growth factor: detection of succinimide and iso-aspartate in place of aspartate. Pharm Res. 1994;11(7):936–944. | ||
Wang YJ, Shahrokh Z, Vemuri S, Eberlein G, Beylin I, Busch M. Characterization, stability, and formulation of basic fibroblast growth factor. In: Pearlman R, Wang YJ, editors. Formulation, Characterization, and Stability of Protein Drugs. New York, NY: Plenum Press; 1996:141–180. | ||
Sharp PE, La Regina MC. The Laboratory Rat. Boca Raton, FL: CRC Press; 1998. | ||
Salama NN, Fasano A, Lu R, Eddington ND. Effect of the biologically active fragment of zonula occludens toxin, delta G, on the intestinal paracellular transport and oral absorption of mannitol. Int J Pharm. 2003;251(1–2):113–121. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.