Back to Journals » International Journal of Nanomedicine » Volume 10 » Issue 1
Para amino benzoic acid-derived self-assembled biocompatible nanoparticles for efficient delivery of siRNA
Authors Reddy TL, Krishnarao PS, Rao GK, Bhimireddy E, Venkateswarlu P, Mohapatra D, Yadav JS, Bhadra U, Pal Bhadra M
Received 7 April 2015
Accepted for publication 23 July 2015
Published 12 October 2015 Volume 2015:10(1) Pages 6411—6424
DOI https://doi.org/10.2147/IJN.S86238
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Thomas Webster
Teegala Lakshminarayan Reddy,1,5 P Sivarama Krishnarao,2 Garikapati Koteswara Rao,1,5 Eswar Bhimireddy,2 P Venkateswarlu,4 Debendra K Mohapatra,2,5 JS Yadav,2 Utpal Bhadra,3 Manika Pal Bhadra1,5
1Centre for Chemical Biology, CSIR-Indian Institute of Chemical Technology, 2Natural Products Chemistry Division, CSIR-Indian Institute of Chemical Technology, 3Functional Genomics and Gene Silencing Group, CSIR-Indian Institute of Chemical Technology, CSIR-Centre for Cellular and Molecular Biology, Hyderabad, 4Department of Chemistry, Sri Venkateswara University, Tirpupati, 5Academy of Scientific and Innovative Research, New Delhi, India
Abstract: A number of diseases can result from abnormal gene expression. One of the approaches for treating such diseases is gene therapy to inhibit expression of a particular gene in a specific cell population by RNA interference. Use of efficient delivery vehicles increases the safety and success of gene therapy. Here we report the development of functionalized biocompatible fluorescent nanoparticles from para amino benzoic acid nanoparticles for efficient delivery of short interfering RNA (siRNA). These nanoparticles were non-toxic and did not interfere with progression of the cell cycle. The intrinsic fluorescent nature of these nanoparticles allows easy tracking and an opportunity for diagnostic applications. Human Bcl-2 siRNA was complexed with these nanoparticles to inhibit expression in cells at both the transcriptional and translational levels. Our findings indicated high gene transfection efficiency. These biocompatible nanoparticles allow targeted delivery of siRNA, providing an efficient vehicle for gene delivery.
Keywords: biocompatibility, nanoparticles, siRNA, cell uptake, endocytosis, gene silencing
Introduction
RNA interference (RNAi) is a cellular process whereby the expression of a particular gene is silenced by small RNAs. These small RNAs, also known as short interfering RNAs (siRNA), are capable of turning off a gene and thereby stopping the production of specific proteins, making them a new class of therapeutic drugs.1 This strategy has significant potential for the treatment of human disease. Considerable effort has gone into developing therapeutics based on RNAi, and a variety of siRNA-based therapeutics has shown great promise for the treatment of disease.2 However, a major obstacle continues to be safe and effective delivery of siRNA to target tissues. Several factors play an important role in delivery of siRNA, including chemical modification of the delivery vehicle and its biological response to the sequence and structure of siRNA.3,4 To achieve successful silencing, the siRNA must not be degraded in the extracellular environment and needs to be able to reach the cell surface and cross the cell membrane to enter the RNA-induced silencing complex.
Although several delivery vehicles have been developed to deliver siRNA therapeutics, they have all shown limitations in terms of target availability, stability, safety, and biodegradability. Delivery of siRNA can have both intended and unintended consequences, such as toxicity and off-target silencing. Viral vectors5 (both retroviral and adenoviral) are the most commonly used gene delivery vehicles and are in clinical trials. These vectors have high efficiency and undergo tissue-specific replication, which is helpful in clinical situations. However, their applicability is limited due to safety issues6 concerning immunogenicity7 and carcinogenicity induced by mutagenesis resulting from insertion of a gene into the host genome.8 They have limited practical utility due to their poor ability to incorporate therapeutic genes, and their high production cost adds an extra burden. Non-viral vectors have been investigated as alternatives, and have useful characteristics, such as easy synthesis enabling large-scale production, enhanced biosafety, and pharmaceutical advantages. Moreover, synthetic vectors show less efficiency for in vivo delivery compared with viral vectors.9 Cationic lipid-based10,11 and cationic polymer-based12,13 delivery vehicles bind electrostatically with the negatively charged phosphate groups of nucleic acids in a reversible and non-reversible manner, which condense the nucleic acid and form virus-like structures protecting them from degradation by nucleases.14
Lipoplexes15 are non-covalent complexes formed between cationic lipids and negatively charged nucleic acids.16 Lipids are generally formed by spontaneous aggregation of cationic surfactants, where positively charged polar head groups and hydrophobic moieties form hydrophobic interiors, resulting in a polycationic surface. After uptake by cells, these lipoplexes are able to lyse into endocytotic vesicles, leading to disruption of the membrane and simultaneously destabilized lipoplexes breakdown to release the nucleic acids efficiently into the cytoplasm.17 However, these cationic lipids are relatively unstable in a physiological (ie serum-containing) environment, which markedly limits their application in vivo. In vitro and in vivo studies published by various research groups in recent years have shown that a wide range of nanoparticles (NPs) can deliver siRNA, resulting in silencing of specific genes.18–20 Several functionalization methods for conjugation of siRNA with the delivery vehicles have also been reported, giving a better understanding of the delivery process and effective silencing.21 Self-assembly of non-toxic biomaterials into nanoscale structures is an efficient way of delivering siRNA to cells. Incorporation of cleavable bonds as part of the functionalization for appropriate release of biological molecules from the nanomaterials is crucial for functionality in the biological milieu.22
Here we present a series of novel self-assembled, biocompatible, nanosized delivery vehicles derived from para amino benzoic acid (PABA), which is a component of folic acid and the vitamin B complex normally present in the human liver. It is also a structural moiety in drugs with a wide range of applications.23 These NPs are non-toxic, biocompatible, and fluorescent in nature. Their ability to fluoresce helps in simultaneous tracking in both in vitro and in vivo environments, making them an appropriate vehicle for diagnostic imaging. Appropriate functionalization of nanomaterials with siRNA enables efficient intracellular transport and delivery of siRNA to target tissue expressing the target gene and achieves highly effective silencing by inhibiting gene expression. In a previous report, we demonstrated the biocompatibility of siRNA in vitro and in vivo.24 This present comparative study using existing delivery vehicles demonstrates even better silencing in in vitro conditions.
Materials and methods
Materials
Ethical approval and informed consent was not sought as the cell lines were procured from a commercial source. N-Boc-protected glycine, 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDCI), hydroxybenzotriazole (HOBt), N,N′-dicyclohexylcarbodiimide, N, N-dimethylpyridin-4-amine, trifluoroacetic acid, and PABA were purchased from Sigma-Aldrich (St Louis, MO, USA). 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) was purchased from Life Technologies (Invitrogen, Carlsbad, CA, USA). siRNA was synthesized and validated by Dharmacon (La Fayette, CO, USA).
Bcl-2 siRNA sequence sense 5′-GUG CCU AUC UGG GCC ACA ATT antisense 5′-UUG UGG CCC AGA UAG GCA CTT. Anti-red fluorescent protein (RFP) siRNA sequence: sense 5′-GGAGUUCAUGCGUUCAAG-dTdT antisense 5′-CUUGAAGCGCAUGAACUCC-dTdT. Cy3-labelled scrambled siRNA. Custom-designed Bcl-2 and β-actin polymerase chain reaction (PCR) primers were synthesized by Eurofins, (New Orleans, LA, USA). Bcl-2 primer sequence forward primer 5′-GGG GAG AAG GT G TTC ATT CA-3′, reverse primer 5′-CAA CTC TTT TCC TCC CAC CA-3′ β-actin primer sequence forward primer 5′-AGA GGG AAA TCG TGC GTG AC-3′, reverse primer 5′-CAATA GTGAT GACCT GGCCGT-3′.
RNeasy kits, QiaShredder spin columns, and negative control RNA were sourced from Qiagen (Hilden, Germany). Superscript III First-Strand synthesis kits, Lipofectamine RNAiMax, Dulbecco’s Modified Eagle’s Medium, Pure Link RNase, RNasin RNase inhibitor, and diethylpyrocarbonate (DEPC)-treated water were obtained from Life Technologies. Power SYBR Green I Mastermix (2×) was purchased from Applied Biosystems (Foster City, CA, USA) and used according to the manufacturer’s instructions. Rabbit monoclonal primary antibody for Bcl-2 and mouse monoclonal antibody β-actin were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA) and Abcam (Cambridge, MA, USA), respectively. Horseradish peroxidase-conjugated anti-rabbit and anti-mouse secondary antibodies were purchased from Santa Cruz Biotechnology. A HeLa human cervical cancer cell line was sourced from the American Type Culture Collection (Manassas, VA, USA) and an RFP-stable MDA-MB-231 reporter cell line was purchased from Cell Biolabs (San Diego, CA, USA).
Synthesis and self-assembly of NPs
We synthesized the target designed molecule 4-decanamido-N-(6-(2-guanidinoacetamido)pyridin-2-yl)benzamide hydrochloride and its analogs starting from commercially available PABA (1). Our method of synthesis involved conversion of PABA (1) to its methyl ester using acetyl chloride in methanol,25 and the amine hydrochloride (2) was coupled with acids of various chain lengths (C10, C12, C14, C16, C18, and C18 unsaturated) using EDCI and HOBt in dichloromethane to produce compound (3a)26 (C10) in 92% yield over two steps (Figure 1). The methyl ester on subsequent hydrolysis with LiOH yielded compound (4a).27 The resulting acid was coupled with 2, 6-diaminopyridine using EDCI and HOBt in dichloromethane to obtain compound (5a)28 in 91% yield over two steps. The free amine was coupled with N-Boc-protected glycine using N,N′-dicyclohexylcarbodiimide and N, N-dimethylpyridin-4-amine in dichloromethane to obtain Boc-protected compound (6a)29 in 89% yield. The Boc group was deprotected using trifluoroacetic acid and the amine was coupled with N, N-bis-tert-butyloxycarbonyl-S-methyl-isothiourea and DIPEA as a base to obtain compound (7a)30,31 in 68% yield. Finally, the designed molecule 4-decanamido-N-(6-(2-guanidinoacetamido) pyridin-2-yl) benzamide hydrochloride (8a) was obtained in 73% yield by deprotection of the Boc groups with bubbling HCl gas in methanol (Figure 1). All compounds were subjected to self-assembly of NP formation. In each case, to obtain a self-assembled nanostructure, 1 mg of compound (8a–f) was added to 2 mL of methanol and heated at 60°C until it dissolved completely. Next, 2 mL of deionized water was mixed slowly at the same temperature to obtain a pure white solution, which on slow cooling at room temperature formed cotton dust-like white aggregates. These aggregates were isolated by centrifugation at 4,500 rpm for 20 minutes, followed by overnight drying at 60°C to afford 0.5 mg of final nanomaterial.24
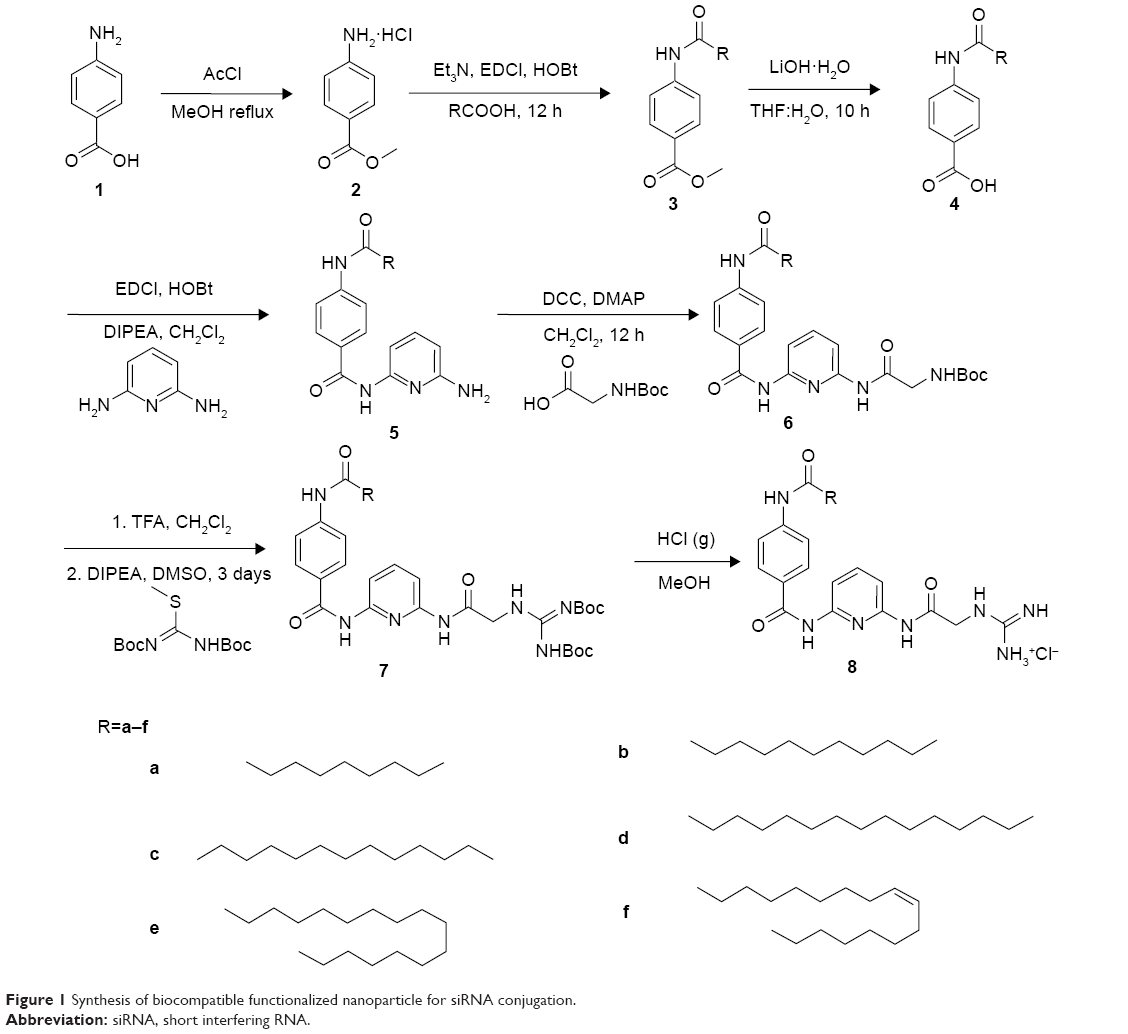 | Figure 1 Synthesis of biocompatible functionalized nanoparticle for siRNA conjugation. |
Particle size distribution and zeta potential
The hydrodynamic radii of the NPs and NP-siRNA complexes (also known as nanoplexes) were analyzed by dynamic light scattering (DLS) performed on a DynaPro Nano DLS system (Wyatt Technologies, Santa Barbara, CA, USA).32–34 The NP solutions were suitably diluted (2 mg/mL) with double-distilled water to analyze the particle size distribution and zeta potential. A Zetasizer (Malvern, UK) was used to determine the zeta potential of the NPs and nanoplexes. All measurements were performed at a fixed angle of 90° at room temperature (25°C). The results were expressed as the size ± standard deviation and zeta potential ± standard deviation.
Scanning electron microscopy
The morphological characteristics of the NPs were studied by scanning electron microscopy (Hitachi S-3400N, Tokyo, Japan). Briefly, a liquid sample was placed on a coverslip, dried, coated with gold, and analyzed under the microscope.
Analysis of cell cycle by flow cytometry
HeLa cells were seeded at a density of 2×105 cells/well of a six-well plate and allowed to attach for 24 hours. The cells were then treated with NPs and incubated for 24 hours at 37°C in a standard incubator. The cells were trypsinized, harvested, and centrifuged using a microcentrifuge at 1,500 rpm for 4 minutes. The cell pellets were washed twice with phosphate-buffered saline (PBS) and fixed in 75% ethanol solution at 4°C followed by centrifugation, and then washed again in PBS. They were again resuspended in PBS containing 5 mg/mL propidium iodide and 50 mg/mL deoxyribonuclease-free ribonuclease A. The suspension was incubated in the dark for 25 minutes, after which the cell cycle patterns were analyzed using flow cytometry.
Apoptosis detection assay
HeLa cells were seeded at a density of 2×105 cells/well of a six-well plate and allowed to attach for 24 hours. The cells were treated with NPs and incubated for 24 hours at 37°C in a standard incubator followed by trypsinization and washed in cold PBS. The cells were resuspended in 200 μL of 1× binding buffer containing 5 μL of Annexin V and 10 μL of propidium iodide for 15 minutes at room temperature in the dark according to the manufacturer’s instructions (ApoAlert™ Annexin V-FITC apoptosis kit; Clontech Laboratories Inc, Mountain View, CA, USA). Apoptotic cells were measured on a flow cytometer (Beckman Coulter, Brea, CA, USA). A total of 10,000 gated events were acquired from each sample. The results are expressed as the percentage of apoptotic cells (propidium iodide-positive and Annexin V-positive) in the gated cell population.
siRNA binding and gel retardation assay
NPs were diluted with DEPC water at different concentrations. Next, 1 μg of siRNA (siBcl-2 or siNonsense) in DEPC water was mixed with an equal volume of NPs by gentle pipetting. The resulting nanoplex was incubated at room temperature for 20 minutes before loading onto the gel. The electrophoretic mobility of the nanoplex was visualized using an ultraviolet illuminator with ethidium bromide staining after electrophoresis on 2% (w/v) agarose gel for 20 minutes at 80 V in TAE buffer (40 mM Tris-HCl, 1% v/v acetic acid, 1 mM EDTA).
RNAse protection assay
To prevent digestion of siRNA by endonucleases, we performed a nuclease protection assay, where samples of siRNA and the siRNA nanoplexes were treated with RNase-A for 30 minutes. The samples were then treated with 8 U of RNase-A inhibitor at 37°C for 15 minutes and run on 2% agarose gel for 20 minutes at 80 V in TAE buffer (40 mM Tris-HCl, 1% v/v acetic acid, 1 mM EDTA). The gel was visualized under an ultraviolet illuminator after staining with ethidium bromide.35
Cell culture
Two human cancer cell lines, HeLa and MDA-MB-231/RFP cells, were selected and grown in Dulbecco’s Modified Eagle’s Medium (Sigma-Aldrich) supplemented with 10% fetal bovine serum and common antibiotics (penicillin, kanamycin, and streptomycin) at a 1× concentration. The cells were routinely maintained in a standard humidified 5% CO2 atmosphere at 37°C and subcultured at 3-day intervals.
Confocal microscopy
Confocal laser scanning microscopy (CLSM) analysis was carried out to identify the presence of NPs within the cells. HeLa cells were grown on coverslips for 24 hours, then transfected with NP-Cy3-labelled siRNA for 12 hours. Next, the cells were washed with PBS and fixed with 4% paraformaldehyde for 20 minutes. The cells were then mounted with Vectashield mounting media (Vector Laboratories Inc, Burlingame, CA, USA) and observed under an FV1000 confocal laser scanning microscope (Olympus, Japan).
siRNA transfection
Nanoplexes were prepared at different NP ratios as described earlier. After incubating the cells (seeded in six-well plates) with the complexes for 4 hours, the medium in the wells was exchanged with fresh medium to remove the free complexes. The cells were then incubated for a further 44 hours, and the cellular levels of Bcl-2 mRNA and Bcl-2 protein were determined using real-time PCR and Western blotting, respectively.
Investigation of uptake mechanism (endocytosis)
Endosomal staining with LysoTracker Red DND-99
Cells were grown on cover slips for 24 hours and then transfected with NPs. Six hours post transfection the medium was removed, and the cells were washed with PBS and incubated for 1 hour with LysoTracker Red DND-99 (50 nmol/L), a marker of endolysosomal compartments. The cells were then washed with PBS and mounted using mounting medium consisting of DAPI nuclear stain and imaged by confocal microscopy (excitation 577 nm, emission 590 nm).
Low temperature incubation at 4°C
After transfection with nanoplexes, the cells were incubated at 4°C instead of the usual 37°C for 4 hours.
Incubation of cells with nanoplexes under ATP depletion
The cells grown on cover slips were preincubated in PBS containing 10 mM NaN3 and 50 mM 2-deoxy-d-glucose for 30 minutes at 37°C, followed by incubation in a solution with NP-siRNA (Cy3-siRNA) complexes.36
Hypertonic incubation
Cells were preincubated for 30 minutes in PBS containing 0.45M sucrose before exposure to NP-siRNA (Cy3-siRNA) complexes at 37°C.36
Potassium-free incubation
Cells were preincubated with potassium-free buffer consisting of HEPES N-(2-hydroxyethyl)piperazine-N-[2-ethanesulfonic acid], NaCl, MgCl2, and d-glucose for 30 minutes at 37°C prior to exposure with BP-siRNA (Cy3-siRNA) complexes.36
Real-time PCR
Each sample was derived from a single well of a six-well plate. Every condition was performed in duplicate to ensure the reproducibility of the results. Total RNA was isolated from each well using the RNeasy kit, and reverse transcribed using oligo-dTs as primers, according to the manufacturer’s instructions. The products were then amplified using gene-specific primers for Bcl-2 (target gene) and β-actin (endogenous control). Custom primers were purchased for both Bcl-2 and β-actin. All primers were used at a concentration of 5 pM, with 2 μL of primer set (sense plus antisense) in 10 μL of PCR reaction solution containing 5 μL of 2× SYBR Green Mastermix, 1 μL of DEPC-treated water, and 2 μL of the sample. Real-time PCR was performed using an AB7900HT system (Applied Biosystems), with cycling conditions as follows: one cycle of 95°C for 10 minutes (to “hot-start” reagents in Mastermix); 40 cycles of 95°C for 30 seconds, 60°C for 30 seconds, 72°C for 30 seconds; one cycle of 72°C for 7 minutes for final extension; ramp from 72°C to 95°C, and 1 degree change per step, with a 5-second interval between steps. Downregulation of Bcl-2 mRNA was determined by comparison of the ratio between Bcl-2 and β-actin mRNA concentrations for the treated samples against that of the untreated sample using the ΔΔCT method.
Western blot analysis
To assay for changes in Bcl-2 protein levels, non-transfected and NP-siRNA complex-transfected cells were collected and lysed in RIPA lysis buffer. The cell lysate was collected by centrifugation. Total protein concentration was determined using a Bradford micro protein assay protocol (Sigma-Aldrich). Next, 30 μg of total protein from each sample were loaded on each well of 10% sodium dodecyl sulfate-polyacrylamide gel and electrophoresed. The proteins were transferred to polyvinylidene difluoride membranes and blocked with 5% non-fat dry milk (Santa Cruz Biotechnology) in Tris-buffered saline with Tween 20 for 1 hour. The blots were hybridized overnight at 4°C with primary monoclonal anti-Bcl-2 (Santa Cruz Biotechnology) and anti-β-actin (Abcam) antibodies, followed by incubation with horseradish peroxidase-conjugated anti-rabbit and anti-mouse secondary antibodies (Santa Cruz Biotechnology). Levels of proteins were detected using Crescendo chemiluminiscent detection reagents (Millipore, Billerica, MA, USA) and visualized using a UVP Biospectrum 810 imaging system (Ultra Violet Products Ltd, Cambridge, UK). Protein expression in each sample was quantified by densitometry using UVP software, normalized to β-actin levels, and then expressed relative to the non-transfected controls. Histograms were drawn.
Results and discussion
Synthesis and characterization of NPs
PABA molecules were synthesized starting with PABA using the afore-mentioned methods, and characterized using 1H-NMR and 13C-NMR (Figures S4 and S5), mass spectroscopy, and infrared spectroscopy. The synthesized compounds were then allowed to self-assemble. The PABA nanomaterials thus obtained from compound (8a–f) were named G-10, G-12, G-14, G-16, G-18, and G-18u respectively, based on the length of the side chains and unsaturated moieties coupled during synthesis. To characterize the physiochemical properties of the functionalized NPs (G10, G12, G14, G16, G18, and G18U) and to examine their structural and spectral properties, freshly prepared NPs were dispersed in water. All the NPs were readily soluble and stable in water. An important consideration in siRNA delivery is the overall size of the NP-siRNA complexes. We performed scanning electron microscopy and DLS to observe the morphology and size of the NPs and when complexed with siRNA. The NPs and NP-siRNA complexes dispersed readily and formed well defined structures (Figure 3). DLS studies confirmed the NPs to have an overall size range of 100–200 nm (Table 1). Confocal microscopic images showed that the six nanostructures containing a guanidine modification and bearing saturated or unsaturated acid side chains exhibited intrinsic green fluorescence.
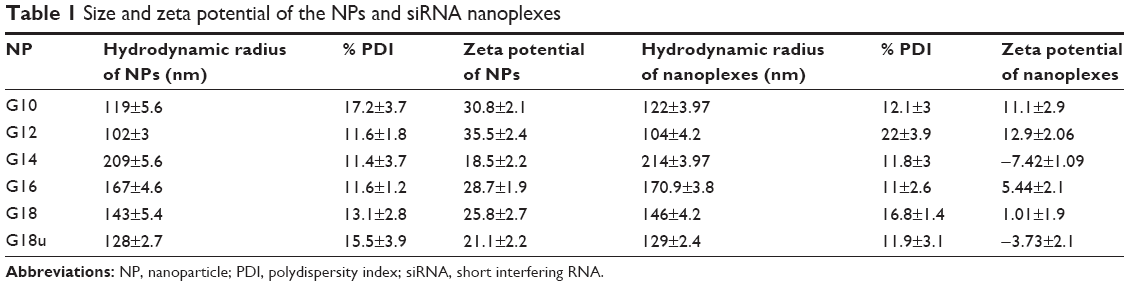 | Table 1 Size and zeta potential of the NPs and siRNA nanoplexes |
As our functionalization scheme was mainly designed to deliver biological material such as siRNA to cells, we performed a series of experiments to confirm electrostatic interaction between siRNA and the NPs. We measured the zeta potential of the NPs and when complexed with siRNA. The NPs were all positively charged (Table 1). However, when the NPs were bound to siRNA, the zeta potential was reduced (Table 1, Figures S1 and S2), confirming interaction of the NPs with siRNA via electrostatic interaction (Table 1).
Binding of siRNA to NPs
To determine the amount of siRNA bound to the NPs, a series of dispersion was prepared using a fixed (1 μg) concentration of siRNA solution in water with increasing concentration of NPs (1–20 μg). A sample of siRNA (without NPs) served as the control (Figure 2A). To confirm the binding of NPs with siRNA, we a performed gel retardation assay. NP-siRNA complexes was prepared (1:20 w/w) and electrophoresed on 2% agarose gel containing ethidium bromide (Figure 2B). The gel was visualized using a Gel Doc system (Bio-Rad Laboratories Inc, Hercules, CA, USA). All the NPs showed significant interaction with siRNA. As expected, the siRNA migrated to the bottom of the gel. In contrast, all of the NPs complexed to fixed amounts of siRNA were blocked in the gel slots at the top, confirming that migration of siRNA was prevented due to formation of complexes with NPs (Figure 2A and 2B). We also quantified the amount of free (unbound) siRNA using Quant-IT Ribogreen reagent and found only trace amounts (<10%).37,38
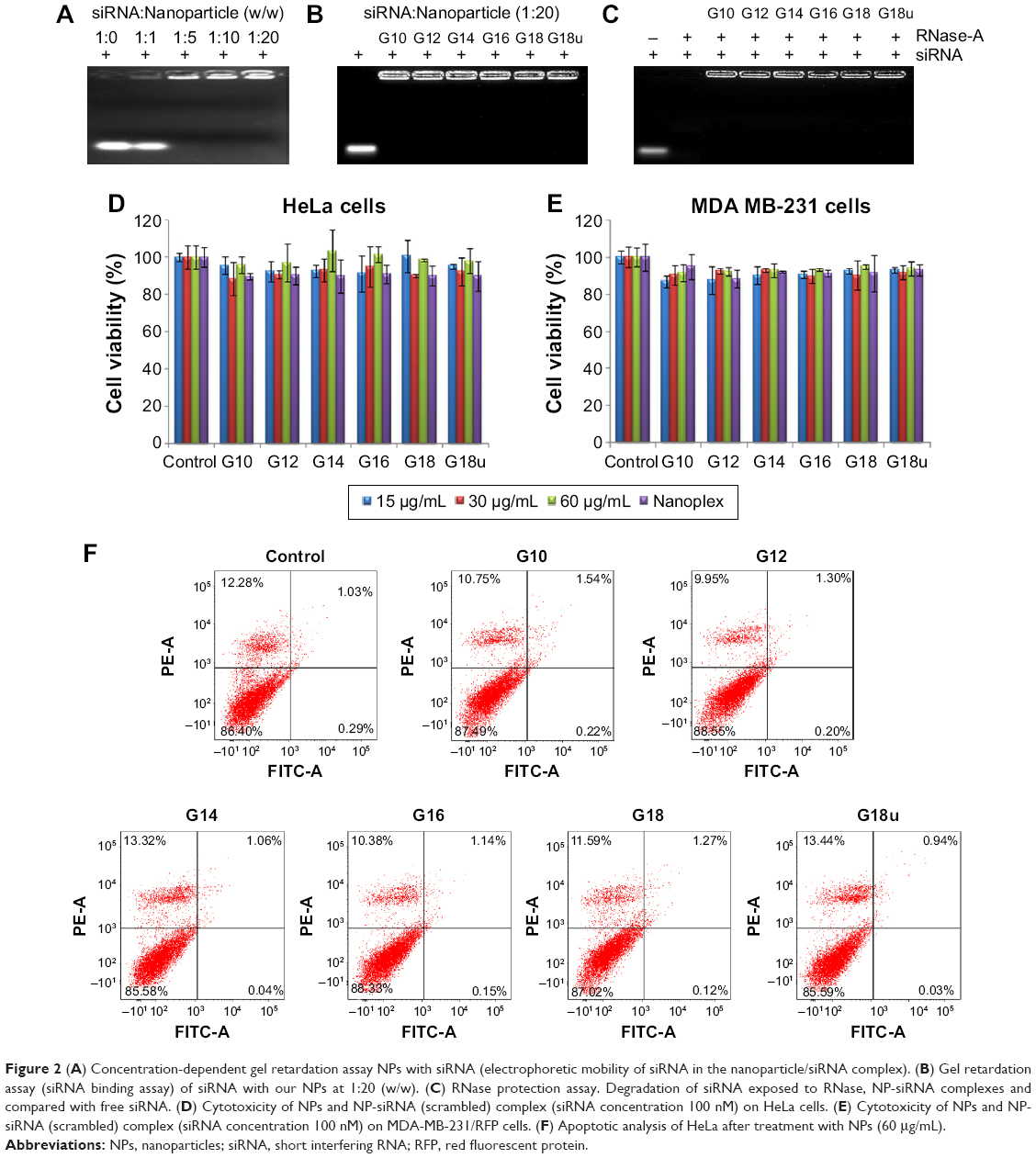 | Figure 2 (A) Concentration-dependent gel retardation assay NPs with siRNA (electrophoretic mobility of siRNA in the nanoparticle/siRNA complex). (B) Gel retardation assay (siRNA binding assay) of siRNA with our NPs at 1:20 (w/w). (C) RNase protection assay. Degradation of siRNA exposed to RNase, NP-siRNA complexes and compared with free siRNA. (D) Cytotoxicity of NPs and NP-siRNA (scrambled) complex (siRNA concentration 100 nM) on HeLa cells. (E) Cytotoxicity of NPs and NP-siRNA (scrambled) complex (siRNA concentration 100 nM) on MDA-MB-231/RFP cells. (F) Apoptotic analysis of HeLa after treatment with NPs (60 μg/mL). |
Protection of siRNA from endonucleases
To prevent digestion of siRNA by endonucleases, we performed a nuclease protection assay where samples of siRNA and NP-siRNA complexes were treated with RNase-A followed by agarose gel electrophoresis. As expected, RNase-A digested the siRNA, showing no visibility in the lane after ethidium bromide staining. This result confirmed that the siRNA in the NP-siRNA complexes is protected from endonucleases (Figure 2C).
Biocompatibility of NPs and nanoplexes
Next, we examined the toxicity of the NPs and NP-siRNA complexes and their effect on cell viability. We treated HeLa and MDA-MB-231/RFP cells with NPs and NP complexed with scrambled siRNA (100 nM) for 24 hours. We initially performed an MTT assay to assess toxicity and also performed an apoptotic assay to confirm that the NPs did not affect cell proliferation (Figure 2D and E). Cell viability was also assessed by phase contrast microscopy (data not shown). Our results showed that the NPs and the NP-siRNA complexes did not cause toxicity when comparison with untreated controls in both cell lines. Annexin V-FITC apoptotic analysis data showed normal cell proliferation in treated cells compared with untreated controls (Figure 2F). Flow cytometry data also showed normal cell cycle progression in treated cells compared with untreated controls (Figure S3A). Taken together, all the data indicated normal cell growth and cell cycle progression in cells treated with NPs when compared with untreated controls.
Cellular internalization of nanoplexes
The next part of this work was to deliver biological molecules into mammalian cells via these functionalized NP transporters to perform biological functions. In order to determine whether the NPs were able to deliver siRNA into the cells, we visualized the pattern of fluorescently labeled siRNA under confocal microscopy. We transfected HeLa cells in a solution containing NP bound to Cy3 labelled-siRNA for 12 hours. The cells were then fixed and mounted in Vectashield mounting media. Confocal microscopic images showed perfect colocalization of the Cy3-labelled siRNA (red) surrounding the cell nucleus and giving rise to yellow-colored spots. Z-section images taken by confocal microscopy at different focal planes confirmed the RNA to have entered the interior of the cell. This indicated that the functionalized NPs were able to transfer siRNA to the cells (Figure 3).
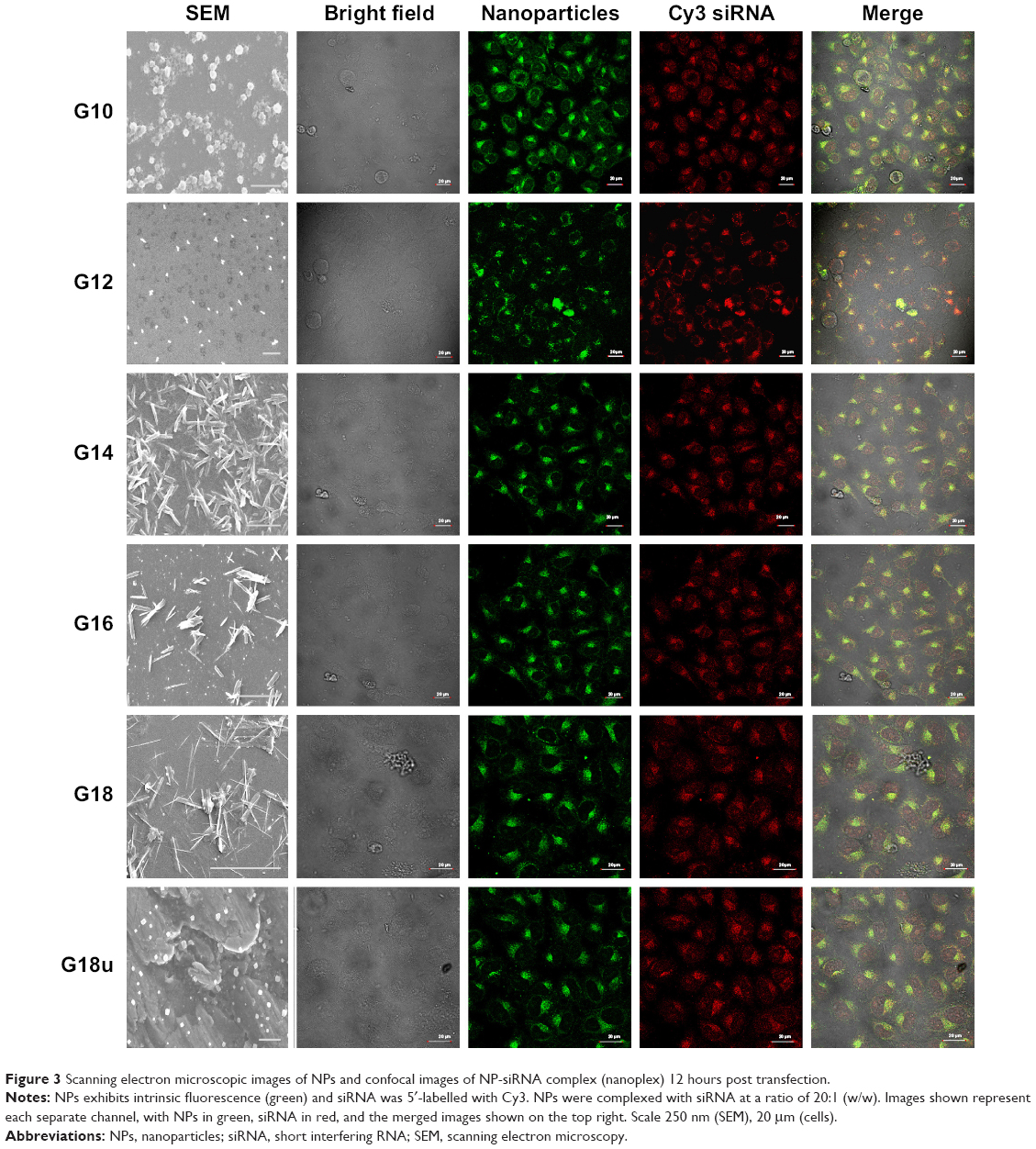 | Figure 3 Scanning electron microscopic images of NPs and confocal images of NP-siRNA complex (nanoplex) 12 hours post transfection. |
Confocal studies also showed that NPs conjugated to Cy3-labelled siRNA were clearly localized to the perinuclear regions of the cytoplasm in the cells, exhibiting the pattern of distribution seen for siRNA transfection (Figure 3). The distribution after uptake was uniform and there was no aggregation after cell uptake. Control studies were also done with a commercially available transfection reagent (Lipofectamine RNAiMAX). The pattern of distribution and cell uptake was similar to that with the NPs, thus confirming high gene transfection efficiency to deliver siRNA in vitro.
Mechanism of cellular uptake
Next we examined the pathway of uptake of the NPs and the NP-siRNA complexes. The mechanism by which cells take up NPs and NP-siRNA complexes has been shown to involve endocytosis via the endosomal pathway.36 To confirm this pathway of uptake, we performed a series of experiments by incubating HeLa cells under different conditions using NPs followed by endosomal staining with LysoTracker Red-DND99. We observed colocalization (indicated by yellow color) of the red and green fluorescence signals in cells treated with NPs (Figure 4A). Of note is that incubation of the NPs not only produced red fluorescence with the LysoTracker Red probe, but also exhibited a diffuse pattern of localization within the cytoplasm. We can therefore hypothesize that NPs first accumulate in the lysosomes, and, as time elapses, they are released into the cytoplasm (Figure 4A). To substantiate our results further, we incubated the cells at 4°C and 37°C; endocytosis is a temperature (energy)-dependent uptake pathway, so uptake was negligible at 4°C (Figure 4B).39 To further verify the mechanism of energy-dependent uptake, we depleted the cellular ATP using NaN3, which is known to disturb the production of ATP in cells, then transfection with nanoplex. The uptake was much less in treated cells when compared with the control, confirming energy-dependent uptake (Figure 4C).40 To assess the role of clathrin in the internalization of the siRNA-nanoplexes, we carried out incubations under conditions that are known to disrupt the formation of clathrin-coated vesicles on the cell membrane. Here the treatment consisted of pretreating the cells with either sucrose (hypertonic treatment) or a K+-depleted medium prior to exposure to the NPs or nanoplexes.41 These pretreatments dramatically reduced the level of cellular uptake of nanoplexes as deduced from confocal images (Figure 4D), suggesting involvement of the clathrin pathway in the endocytotic cellular uptake of NPs and nanoplexes. The results confirmed an endosome-mediated mechanism (Figure 4E).
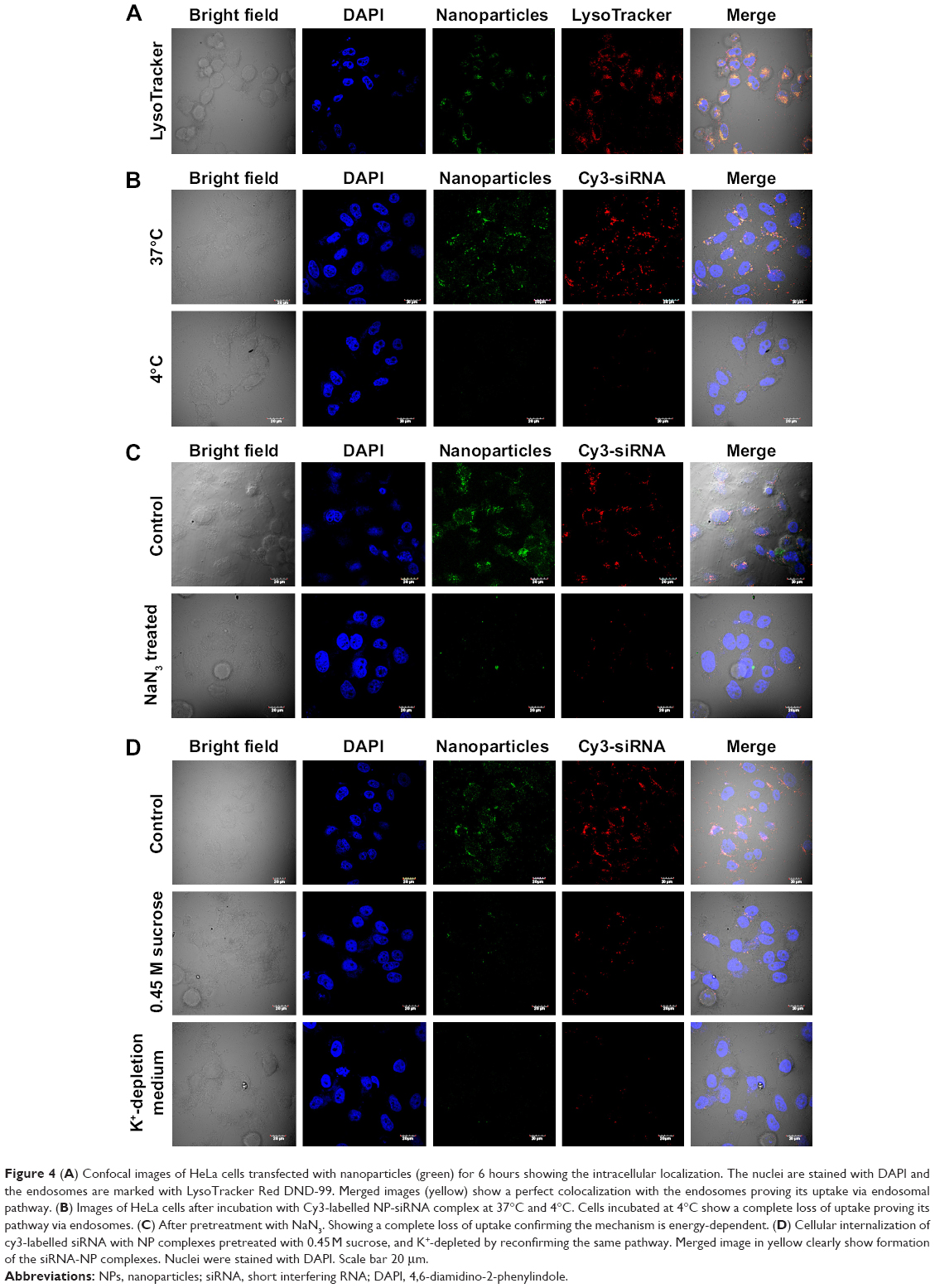 | Figure 4 (A) Confocal images of HeLa cells transfected with nanoparticles (green) for 6 hours showing the intracellular localization. The nuclei are stained with DAPI and the endosomes are marked with LysoTracker Red DND-99. Merged images (yellow) show a perfect colocalization with the endosomes proving its uptake via endosomal pathway. (B) Images of HeLa cells after incubation with Cy3-labelled NP-siRNA complex at 37°C and 4°C. Cells incubated at 4°C show a complete loss of uptake proving its pathway via endosomes. (C) After pretreatment with NaN3. Showing a complete loss of uptake confirming the mechanism is energy-dependent. (D) Cellular internalization of cy3-labelled siRNA with NP complexes pretreated with 0.45M sucrose, and K+-depleted by reconfirming the same pathway. Merged image in yellow clearly show formation of the siRNA-NP complexes. Nuclei were stained with DAPI. Scale bar 20 μm. |
Gene silencing efficiency of siRNA delivered with NPs
To examine whether the NPs could deliver the siRNA to their target and silence the gene of interest at the mRNA level, we performed real time-PCR analysis after transfecting HeLa cells for 24 hours with NPs complexed with siRNA against Bcl-2 (conc. of siRNA 100 nM). Bcl-2 was chosen as it exhibits anti-apoptotic activity and is overexpressed in several types of cancer. Bcl-2 protein blocks the release of cytochrome-C, which in turn prevents the propagation of death signals and promotes cell survival. Earlier studies have shown that apoptosis in cancer cells can be induced by silencing this gene. Real time-PCR was performed and the amount of mRNA expression was calculated using the ΔΔCT method. Downregulation of Bcl-2 mRNA was determined by comparison of the ratio between Bcl-2 and endogenous control (β-actin) mRNA expression for the transfected samples against the non-transfected samples. NP-Bcl-2-siRNA complexes were able to silence Bcl-2 mRNA levels by 80%–90% of the normal expression level (G10–G18U) when compared with the control (Figure 5A). Simultaneous experiments with scrambled siRNA confirmed that the NPs themselves do not contribute to silencing the target gene because there was no significant change in expression of mRNA of the target gene when compared with untreated controls (Figure S3B).
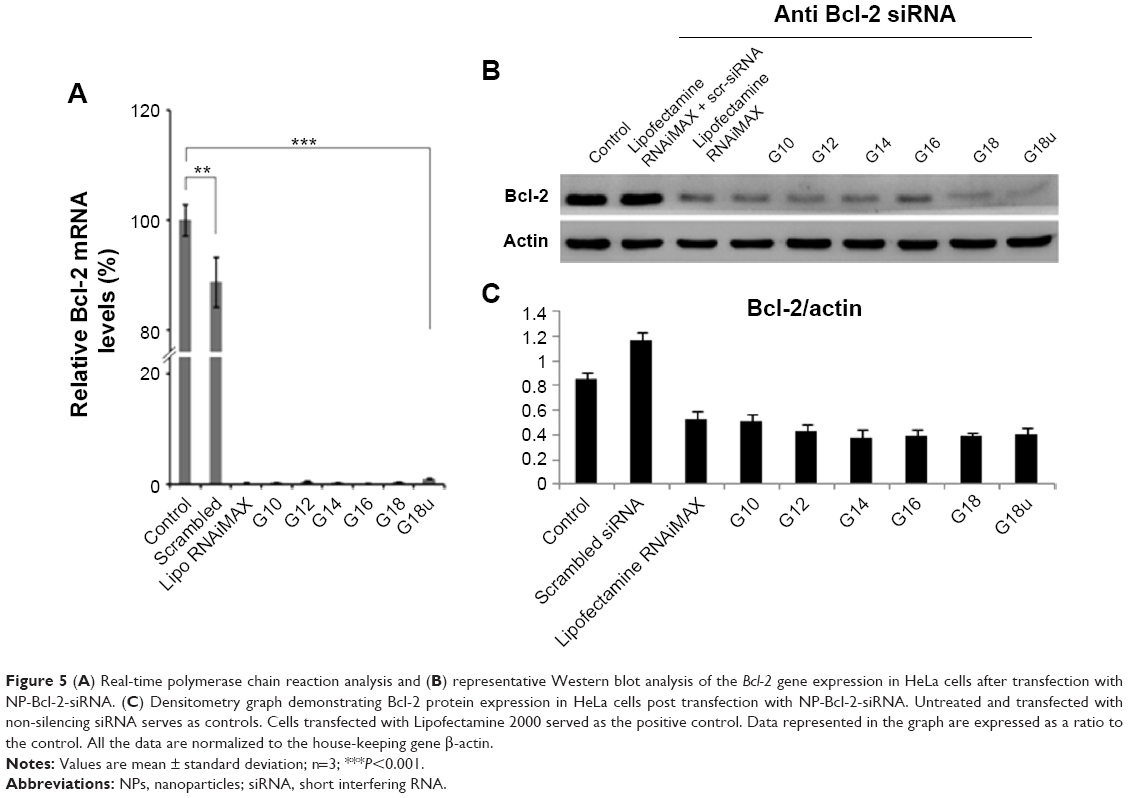 | Figure 5 (A) Real-time polymerase chain reaction analysis and (B) representative Western blot analysis of the Bcl-2 gene expression in HeLa cells after transfection with NP-Bcl-2-siRNA. (C) Densitometry graph demonstrating Bcl-2 protein expression in HeLa cells post transfection with NP-Bcl-2-siRNA. Untreated and transfected with non-silencing siRNA serves as controls. Cells transfected with Lipofectamine 2000 served as the positive control. Data represented in the graph are expressed as a ratio to the control. All the data are normalized to the house-keeping gene β-actin. |
To estimate the extent of downregulation of Bcl-2 at the protein level, we performed Western blot analysis. Band intensities were quantified using UVP software after normalizing to endogenous control (β-actin) levels and to non-transfected cells. Bcl-2 protein levels transfected with siRNA complexed with Lipofectamine RNAiMAX, a well-known commercial transfection reagent, served as positive controls and non-transfected as negative control. Consistent with downregulation of Bcl-2 mRNA levels, expression of Bcl-2 protein showed significant downregulation in cells treated with NP-Bcl-2-siRNA compared with non-transfected controls (Figure 5B). The marked reduction in Bcl-2 mRNA expression in comparison with untreated cells consistently correlated with the protein level. To reconfirm that the effect of downregulating the expression of the targeted gene is only produced by the siRNA specific to the particular gene sequence, NPs transfected with scrambled siRNA as well as scrambled siRNA transfected with Lipofectamine RNAiMAX were studied at both the mRNA and protein levels, As expected, no significant change was observed in the expression of the gene at the mRNA or protein level when compared with untreated controls (Figures S3B and C).
To further visualize the silencing efficiency of the NP-siRNA complexes, we transfected MDA-MB-231/RFP cells with NPs complexed siRNA against RFP. Forty-eight hours post transfection, the cells were visualized under confocal microscopy. The amount of silencing was compared with non-transfected controls. Simultaneously, Lipofectamine RNAiMAX complexed siRNA against RFP was taken as positive control. All the NPs complexed to RFP-siRNA (100 nM) significantly and potently silenced RFP expression by more than 80% when compared with untreated controls (Figure 6).
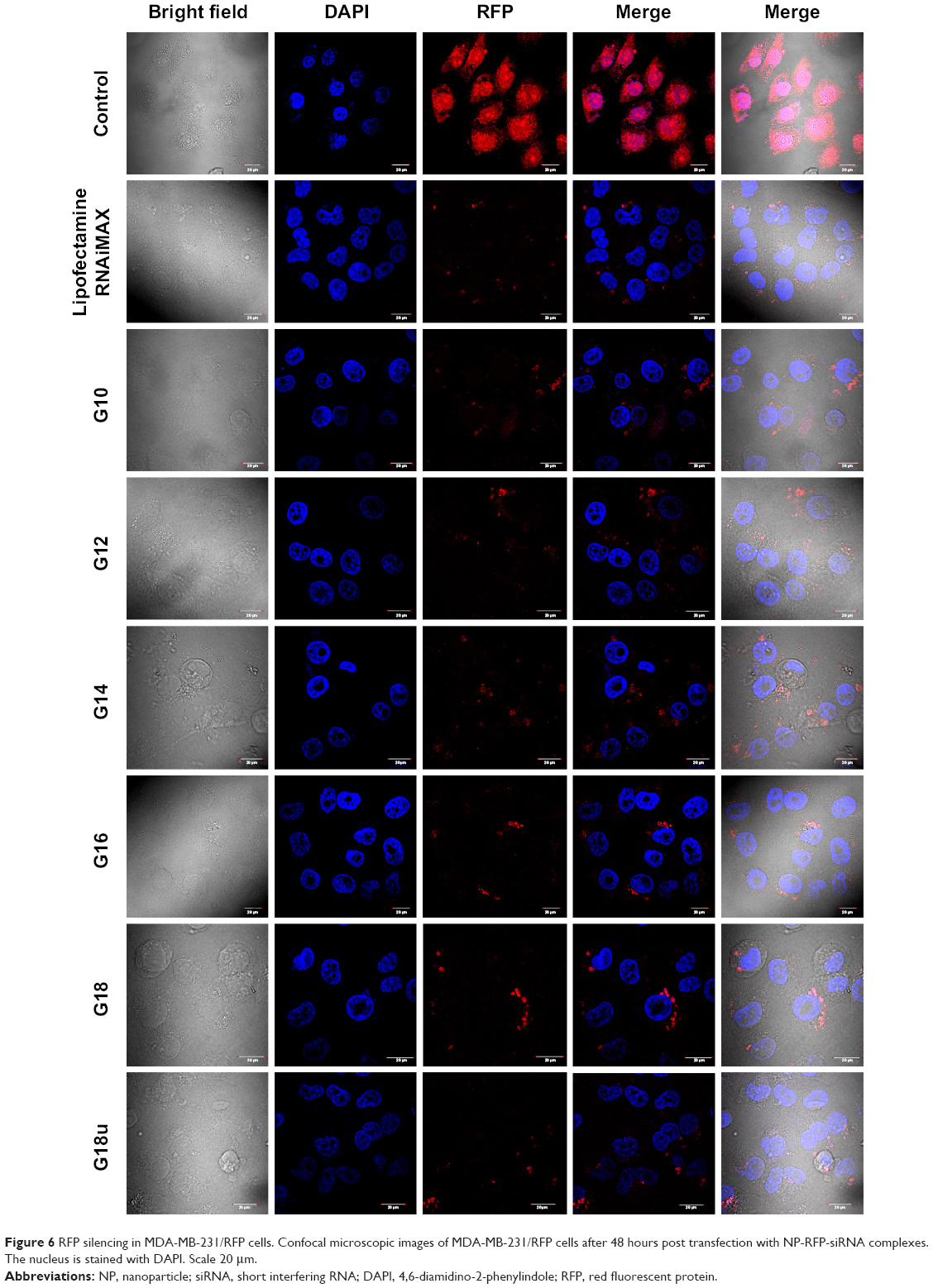 | Figure 6 RFP silencing in MDA-MB-231/RFP cells. Confocal microscopic images of MDA-MB-231/RFP cells after 48 hours post transfection with NP-RFP-siRNA complexes. The nucleus is stained with DAPI. Scale 20 μm. |
Conclusion
A major challenge for siRNA therapeutics is their intracellular delivery to specific tissues and organs where the target gene is expressed. In vivo, free siRNA is cleared through the kidneys. Thus, several methods have been proposed for conjugating siRNA to cholesterol, liposomes, and aptamers, as well as antibody targeting. In the present study we functionalized PABA NPs with guanidine to produce well defined and charged NPs that are biocompatible as well as being stable in water. These NPs contain amino groups that allow efficient assembly of siRNA. The NP-siRNA complexes are small and uniform in size and have no cytotoxicity. They are also biodegradable. These NPs efficiently delivered siRNA to silence the target genes at the transcriptional and post transcriptional level. Delivery of siRNA using NPs showed better efficiency than a commercially available transfecting agent. There was a clear increase in transfection when compared with transfection by Lipofectamine. Overall, this functionalization shows nuclear translocation of RNA oligos as transporters, shows improved efficiency for delivering siRNA, and provides more potent RNAi.
Acknowledgments
This work was supported by DBT GAP 0239. TLR, PSR, EB, and GKR thank UGC and Council of Scientific and Industrial Research for fellowship support. The authors thank Y Suresh for helping microscopy and flow cytometry studies.
Disclosure
The authors report no conflicts of interest in this work.
References
Elbashir SM, Harborth J, Lendeckel W, Yalcin A, Weber K, Tuschl T. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature. 2001;411(6836):494–498. | ||
Dykxhoorn DM, Palliser D, Lieberman J. The silent treatment: siRNAs as small molecule drugs. Gene Ther. 2006;13(6):541–552. | ||
Whitehead KA, Langer R, Anderson DG. Knocking down barriers: advances in siRNA delivery. Nat Rev Drug Discov. 2009;8(2):129–138. | ||
Kanasty R, Dorkin JR, Vegas A, Anderson D. Delivery materials for siRNA therapeutics. Nat Mater. 2013;12(11):967–977. | ||
Jager L, Ehrhardt A. Emerging adenoviral vectors for stable correction of genetic disorders. Curr Gene Ther. 2007;7(4):272–283. | ||
Nair V. Retrovirus-induced oncogenesis and safety of retroviral vectors. Curr Opin Mol Ther. 2008;10(5):431–438. | ||
Odom GL, Gregorevic P, Chamberlain JS. Viral-mediated gene therapy for the muscular dystrophies: successes, limitations and recent advances. Biochim Biophys Acta. 2007;1772(2):243–262. | ||
Check E. Regulators split on gene therapy as patient shows signs of cancer. Nature. 2002;419(6907):545–546. | ||
Li SD, Huang L. Non-viral is superior to viral gene delivery. J Control Release. 2007;123(3):181–183. | ||
Li W, Szoka FC Jr. Lipid-based nanoparticles for nucleic acid delivery. Pharm Res. 2007;24(3):438–449. | ||
Gu L, Nusblat LM, Tishbi N, et al. Cationic amphiphilic macromolecule (CAM)-lipid complexes for efficient siRNA gene silencing. J Control Release. 2014;184:28–35. | ||
Schaffert D, Wagner E. Gene therapy progress and prospects: synthetic polymer-based systems. Gene Ther. 2008;15(16):1131–1138. | ||
Qi L, Wu L, Zheng S, Wang Y, Fu H, Cui D. Cell-penetrating magnetic nanoparticles for highly efficient delivery and intracellular imaging of siRNA. Biomacromolecules. 2012;13(9):2723–2730. | ||
Wagner E, Cotten M, Foisner R, Birnstiel ML. Transferrin-polycation-DNA complexes: the effect of polycations on the structure of the complex and DNA delivery to cells. Proc Natl Acad Sci U S A. 1991;88(10):4255–4259. | ||
Felgner PL, Barenholz Y, Behr JP, et al. Nomenclature for synthetic gene delivery systems. Hum Gene Ther. 1997;8(5):511–512. | ||
Li W, Huang Z, MacKay JA, Grube S, Szoka FC Jr. Low-pH-sensitive poly(ethylene glycol) (PEG)-stabilized plasmid nanolipoparticles: effects of PEG chain length, lipid composition and assembly conditions on gene delivery. J Gene Med. 2005;7(1):67–79. | ||
Hoekstra D, Rejman J, Wasungu L, Shi F, Zuhorn I. Gene delivery by cationic lipids: in and out of an endosome. Biochem Soc Trans. 2007;35 Pt 1:68–71. | ||
Jeong JH, Mok H, Oh YK, Park TG. siRNA conjugate delivery systems. Bioconjug Chem. 2009;20(1):5–14. | ||
Bumcrot D, Manoharan M, Koteliansky V, Sah DW. RNAi therapeutics: a potential new class of pharmaceutical drugs. Nat Chem Biol. 2006;2(12):711–719. | ||
Nishina K, Unno T, Uno Y, et al. Efficient in vivo delivery of siRNA to the liver by conjugation of alpha-tocopherol. Mol Ther. 2008;16(4):734–740. | ||
Dalby B, Cates S, Harris A, et al. Advanced transfection with Lipofectamine 2000 reagent: primary neurons, siRNA, and high-throughput applications. Methods. 2004;33(2):95–103. | ||
Kam NW, Liu Z, Dai H. Functionalization of carbon nanotubes via cleavable disulfide bonds for efficient intracellular delivery of siRNA and potent gene silencing. J Am Chem Soc. 2005;127(36):12492–12493. | ||
Yadav JS, Lavanya MP, Das PP, et al. 4-N-pyridin-2-yl-benzamide nanotubes compatible with mouse stem cell and oral delivery in Drosophila. Nanotechnology. 2010;21(15):155102. | ||
Yadav JS, Das PP, Reddy TL, et al. Sub-cellular internalization and organ specific oral delivery of PABA nanoparticles by side chain variation. J Nanobiotechnol. 2011;9:10. | ||
Chowdhury MA, Huang Z, Abdellatif KR, et al. Synthesis and biological evaluation of indomethacin analogs possessing a N-difluoromethyl-1,2-dihydropyrid-2-one ring system: a search for novel cyclooxygenase and lipoxygenase inhibitors. Bioorg Med Chem Lett. 2010;20(19):5776–5780. | ||
Dehnhardt CM, Venkatesan AM, Delos Santos E, et al. Lead optimization of N-3-substituted 7-morpholinotriazolopyrimidines as dual phosphoinositide 3-kinase/mammalian target of rapamycin inhibitors: discovery of PKI-402. J Med Chem. 2010;53(2):798–810. | ||
Liu KG, Lambert MH, Leesnitzer LM, et al. Identification of a series of PPAR gamma/delta dual agonists via solid-phase parallel synthesis. Bioorg Med Chem Lett. 2001;11(22):2959–2962. | ||
Kalgutkar AS, Crews BC, Saleh S, Prudhomme D, Marnett LJ. Indolyl esters and amides related to indomethacin are selective COX-2 inhibitors. Bioorg Med Chem. 2005;13(24):6810–6822. | ||
Hamada Y, Ohtake J, Sohma Y, Kimura T, Hayashi Y, Kiso Y. New water-soluble prodrugs of HIV protease inhibitors based on O – >N intramolecular acyl migration. Bioorg Med Chem. 2002;10(12):4155–4167. | ||
Nicolaou KC, Zak M, Safina BS, Lee SH, Estrada AA. Total synthesis of thiostrepton, part 2: construction of the quinaldic acid macrocycle and final stages of the synthesis. Angew Chem Int Ed Engl. 2004;43(38):5092–5097. | ||
Hong F, Zaidi J, Cusack B, Richelson E. Synthesis and biological studies of novel neurotensin(8–13) mimetics. Bioorg Med Chem. 2002;10(12):3849–3858. | ||
Muroski ME, Morgan TJ, Levenson CW, Strouse GF. A gold nanoparticle pentapeptide: gene fusion to induce therapeutic gene expression in mesenchymal stem cells. J Am Chem Soc. 2014;136(42):14763–14771. | ||
Abbas S, Lodge TP. Depletion interactions: effects of added homopolymer on ordered phases formed by spherical block copolymer micelles. Macromolecules. 2008;41(22):8895–8902. | ||
Mukherjee S, Chowdhury D, Kotcherlakota R, et al. Potential theranostics application of bio-synthesized silver nanoparticles (4-in-1 system). Theranostics. 2014;4(3):316–335. | ||
Tagalakis AD, He L, Saraiva L, Gustafsson KT, Hart SL. Receptor-targeted liposome-peptide nanocomplexes for siRNA delivery. Biomaterials. 2011;32(26):6302–6315. | ||
Kam NW, Liu Z, Dai H. Carbon nanotubes as intracellular transporters for proteins and DNA: an investigation of the uptake mechanism and pathway. Angew Chem Int Ed Engl. 2006;45(4):577–581. | ||
Yu B, Wang X, Zhou C, et al. Insight into mechanisms of cellular uptake of lipid nanoparticles and intracellular release of small RNAs. Pharm Res. 2014;31(10):2685–2695. | ||
Wang X, Yu B, Ren W, et al. Enhanced hepatic delivery of siRNA and microRNA using oleic acid based lipid nanoparticle formulations. J Control Release. 2013;172(3):690–698. | ||
Marsh M, McMahon HT. The structural era of endocytosis. Science. 1999;285(5425):215–220. | ||
Schmid SL, Carter LL. ATP is required for receptor-mediated endocytosis in intact cells. J Cell Biol. 1990;111(6 Pt 1):2307–2318. | ||
Qaddoumi MG, Gukasyan HJ, Davda J, Labhasetwar V, Kim KJ, Lee VH. Clathrin and caveolin-1 expression in primary pigmented rabbit conjunctival epithelial cells: role in PLGA nanoparticle endocytosis. Mol Vis. 2003;9:559–568. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.