Back to Journals » Journal of Inflammation Research » Volume 19
PANoptosis as a Potential Therapeutic Target in Drug-Induced Liver Injury: A Narrative Review
Authors Zhang X, Zhou Z ![]() , Xie Z
, Xie Z ![]() , Chen J, Deng Y
, Chen J, Deng Y ![]() , Fang Y, Deng J, Ye X
, Fang Y, Deng J, Ye X
Received 29 September 2025
Accepted for publication 23 December 2025
Published 8 January 2026 Volume 2026:19 569771
DOI https://doi.org/10.2147/JIR.S569771
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Editor who approved publication: Dr Fatih Türker
Xinru Zhang,1 Zhipin Zhou,1 Zhuohua Xie,2 Jieyi Chen,1 Yanting Deng,2 Yibin Fang,1 Jiasheng Deng,2 Xiaoxue Ye1
1Liuzhou People’s Hospital Affiliated to Guangxi Medical University, Guangxi, 545006, People’s Republic of China; 2School of Pharmacy, Guangxi University of Chinese Medicine, Nanning, Guangxi, 530200, People’s Republic of China
Correspondence: Zhipin Zhou, Liuzhou People’s Hospital Affiliated to Guangxi Medical University, 8 Wenchang Road, Cheng Zhong District, Liuzhou, Guangxi, People’s Republic of China, Tel +86− 18776921947, Email [email protected]
Abstract: PANoptosis is a recently characterized form of programmed cell death defined by the coordinated regulation of pyroptosis, apoptosis, and necroptosis mediated by the multi-protein PANoptosome complex, a phenomenon that cannot be fully explained by any individual pathway. The initiation of PANoptosis relies on the assembly and activation of the PANoptosome, a process that has been closely linked to the onset and progression of multiple disorders, including inflammatory and neurodegenerative diseases, cancer, and pulmonary conditions. However, the contribution of PANoptosis to drug-induced liver injury (DILI) has not been thoroughly evaluated. DILI is a frequently encountered clinical adverse drug reaction, and its development is strongly associated with hepatocyte death. Therefore, this review aims to summarize the molecular mechanisms underlying PANoptosis and the assembly of the PANoptosome complex, elucidate the interactions among apoptosis, necroptosis, and pyroptosis, and assess the potential involvement of PANoptosis in DILI. Furthermore, investigating natural products that inhibit PANoptosome assembly or target key regulators such as ZBP1, AIM2, and NLRP3 to modulate PANoptosis may offer promising therapeutic opportunities for the management of DILI.
Keywords: drug-induced liver injury, PANoptosis, PANoptosome, therapeutic targets
Introduction
Programmed cell death is essential for maintaining organismal homeostasis and defending against pathogenic infections, and its dysregulation is closely linked to the onset and progression of infectious diseases, cancer, and organ injury. According to the international standards established by the Nomenclature Committee on Cell Death (NCCD), programmed cell death can be categorized into several types based on morphological, biochemical, and functional features, including intrinsic and extrinsic apoptosis, necroptosis, ferroptosis, and pyroptosis.1 In recent years, PANoptosis has emerged as a novel pro-inflammatory form of programmed cell death, characterized by the PANoptosome-mediated coordination of pyroptosis, apoptosis, and necroptosis.2 Increasing evidence indicates that Z-DNA-binding protein 1 (ZBP1) functions as a key molecular sensor that initiates inflammasome activation and drives PANoptosis.3 Nearly all pathological processes of PANoptosis depend on the assembly of the PANoptosome, as it integrates diverse cell-death signals and detects danger cues, including pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs).
PANoptosis is closely linked to the pathogenesis and progression of a wide range of diseases, including infectious, inflammatory, neurological, neoplastic, and pulmonary disorders.4–6 It also plays a significant role in liver diseases.7,8 Drug-induced liver injury (DILI) refers to hepatic damage caused by a wide range of over-the-counter and prescription medications, including herbal products, dietary supplements, anti-infective agents, nonsteroidal anti-inflammatory drugs, and chemotherapeutic agents.9,10 A two-year, population-based prospective study conducted in Iceland reported an incidence of 19.1 cases per 100,000 inhabitants,11 whereas a retrospective study from mainland China documented an annual incidence of 23.80 cases per 100,000 individuals—higher than that reported in Iceland, France, and other Western countries.12 Diagnosing DILI remains challenging due to the lack of specific biomarkers, resulting in substantial underdiagnosis and misdiagnosis, as differentiation from other liver diseases often relies on exclusion.13–15 Moreover, some primary diseases requiring pharmacological treatment may themselves induce hepatic dysfunction, further complicating causal inference.14 Hepatocyte death is one of the defining features of DILI. The underlying mechanisms involve well-characterized forms of cell death, including apoptosis, necroptosis, autophagy, and necrosis, as well as more recently identified processes such as inflammatory cell death and ferroptosis.16 The specific cell death subroutine activated largely depends on the primary drug.17 However, in certain cases, targeting a single pathway of pyroptosis, apoptosis, or necroptosis fails to ameliorate DILI.18–20 Given the pivotal roles of apoptosis, necroptosis, and pyroptosis in DILI, we propose that PANoptosis is critically involved in the onset and progression of this condition. Nevertheless, the molecular mechanisms regulating PANoptosis in DILI remain poorly understood. Accordingly, this review summarizes the mechanisms and recent advances related to PANoptosis, highlights the distinctions and interconnections among these three forms of programmed cell death, and discusses the role of PANoptosis in DILI and potential therapeutic targets, thereby providing new strategies and theoretical frameworks for targeted intervention.
Apoptosis
Apoptosis is a normal biological process within organisms and is commonly referred to as programmed cell death. It constitutes a critical component of cellular self-regulation, guiding cells toward an orchestrated form of death through the activation of defined signaling pathways. Mechanistically, the release of cytochrome c from mitochondria initiates two primary downstream pathways: the intrinsic (mitochondrial) pathway and the extrinsic (death receptor–mediated) pathway (Figure 1).
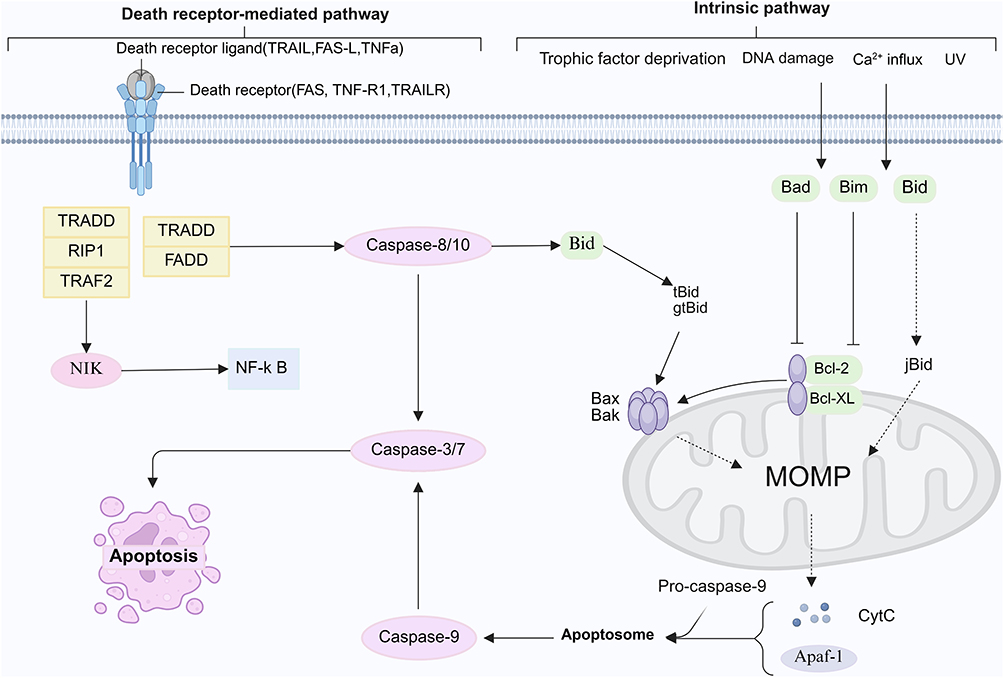 |
Figure 1 Apoptosis pathway. The apoptosis pathway comprises both the intrinsic and extrinsic pathways. In the intrinsic pathway, cellular stressors such as DNA damage, hypoxia, elevated cytosolic Ca2⁺ levels, or metabolic stress initiate the translocation and oligomerization of Bax and Bak in the mitochondrial outer membrane. This process forms membrane pores that enable the release of pro-apoptotic factors—such as cytochrome c, Smac, and endonuclease G—from the mitochondrial intermembrane space. Cytochrome c subsequently binds to the scaffold protein Apaf-1, inducing conformational changes that drive the formation of a seven-spoked apoptosome. The apoptosome subsequently recruits and activates pro-caspase-9, which then triggers the activation of downstream effector caspases to execute apoptosis. In the extrinsic pathway, engagement of death receptors—including TNFR, FAS (CD95), DR3, and TRAIL-R1/R2—by their respective ligands recruits adaptor proteins such as FADD, ultimately resulting in effector caspase activation and the induction of apoptotic cell death. By BioRender (https://BioRender.com/y09b871). |
The intrinsic pathway is triggered by diverse intracellular stressors—such as DNA damage, nutrient deprivation, cytosolic Ca2⁺ overload, reactive oxygen species (ROS), and the accumulation of misfolded or unfolded proteins,21 and is tightly regulated by pro- and anti-apoptotic members of the Bcl-2 family.22,23 As a central organelle responsible for cellular energy production, mitochondria play a pivotal role in maintaining intracellular homeostasis and are major targets of oxidative stress.24 Bcl-2 family proteins localize to the outer mitochondrial membrane, where they modulate membrane permeability, thereby governing cytochrome c release and subsequent activation of the caspase cascade.25,26 Upon release into the cytosol, cytochrome c associates with the C-terminal WD-40 domains of Apaf-1 in an ATP/dATP-dependent manner to assemble the apoptosome, which facilitates the homotypic interaction and autoactivation of pro-caspase-9,27 ultimately activating downstream caspase-3/7 to execute apoptosis.28 Mitochondrial stress, such as the accumulation of misfolded proteins, can further disrupt membrane permeability, leading to excessive ATP depletion, increased production of ROS, and perturbations in calcium homeostasis.29 Elevated mitochondrial calcium exacerbates oxidative stress and facilitates cytochrome c release, while aberrant calcium signaling may induce endoplasmic reticulum stress, activate calpains, and initiate caspase-dependent apoptosis.21
In the extrinsic pathway, ligand engagement with death receptors—including TNFR, FAS (CD95), DR3, and TRAIL-R1/R2—recruits adaptor proteins such as FADD, thereby activating caspase-8, which in turn cleaves and activates downstream caspase-3/7 to execute apoptotic signaling.30
Necroptosis
Necroptosis is a recently defined form of regulated cell death. RIPK1 and RIPK3 function as key regulators of both apoptosis and necroptosis. Activated caspase-8 degrades RIPK1 and RIPK3, which suppresses the necroptotic signaling pathway and promotes apoptosis.31,32 When caspase-8 activity is inhibited, RIPK1 recruits and phosphorylates RIPK3, resulting in its activation and oligomerization and ultimately leading to the formation of the necrosome.33–35 In addition to RIPK1 and RIPK3, the mixed lineage kinase domain-like protein MLKL also participates in necroptosis, and phosphorylation of RIPK3 at Ser 232 enhances the recruitment of MLKL.36 RIPK3 subsequently recruits and phosphorylates MLKL, promoting its oligomerization and conformational changes that enable its translocation to the plasma membrane, where it increases membrane permeability and initiates inflammatory responses.37,38
ROS are key mediators of necroptosis and enhance TNFα-induced necroptosis by activating RIPK1 and RIPK3 through promoting RIPK1 autophosphorylation at the S161 site.39,40 However, the functional role of ROS in necroptosis remains a subject of debate. Studies have shown that mitochondrial depletion suppresses TNF-induced ROS production but does not alter the progression of necroptosis.41 Furthermore, even when mitochondria are impaired, activated RIPK3 can still induce necroptosis independently of ROS.41 These findings underscore the need for further study of the interactions and underlying mechanisms involving ROS and RIPKs across various cell death pathways. In addition, pattern recognition receptor-mediated necroptosis represents an important mode of cell death, and Toll-like receptors together with ZBP1 function as essential mediators in this process. TLR3 recognizes viral double-stranded RNA and TLR4 recognizes lipopolysaccharide, and upon activation, both receptors recruit TRIF and interact with RIPK3 to initiate necroptosis (Figure 2).42 Overall, the induction and regulation of necroptosis constitute highly complex processes that depend on the coordinated activities of multiple signaling molecules.
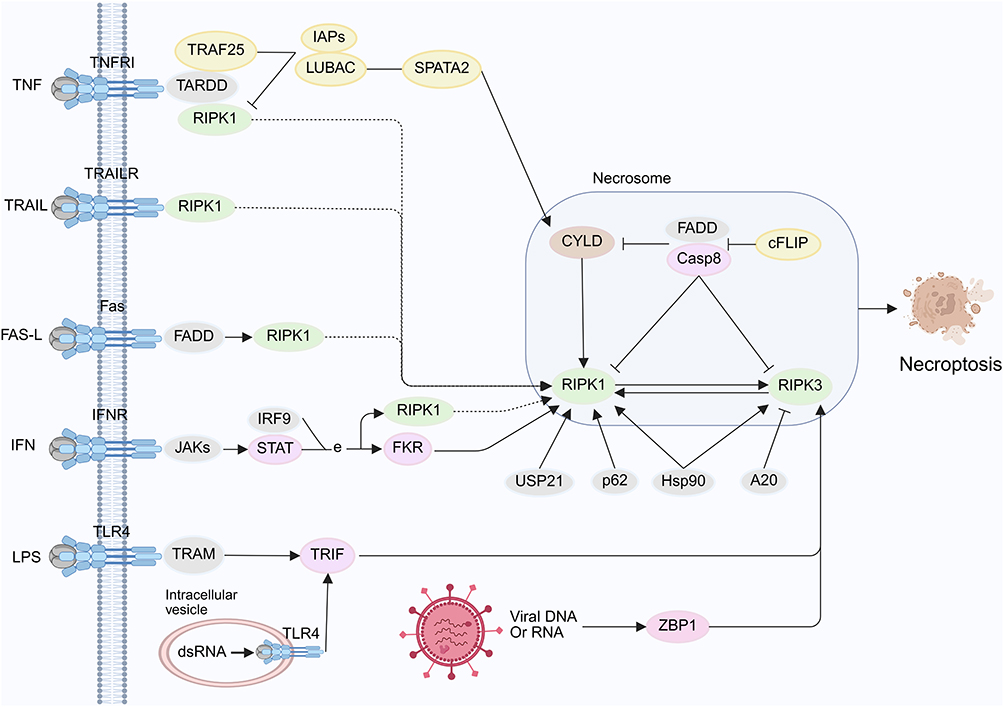 |
Figure 2 Necroptosis pathway. The necroptotic pathway is initiated upon activation of TNFR1 by TNFα. Activation of additional cellular receptors can also induce necroptosis, including death receptors such as Fas, Toll-like receptors such as TLR4, tumor necrosis factor-related apoptosis-inducing ligand receptors such as TRAIL-R, and interferon receptors such as IFNR. Downstream of these receptors, activated RIPK1 is recruited into a complex that includes FADD, caspase-8, and caspase-10. In the absence of caspase-8 activity, RIPK1 recruits and phosphorylates RIPK3, which leads to the formation of the necrosome. By BioRender (https://BioRender.com/n56j719). |
Pyroptosis
Pyroptosis is an inflammatory form of programmed cell death that was initially recognized to eliminate intracellular pathogens through caspase-1 activation, which promotes plasma membrane pore formation and results in membrane rupture or lysis.43 Recent studies have shown that in addition to caspase-1, other cysteine proteases, including caspase-4, caspase-5, caspase-12, caspase-11 in mice, and caspase-8, also participate in this process.44,45 Activated inflammatory caspases cleave Gasdermin D(GSDMD) and release it from its inhibitory state. The activated GSDMD subsequently translocates to the plasma membrane, binds to phospholipids, and initiates pore formation, which disrupts membrane integrity, causes cytoplasmic swelling, and promotes the release of intracellular contents, ultimately leading to cell death.45–47
Inflammasomes, such as absent in melanoma 2 (AIM2) and NLR family pyrin domain containing 3(NLRP3), can activate caspase-8 and caspase-1, thereby inducing apoptosis and promoting the classical pyroptotic pathway.48 In contrast, the non-classical pathway occurs when cytosolic lipopolysaccharide forms an inflammasome complex that activates caspase-4, caspase-5, and caspase-11, which induce pyroptosis through GSDMD cleavage (Figure 3).49 Activation of caspase-1 under non-classical stimulation requires NLRP3 and apoptosis-associated speck-like protein containing a CARD(ASC), whereas caspase-11-mediated activation and the resulting cell death proceed independently of these components.50 As research progresses, additional insights into the roles and interactions of caspases in pyroptosis are anticipated.
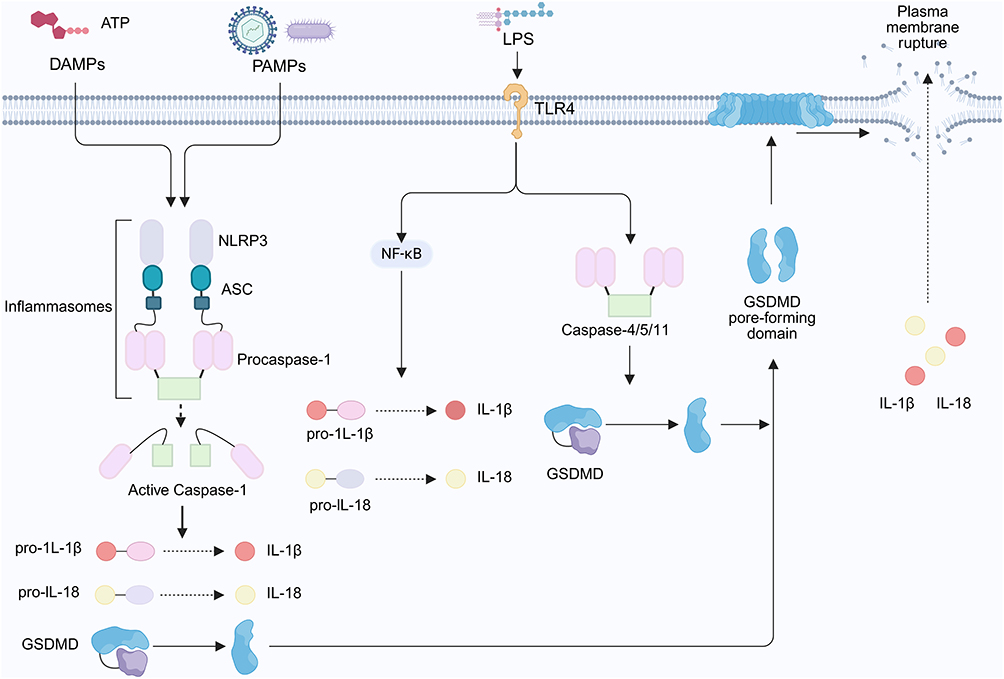 |
Figure 3 Pyroptosis pathway. DAMPs and PAMPs activate inflammasomes, such as AIM2 and NLRP3, which initiate the classical pyroptotic pathway through the activation of caspase-8 and caspase-1. In the non-classical pathway, cytosolic lipopolysaccharide forms an inflammasome complex that activates caspase-4, caspase-5, and caspase-11. These enzymes induce pyroptosis by cleaving GSDMD and generating its activated form, which subsequently translocates to the plasma membrane, associates with phospholipids, and initiates pore formation. This series of events compromises membrane integrity, induces cytoplasmic swelling, and promotes the release of intracellular contents, ultimately resulting in cell death. By BioRender (https://BioRender.com/h37i302). |
PANoptosis
PANoptosis is a recently identified form of programmed cell death that integrates the defining features of pyroptosis, apoptosis and necroptosis, and the cell death processes mediated by these pathways are not completely independent. A study conducted in 2016 first reported the simultaneous activation of these three forms of cell death in macrophages infected with influenza A virus, demonstrating close molecular interactions among the different programmed cell death pathways.51 Current research in this field mainly centers on pathogen infection models, where the core regulatory mechanism involves a multiprotein complex termed the PANoptosome. This complex plays a central role in recognizing danger signals, including DAMPs and PAMPs, and in initiating the cell death process.52–55 The molecular composition of the PANoptosome displays substantial plasticity, with its components changing in response to different activating signals and comprising cytosolic innate immune sensors such as ZBP1, AIM2, RIPK3, RIPK1, ASC, FADD, members of the caspase family, and key effector molecules that execute pyroptosis, apoptosis, and necroptosis.53,54,56,57 Activation of these sensor molecules induces inflammatory responses and regulated forms of cell death, including apoptosis, inflammatory cell death, necroptosis, and PANoptosis (Figure 4).58
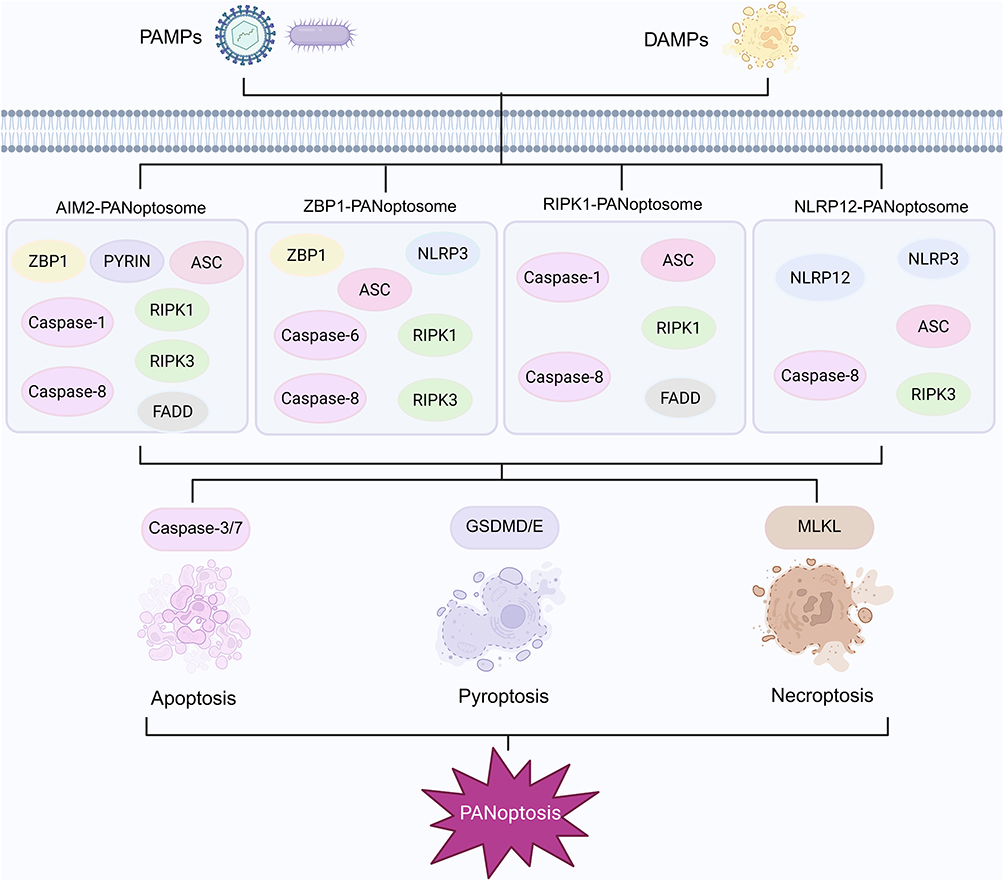 |
Figure 4 Assembly of different PANoptosome complexes. Upon activation by PAMPs or DAMPs, sensor proteins interact with adaptor proteins to form specific PANoptosome complexes, such as the AIM2 PANoptosome composed of ZBP1, PYRIN, ASC, caspase-1, caspase-8, RIPK1, RIPK3, and FADD, the ZBP1 PANoptosome consisting of ZBP1, NLRP3, ASC, caspase-6, caspase-8, RIPK1 and RIPK3, the RIPK1 PANoptosome containing RIPK1, caspase-1, caspase-8, ASC and FADD, and the NLRP12 PANoptosome comprising NLRP12, NLRP3, ASC, caspase-8 and RIPK3. These complexes integrate the essential signaling pathways that mediate apoptosis, pyroptosis, and necroptosis. Caspase-1 activates GSDMD or GSDME to initiate pyroptosis, while RIPK3 interacts with MLKL to promote necroptosis. In addition, caspase-8, together with caspase-3 or caspase-7, mediates apoptosis, and the integration of these pathways culminates in the coordinated cell death modality termed PANoptosis. By BioRender (https://BioRender.com/yks3w20). |
In recent years, several PANoptosome complexes have been identified, among which the ZBP1 PANoptosome is considered the most representative. Following activation by influenza A virus, ZBP1 interacts with RIPK1 to form the ZBP1 RIPK1 complex, which subsequently promotes the assembly of the ZBP1 PANoptosome composed of ZBP1, RIPK3, RIPK1, FADD, and caspase-8.59,60 The formation of this complex initiates downstream molecular events, including the activation of GSDMD and GSDME, which promotes the secretion of IL-1β and IL-18, and the activation of caspase-3/7 and the MLKL pathway.58,59 These events collectively induce pyroptosis, apoptosis, and necroptosis, ultimately resulting in the execution of PANoptosis (Figure 4).58,59 Wang et al developed an expansion microscopy approach that enables visualization of the spatial organization of ASC, caspase-8, and RIPK3 within the PANoptosome. This optimized protocol exhibits broad applicability because it validates PANoptosome assembly at the single-cell level and facilitates the study of other multiprotein complexes and cell death inducers, indicating considerable potential for future research.61 To date, the molecular features of AIM2, RIPK1, and NLRP12 associated PANoptosomes have also been elucidated (Figure 4).53,54,57 In summary, the concept of PANoptosis is expected to continue expanding and advancing with ongoing scientific investigation. In the following sections, we outline the distinctions and interrelationships among PANoptosis, apoptosis, necroptosis, and pyroptosis.
The Interactions Between Apoptosis, Necroptosis, and Pyroptosis
ZBP1 has been identified as a sensor of double-stranded DNA and functions as both a key initiator of innate immune responses and a central regulatory hub within the PANoptosis signaling network. Its mechanism of action involves the recognition of the NP and PB1 proteins of influenza A virus and the Z nucleic acids produced during viral replication, thereby initiating downstream signaling cascades.51,62 This recognition event primarily activates the RIPK1-RIPK3-caspase-8 axis, which mediates both cell death and inflammatory responses. Through the regulation of RIPK3, caspase-8, and the NLRP3 inflammasome, this pathway concurrently induces pyroptosis, necroptosis, and apoptosis and enhances inflammatory responses.51,63 The Zα2 domain of ZBP1 is critical for inflammasome assembly and for initiating PANoptosis.3 Evidence from influenza A virus infection models indicates that deletion of the Zα2 domain abolishes influenza A virus-induced PANoptosis and activation of the NLRP3 inflammasome.63 In these models, interferon regulatory factor one functions as an upstream regulatory element. It promotes PANoptosome activation by transcriptionally regulating the expression of key molecules, including ZBP1, AIM2, RIPK1, and NLRP12.64,65 Loss of interferon regulatory factor one markedly reduces ZBP1 expression, suppresses activation of the NLRP3 inflammasome, diminishes the execution of apoptosis and necroptosis, and ultimately compromises viral clearance.64
The ZBP1 PANoptosome complex is composed of the sensor molecules ZBP1, NLRP3, caspase-8, caspase-6, ASC, RIPK1, and RIPK3.52,56,66 Its assembly is linked to the activation of TNFR or TLR4 signaling and the suppression of MAPK and NF-kappa-B pathways. Notably, ZBP1 interacts with the NLRP3 inflammasome to form the ZBP1 NLRP3 complex, which functions as a central structural unit essential for PANoptosome assembly.62 Additional evidence indicates that ASC, caspase-8, and RIPK1 colocalize in a ZBP1-dependent manner during PANoptosis.61 Moreover, the lack of ASC speck formation in ZBP1-deficient cells underscores the essential role of ZBP1 in this process.61 AIM2 acts as a cytosolic sensor of double-stranded DNA originating from pathogens or damaged organelles. By recruiting ASC and caspase-1 to assemble the AIM2 inflammasome, AIM2 activates caspase-1 and triggers inflammatory responses through cytokine maturation and pyroptosis.67 Activation of AIM2 and NLRP3 promotes the formation of a shared cytosolic inflammasome platform that contains the adaptor ASC along with caspase-1 and caspase-8, thereby establishing a dual cytosolic surveillance system.68 AIM2 also modulates the innate immune sensors Pyrin and ZBP1 to promote inflammatory signaling and PANoptosis.53 AIM2, Pyrin, and ZBP1 interact with ASC to form a multiprotein complex that includes ASC, caspase-1, caspase-8, RIPK3, RIPK1, and FADD, which collectively mediate inflammatory cell death (Figure 4).53 Activation of this cooperative cell death mechanism depends on both the direct detection of viral components and the sensing of cellular stress conditions, although the specific mechanisms and functions of AIM2 in PANoptosis have yet to be fully elucidated. Although the composition of PANoptosomes varies depending on the stimulus, all forms ultimately induce necroptosis, apoptosis, and pyroptosis.
The caspase family coordinates the crosstalk among apoptosis, pyroptosis, and necroptosis during PANoptosis. Initiator caspases, including caspase-2, caspase-8, caspase-9, and caspase-10, act as proteolytic signal amplifiers that activate effector caspases, while effector caspases such as caspase-3, caspase-6, and caspase-7 cleave specific cellular substrates to promote apoptosis.69 Among these enzymes, caspase-8 serves as a molecular switch that regulates apoptosis, necroptosis, and pyroptosis.70 Following viral infection, RIPK1 and caspase-8 are recruited to the retinoic acid-inducible gene I (RIG-I) signaling complex, where ubiquitination of RIPK1 promotes complex assembly and enhances phosphorylation of interferon regulatory factor 3 (IRF3).71 This modification also exposes the RIPK1 cleavage site, allowing caspase-8 to process it into an inhibitory fragment.71 Caspase-8 additionally suppresses type I interferon production by inhibiting the RIPK1-TBK1 axis, and its loss or functional impairment enhances RIPK1-TBK1 interaction, markedly elevates interferon levels, and exacerbates inflammatory injury.72 Enzymatically inactive caspase-8 induces embryonic lethality and inflammatory damage in mice by triggering necroptosis and pyroptosis, while activated caspase-8 mediates intrinsic apoptosis and cleaves GSDMD and GSDME to induce macrophage pyroptosis.73,74 Furthermore, the scaffold functions of caspase-8 and MLKL contribute to the regulation of NLRP3 inflammasome activation downstream of TLR3.75 Caspase-1-mediated apoptosis proceeds through the Bid caspase-9/3 axis and may subsequently transition into GSDME-dependent secondary necrosis or pyroptosis.76 Caspase-1 induces pyroptosis when GSDMD is present, yet in GSDMD-deficient cells, it promotes apoptosis by activating caspase-3/7.77 Caspase-3 also cleaves GSDME to generate a pore-forming fragment that induces pyroptosis,78 and restrains excessive cytokine production by processing cyclic GMP-AMP synthase (cGAS), mitochondrial antiviral signaling protein (MAVS), and IRF3.79 Caspase-6 plays a central role in host defense against influenza A virus by promoting RHIM-dependent binding between RIPK3 and ZBP1, which leads to inflammasome activation and coordination of PANoptosis.59 Additionally, apoptosis mediated by Bak and Bax promotes interferon production through mitochondrial DNA release and activation of the cGAS/STING pathway, while the caspase-9/3/7 cascade restricts interferon beta secretion by suppressing this pathway.80 Collectively, apoptosis, pyroptosis, and necroptosis all depend on the activation of caspase family members, indicating that these cell death programs are interdependent rather than isolated processes.
RIPK1 is a central regulator of innate immune signaling, and its activation state governs TNF-mediated apoptosis, necroptosis, and inflammatory responses.81 RIPK1 undergoes post-translational regulation through ubiquitination, phosphorylation, and caspase-8-mediated cleavage.82 FADD and caspase-8 may cooperate to suppress RIPK1 by promoting its cleavage and inactivation, thereby inhibiting RIPK1-mediated necroptosis.83 In the absence of RIPK1, cells exhibit increased sensitivity to TNF-induced TNFR1-FADD-caspase-8-mediated apoptosis and to TLR ligation or interferon-induced RIPK3-dependent necrosis.84 RIPK3 functions as a critical regulator of apoptosis and necroptosis and redirects TNF-triggered cell death from apoptosis to necroptosis in a kinase-dependent manner.85 Therefore, RIPK1 and RIPK3 are essential mediators of PANoptosis and host defense. TAK1 functions as a regulatory switch that governs PANoptosome assembly. In the absence of TAK1, macrophages undergo spontaneous activation of the NLRP3 inflammasome, and RIPK1 recruits NLRP3 and ASC to form a death-inducing complex that activates the inflammasome, caspase-8, and GSDMD, thereby promoting pyroptosis and apoptosis.86,87 The RIPK1-mediated PANoptosome primarily consists of RIPK1, ASC, caspase-1, caspase-8, and FADD, and it was initially described by the Malireddi group.57 Moreover, the RIPK3-MLKL pathway can be activated independently of RIPK1 kinase activity.85,88 Collectively, these mechanisms highlight the central roles of RIPKs in the regulation of PANoptosis and in the coordination of cell death and inflammatory processes.
Activation of the NLRP3 inflammasome and GSDMD-mediated pyroptosis plays a central role in regulating apoptosis and necroptosis. Both pyroptotic and necroptotic pathways can activate the NLRP3/IL-1β signaling axis. During necroptosis, RIPK3-MLKL activates the NLRP3 inflammasome to induce caspase-1-mediated IL-1β maturation, and the cytokine is released through membrane pores formed by GSDMD and MLKL, thereby establishing a positive inflammatory feedback loop.89 The MLKL inhibitor necrosulfonamide targets GSDMD to suppress pyroptosis, highlighting the MLKL-GSDMD interaction as a critical regulatory node that links pyroptosis and necroptosis.90 In addition, GSDMD disrupts mitochondrial membrane permeability, which enhances apoptosis-related caspase-3 activation and inflammasome signaling to promote cooperation between pyroptosis and apoptosis.91 Beyond the canonical NLRP3 inflammasome, the NLRC4 inflammasome also plays a significant role in PANoptosis. Upon sensing T3SS proteins, NAIP binds to NLRC4 to activate the NAIP-NLRC4 inflammasome, which subsequently activates caspase-1 and induces GSDMD-mediated pyroptosis.92 NLRC4 also activates caspase-8, which subsequently activates caspase-3/7 to trigger apoptosis.92 During Pseudomonas aeruginosa infection, MLKL-dependent cell death is enhanced in NLRC4-deficient cells, which promotes necroptosis, and inhibition of pyroptosis does not fully prevent cell death.93 It is suggested that this pathogen induces PANoptosis and activates MLKL as a compensatory mechanism when inflammasome signaling is impaired. The sensor NLRP12 is associated with infectious and inflammatory diseases and interacts with cell death molecules to assemble a protein PANoptosome that promotes inflammatory cell death.54 By coordinating a PANoptosome composed of ASC, NLRP3, caspase-8, and RIPK3, NLRP12 induces caspase-1, caspase-8, and RIPK3-dependent PANoptosis, thereby promoting inflammatory cell death (Figure 4).54 Notably, during coronavirus infection, impaired NLRP3 inflammasome activation and reduced pyroptosis lead to pronounced inflammatory cell death mediated by caspase-8 and RIPK3.94 Further studies are required to determine whether NLRP12-mediated cell death pathways operate in non-immune cells such as epithelial cells and to define the breadth of their biological functions.
Mitochondrial reactive oxygen species have long been recognized as essential signaling molecules in pyroptosis.95 GSDMD rapidly disrupts the inner and outer mitochondrial membranes, leading to reduced mitochondrial abundance, increased mitophagy, and elevated ROS production. GSDMD-mediated mitochondrial permeabilization facilitates cytochrome c release and activates caspase-3, thereby establishing extensive crosstalk between pyroptosis and mitochondrial apoptosis.91 Furthermore, GSDMD pores formed on the mitochondrial membrane promote the release of mitochondrial reactive oxygen species, thereby shifting cell death toward RIPK1, RIPK3, and MLKL-dependent necroptosis.96 These findings indicate that oxidative stress modulates mitochondrial membrane permeability and signal transduction to regulate the activation and interaction among pyroptosis, apoptosis, and necroptosis. As a key initiator of mitochondrial death pathways, oxidative stress has been shown to contribute to PANoptosis in models of hepatic and toxic injury. Excessive acetaminophen-induced liver injury stimulates mitochondrial reactive oxygen species production and mitochondrial DNA leakage, and mitochondrial DNA subsequently activates the TLR9 pathway to promote neutrophil extracellular trap formation, thereby inducing PANoptosis.97 HDN attenuates deoxynivalenol-induced liver injury and associated PANoptosis by inhibiting mitochondrial damage mediated through the ROS-P53-PGC-1α pathway and by reducing excessive mitochondrial reactive oxygen species production.98 Similarly, oxidative stress triggered by SiO2 exposure leads to mitochondrial dysfunction and mitochondrial DNA leakage, which activates ZBP1-mediated PANoptosis, whereas quercetin suppresses this process by modulating the Nrf2/ROS/Drp1 axis.99
PANoptosis in Drug-Induced Liver Injury
Acetaminophen
Excessive administration of acetaminophen (APAP) can cause severe liver injury, and nearly half of DILI cases are associated with its use. Previous investigations have shown that APAP-induced hepatotoxicity is characterized by extensive hepatocellular necrosis and pronounced inflammatory responses. However, the specific mode of cell death triggered by APAP remains a subject of debate. Although a small number of studies have suggested the involvement of apoptosis, the absence of detectable caspase activation raises questions regarding whether APAP truly induces apoptotic cell death.100
A recent study reported that APAP promotes caspase-1-mediated pyroptosis by activating the NLRP3 inflammasome. Mice with hepatocyte-specific deletion of NLRP3 exhibited reduced liver injury and lower mortality after APAP exposure, accompanied by diminished inflammatory cell infiltration and attenuated inflammatory responses. Pharmacological inhibition of NLRP3 or GSDMD using MCC950 or disulfiram substantially ameliorated liver injury and reduced hepatocyte death.101 Pyroptosis is characterized by activation of the NLRP3 inflammasome, which leads to the generation of caspase-1.102 However, several findings challenge the concept that APAP induces acute liver injury through pyroptosis. Treatment of C57BL/6 mice with a pan-caspase inhibitor to suppress caspase-1-mediated IL-1β maturation during APAP overdose did not modify APAP-induced liver injury or neutrophil infiltration.103 Moreover, genetic deletion of GSDMD or GSDME did not prevent APAP-induced liver injury.104 Necroptosis, also known as receptor-interacting protein kinase-dependent necrosis, is regulated by RIPK1 and RIPK3. Although activation of RIPK1 and RIPK3 has been observed in APAP-induced liver injury and inhibition of either kinase attenuates liver injury,105–107 other studies show that deletion of RIPK3 or MLKL does not provide protection.108 These observations suggest that MLKL is not required for APAP-induced acute liver injury.
Based on the available evidence, Dr. Hartmut Jaeschke proposed that APAP-induced hepatocyte death does not occur through apoptosis, pyroptosis, or necroptosis, and that morphological observations instead support a necrotic pattern of cell death.100 However, recent studies indicate that hepatocellular injury induced by excessive APAP involves several forms of programmed cell death, including pyroptosis, apoptosis, and necroptosis.
Cells can die in multiple ways, but does it matter which way they die? The occurrence of PANoptosis may reflect the coordinated effects of various stimuli acting on the cell. Shi et al reported that the mode of cell death is determined by three major factors, including the nature of the stimulus, the cellular state, and the expression of cell death-related genes.19 Different stimuli can activate distinct intracellular signaling pathways, thereby inducing multiple forms of programmed cell death. For example, in the APAP-induced liver injury model, loss of GSDMD leads to differential upregulation of apoptosis and necroptosis.109 The severity of necrosis may further influence the choice of death modality, with necroptosis occurring only when the injury is more severe. Caspase-8, a central regulator of apoptosis and necroptosis, also plays an essential role in PANoptosis. Activated caspase-8 mediates intrinsic apoptosis and also cleaves GSDMD and GSDME to trigger pyroptosis in macrophages.73,74 Moreover, APAP exposure has revealed an alternative pathway in which GSDMD modulates caspase-8, as caspase-8 is elevated in the absence of GSDMD and subsequently regulates apoptosis and necroptosis.109 As previously noted, cells under stress may preferentially initiate faster forms of death such as pyroptosis. Ultimately, the choice of a specific death pathway is also influenced by the expression of genes associated with programmed cell death.19 Based on these observations, we speculate that during APAP-induced liver injury, APAP may regulate the interactions among apoptosis, necroptosis, and pyroptosis by modulating the activity and expression of key molecules such as GSDMD and caspase-8, thereby contributing to the regulation of PANoptosis.
Studies have shown that in mouse models of APAP-induced acute liver injury, markers of neutrophil extracellular traps in both liver tissue and serum are markedly elevated, along with activation of GSDMD, caspase-3, and MLKL, which collectively initiate PANoptosis.97 Administration of DNase1 significantly suppresses the formation of neutrophil extracellular traps, decreases PANoptotic cell death and mitigates APAP-induced liver injury in this model.97 Furthermore, mice deficient in AIM2 exhibit markedly attenuated hepatic injury after APAP exposure and show a reduced occurrence of PANoptosis.97 These findings suggest that PANoptosis contributes to APAP-induced liver injury through a mechanism in which DNA contained within neutrophil extracellular traps activates the DNA sensor AIM2 in hepatocytes, leading to AIM2-dependent assembly of the PANoptosome. Human umbilical cord mesenchymal stromal cells (hucMSCs) and hucMSCs-exosomes (MSCs-Exo) also markedly ameliorate APAP-induced acute liver failure in mice. Mechanistic analyses reveal that miR-423-5p enriched in these extracellular vesicles exerts anti-inflammatory effects by targeting and suppressing ZBP1, a key component of the ZBP1-associated PANoptosome complex that is essential for the execution of PANoptosis.110
However, another study reported that deletion of the AIM2 gene exacerbates APAP-induced acute liver injury in mice, which is characterized by impaired mitochondrial stability, increased glutathione depletion and diminished autophagy, although AIM2 expression is modulated in an age-dependent manner.111 AIM2 expression in the liver progressively increases with age, and AIM2 facilitates autophagy and confers protection during APAP-induced hepatic injury. Nevertheless, AIM2 serves as a critical component of the AIM2 PANoptosome and plays a central role in the pathogenesis of APAP-induced liver injury.
Overall, current evidence suggests that PANoptosis may participate in the progression of APAP-induced liver injury. Therefore, the specific regulatory mechanisms underlying PANoptosis require further in-depth investigation and refinement, particularly in relation to its potential as a therapeutic target.
Triptolide
Triptolide is an active diterpenoid lactone isolated from Tripterygium wilfordii and exhibits distinct immunomodulatory and antitumor pharmacological properties, although its clinical application is limited by adverse reactions. It shows substantial cytotoxicity toward macrophages and is capable of inducing multiple forms of regulated cell death. Notably, inhibitors that target individual cell death pathways are unable to fully suppress triptolide-induced cytotoxicity. Studies by Zhang et al showed that triptolide induces PANoptosis, as demonstrated by the simultaneous activation of pyroptotic, apoptotic, and necroptotic markers, as well as ASC specks that colocalize with RIPK3 or caspase-8 and interact with these proteins, suggesting the formation of a PANoptosome.18 The study further indicated that triptolide-induced renal and hepatic injuries may be associated with its capacity to trigger PANoptosis. Mechanistically, triptolide binds to the VAL27 site of caspase-3 and promotes its cleavage, and the cleaved enzyme subsequently induces pyroptosis in Kupffer cells through GSDME cleavage.112 Yuan et al reported that mice treated with triptolide exhibited liver injury and NLRP3 inflammasome activation accompanied by increased neutrophil infiltration, further supporting the contribution of pyroptosis to triptolide mediated hepatic injury.113 Notably, deletion of GSDMD in Kupffer cells suppresses triptolide-induced pyroptosis, suggesting that triptolide acts primarily on caspase-3 rather than GSDMD and may impair cellular viability through additional signaling pathways.112 Moreover, triptolide-induced liver injury may also involve apoptotic signaling and is associated with altered expression of apoptosis related genes including Bcl-2, Bax, caspase-9, and caspase-3.114
Caspase-8, caspase-3, NLRP3, and GSDMD constitute essential components of the PANoptosome. These findings suggest that triptolide-induced liver injury may be associated with several modes of cell death, including apoptosis, pyroptosis, and necroptosis. Although the presence of the PANoptosome has not yet been clearly established in liver injury models, further studies are needed to clarify whether triptolide can induce its formation in the liver and to identify the key molecules that regulate PANoptosis.
Isoniazid/Rifampicin
Tuberculosis continues to represent a significant global health burden. Although anti-tuberculosis drugs provide therapeutic benefits, they are accompanied by various toxic side effects, most notably DILI. However, the precise mechanisms underlying these hepatic injuries remain incompletely understood. Previous studies have demonstrated that antituberculosis drugs can induce hepatocyte apoptosis in liver tissue. For example, treatment with isoniazid and rifampicin downregulates the antiapoptotic protein Bcl-2 and enhances the expression of the proapoptotic protein Bax.115 In a zebrafish larval model of isoniazid-induced hepatotoxicity, increased expression of apoptosis-related genes, including caspase-3, caspase-8, and caspase-9, has also been observed.116 Notably, isoniazid and rifampicin provoke a marked inflammatory response in rat liver tissue through activation of the NLRP3 inflammasome, whereas treatment with inflammasome inhibitors such as INF39 or CP-456773 significantly attenuates the hepatotoxicity induced by these agents.117 Caspase-3, caspase-8, and NLRP3 represent essential components of the PANoptosome. Although hepatic PANoptosome formation has not yet been demonstrated in response to isoniazid or rifampicin, the current evidence provides a foundational basis for the potential involvement of PANoptosis and PANoptosome-associated mechanisms in this process.
Methotrexate
Methotrexate (MTX) is an effective chemotherapeutic and immunosuppressive agent, although its clinical application is limited by significant hepatotoxicity. Pronounced hepatocyte apoptosis has been consistently observed in animal models of MTX-induced toxicity. MTX-induced liver injury is characterized by increased immunoexpression of Bax, elevated levels of Bax and caspase-3, and decreased expression of Bcl-2.118 The metabolite MTX polyglutamate activates caspase-3 through the intrinsic apoptotic pathway and thereby initiates the classical apoptotic cascade.119 Moreover, MTX triggers inflammatory responses by upregulating the TLR4/NF-κB signaling pathway and subsequently activates the downstream NLRP3caspase-1 inflammasome axis, which further exacerbates hepatotoxicity.120 MTX also activates caspase-3 and GSDME through the TLR4/NF-κB pathway, thereby promoting pyroptosis.121 Notably, AIM2 expression is elevated in MTX-induced liver injury models and shows a positive correlation with the expression of caspase-1 and IL-1β.122 As a key component of the AIM2 PANoptosome, AIM2 may contribute to the inflammatory processes associated with MTX-induced liver injury. Collectively, these findings indicate that MTX-induced liver injury may involve PANoptosis, a recently recognized form of programmed cell death, although its precise mechanisms require further elucidation.
Cisplatin
Cisplatin-induced hepatotoxicity is closely associated with apoptosis. Evidence indicates that cisplatin markedly increases the expression of P53 and Bax in liver tissue while reducing Bcl-2 expression, thereby promoting apoptotic progression.123 Consistent with these observations, cisplatin significantly activates caspase-8 and caspase-9.123 Moreover, exposure to cisplatin results in a dose-dependent increase in the expression of NLRP3, caspase-1, and IL-1β.124 Additional studies have shown that cisplatin promotes GSDME activation in vivo and in vitro and induces Bax-mediated caspase-3 cleavage, suggesting that caspase-3 and GSDME-dependent pyroptosis also contributes to cisplatin-induced liver injury.125 Although direct evidence linking cisplatin-induced hepatotoxicity to PANoptosis is still lacking, existing data suggest that PANoptosis may play a potential role in this process.
Aurantio-Obtusin
Previous studies have shown that aurantio-obtusin, a major anthraquinone component derived from Cassia obtusifolia seeds, exhibits diverse biological activities in preclinical research and clinical practice. However, this compound can also induce hepatotoxicity, which is characterized by reduced hepatocyte viability and impaired biochemical functions. Its hepatotoxic effects are associated with apoptosis, as indicated by increased expression of caspase-3 and cleaved caspase-3, enhanced cytochrome c release, elevated Bax expression, and reduced Bcl-2 expression.126 Furthermore, aurantio-obtusin has been reported to induce hepatic inflammation in zebrafish larvae and in female mice. In female mice, it activates the NOD-like receptor signaling pathway, resulting in increased expression of NLRP3, caspase-1, pro-IL-1β, IL-1β, and IL-18, which suggests that pyroptosis may contribute to its hepatotoxicity.127 Given that multiple cell death pathways may jointly contribute to aurantio-obtusin-induced liver injury, identifying key targets related to PANoptosis may offer new strategies for the treatment of DILI. The drugs, components, and mechanisms of the PANoptosome involved in DILI are summarized in Table 1.
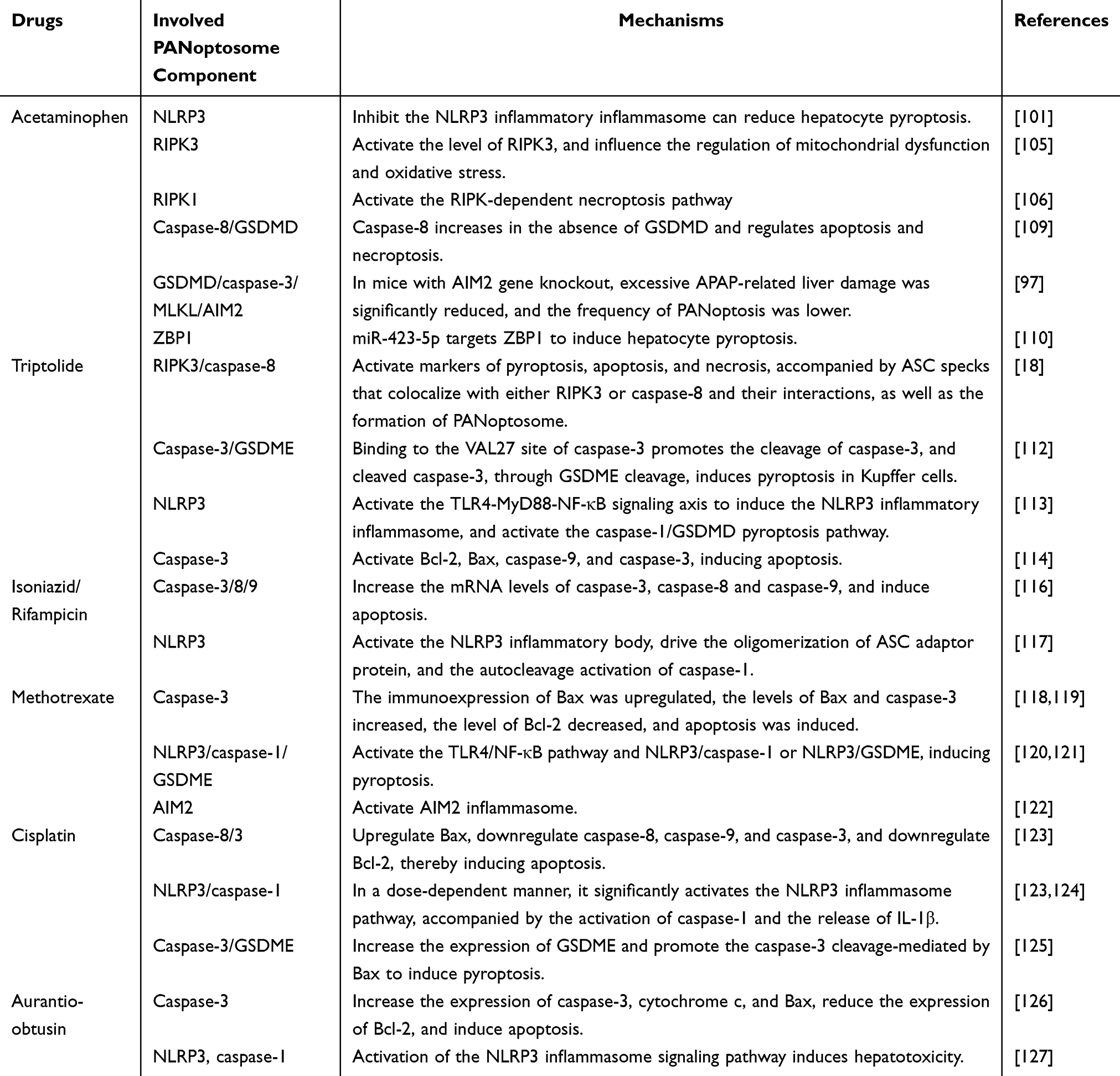 |
Table 1 Components and Mechanisms of the PANoptosome Involved in DILI |
Inhibit PANoptosis and Alleviate Liver Damage
Baicalin is a natural flavonoid compound isolated from the root of Scutellaria baicalensis, and it possesses anti-inflammatory, antioxidant, and antimicrobial properties. It is one of the major active constituents of this traditional medicinal herb and is widely used in herbal formulations and modern drug development. Numerous studies have demonstrated that baicalin inhibits multiple forms of regulated cell death, including apoptosis-associated secondary necrosis, pyroptosis, and necroptosis, thereby mitigating inflammatory responses. Additional evidence indicates that baicalin alleviates liver injury by suppressing PANoptosis. Specifically, baicalin inhibits the formation of mitochondrial Z-DNA and the assembly of the ZBP1 PANoptosome complex, thereby suppressing PANoptosis in macrophages.128 In a mouse model of hemophagocytic lymphohistiocytosis, baicalin markedly reduced hepatic injury and the expression of PANoptosis markers in Kupffer cells, suggesting that its hepatoprotective effects are mediated through modulation of the PANoptosis pathway.128 Hesperidin similarly mitigates deoxynivalenol (DON)-induced liver injury in mice, and this protective effect is closely associated with its ability to attenuate oxidative stress and PANoptosis-related cell death. Mechanistic studies show that hesperidin inhibits ROS-induced DNA damage and decreases p53 expression, which leads to increased PGC-1α levels, improved mitochondrial dynamics, reduced mitochondrial ROS, and suppression of PANoptosis, ultimately mitigating DON-induced liver injury.98 Moreover, the development of therapeutic agents targeting PANoptosome sensors and other components of the PANoptosis pathway may provide important opportunities for the treatment of DILI.129
Summary and Outlook
PANoptosis is a recently identified mode of cell death that arises from the coordinated action of multiple factors and underscores the close interconnections and reciprocal regulation among pyroptosis, apoptosis, and necroptosis. Various stimuli can activate distinct intracellular signaling pathways, ultimately initiating specific cell death programs. Under stress conditions, cells often preferentially activate rapid death mechanisms, and the specific pathway adopted is largely determined by the expression profile of death-related genes. In DILI models, activation of GSDMD, caspase-3, and MLKL has been observed, along with the colocalization of ASC specks with RIPK3 or caspase-8, thereby providing additional evidence supporting the involvement of PANoptosis. Moreover, miR-423-5p enriched in hucMSCs-Exo attenuates hepatocellular inflammation and cell death by suppressing ZBP1 expression and disrupting ZBP1 PANoptosome assembly, suggesting a promising therapeutic strategy based on exosomal miRNA-mediated targeting of ZBP1. Interventions targeting various nodes within the PANoptosis pathway, such as NETs, AIM2, and ZBP1, demonstrate considerable therapeutic potential.
However, it should be noted that current insights into the mechanisms of DILI remain largely speculative and are predominantly based on experimental observations, whereas direct evidence demonstrating that specific drugs activate the PANoptosome complex in vivo and regulate multiple cell death pathways remains limited. Consequently, critical questions, including whether PANoptosomes are present in DILI-induced by different drugs and how to rationally select appropriate inhibitors or inducers, require further investigation. Comprehensive investigation of natural compounds capable of inhibiting PANoptosome assembly, along with the development of therapeutic agents that target PANoptosome sensors and other components of the PANoptosis pathway, may offer substantial opportunities for advancing the treatment of DILI.
In summary, PANoptosis, as a newly identified form of programmed cell death, plays a pivotal role in the initiation and progression of DILI. Therefore, elucidating the regulatory mechanisms that govern PANoptosis in DILI is essential for deepening our understanding of drug-induced hepatotoxicity and provides new opportunities for the development of targeted therapeutic strategies.
Data Sharing Statement
Data sharing is not applicable to this article as no data were created or analysed in this study.
Author Contributions
Zhang Xinru: Conceptualization, Writing – original draft.
Zhou Zhipin: Conceptualization, Supervision, Writing – review & editing.
Xie Zhuohua, Chen Jieyi, Deng Yanting: Methodology.
Fang Yibin, Deng Jiasheng, Ye Xiaoxue: Investigation, Resources.
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This work was supported by grants of the National Natural Scientific Foundation of China (No.81760751), Guangxi Provincial Natural Scientific Foundation (No. 2021GXNSFAA075020).
Disclosure
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
1. Galluzzi L, Vitale I, Aaronson SA, et al. Molecular mechanisms of cell death: recommendations of the nomenclature committee on cell death 2018. Cell Death Differ. 2018;25(3):486–18. doi:10.1038/s41418-017-0012-4
2. Samir P, Malireddi RKS, Kanneganti TD. The PANoptosome: a deadly protein complex driving pyroptosis, apoptosis, and necroptosis (PANoptosis). Front Cell Infect Microbiol. 2020;10:238. doi:10.3389/fcimb.2020.00238
3. Banoth B, Tuladhar S, Karki R, et al. ZBP1 promotes fungi-induced inflammasome activation and pyroptosis, apoptosis, and necroptosis (PANoptosis). J Biol Chem. 2020;295(52):18276–18283. doi:10.1074/jbc.RA120.015924
4. Zhu P, Ke ZR, Chen JX, Li SJ, Ma TL, Fan XL. Advances in mechanism and regulation of PANoptosis: prospects in disease treatment. Front Immunol. 2023;14:1120034. doi:10.3389/fimmu.2023.1120034
5. Chen S, Jiang J, Li T, Huang L. PANoptosis: mechanism and role in pulmonary diseases. Int J Mol Sci. 2023;24(20):15343. doi:10.3390/ijms242015343
6. Rajesh Y, Kanneganti TD. Innate Immune Cell Death in Neuroinflammation and Alzheimer’s Disease. Cells. 2022;11(12):1885. doi:10.3390/cells11121885
7. Liu M, Lu J, Hu J, et al. Sodium sulfite triggered hepatic apoptosis, necroptosis, and pyroptosis by inducing mitochondrial damage in mice and AML-12 cells. J Hazard Mater. 2024;467:133719. doi:10.1016/j.jhazmat.2024.133719
8. Ye Q, Wang H, Chen Y, et al. PANoptosis-like death in acute-on-chronic liver failure injury. Sci Rep. 2024;14(1):392. doi:10.1038/s41598-023-50720-1
9. Zheng E, Sandhu N, Navarro V. Drug-induced liver injury secondary to herbal and dietary supplements. Clin Liver Dis. 2020;24(1):141–155. doi:10.1016/j.cld.2019.09.009
10. Li X, Tang J, Mao Y. Incidence and risk factors of drug-induced liver injury. Liver Int. 2022;42(9):1999–2014. doi:10.1111/liv.15262
11. Björnsson ES, Bergmann OM, Björnsson HK, Kvaran RB, Olafsson S. Incidence, presentation, and outcomes in patients with drug-induced liver injury in the general population of Iceland. Gastroenterology. 2013;144(7):
12. Shen T, Liu Y, Shang J, et al. Incidence and etiology of drug-induced liver injury in Mainland China. Gastroenterology. 2019;156(8):2230–2241.e11. doi:10.1053/j.gastro.2019.02.002
13. Allison R, Guraka A, Shawa IT, Tripathi G, Moritz W, Kermanizadeh A. Drug induced liver injury - a 2023 update. J Toxicol Environ Health B Crit Rev. 2023;26(8):442–467. doi:10.1080/10937404.2023.2261848
14. Devarbhavi H, Aithal G, Treeprasertsuk S, et al. Drug-induced liver injury: Asia Pacific Association of Study of Liver consensus guidelines. Hepatol Int. 2021;15(2):258–282. doi:10.1007/s12072-021-10144-3
15. Suzuki A, MinjunChen. Epidemiology and risk determinants of drug-induced liver injury: current knowledge and future research needs. Liver Int. 2025;45(4):e16146. doi:10.1111/liv.16146
16. Iorga A, Dara L. Cell death in drug-induced liver injury. Adv Pharmacol. 2019;85:31–74. doi:10.1016/bs.apha.2019.01.006
17. Iorga A, Dara L, Kaplowitz N. Drug-induced liver injury: cascade of events leading to cell death, apoptosis or necrosis. Int J Mol Sci. 2017;18(5):1018. doi:10.3390/ijms18051018
18. Zhang HR, Li YP, Shi ZJ, et al. Triptolide induces PANoptosis in macrophages and causes organ injury in mice. Apoptosis. 2023;28(11–12):1646–1665. doi:10.1007/s10495-023-01886-6
19. Shi C, Cao P, Wang Y, et al. PANoptosis: a cell death characterized by pyroptosis, apoptosis, and necroptosis. J Inflamm Res. 2023;16:1523–1532. doi:10.2147/JIR.S403819
20. Liu LX, Heng JH, Deng DX, et al. Sulconazole induces PANoptosis by triggering oxidative stress and inhibiting glycolysis to increase radiosensitivity in esophageal cancer. Mol Cell Proteomics. 2023;22(6):100551. doi:10.1016/j.mcpro.2023.100551
21. Lebeaupin C, Blanc M, Vallée D, Keller H, Bailly-Maitre B. BAX inhibitor-1: between stress and survival. FEBS J. 2020;287(9):1722–1736. doi:10.1111/febs.15179
22. Nicholson DW, Thornberry NA. Caspases: killer proteases. Trends Biochem Sci. 1997;22(8):299–306. doi:10.1016/s0968-0004(97)01085-2
23. Bertheloot D, Latz E, Franklin BS. Necroptosis, pyroptosis and apoptosis: an intricate game of cell death. Cell Mol Immunol. 2021;18(5):1106–1121. doi:10.1038/s41423-020-00630-3
24. Marciniak SJ, Chambers JE, Ron D. Pharmacological targeting of endoplasmic reticulum stress in disease. Nat Rev Drug Discov. 2022;21(2):115–140. doi:10.1038/s41573-021-00320-3
25. Lindsay J, Esposti MD, Gilmore AP. Bcl-2 proteins and mitochondria--specificity in membrane targeting for death. Biochim Biophys Acta. 2011;1813(4):532–539. doi:10.1016/j.bbamcr.2010.10.017
26. Tsujimoto Y. Role of Bcl-2 family proteins in apoptosis: apoptosomes or mitochondria? Genes Cells. 1998;3(11):697–707. doi:10.1046/j.1365-2443.1998.00223.x
27. Bock FJ, Tait SWG. Mitochondria as multifaceted regulators of cell death. Nat Rev Mol Cell Biol. 2020;21(2):85–100. doi:10.1038/s41580-019-0173-8
28. Acehan D, Jiang X, Morgan DG, Heuser JE, Wang X, Akey CW. Three-dimensional structure of the apoptosome: implications for assembly, procaspase-9 binding, and activation. Mol Cell. 2002;9(2):423–432. doi:10.1016/s1097-2765(02)00442-2
29. Pu S, Pan Y, Zhang Q, et al. Endoplasmic reticulum stress and mitochondrial stress in drug-induced liver injury. Molecules. 2023;28(7):3160. doi:10.3390/molecules28073160
30. D’Arcy MS. Cell death: a review of the major forms of apoptosis, necrosis and autophagy. Cell Biol Int. 2019;43(6):582–592. doi:10.1002/cbin.11137
31. Kikuchi M, Kuroki S, Kayama M, Sakaguchi S, Lee KK, Yonehara S. Protease activity of procaspase-8 is essential for cell survival by inhibiting both apoptotic and nonapoptotic cell death dependent on receptor-interacting protein kinase 1 (RIP1) and RIP3. J Biol Chem. 2012;287(49):41165–41173. doi:10.1074/jbc.M112.419747
32. Li X, Zhong CQ, Wu R, et al. RIP1-dependent linear and nonlinear recruitments of caspase-8 and RIP3 respectively to necrosome specify distinct cell death outcomes. Protein Cell. 2021;12(11):858–876. doi:10.1007/s13238-020-00810-x
33. Vanden Berghe T, Hassannia B, Vandenabeele P. An outline of necrosome triggers. Cell Mol Life Sci. 2016;73(11–12):2137–2152. doi:10.1007/s00018-016-2189-y
34. Zhang J, Yang Y, He W, Sun L. Necrosome core machinery: MLKL. Cell Mol Life Sci. 2016;73(11–12):2153–2163. doi:10.1007/s00018-016-2190-5
35. Li J, McQuade T, Siemer AB, et al. The RIP1/RIP3 necrosome forms a functional amyloid signaling complex required for programmed necrosis. Cell. 2012;150(2):339–350. doi:10.1016/j.cell.2012.06.019
36. C W, Z Z, L L, et al. Diverse sequence determinants control human and mouse receptor interacting protein 3 (RIP3) and mixed lineage kinase domain-like (MLKL) interaction in necroptotic signaling. J Biol Chem. 2013;288(23). doi:10.1074/jbc.M112.435545
37. Sun L, Wang H, Wang Z, et al. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell. 2012;148(1–2):213–227. doi:10.1016/j.cell.2011.11.031
38. Petrie EJ, Sandow JJ, Jacobsen AV, et al. Conformational switching of the pseudokinase domain promotes human MLKL tetramerization and cell death by necroptosis. Nat Commun. 2018;9(1):2422. doi:10.1038/s41467-018-04714-7
39. Zhang Y, Su SS, Zhao S, et al. RIP1 autophosphorylation is promoted by mitochondrial ROS and is essential for RIP3 recruitment into necrosome. Nat Commun. 2017;8:14329. doi:10.1038/ncomms14329
40. Lu B, Gong X, Wang ZQ, et al. Shikonin induces glioma cell necroptosis in vitro by ROS overproduction and promoting RIP1/RIP3 necrosome formation. Acta Pharmacol Sin. 2017;38(11):1543–1553. doi:10.1038/aps.2017.112
41. Tait SWG, Oberst A, Quarato G, et al. Widespread mitochondrial depletion via mitophagy does not compromise necroptosis. Cell Rep. 2013;5(4):878–885. doi:10.1016/j.celrep.2013.10.034
42. Kaiser WJ, Sridharan H, Huang C, et al. Toll-like receptor 3-mediated necrosis via TRIF, RIP3, and MLKL. J Biol Chem. 2013;288(43):31268–31279. doi:10.1074/jbc.M113.462341
43. Fink SL, Bergsbaken T, Cookson BT. Anthrax lethal toxin and Salmonella elicit the common cell death pathway of caspase-1-dependent pyroptosis via distinct mechanisms. Proc Natl Acad Sci U S A. 2008;105(11):4312–4317. doi:10.1073/pnas.0707370105
44. Aglietti RA, Dueber EC. Recent insights into the molecular mechanisms underlying pyroptosis and gasdermin family functions. Trends Immunol. 2017;38(4):261–271. doi:10.1016/j.it.2017.01.003
45. Orning P, Weng D, Starheim K, et al. Pathogen blockade of TAK1 triggers caspase-8-dependent cleavage of gasdermin D and cell death. Science. 2018;362(6418):1064–1069. doi:10.1126/science.aau2818
46. Kayagaki N, Stowe IB, Lee BL, et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature. 2015;526(7575):666–671. doi:10.1038/nature15541
47. Shi J, Zhao Y, Wang K, et al. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature. 2015;526(7575):660–665. doi:10.1038/nature15514
48. Sagulenko V, Thygesen SJ, Sester DP, et al. AIM2 and NLRP3 inflammasomes activate both apoptotic and pyroptotic death pathways via ASC. Cell Death Differ. 2013;20(9):1149–1160. doi:10.1038/cdd.2013.37
49. Shi J, Zhao Y, Wang Y, et al. Inflammatory caspases are innate immune receptors for intracellular LPS. Nature. 2014;514(7521):187–192. doi:10.1038/nature13683
50. Kayagaki N, Warming S, Lamkanfi M, et al. Non-canonical inflammasome activation targets caspase-11. Nature. 2011;479(7371):117–121. doi:10.1038/nature10558
51. Kuriakose T, Man SM, Malireddi RKS, et al. ZBP1/DAI is an innate sensor of influenza virus triggering the NLRP3 inflammasome and programmed cell death pathways. Sci Immunol. 2016;1(2):aag2045. doi:10.1126/sciimmunol.aag2045
52. Christgen S, Zheng M, Kesavardhana S, et al. Identification of the PANoptosome: a molecular platform triggering pyroptosis, apoptosis, and necroptosis (PANoptosis). Front Cell Infect Microbiol. 2020;10:237. doi:10.3389/fcimb.2020.00237
53. Lee S, Karki R, Wang Y, Nguyen LN, Kalathur RC, Kanneganti TD. AIM2 forms a complex with pyrin and ZBP1 to drive PANoptosis and host defence. Nature. 2021;597(7876):415–419. doi:10.1038/s41586-021-03875-8
54. Sundaram B, Pandian N, Mall R, et al. NLRP12-PANoptosome activates PANoptosis and pathology in response to heme and PAMPs. Cell. 2023;186(13):2783–2801.e20. doi:10.1016/j.cell.2023.05.005
55. Karki R, Lee S, Mall R, et al. ZBP1-dependent inflammatory cell death, PANoptosis, and cytokine storm disrupt IFN therapeutic efficacy during coronavirus infection. Sci Immunol. 2022;7(74):eabo6294. doi:10.1126/sciimmunol.abo6294
56. Gullett JM, Tweedell RE, Kanneganti TD. It’s all in the PAN: crosstalk, plasticity, redundancies, switches, and interconnectedness encompassed by panoptosis underlying the totality of cell death-associated biological effects. Cells. 2022;11(9):1495. doi:10.3390/cells11091495
57. Malireddi RKS, Kesavardhana S, Karki R, Kancharana B, Burton AR, Kanneganti TD. RIPK1 distinctly regulates yersinia-induced inflammatory cell death, PANoptosis. Immunohorizons. 2020;4(12):789–796. doi:10.4049/immunohorizons.2000097
58. Chen W, Gullett JM, Tweedell RE, Kanneganti TD. Innate immune inflammatory cell death: pANoptosis and PANoptosomes in host defense and disease. European Journal of Immunology. 2023;53(11):e2250235. doi:10.1002/eji.202250235
59. Zheng M, Karki R, Vogel P, Kanneganti TD. Caspase-6 is a key regulator of innate immunity, inflammasome activation, and host defense. Cell. 2020;181(3):674–687.e13. doi:10.1016/j.cell.2020.03.040
60. Thapa RJ, Ingram JP, Ragan KB, et al. DAI senses influenza a virus genomic RNA and Activates RIPK3-dependent cell death. Cell Host Microbe. 2016;20(5):674–681. doi:10.1016/j.chom.2016.09.014
61. Wang Y, Pandian N, Han JH, et al. Single cell analysis of PANoptosome cell death complexes through an expansion microscopy method. Cell Mol Life Sci. 2022;79(10):531. doi:10.1007/s00018-022-04564-z
62. Zheng M, Kanneganti TD. The regulation of the ZBP1-NLRP3 inflammasome and its implications in pyroptosis, apoptosis, and necroptosis (PANoptosis). Immunol Rev. 2020;297(1):26–38. doi:10.1111/imr.12909
63. Kesavardhana S, Malireddi RKS, Burton AR, et al. The Zα2 domain of ZBP1 is a molecular switch regulating influenza-induced PANoptosis and perinatal lethality during development. J Biol Chem. 2020;295(24):8325–8330. doi:10.1074/jbc.RA120.013752
64. Kuriakose T, Zheng M, Neale G, Kanneganti TD. IRF1 is a transcriptional regulator of ZBP1 promoting NLRP3 inflammasome activation and cell death during influenza virus infection. J Immunol. 2018;200(4):1489–1495. doi:10.4049/jimmunol.1701538
65. Sharma BR, Karki R, Rajesh Y, Kanneganti TD. Immune regulator IRF1 contributes to ZBP1-, AIM2-, RIPK1-, and NLRP12-PANoptosome activation and inflammatory cell death (PANoptosis). J Biol Chem. 2023;299(9):105141. doi:10.1016/j.jbc.2023.105141
66. Zheng M, Kanneganti TD. Newly identified function of caspase-6 in ZBP1-mediated innate immune responses, NLRP3 inflammasome activation, PANoptosis, and host defense. J Cell Immunol. 2020;2(6):341–347. doi:10.33696/immunology.2.064
67. Wang B, Tian Y, Yin Q. AIM2 inflammasome assembly and signaling. Adv Exp Med Biol. 2019;1172:143–155. doi:10.1007/978-981-13-9367-9_7
68. Karki R, Man SM, Malireddi RKS, et al. Concerted activation of the AIM2 and NLRP3 inflammasomes orchestrates host protection against Aspergillus infection. Cell Host Microbe. 2015;17(3):357–368. doi:10.1016/j.chom.2015.01.006
69. K S, Rks M, Td K. Caspases in cell death, inflammation, and pyroptosis. Ann Rev Immunol. 2020;38. doi:10.1146/annurev-immunol-073119-095439
70. Fritsch M, Günther SD, Schwarzer R, et al. Caspase-8 is the molecular switch for apoptosis, necroptosis and pyroptosis. Nature. 2019;575(7784):683–687. doi:10.1038/s41586-019-1770-6
71. Rajput A, Kovalenko A, Bogdanov K, et al. RIG-I RNA helicase activation of IRF3 transcription factor is negatively regulated by caspase-8-mediated cleavage of the RIP1 protein. Immunity. 2011;34(3):340–351. doi:10.1016/j.immuni.2010.12.018
72. Wang Y, Karki R, Mall R, et al. Molecular mechanism of RIPK1 and caspase-8 in homeostatic type I interferon production and regulation. Cell Rep. 2022;41(1):111434. doi:10.1016/j.celrep.2022.111434
73. Jiang M, Qi L, Li L, Wu Y, Song D, Li Y. Caspase-8: a key protein of cross-talk signal way in “PANoptosis” in cancer. Int, J, Cancer. 2021;149(7):1408–1420. doi:10.1002/ijc.33698
74. Sarhan J, Liu BC, Muendlein HI, et al. Caspase-8 induces cleavage of gasdermin D to elicit pyroptosis during Yersinia infection. Proc Natl Acad Sci U S A. 2018;115(46):E10888–E10897. doi:10.1073/pnas.1809548115
75. Kang S, Fernandes-Alnemri T, Rogers C, et al. Caspase-8 scaffolding function and MLKL regulate NLRP3 inflammasome activation downstream of TLR3. Nat Commun. 2015;6:7515. doi:10.1038/ncomms8515
76. Tsuchiya K, Nakajima S, Hosojima S, et al. Caspase-1 initiates apoptosis in the absence of gasdermin D. Nat Commun. 2019;10(1):2091. doi:10.1038/s41467-019-09753-2
77. Taabazuing CY, Okondo MC, Bachovchin DA. Pyroptosis and apoptosis pathways engage in bidirectional crosstalk in monocytes and macrophages. Cell Chem Biol. 2017;24(4):507–514.e4. doi:10.1016/j.chembiol.2017.03.009
78. Wang Y, Gao W, Shi X, et al. Chemotherapy drugs induce pyroptosis through caspase-3 cleavage of a gasdermin. Nature. 2017;547(7661):99–103. doi:10.1038/nature22393
79. Ning X, Wang Y, Jing M, et al. Apoptotic caspases suppress type i interferon production via the cleavage of cGAS, MAVS, and IRF3. Mol Cell. 2019;74(1):19–31.e7. doi:10.1016/j.molcel.2019.02.013
80. White MJ, McArthur K, Metcalf D, et al. Apoptotic caspases suppress mtDNA-induced STING-mediated type I IFN production. Cell. 2014;159(7):1549–1562. doi:10.1016/j.cell.2014.11.036
81. Tao P, Sun J, Wu Z, et al. A dominant autoinflammatory disease caused by non-cleavable variants of RIPK1. Nature. 2020;577(7788):109–114. doi:10.1038/s41586-019-1830-y
82. Lalaoui N, Boyden SE, Oda H, et al. Mutations that prevent caspase cleavage of RIPK1 cause autoinflammatory disease. Nature. 2020;577(7788):103–108. doi:10.1038/s41586-019-1828-5
83. Zhang H, Zhou X, McQuade T, Li J, Chan FKM, Zhang J. Functional complementation between FADD and RIP1 in embryos and lymphocytes. Nature. 2011;471(7338):373–376. doi:10.1038/nature09878
84. Dillon CP, Weinlich R, Rodriguez DA, et al. RIPK1 blocks early postnatal lethality mediated by caspase-8 and RIPK3. Cell. 2014;157(5):1189–1202. doi:10.1016/j.cell.2014.04.018
85. Zhang DW, Shao J, Lin J, et al. RIP3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. Science. 2009;325(5938):332–336. doi:10.1126/science.1172308
86. Malireddi RKS, Gurung P, Mavuluri J, et al. TAK1 restricts spontaneous NLRP3 activation and cell death to control myeloid proliferation. J Exp Med. 2018;215(4):1023–1034. doi:10.1084/jem.20171922
87. Malireddi RKS, Kesavardhana S, Kanneganti TD. ZBP1 and TAK1: master Regulators of NLRP3 inflammasome/pyroptosis, apoptosis, and necroptosis (PAN-optosis). Front Cell Infect Microbiol. 2019;9:406. doi:10.3389/fcimb.2019.00406
88. Malireddi RKS, Gurung P, Kesavardhana S, et al. Innate immune priming in the absence of TAK1 drives RIPK1 kinase activity-independent pyroptosis, apoptosis, necroptosis, and inflammatory disease. J Exp Med. 2020;217(3):
89. Frank D, Vince JE. Pyroptosis versus necroptosis: similarities, differences, and crosstalk. Cell Death Differ. 2019;26(1):99–114. doi:10.1038/s41418-018-0212-6
90. Rathkey JK, Zhao J, Liu Z, et al. Chemical disruption of the pyroptotic pore-forming protein gasdermin D inhibits inflammatory cell death and sepsis. Sci Immunol. 2018;3(26):eaat2738. doi:10.1126/sciimmunol.aat2738
91. Rogers C, Erkes DA, Nardone A, Aplin AE, Fernandes-Alnemri T, Alnemri ES. Gasdermin pores permeabilize mitochondria to augment caspase-3 activation during apoptosis and inflammasome activation. Nat Commun. 2019;10(1):1689. doi:10.1038/s41467-019-09397-2
92. Sundaram B, Kanneganti TD. Advances in Understanding Activation and Function of the NLRC4 Inflammasome. Int J Mol Sci. 2021;22(3):1048. doi:10.3390/ijms22031048
93. Sundaram B, Karki R, Kanneganti TD. NLRC4 deficiency leads to enhanced phosphorylation of MLKL and necroptosis. Immunohorizons. 2022;6(3):243–252. doi:10.4049/immunohorizons.2100118
94. Zheng M, Williams EP, Malireddi RKS, et al. Impaired NLRP3 inflammasome activation/pyroptosis leads to robust inflammatory cell death via caspase-8/RIPK3 during coronavirus infection. J Biol Chem. 2020;295(41):14040–14052. doi:10.1074/jbc.RA120.015036
95. Miao R, Jiang C, Chang WY, et al. Gasdermin D permeabilization of mitochondrial inner and outer membranes accelerates and enhances pyroptosis. Immunity. 2023;56(11):2523–2541.e8. doi:10.1016/j.immuni.2023.10.004
96. Weindel CG, Martinez EL, Zhao X, et al. Mitochondrial ROS promotes susceptibility to infection via gasdermin D-mediated necroptosis. Cell. 2022;185(17):3214–3231.e23. doi:10.1016/j.cell.2022.06.038
97. Zeng F, Zhang Y, Wang ZH, et al. Neutrophil extracellular traps promote Acetaminophen-induced acute liver injury in mice via AIM2. Acta Pharmacol Sin. 2024;45(8):1660–1672. doi:10.1038/s41401-024-01239-2
98. Wang X, Nie T, Li A, Ma J. Hesperidin mitigated deoxynivalenol-induced liver injury by inhibiting ROS/P53/PGC-1α-mediated disruption of mitochondrial dynamics and PANoptosis. Phytomedicine. 2025;142:156747. doi:10.1016/j.phymed.2025.156747
99. Li K, Yang X, Xu T, Shi X, Xu S. Quercetin protects against silicon dioxide particles-induced spleen ZBP1-mediated PANoptosis by regulating the Nrf2/Drp1/mtDNA axis. Int Immunopharmacol. 2024;143(Pt 3):113546. doi:10.1016/j.intimp.2024.113546
100. Jaeschke H, Ramachandran A. Acetaminophen hepatotoxicity: paradigm for understanding mechanisms of drug-induced liver injury. Annu Rev Pathol. 2024;19:453–478. doi:10.1146/annurev-pathmechdis-051122-094016
101. Yuan X, Chen P, Luan X, et al. NLRP3 deficiency protects against Acetaminophen‑induced liver injury by inhibiting hepatocyte pyroptosis. Mol Med Rep. 2024;29(4):61. doi:10.3892/mmr.2024.13185
102. Shi J, Gao W, Shao F. Pyroptosis: gasdermin-mediated programmed necrotic cell death. Trends Biochem Sci. 2017;42(4):245–254. doi:10.1016/j.tibs.2016.10.004
103. Williams CD, Farhood A, Jaeschke H. Role of caspase-1 and interleukin-1beta in Acetaminophen-induced hepatic inflammation and liver injury. Toxicol Appl Pharmacol. 2010;247(3):169–178. doi:10.1016/j.taap.2010.07.004
104. Li Z, Wang H, Zhu J, et al. Inhibition of TWEAK/Tnfrsf12a axis protects against acute liver failure by suppressing RIPK1-dependent apoptosis. Cell Death Discov. 2022;8(1):328. doi:10.1038/s41420-022-01123-0
105. Ramachandran A, McGill MR, Xie Y, Ni HM, Ding WX, Jaeschke H. Receptor interacting protein kinase 3 is a critical early mediator of Acetaminophen-induced hepatocyte necrosis in mice. Hepatology. 2013;58(6):2099–2108. doi:10.1002/hep.26547
106. Takemoto K, Hatano E, Iwaisako K, et al. Necrostatin-1 protects against reactive oxygen species (ROS)-induced hepatotoxicity in Acetaminophen-induced acute liver failure. FEBS Open Bio. 2014;4:777–787. doi:10.1016/j.fob.2014.08.007
107. Li JX, Feng JM, Wang Y, et al. The B-Raf(V600E) inhibitor dabrafenib selectively inhibits RIP3 and alleviates Acetaminophen-induced liver injury. Cell Death Dis. 2014;5(6):e1278. doi:10.1038/cddis.2014.241
108. Dara L, Johnson H, Suda J, et al. Receptor interacting protein kinase 1 mediates murine Acetaminophen toxicity independent of the necrosome and not through necroptosis. Hepatology. 2015;62(6):1847–1857. doi:10.1002/hep.27939
109. Yang C, Sun P, Deng M, et al. Gasdermin D protects against noninfectious liver injury by regulating apoptosis and necroptosis. Cell Death Dis. 2019;10(7):481. doi:10.1038/s41419-019-1719-6
110. Xie D, Yu L, Wang Z, Qiu G, Ouyang S. Exosomes derived from human umbilical cord mesenchymal stem cells inhibit hepatocyte pyroptosis via miR-423-5p/ZBP1 in acute liver failure. Hum Cell. 2025;38(5):124. doi:10.1007/s13577-025-01248-1
111. Hu C, Li M, Chen Y, et al. AIM2 regulates autophagy to mitigate oxidative stress in aged mice with acute liver injury. Cell Death Discov. 2024;10(1):107. doi:10.1038/s41420-024-01870-2
112. Han C, Pei H, Sheng Y, et al. Toxicological mechanism of triptolide-induced liver injury: caspase3-GSDME-mediated pyroptosis of Kupffer cell. Ecotoxicol Environ Saf. 2023;258:114963. doi:10.1016/j.ecoenv.2023.114963
113. Yuan Z, Hasnat M, Liang P, et al. The role of inflammasome activation in Triptolide-induced acute liver toxicity. Int Immunopharmacol. 2019;75:105754. doi:10.1016/j.intimp.2019.105754
114. Huo J, Yu Q, Zhang Y, et al. Triptolide-induced hepatotoxicity via apoptosis and autophagy in zebrafish. J Appl Toxicol. 2019;39(11):1532–1540. doi:10.1002/jat.3837
115. Ezhilarasan D. Antitubercular drugs induced liver injury: an updated insight into molecular mechanisms. Drug Metab. Rev. 2023;55(3):239–253. doi:10.1080/03602532.2023.2215478
116. Jia ZL, Cen J, Wang JB, et al. Mechanism of isoniazid-induced hepatotoxicity in zebrafish larvae: activation of ROS-mediated ERS, apoptosis and the Nrf2 pathway. Chemosphere. 2019;227:541–550. doi:10.1016/j.chemosphere.2019.04.026
117. Su Q, Kuang W, Hao W, et al. Antituberculosis drugs (Rifampicin and Isoniazid) induce liver injury by regulating NLRP3 inflammasomes. Mediators Inflamm. 2021;2021:8086253. doi:10.1155/2021/8086253
118. Abo-Haded HM, Elkablawy MA, Al-Johani Z, Al-Ahmadi O, El-Agamy DS. Hepatoprotective effect of sitagliptin against methotrexate induced liver toxicity. PLoS One. 2017;12(3):e0174295. doi:10.1371/journal.pone.0174295
119. Ezhilarasan D. Hepatotoxic potentials of methotrexate: understanding the possible toxicological molecular mechanisms. Toxicology. 2021;458:152840. doi:10.1016/j.tox.2021.152840
120. Matouk AI, Awad EM, El-Tahawy NFG, El-Sheikh AAK, Waz S. Dihydromyricetin alleviates methotrexate-induced hepatotoxicity via suppressing the TLR4/NF-κB pathway and NLRP3 inflammasome/caspase 1 axis. Biomed Pharmacother. 2022;155:113752. doi:10.1016/j.biopha.2022.113752
121. Alsaab J, Sarawi WS, Alhusaini AM, et al. Procyanidin B2 mitigates methotrexate-induced hepatic pyroptosis by suppressing TLR4/NF-κB and caspase-3/GSDME pathways. Food Chem Toxicol. 2025;199:115341. doi:10.1016/j.fct.2025.115341
122. Chen C, Liu YH, Cheng SB, Wu SL, Zhai XJ. The hepatoprotective effects of XCHD and MgIG against methotrexate-induced liver injury and inflammation in rats through suppressing the activation of AIM2 inflammasomes. Pathol Res Pract. 2020;216(4):152875. doi:10.1016/j.prp.2020.152875
123. Eisa NH, El-Sherbiny M, Abo El-Magd NF. Betulin alleviates cisplatin-induced hepatic injury in rats: targeting apoptosis and Nek7-independent NLRP3 inflammasome pathways. Int Immunopharmacol. 2021;99:107925. doi:10.1016/j.intimp.2021.107925
124. Qu X, Gao H, Tao L, et al. Autophagy inhibition-enhanced assembly of the NLRP3 inflammasome is associated with cisplatin-induced acute injury to the liver and kidneys in rats. J Biochem Mol Toxicol. 2018:e22208. doi:10.1002/jbt.22228
125. Wu PP, Shen XJ, Zheng SS. Cisplatin induces acute liver injury by triggering caspase-3/GSDME-mediated cell pyroptosis. Hepatobiliary Pancreat Dis Int. 2024:
126. Hu M, Zhong Y, Liu J, et al. An adverse outcome pathway-based approach to assess aurantio-obtusin-induced hepatotoxicity. Toxicology. 2022;478:153293. doi:10.1016/j.tox.2022.153293
127. Hu M, Lin L, Liu J, et al. Aurantio-obtusin induces hepatotoxicity through activation of NLRP3 inflammasome signaling. Toxicol Lett. 2022;354:1–13. doi:10.1016/j.toxlet.2021.10.011
128. You YP, Yan L, Ke HY, et al. Baicalin inhibits PANoptosis by blocking mitochondrial Z-DNA formation and ZBP1-PANoptosome assembly in macrophages. Acta Pharmacol Sin. 2025;46(2):430–447. doi:10.1038/s41401-024-01376-8
129. Pandeya A, Kanneganti TD. Therapeutic potential of PANoptosis: innate sensors, inflammasomes, and RIPKs in PANoptosomes. Trends Mol Med. 2024;30(1):74–88. doi:10.1016/j.molmed.2023.10.001
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.