Back to Archived Journals » Gastrointestinal Cancer: Targets and Therapy » Volume 6
Pancreatic cancer vaccine: a unique potential therapy
Authors Cappello P, Principe M, Novelli F
Received 10 September 2015
Accepted for publication 9 October 2015
Published 21 December 2015 Volume 2016:6 Pages 1—11
DOI https://doi.org/10.2147/GICTT.S55850
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Eileen O'Reilly
Paola Cappello, Moitza Principe, Francesco Novelli
Department of Molecular Biotechnologies and Health Sciences, Center for Experimental Research and Medical Studies, AOU Città della Salute e della Scienza di Torino, University of Turin, Turin, Italy
Abstract: Pancreatic ductal adenocarcinoma (PDA) is a lethal disease and is one of the cancers that is most resistant to traditional therapies. Historically, neither chemotherapy nor radiotherapy has provided any significant increase in the survival of patients with PDA. Despite intensive efforts, any attempts to improve the survival in the past 15 years have failed. This holds true even after the introduction of molecularly targeted agents, chosen on the basis of their involvement in pathways that are considered to be important in PDA development and progression. Recently, however, FOLFIRINOX (5-fluorouracil, leucovorin, irinotecan, and oxaliplatin) treatment has provided a limited survival advantage in patients with advanced PDA. Therefore, effective therapeutic strategies are urgently needed to improve the survival rate of patients with PDA. Results from the last 10 years of research in the field of PDA have helped to identify new immunological targets and develop new vaccines that are capable of stimulating an immune response. In addition, the information obtained about the role of the tumor microenvironment in suppressing the immune response and the possibility of targeting PDA microenvironment to limit immune suppression and enhance the response of effector T-cells has opened new avenues for treating this incurable disease. The time is ripe for developing new therapeutic approaches that are able to effectively counteract the progression and spreading of PDA. This review discusses the potential prospects in the care of patients with pancreatic cancer through vaccination and its combination therapy with surgery, chemotherapy, targeting of the tumor microenvironment, and inhibition of immunological checkpoints.
Keywords: pancreatic cancer, vaccines, T-cells, antibody, immunotherapy
Pancreatic cancer
Although lung, breast, prostate, and colorectal cancers are considered to be the “big four” cancer types in the USA, pancreas and liver cancers are expected to surpass breast, prostate, and colorectal cancers to become the second and third leading causes of cancer-related deaths by 2030, respectively.1 Pancreatic ductal adenocarcinomas (PDAs) arise from the exocrine pancreas and account for 95% of pancreatic cancers.2 In the USA, there were an estimated 48,960 cases of new-onset pancreatic cancer in 2015, which led to 40,560 deaths, with a 5-year survival rate of ~7%.3 PDA is nearly universally lethal; <20% of patients are suitable candidates for surgery at the time of diagnosis, and the median survival rate is 3.5 months and 12.6 months for nonresected patients and resected patients, respectively.2,4 PDA evolves from noninvasive precursor lesions that do not invade the basement membrane.5–7 Three precursors have been characterized, namely, pancreatic intraepithelial neoplasias (PanINs), intraductal papillary mucinous neoplasms, and mucinous cystic neoplasms.8
The most frequent genetic abnormalities in PDA are mutational activation of the KRAS oncogene, inactivation of tumor suppressor genes, including CDKN2A, TP53, SMAD4, and BRCA2,9 widespread chromosomal losses, gene amplifications,10 and telomere shortening.11
The early stages of pancreatic cancer are usually asymptomatic, and therefore, most patients with PDA present an advanced-stage disease at diagnosis.12 Currently, no early detection tests are available, and as a result, most patients (80%–85%) are not diagnosed until the later stage of the disease.13–15 For the diagnosis of PDA, there are many potential serum biomarkers that allow stratifying patients according to the prognosis and for monitoring the therapy.16 CA 19-9 is the only biomarker with clinical usefulness in predicting the patient’s response to chemotherapy,17,18 preoperative prognosis, as well as assessing treatment response, overall survival, and recurrence.19–23 However, CA 19-9 has several limitations; it is not a specific biomarker for PDA and may also be elevated in cholestasis. In addition, patients who are negative for Lewis antigen A or B (~10% of patients with PDA) do not synthesize CA 19-9 and have undetectable levels, even in the advanced stages of the disease.24
A promising discovery of biomarkers for the early diagnosis of PDA came from a serological proteome approach, which identified autoantibodies as a potential diagnostic marker in patients with PDA, in a genetically engineered mouse (GEM) model of PDA and in a prediagnostic cohort. These autoantibodies recognize the cytoskeletal protein Ezrin25 and the phosphorylated α-enolase (ENO1), a glycolytic enzyme, which also functions as a plasminogen receptor.26 Combined detection of antibodies to Ezrin, phosphorylated ENO1, and CA 19-9 correctly discriminates PDA from control patients with high sensitivity and specificity.25 A cell surface proteoglycan, glypican-1, specifically enriched on PDA cell-derived exosomes, was found to be able to discriminate early stages of pancreatic cancer.27 Validating these newly discovered markers in large cohorts of early and advanced patients with PDA, as well as in selected risk populations, and developing easy and convenient clinical tests for their detection will greatly facilitate curative surgical and immune-based therapies.
Conventional therapies
Currently, surgical resection offers the only hope of curative therapy for PDA. Nevertheless, this procedure is only considered in patients with resectable PDA, which represents ~20% of patients.28,29 Although additional palliative care is often utilized, controversy surrounds the potential benefits. Several poor predictors for successful resection have been identified, including lymph node involvement,30 high tumor grade,31 large tumor size,32 elevated CA 19-9 levels,32 and positive margins of tumors following resection.33 These same factors are also indicative of recurrence of pancreatic tumors. Even with complete and successful surgical resection, 5-year survival rates remain dismal, at ~20% following surgery.34 Therefore, postoperative chemotherapy involving gemcitabine or 5-fluorouracil or chemoradiation is almost always incorporated into the therapeutic regimen. Developing other complementary agents to enhance chemotherapeutic effects such as Hedgehog signaling inhibitors,35 second mitochondria-derived activator of caspase mimetics,36 microRNAs,37 resveratrol,38 capecitabine,39 thymoquinone,40 heat-shock protein complements,41 or curcumin42 are currently under review.13 Certain patients might receive neoadjuvant (preoperative) gemcitabine-based therapy that increased survival rates.43,44 Chemotherapy with radiation has also been shown to improve survival, but not stage, of locally invasive tumor patients without metastasis.45
Due to the difficulties of making an early diagnosis, most patients present metastatic PDA upon initial diagnosis. In the advanced stage of disease, PDA causes imminent mortality. Gemcitabine was once considered as the chemotherapeutic standard of care for advanced PDA, which improved median overall survival as well as 1-year survival (18%).46 At present, FOLFIRINOX (5-fluorouracil, leucovorin, irinotecan, and oxaliplatin) is considered as the new standard in the treatment of advanced PDA.47 Compared to gemcitabine alone, FOLFIRINOX has been shown to result in a better objective response rate (32% vs 9%), improved median progression-free survival (6.4 months vs 3.3 months), and increased overall survival (11.1 months vs 6.8 months). While the toxicity levels associated with FOLFIRINOX are greater than those caused by gemcitabine, the effects did not have a significant impact on the quality of life, and very few deaths correlated with toxicity have been reported. However, gemcitabine-based therapies in patients who do not tolerate FOLFIRINOX as a first-line therapy have been demonstrated to be beneficial.47
In the recent years, targeted therapies have been tested in metastatic pancreatic cancer patients: Erlotinib, an inhibitor of the epidermal growth factor receptor (EGFR) which, in combination with gemcitabine, significantly decreased tumor progression and increased overall survival rates.48 This was especially true in patients with EGFR mutations compared to wild-type EGFR (ClinicalTrials.gov number, NCT01608841).49
In addition, nab-paclitaxel (microparticles of albumin-containing paclitaxel that diminishes the stromal tissue associated with PDA)50,51 in combination with gemcitabine improved the overall and progression-free survival, as well as the response rate in patients with metastatic PDA.52
Mechanisms and major players of suppression of the immune response in PDA
Tumors engage the immune system response from their inception and this initially involves mainly macrophages and mast cells. However, even early on, the engagement of T-cells is evident.53 Immune cells (principally innate cells) are able to recognize tumor cells as “different” and fight them by inducing an inflammatory response. However, inflammation creates a tumor mutagenic environment, either directly through free radical generation or indirectly via alterations in the microbiome and barrier functions, which enable access of genotoxic bacteria to the epithelial cells.54
Inflammation can promote the formation of premalignant lesions and accelerate the PDA development. Indeed, PDA is characterized by an immunosuppressive environment, which favors tumor progression and invasion. Pancreatitis, or inflammation of the pancreas, often leads to pancreatic damage and promotes the development of neoplastic disease.
In a GEM model that spontaneously develops PDA comparable to the human disease, 50% of the tumor was composed of leukocytes, particularly immunosuppressive cells, including tumor-associated macrophages (TAM), myeloid-derived suppressor cells (MDSCs), and regulatory T-cells (Tregs), as well as a small percentage of effector T-cells. At early stages of carcinogenesis, for example, during PanIN development, Tregs and MDSC dominate the tumor infiltrate. Upon disease progression to PDA, CD4+ and CD8+ T-cells are inconsistently found to be associated with the tumor, and these CD8+ T-cells associated with the tumor appear to be inactivated, suggesting a suppressed immune environment.55 In all stages of the disease, there is a strong inverse correlation between MDSC and CD8+ T-cells, suggesting that MDSCs are the mediators of the immunosuppression.55 Other preclinical models have demonstrated the role of chronic inflammation in PDA progression56 and of infiltrating neutrophils in mutant Kras-driven tumorigenesis and evasion from the immune system by promoting early angiogenesis.57,58
Many studies have reported that targeting specific tumor microenvironment components (Table 1), especially in conjunction with chemotherapy, unleashes the T-cell response against PDA. Altering the tumor stroma with an anti-CD40 agonist, which activated macrophages, showed therapeutical efficacy both in human and mouse PDA. The therapeutic effect of anti-CD40-dependent macrophage activation does not require CD4+ and CD8+ T-cells.59 However, when the anti-CD40 agonist and gemcitabine treatment are combined with clodronate-encapsulated liposomes that deplete macrophages, a T-cell-dependent regression of the subcutaneous tumor is elicited in GEM.60 Ly6ClowF4/80+ macrophages appear to be responsible for this effect, and they reside outside the tumor microenvironment but are able to inhibit the infiltration of CD4+ and CD8+ T-cells into the tumor site, creating a site of acquired T-cell “immune privilege” in PDA. These data demonstrate that a T-cell response capable of inducing tumor regression can be invoked against spontaneously arising PDA and indicate a strategy that targets macrophages for enhancing the efficacy of T-cell-based immunotherapy in PDA. In a similar manner, the depletion of TAM by inhibitors of the chemokine receptor CCR2 or the colony stimulating factor 1 receptor, in conjunction with gemcitabine, enhanced the infiltration of CD4+ and CD8+ T-cells, reduced the number of Treg cells, and inhibited the growth and spreading of GEM-derived PDA cells orthotopically injected into syngeneic mice.61–63 Notably, the blockade of colony stimulating factor 1 (CSF1) or CSF1 receptor (CSF1R) significantly enhanced the responses to immune checkpoint-based immunotherapy.64,65 Similar results were obtained by the depletion of myeloid cells, which were actively recruited and maintained by two cytokines, namely, granulocyte-colony stimulating factor and granulocyte and monocyte–colony stimulating factor (GM-CSF), principally secreted by PDA cells harboring Kras mutations.66–68 Therefore, targeting the TAM or macrophages that are responsible for inhibiting the T-cell immune response is a feasible option effective, especially in combination with chemotherapy, for treating PDA. A well-known feature of PDA is the rapid development of desmoplasia, which is derived from pancreatic stellate cells that are activated to proliferate and produce collagens, laminin, and fibronectin, that differentially shape the stroma.69 The stroma also contributes to tumor hypoperfusion and hypoxia and harbors suppressive infiltrative macrophages and inflammatory cells. Therefore, limiting either the inflammatory response or the stroma depletion could enhance drug delivery to the cancer cells within the tumor mass while disrupting deleterious stroma–cancer cell interactions. A lot of data seem to support this hypothesis, for example, ablation of cancer-associated fibroblasts or inhibition of cancer-associated fibroblasts-secreted CXCL12 chemokines led to a T-cell infiltration of the tumor site and synergized with other immunotherapeutic strategies to greatly diminish cancer cells.70,71 New insights have been revealed from two studies that have demonstrated that 1) the stroma in PDA is protective, and this action is already exerted at the PanIN stage and72 2) targeting the stroma not only can lead to a more aggressive and proliferating form of PDA cells, but also can “prime” the tumor to more efficiently respond to immune checkpoint blockade and to antiangiogenic therapy;73 therefore, it is important to reassess various therapeutic approaches bearing in mind that the actions of the stroma in PDA may be context dependent.74
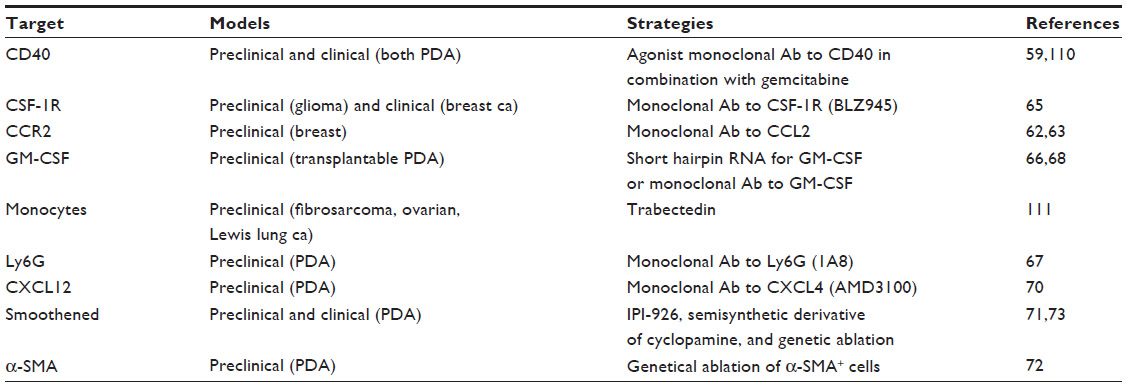 | Table 1 Strategies to modulate tumor microenvironment |
All these data have corroborated the original idea that PDA is poorly immunogenic or an “immune privileged” site. However, the presence of T-cells in PDA and their ability to be activated by tumor antigens have been documented. Higher levels of tumor-infiltrating CD4+ and CD8+ cells and dendritic cells (DCs) positively correlate with longer survival after surgical resection.75 Indeed, clinically, CD8+-cells are elevated in the circulation of patients with PDA,76 and leukocytes, the majority of which are T-cells, surround the pancreatic lesion.77 T-cells are more frequently found in the fibrotic interstitial tissue than in the epithelial area of PDA.78 Notably, PDA-infiltrating lymphocytes specifically recognize PDA-associated antigens but are often frustrated in their effector functions by the presence of Tregs.79
Vaccination in PDA
The prospect of effective immunotherapies for patients with PDA is now becoming a clinical reality, and the identification of tumor-associated antigens has been crucial in creating a springboard to accelerate this reality. The US FDA approval of PROVENGE (Sipuleucel-T), an autologous cellular vaccine, in 2010, as well as the anticytotoxic T-lymphocyte-associated antigen 4 (CTLA-4) monoclonal antibody (mAB) ipilimumab in 2011, represents the success and great potential of immunotherapy in treating cancers.
The idea of training the immune system in vivo to recognize and kill cancer cells is the basis of the hypothesis for using vaccines in patients with cancer. In addition, any lessons from antiviral immune responses and vaccinations against infectious disease have been useful to develop antitumor vaccines. Types of immunotherapy that are currently being tested in clinical trials for PDA include whole cell, peptide/DNA, antigen pulsed-DC vaccines, and mAb treatments.
Whole-cancer cell vaccines
Cancer vaccines are preparations aimed at boosting the patient’s immune system against the administered antigen, which is usually specific for a particular tumor type, in order to protect the patient. There are several ways to deliver vaccines: whole-cell recombinant vaccines, DC vaccines that combine antigens with DC to present to leukocytes, DNA vaccines that act by inserting viral or bacterial DNA into animal or human cells, or peptide vaccines to modulate T-cell immunity.
Whole-cell vaccines typically use irradiated PDA cells as immunogens. These cells have the potential to elicit a robust immune response because they express the full repertoire of tumor-associated antigens. Algenpantucel-L is one of the most clinically advanced and promising immunotherapies; it is an irradiated, live combination of two human allogeneic PDA cell lines that express the murine enzyme α-1,3-galactosyl transferase, which directs the synthesis of α-galactosyl epitopes, usually absent in humans and, therefore, has the potential to be strongly recognized by the immune system. Algenpantucel-L causes a hyperacute rejection of such allografts in humans, which is thought to trigger an immune response against cancer cells.80 A multicenter Phase II trial (NLG0205) (ClinicalTrials.gov identifier: NCT00569387) of algenpantucel-L combined with adjuvant chemotherapy using gemcitabine and 5-fluorouracil/radiation after resection showed the induction of eosinophilia for up to 2 years; 62% of patients were disease free after 1 year, with a 1 year overall survival rate of 86% (vs 69% of patients who received the same chemoradiation regimen without the vaccine).81 Another whole-cell vaccine is GVAX, which consists of irradiated tumor cells expressing the murine GM-CSF. This caused a potent, long-lasting antitumor response requiring both CD4+ and CD8+ T-cells in the melanoma system.82 Phase I83 and single-institution Phase II84 clinical trials have also shown promising results for patients with PDA, even if only surgically treated patients were enrolled. Further stage II/III investigations are needed in patients with advanced or metastatic disease, and current clinical trials will provide important information (ClinicalTrials.gov identifier: NCT01088789, NCT01417000, NCT00727441).
Peptide vaccines and adoptive transfer therapy
The first peptide vaccine applied to PDA in a clinical trial was the synthetic Ras-peptide vaccine, which was proven to be safe.85 Combination with GM-CSF induced a specific immune response in 25 out of 43 patients and those with advanced cancer and immune response to the vaccine showed prolonged survival compared to nonresponders.86 Other peptide vaccines investigated in clinical trials with PDA patients include the telomerase peptide vaccine (GV1001), the carcinoembryonic antigen (CEA), alone or in combination with mucin-1 (MUC-1), and survivin, in a case report of a 77-year-old patient with metastatic PDA refractory to gemcitabine.87 A study was conducted to investigate the safety and immunogenicity in patients with nonresectable pancreatic carcinoma using GV1001 for vaccination in combination with GM-CSF and gemcitabine as a first-line treatment. Mild adverse events were observed, and a GV1001-specific immune response, albeit transient, was reported in 75% of patients who contextually received GV1001, GM-CSF, and gemcitabine.88 The multicenter Phase III trial of the GV1001 vaccine in advanced and metastatic PDA recruited 1,062 patients in 52 centers throughout the UK. There was no significant difference in the overall survival between the groups that received the vaccine and the control group receiving chemotherapy, but two biomarkers that defined responders to the vaccine were identified.89 A viral-based vaccine was used to target the CEA antigen in combination with a poxvirus-based vaccine containing three T-cell costimulatory molecules such as B7-1 (CD80), intracellular adhesion molecule 1, and leukocyte function associated antigen-3. Phase I trials have demonstrated that CEA-T-cell costimulatory molecules vaccine is safe, is able to generate a significant anti-CEA immune response, and has some clinical benefit in patients with advanced cancer.90 The addition of MUC-1 and of a boost with a different viral-expressing vaccine (PANVAC-V and vaccinia virus to immunize and PANCAV-F and fowl-pox virus to boost) in combination with GM-CSF showed an induction of antibodies in some patients, with a consequent clinical benefit. However, a Phase III trial failed to improve the overall survival compared to the canonical chemotherapy.91 CEA and MUC-1 antigens were also used to pulse DC purified from patients and reinfused after in vitro expansion and loading. Both these DC-based vaccines were demonstrated to be safe, well tolerated, and elicited remarkable T-cell responses.92,93 To our knowledge, no major trials have been carried out or are ongoing; thus, both strategies would require further investigations to translate into clinical efficacy. MUC-1 RNA was also used to transfect autologously purified DC, which were intradermally injected into unresectable or recurrent patients with PDA, who also received gemcitabine and in vitro expanded cytotoxic T-cells in the presence of a PDA cell line plus IL-2. The median survival rate was 13.9 months and the 1 year survival rate was 51.1%, with a disease control ratio of 61.9%. Liver metastasis occurred in just five patients of the 35 patients investigated with no liver metastasis before treatment, and there were no severe toxicities associated with the adoptive immunotherapy.94
Adoptive T-cell therapy, which aims at in vitro expansion and “education” of T-cells from patients with cancer, for subsequent reinfusion to fight cancer cells in vivo, has not been widely explored in PDA. One Phase I clinical trial (ClinicalTrials.gov identifier: NCT02465983) is recruiting patients and involves the transfection of patient T-cells with a specific receptor recognizing a mesothelin peptide (CART-meso) and the protein CD19 (CART-19) expressed by B-cells. The latter T-cells should kill patient B-lymphocytes by preventing the production of antibodies against CART-meso cells. Mesothelin is an antigen that has been initially characterized in ovarian cancer and PDA95 and has been observed to induce the CD8+ T-cell response in patients treated with GVAX and cyclophosphamide, either with or without live attenuated-Listeria monocytogene-expressing mesothelin.96 In this study, the prime/boost strategy with GVAX and L. monocytogene-expressing mesothelin extended survival of patients with pancreatic cancer and enhanced mesothelin-specific CD8+ T-cell responses and was associated with a longer overall survival, regardless of the treated arm.97
Wilms tumor (WT-1) peptide-based vaccine in combination with gemcitabine is another strategy that was shown to be well tolerated in unresectable patients with advanced PDA. The Phase I clinical trial demonstrated its safety and revealed a statistically significant correlation between longer survival and positive delayed-type hypersensitivity to WT-1. Moreover, longer survivors displayed a higher frequency of memory WT-1-specific cytotoxic T-cells both before and after treatment.98
Cancer testis (CT) antigens were the first tumor-associated antigens proposed for antitumor vaccination strategies because of their specific expression in tumors but not normal cells of different histological origin. Several preclinical and clinical studies demonstrated CT-based immunotherapies as being effective in inducing a specific immune response and occasionally clinical responses in different types of tumors.99 A peptide vaccine consisting of four distinct nonamers from CT and vascular endothelial growth factor receptor 1 (VEGF-R1) and 2 (VEGF-R2) proteins has been shown to be well tolerated and able to elicit a peptide-specific T-cell response in all the nine patients with advanced PDA refractory to standard chemotherapy enrolled in a Phase I clinical trial. Notably, clinical benefits were observed in four out of nine patients, with a disease-free period of 90 days and an overall survival of 207 days.100
Passive immunotherapy combined with antitumor vaccines
Several preclinical studies are in the process of investigating the potential of Treg suppression and immune checkpoint blockade by mAbs alone or in combination with chemotherapy to boost the patient’s immune system in recognizing and destroying tumors. Administering ipilimumab in combination with the GVAX vaccine (ClinicalTrials.gov identifier: NCT00836407) and the administration of nivolumab in combination with the GVAX vaccine and cyclophosphamide (ClinicalTrials.gov identifier: NCT02451982) are two representative clinical trials. Ipilimumab and nivolumab are mAb against CTLA-4 and programmed cell death-1 (PD-1), respectively. Both molecules are protein receptors expressed on the surface of activated T-cells and are known as checkpoints due to their inhibitory function. After binding of CTLA-4 with the costimulatory molecule CD80 and of PD-1 with the ligand PD-L1, an inhibitory signal to T-lymphocytes is transmitted. The success obtained in autoimmune disease with the anti-CTLA-4 antibody treatment has prompted scientists to also employ this antibody in patients with cancer. The notion of blocking the inhibitory signal in order to sustain the antitumor T-cell response for longer was straight forward and insightful, and it has been demonstrated to be effective in potentiating the antitumor response and in unrestrained T-cell proliferation in different types of cancer. In a first small study (30 patients with PDA previously treated), the combination of ipilimumab with the GVAX vaccine showed a potential clinical benefit, which needs to be confirmed in a larger cohort. Among the patients with increased overall survival, there was an increase in the peak of mesothelin-specific T-lymphocytes and an enhancement of the T-cell repertoire.101 Although not without side effects, the blockade of checkpoint molecules displays a great therapeutical potential.
In conclusion, researchers have long considered PDA as being nonamenable to immunotherapy, owing to an immunosuppressive tumor microenvironment. However, preclinical and clinical studies have shown the reestablishing of tumor immunosurveillance by activating T-cells and macrophages, and several clinical trials are following this direction (Table 2).
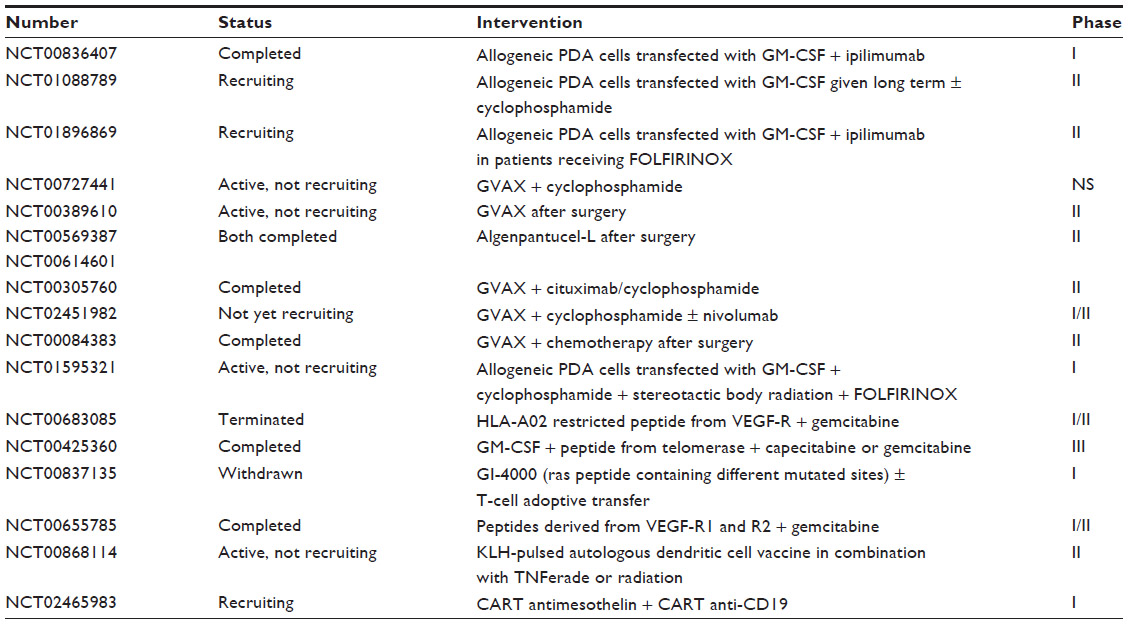 | Table 2 Clinical trials consisting in vaccine strategiesa |
Novel DNA vaccine promising strategy for PDA therapy
Among vaccines, DNA vaccines display several advantages such as they are stable, do not contain viral proteins that could downregulate the immune system or elicit neutralizing antibodies, and are safe, as several studies have shown that mutations arising from a putative integration event are extremely rare.102 In turn, DNA vaccination usually fails to mount a strong immune response and requires additional adjuvant strategies.
For several years, our group has focused on the identification of novel PDA-associated targets to use for developing immunotherapeutic strategies to be applied in treating this dismal disease. Through a serological approach, we have identified 12 PDA-associated proteins,103 which we have investigated for their ability to activate a T-cell immune response both in vitro and in vivo. Among these, ENO1 has been shown to be a promising candidate target as it was recognized by the autoantibodies present in >60% of patients with PDA.26,104 ENO1 is coded by the ENO1 gene and is overexpressed in the cytoplasm of PDA cells being membrane expressed.104,105 In the cytoplasm, ENO1 acts as a glycolytic enzyme, whereas membrane-expressed ENO1 behaves as a plasminogen receptor and plays an important role in the cell migration.106 The ENO1 antibody effectively inhibits the spreading and invasion of PDA in vivo and in vitro.105 Patients with PDA with autoantibodies to ENO1 also present an ENO1-specific T-cell response, which is not observed in patients with no ENO1 autoantibodies.104 Upon transfer into immunocompromised mice, ENO1-specific T-cells inhibit the growth of xeno-transplanted human pancreatic tumors. Despite the ubiquitous presence of ENO1 in all mammalian cells, normal cells expressing low levels of ENO1 are spared by ENO1-specific cytotoxic T-lymphocytes.104 Anti-ENO1-specific T-cells were found to infiltrate tumor tissue in patient with resected PDA.79 These results led us to develop a DNA-based vaccine to assess the ability of an ENO1-specific antitumor response to inhibit the tumor growth. To do this, we exploited two of the most sophisticated murine models that spontaneously develop autochthonous PDA (known as KC and KPC mice). GEM that develops spontaneous PDA was vaccinated with a plasmid encoding human ENO1, which displays >95% identity (99% homology) with the mouse ortholog. The ENO1 DNA vaccine significantly induced a specific immune response that prolonged the survival of both PDA mouse models: from 336 days to 474 days of age for KC mice, the longest overall survival ever reported, and from 203 days to 245 days for KPC mice. The ENO1 DNA vaccine elicited several protective immunological mechanisms, namely, high levels of anti-ENO1 IgG, activation of specific Th1 and Th17 cells, and a large recruitment of CD3 cells into the tumor area. Notably, anti-ENO1 IgG was able to bind to the cell surface of murine PDA cells and induce their killing by complement-dependent cytotoxicity. Th1/Th17 cytokines favor the switching to effector subclasses of anti-ENO1 antibodies. An additional effect of the ENO1 DNA vaccine is the decrease in cells with suppressive activity, typically found in the tumor microenvironment, namely, MDSC and Tregs. Of clinical relevance, the therapeutic efficacy of the ENO1 DNA vaccine was observed to be very promising when the administration protocol started at 8 months to 9 months of age.107 In these mice, the ENO1 DNA vaccine also induced the formation of intratumoral tertiary lymphoid tissue (TLT) with active germinal centers and correlated with increased recruitment of T-cells.108 Analysis of human PDA tissues by immunohistochemistry revealed two distinct patterns of B-cell distribution, either as randomly infiltrating cells (CD20-tumor-infiltrating lymphocytes) or strategically located within organized TLT (CD20-TLT). Notably, high density of CD20-TLT correlated with CD8+ T-cell infiltration and a more favorable prognostic value.108 Overall, data on ENO1 vaccination indicate that it is a promising candidate target to design adjuvant therapies to prevent recurrences in resected patients or to prolong the survival of untreatable patients.
Conclusion
Despite several successes in curing solid tumors, PDA remains the only tumor to have had the same mortality rate for the last 15 years. To date, the only curative treatment is represented by surgical resection, only applicable, however, to a small percentage of patients. Recent advances have been raised with the use of gemcitabine in a neoadjuvant setting to increase the resectable percentage of patients with PDA (Figure 1). The time is ripe to transfer the information obtained to elicit an active immune response against PDA to clinical trials that effectively prolong the survival of patients with PDA. The right drug combination could transform encouraging vaccine preclinical and clinical results into an effective protocol able to cure PDA (Figure 1). A lot of evidence has shown that several anticancer agents, including classic chemotherapeutics and targeted compounds, stimulate tumor-specific immune responses either by inducing immunogenic cell death or by engaging immune effector mechanisms.109 Their combination with vaccination against selected immunological targets (eg, ENO1 and mesothelin) and with different strategies (whole cells, DC, peptides, or DNA) will allow the design of novel therapies to elicit effective immune responses in resected patients to prevent recurrences or to prolong survival of unresectable patients (Figure 1). In addition, the novel strategies for modulating the tumor microenvironment and immunosuppressive mechanisms through mAb may be combined to further enhance the antitumor response induced by vaccination following surgery, chemo- or radiotherapy (Figure 1). The greatest challenge in future studies will be to establish the most effective timeline and the best combinations of different treatments.
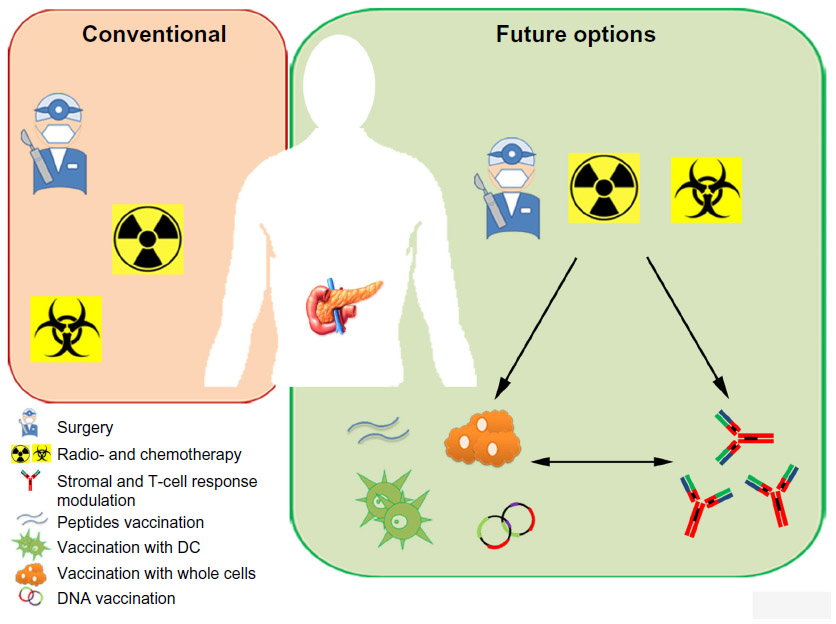 | Figure 1 Conventional and future therapies for PDA. |
Acknowledgments
We thank Dr Radhika Srinivasan for critical reading of the manuscript. This study was supported by Associazione Italiana Ricerca sul Cancro (5 no 12182) and (IG no 15257); Ministry of Health (RF-2013-02354892), University of Turin-Progetti Ateneo 2014-Compagnia di San Paolo (PC-METAIMMUNOTHER and PANTHER); Fondazione Ricerca Molinette; and Fondazione Ursula e Giorgio Cytron.
Disclosure
The authors report no conflicts of interest in this work.
References
Rahib L, Smith BD, Aizenberg R, Rosenzweig AB, Fleshman JM, Matrisian LM. Projecting cancer incidence and deaths to 2030: the unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014;74(11):2913–2921. | |
Becker AE, Hernandez YG, Frucht H, Lucas AL. Pancreatic ductal adenocarcinoma: risk factors, screening, and early detection. World J Gastroenterol. 2014;20(32):11182–11198. | |
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin. 2015;65(1):5–29. | |
Bilimoria KY, Bentrem DJ, Ko CY, et al. Validation of the 6th edition AJCC pancreatic cancer staging system: report from the National Cancer Database. Cancer. 2007;110(4):738–744. | |
Distler M, Aust D, Weitz J, Pilarsky C, Grutzmann R. Precursor lesions for sporadic pancreatic cancer: PanIN, IPMN, and MCN. Biomed Res Int. 2014;2014:474905. | |
Hruban RH, Adsay NV, Albores-Saavedra J, et al. Pancreatic intraepithelial neoplasia: a new nomenclature and classification system for pancreatic duct lesions. Am J Surg Pathol. 2001;25(5):579–586. | |
Hruban RH, Takaori K, Klimstra DS, et al. An illustrated consensus on the classification of pancreatic intraepithelial neoplasia and intraductal papillary mucinous neoplasms. Am J Surg Pathol. 2004;28(8):977–987. | |
Macgregor-Das AM, Iacobuzio-Donahue CA. Molecular pathways in pancreatic carcinogenesis. J Surg Oncol. 2013;107(1):8–14. | |
Vincent A, Herman J, Schulick R, Hruban RH, Goggins M. Pancreatic cancer. Lancet. 2011;378(9791):607–620. | |
Jones S, Zhang X, Parsons DW, et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science. 2008;321(5897):1801–1806. | |
van Heek NT, Meeker AK, Kern SE, et al. Telomere shortening is nearly universal in pancreatic intraepithelial neoplasia. Am J Pathol. 2002;161(5):1541–1547. | |
Garrido-Laguna I, Hidalgo M. Pancreatic cancer: from state-of-the-art treatments to promising novel therapies. Nat Rev Clin Oncol. 2015;12(6):319–334. | |
Rossi ML, Rehman AA, Gondi CS. Therapeutic options for the management of pancreatic cancer. World J Gastroenterol. 2014;20(32):11142–11159. | |
Stathis A, Moore MJ. Advanced pancreatic carcinoma: current treatment and future challenges. Nat Rev Clin Oncol. 2010;7(3):163–172. | |
Wolfgang CL, Herman JM, Laheru DA, et al. Recent progress in pancreatic cancer. CA Cancer J Clin. 2013;63(5):318–348. | |
Harsha HC, Kandasamy K, Ranganathan P, et al. A compendium of potential biomarkers of pancreatic cancer. PLoS Med. 2009;6(4):e1000046. | |
Chung MH, Gupta RK, Bilchik AJ, Ye W, Yee R, Morton DL. Preoperative serum TA90-IC as an adjunct to serum CA 19-9 in the diagnosis of pancreatic malignancy: a pilot study. Curr Surg. 2002;59(2):194–198. | |
Goonetilleke KS, Siriwardena AK. Systematic review of carbohydrate antigen (CA 19-9) as a biochemical marker in the diagnosis of pancreatic cancer. Eur J Surg Oncol. 2007;33(3):266–270. | |
Berger AC, Garcia M Jr, Hoffman JP, et al. Postresection CA 19-9 predicts overall survival in patients with pancreatic cancer treated with adjuvant chemoradiation: a prospective validation by RTOG 9704. J Clin Oncol. 2008;26(36):5918–5922. | |
Halm U, Schumann T, Schiefke I, Witzigmann H, Mössner J, Keim V. Decrease of CA 19-9 during chemotherapy with gemcitabine predicts survival time in patients with advanced pancreatic cancer. Br J Cancer. 2000;82(5):1013–1016. | |
Ishii H, Okada S, Sato T, et al. CA 19-9 in evaluating the response to chemotherapy in advanced pancreatic cancer. Hepatogastroenterology. 1997;44(13):279–283. | |
Montgomery RC, Hoffman JP, Riley LB, Rogatko A, Ridge JA, Eisenberg BL. Prediction of recurrence and survival by post-resection CA 19-9 values in patients with adenocarcinoma of the pancreas. Ann Surg Oncol. 1997;4(7):551–556. | |
Wong D, Ko AH, Hwang J, Venook AP, Bergsland EK, Tempero MA. Serum CA19-9 decline compared to radiographic response as a surrogate for clinical outcomes in patients with metastatic pancreatic cancer receiving chemotherapy. Pancreas. 2008;37(3):269–274. | |
Frebourg T, Bercoff E, Manchon N, et al. The evaluation of CA 19-9 antigen level in the early detection of pancreatic cancer. A prospective study of 866 patients. Cancer. 1988;62(11):2287–2290. | |
Capello M, Cappello P, Linty FC, et al. Autoantibodies to Ezrin are an early sign of pancreatic cancer in humans and in genetically engineered mouse models. J Hematol Oncol. 2013;6:67. | |
Tomaino B, Cappello P, Capello M, et al. Circulating autoantibodies to phosphorylated alpha-enolase are a hallmark of pancreatic cancer. J Proteome Res. 2011;10(1):105–112. | |
Melo SA, Luecke LB, Kahlert C, et al. Glypican-1 identifies cancer exosomes and detects early pancreatic cancer. Nature. 2015;523(7559):177–182. | |
Li D, Xie K, Wolff R, Abbruzzese JL. Pancreatic cancer. Lancet. 2004;363(9414):1049–1057. | |
Wagner M, Redaelli C, Lietz M, Seiler CA, Friess H, Büchler MW. Curative resection is the single most important factor determining outcome in patients with pancreatic adenocarcinoma. Br J Surg. 2004; 91(5):586–594. | |
Riediger H, Keck T, Wellner U, et al. The lymph node ratio is the strongest prognostic factor after resection of pancreatic cancer. J Gastrointest Surg. 2009;13(7):1337–1344. | |
Wasif N, Ko CY, Farrell J, et al. Impact of tumor grade on prognosis in pancreatic cancer: should we include grade in AJCC staging? Ann Surg Oncol. 2010;17(9):2312–2320. | |
Fortner JG, Klimstra DS, Senie RT, Maclean BJ. Tumor size is the primary prognosticator for pancreatic cancer after regional pancreatectomy. Ann Surg. 1996;223(2):147–153. | |
Hidalgo M. Pancreatic cancer. N Engl J Med. 2010;362(17):1605–1617. | |
Maitra A, Hruban RH. Pancreatic cancer. Annu Rev Pathol. 2008;3:157–188. | |
Tian H, Callahan CA, DuPree KJ, et al. Hedgehog signaling is restricted to the stromal compartment during pancreatic carcinogenesis. Proc Natl Acad Sci U S A. 2009;106(11):4254–4259. | |
Dineen SP, Roland CL, Greer R, et al. Smac mimetic increases chemotherapy response and improves survival in mice with pancreatic cancer. Cancer Res. 2010;70(7):2852–2861. | |
Hwang JH, Voortman J, Giovannetti E, et al. Identification of microRNA-21 as a biomarker for chemoresistance and clinical outcome following adjuvant therapy in resectable pancreatic cancer. PLoS One. 2010;5(5):e10630. | |
Harikumar KB, Kunnumakkara AB, Sethi G, et al. Resveratrol, a multitargeted agent, can enhance antitumor activity of gemcitabine in vitro and in orthotopic mouse model of human pancreatic cancer. Int J Cancer. 2010;127(2):257–268. | |
Cunningham D, Chau I, Stocken DD, et al. Phase III randomized comparison of gemcitabine versus gemcitabine plus capecitabine in patients with advanced pancreatic cancer. J Clin Oncol. 2009;27(33):5513–5518. | |
Banerjee S, Kaseb AO, Wang Z, et al. Antitumor activity of gemcitabine and oxaliplatin is augmented by thymoquinone in pancreatic cancer. Cancer Res. 2009;69(13):5575–5583. | |
Baylot V, Andrieu C, Katsogiannou M, et al. OGX-427 inhibits tumor progression and enhances gemcitabine chemotherapy in pancreatic cancer. Cell Death Dis. 2011;2:e221. | |
Kanai M, Yoshimura K, Asada M, et al. A phase I/II study of gemcitabine-based chemotherapy plus curcumin for patients with gemcitabine-resistant pancreatic cancer. Cancer Chemother Pharmacol. 2011;68(1):157–164. | |
Gillen S, Schuster T, Meyer Zum Buschenfelde C, Friess H, Kleeff J. Preoperative/neoadjuvant therapy in pancreatic cancer: a systematic review and meta-analysis of response and resection percentages. PLoS Med. 2010;7(4):e1000267. | |
Lemmens VE, Bosscha K, van der Schelling G, Brenninkmeijer S, Coebergh JW, de Hingh IH. Improving outcome for patients with pancreatic cancer through centralization. Br J Surg. 2011;98(10):1455–1462. | |
Massucco P, Capussotti L, Magnino A, et al. Pancreatic resections after chemoradiotherapy for locally advanced ductal adenocarcinoma: analysis of perioperative outcome and survival. Ann Surg Oncol. 2006; 13(9):1201–1208. | |
Burris HA 3rd, Moore MJ, Andersen J, et al. Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: a randomized trial. J Clin Oncol. 1997; 15(6):2403–2413. | |
Conroy T, Desseigne F, Ychou M, et al. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N Engl J Med. 2011; 364(19):1817–1825. | |
Moore MJ, Goldstein D, Hamm J, et al; National Cancer Institute of Canada Clinical Trials Group. Erlotinib plus gemcitabine compared with gemcitabine alone in patients with advanced pancreatic cancer: a phase III trial of the National Cancer Institute of Canada Clinical Trials Group. J Clin Oncol. 2007;25(15):1960–1966. | |
Wang JP, Wu CY, Yeh YC, et al. Erlotinib is effective in pancreatic cancer with epidermal growth factor receptor mutations: a randomized, open-label, prospective trial. Oncotarget. 2015;6(20):18162–18173. | |
Edmonds C, Cengel KA. Tumor-stroma interactions in pancreatic cancer: will this SPARC prove a raging fire? Cancer Biol Ther. 2008; 7(11):1816–1817. | |
Infante JR, Matsubayashi H, Sato N, et al. Peritumoral fibroblast SPARC expression and patient outcome with resectable pancreatic adenocarcinoma. J Clin Oncol. 2007;25(3):319–325. | |
Von Hoff DD, Ervin T, Arena FP, et al. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N Engl J Med. 2013;369(18):1691–1703. | |
Gajewski TF, Schreiber H, Fu YX. Innate and adaptive immune cells in the tumor microenvironment. Nat Immunol. 2013;14(10):1014–1022. | |
Jobin C. Colorectal cancer: looking for answers in the microbiota. Cancer Discov. 2013;3(4):384–387. | |
Clark CE, Beatty GL, Vonderheide RH. Immunosurveillance of pancreatic adenocarcinoma: insights from genetically engineered mouse models of cancer. Cancer Lett. 2009;279(1):1–7. | |
Guerra C, Collado M, Navas C, et al. Pancreatitis-induced inflammation contributes to pancreatic cancer by inhibiting oncogene-induced senescence. Cancer Cell. 2011;19(6):728–739. | |
Gregory AD, Houghton AM. Tumor-associated neutrophils: new targets for cancer therapy. Cancer Res. 2011;71(7):2411–2416. | |
Sparmann A, Bar-Sagi D. Ras-induced interleukin-8 expression plays a critical role in tumor growth and angiogenesis. Cancer Cell. 2004;6(5):447–458. | |
Beatty GL, Chiorean EG, Fishman MP, et al. CD40 agonists alter tumor stroma and show efficacy against pancreatic carcinoma in mice and humans. Science. 2011;331(6024):1612–1616. | |
Beatty GL, Winograd R, Evans RA, et al. Exclusion of T cells from pancreatic carcinomas in mice is regulated by Ly6C(low) F4/80(+) extratumoral macrophages. Gastroenterology. 2015;149(1):201–210. | |
Mitchem JB, Brennan DJ, Knolhoff BL, et al. Targeting tumor-infiltrating macrophages decreases tumor-initiating cells, relieves immunosuppression, and improves chemotherapeutic responses. Cancer Res. 2013;73(3):1128–1141. | |
Qian BZ, Li J, Zhang H, et al. CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature. 2011;475(7355):222–225. | |
Kitamura T, Qian BZ, Soong D, et al. CCL2-induced chemokine cascade promotes breast cancer metastasis by enhancing retention of metastasis-associated macrophages. J Exp Med. 2015;212(7):1043–1059. | |
Zhu Y, Knolhoff BL, Meyer MA, et al. CSF1/CSF1R blockade reprograms tumor-infiltrating macrophages and improves response to T-cell checkpoint immunotherapy in pancreatic cancer models. Cancer Res. 2014;74(18):5057–5069. | |
Pyonteck SM, Akkari L, Schuhmacher AJ, et al. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat Med. 2013;19(10):1264–1272. | |
Bayne LJ, Beatty GL, Jhala N, et al. Tumor-derived granulocyte-macrophage colony-stimulating factor regulates myeloid inflammation and T cell immunity in pancreatic cancer. Cancer Cell. 2012;21(6):822–835. | |
Stromnes IM, Brockenbrough JS, Izeradjene K, et al. Targeted depletion of an MDSC subset unmasks pancreatic ductal adenocarcinoma to adaptive immunity. Gut. 2014;63(11):1769–1781. | |
Pylayeva-Gupta Y, Lee KE, Hajdu CH, Miller G, Bar-Sagi D. Oncogenic Kras-induced GM-CSF production promotes the development of pancreatic neoplasia. Cancer Cell. 2012;21(6):836–847. | |
Apte MV, Wilson JS, Lugea A, Pandol SJ. A starring role for stellate cells in the pancreatic cancer microenvironment. Gastroenterology. 2013;144(6):1210–1219. | |
Feig C, Jones JO, Kraman M, et al. Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti-PD-L1 immunotherapy in pancreatic cancer. Proc Natl Acad Sci U S A. 2013;110(50):20212–20217. | |
Olive KP, Jacobetz MA, Davidson CJ, et al. Inhibition of hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science. 2009;324(5933):1457–1461. | |
Özdemir BC, Pentcheva-Hoang T, Carstens JL, et al. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell. 2014;25(6):719–734. | |
Rhim AD, Oberstein PE, Thomas DH, et al. Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer Cell. 2014;25(6):735–747. | |
Gore J, Korc M. Pancreatic cancer stroma: friend or foe? Cancer Cell. 2014;25(6):711–712. | |
Fukunaga A, Miyamoto M, Cho Y, et al. CD8+ tumor-infiltrating lymphocytes together with CD4+ tumor-infiltrating lymphocytes and dendritic cells improve the prognosis of patients with pancreatic adenocarcinoma. Pancreas. 2004;28(1):e26–e31. | |
Fogar P, Sperti C, Basso D, et al. Decreased total lymphocyte counts in pancreatic cancer: an index of adverse outcome. Pancreas. 2006;32(1):22–28. | |
Emmrich J, Sparmann G, Hopt U, Lohr M, Liebe S. Typing of leukocytes in pancreatic tissue surrounding human pancreatic carcinoma. Ann N Y Acad Sci. 1999;880:171–174. | |
Ademmer K, Ebert M, Müller-Ostermeyer F, et al. Effector T lymphocyte subsets in human pancreatic cancer: detection of CD8+CD18+ cells and CD8+CD103+ cells by multi-epitope imaging. Clin Exp Immunol. 1998;112(1):21–26. | |
Amedei A, Niccolai E, Benagiano M, et al. Ex vivo analysis of pancreatic cancer-infiltrating T lymphocytes reveals that ENO-specific Tregs accumulate in tumor tissue and inhibit Th1/Th17 effector cell functions. Cancer Immunol Immunother. 2013;62(7):1249–1260. | |
Rossi GR, Mautino MR, Unfer RC, Seregina TM, Vahanian N, Link CJ. Effective treatment of preexisting melanoma with whole cell vaccines expressing alpha(1,3)-galactosyl epitopes. Cancer Res. 2005;65(22):10555–10561. | |
Gunturu KS, Rossi GR, Saif MW. Immunotherapy updates in pancreatic cancer: are we there yet? Ther Adv Med Oncol. 2013;5(1):81–89. | |
Dranoff G, Jaffee E, Lazenby A, et al. Vaccination with irradiated tumor cells engineered to secrete murine granulocyte-macrophage colony-stimulating factor stimulates potent, specific, and long-lasting anti-tumor immunity. Proc Natl Acad Sci U S A. 1993;90(8):3539–3543. | |
Jaffee EM, Hruban RH, Biedrzycki B, et al. Novel allogeneic granulocyte-macrophage colony-stimulating factor-secreting tumor vaccine for pancreatic cancer: a phase I trial of safety and immune activation. J Clin Oncol. 2001;19(1):145–156. | |
Lutz E, Yeo CJ, Lillemoe KD, et al. A lethally irradiated allogeneic granulocyte-macrophage colony stimulating factor-secreting tumor vaccine for pancreatic adenocarcinoma. A phase II trial of safety, efficacy, and immune activation. Ann Surg. 2011;253(2):328–335. | |
Gjertsen MK, Bakka A, Breivik J, et al. Vaccination with mutant ras peptides and induction of T-cell responsiveness in pancreatic carcinoma patients carrying the corresponding RAS mutation. Lancet. 1995;346(8987):1399–1400. | |
Gjertsen MK, Buanes T, Rosseland AR, et al. Intradermal ras peptide vaccination with granulocyte-macrophage colony-stimulating factor as adjuvant: clinical and immunological responses in patients with pancreatic adenocarcinoma. Int J Cancer. 2001;92(3):441–450. | |
Wobser M, Keikavoussi P, Kunzmann V, Weininger M, Andersen MH, Becker JC. Complete remission of liver metastasis of pancreatic cancer under vaccination with a HLA-A2 restricted peptide derived from the universal tumor antigen survivin. Cancer Immunol Immunother. 2006;55(10):1294–1298. | |
Staff C, Mozaffari F, Frodin JE, Mellstedt H, Liljefors M. Telomerase (GV1001) vaccination together with gemcitabine in advanced pancreatic cancer patients. Int J Oncol. 2014;45(3):1293–1303. | |
Middleton G, Silcocks P, Cox T, et al. Gemcitabine and capecitabine with or without telomerase peptide vaccine GV1001 in patients with locally advanced or metastatic pancreatic cancer (TeloVac): an open-label, randomised, phase 3 trial. Lancet Oncol. 2014;15(8):829–840. | |
Marshall JL, Gulley JL, Arlen PM, et al. Phase I study of sequential vaccinations with fowlpox-CEA(6D)-TRICOM alone and sequentially with vaccinia-CEA(6D)-TRICOM, with and without granulocyte-macrophage colony-stimulating factor, in patients with carcinoembryonic antigen-expressing carcinomas. J Clin Oncol. 2005;23(4):720–731. | |
Madan RA, Bilusic M, Heery C, Schlom J, Gulley JL. Clinical evaluation of TRICOM vector therapeutic cancer vaccines. Semin Oncol. 2012;39(3):296–304. | |
Morse MA, Nair SK, Boczkowski D, et al. The feasibility and safety of immunotherapy with dendritic cells loaded with CEA mRNA following neoadjuvant chemoradiotherapy and resection of pancreatic cancer. Int J Gastrointest Cancer. 2002;32(1):1–6. | |
Rong Y, Qin X, Jin D, et al. A phase I pilot trial of MUC1-peptide-pulsed dendritic cells in the treatment of advanced pancreatic cancer. Clin Exp Med. 2012;12(3):173–180. | |
Shindo Y, Hazama S, Maeda Y, et al. Adoptive immunotherapy with MUC1-mRNA transfected dendritic cells and cytotoxic lymphocytes plus gemcitabine for unresectable pancreatic cancer. J Transl Med. 2014;12:175. | |
Muminova ZE, Strong TV, Shaw DR. Characterization of human mesothelin transcripts in ovarian and pancreatic cancer. BMC Cancer. 2004;4:19. | |
Laheru D, Lutz E, Burke J, et al. Allogeneic granulocyte macrophage colony-stimulating factor-secreting tumor immunotherapy alone or in sequence with cyclophosphamide for metastatic pancreatic cancer: a pilot study of safety, feasibility, and immune activation. Clin Cancer Res. 2008;14(5):1455–1463. | |
Le DT, Wang-Gillam A, Picozzi V, et al. Safety and survival with GVAX pancreas prime and Listeria Monocytogenes-expressing mesothelin (CRS-207) boost vaccines for metastatic pancreatic cancer. J Clin Oncol. 2015;33(12):1325–1333. | |
Nishida S, Koido S, Takeda Y, et al. Wilms tumor gene (WT1) peptide-based cancer vaccine combined with gemcitabine for patients with advanced pancreatic cancer. J Immunother. 2014;37(2):105–114. | |
Gjerstorff MF, Andersen MH, Ditzel HJ. Oncogenic cancer/testis antigens: prime candidates for immunotherapy. Oncotarget. 2015; 6(18):15772–15787. | |
Okuyama R, Aruga A, Hatori T, Takeda K, Yamamoto M. Immunological responses to a multi-peptide vaccine targeting cancer-testis antigens and VEGFRs in advanced pancreatic cancer patients. Oncoimmunology. 2013;2(11):e27010. | |
Le DT, Lutz E, Uram JN, et al. Evaluation of ipilimumab in combination with allogeneic pancreatic tumor cells transfected with a GM-CSF gene in previously treated pancreatic cancer. J Immunother. 2013;36(7):382–389. | |
Wang Z, Troilo PJ, Wang X, et al. Detection of integration of plasmid DNA into host genomic DNA following intramuscular injection and electroporation. Gene Ther. 2004;11(8):711–721. | |
Tomaino B, Cappello P, Capello M, et al. Autoantibody signature in human ductal pancreatic adenocarcinoma. J Proteome Res. 2007; 6(10):4025–4031. | |
Cappello P, Tomaino B, Chiarle R, et al. An integrated humoral and cellular response is elicited in pancreatic cancer by alpha-enolase, a novel pancreatic ductal adenocarcinoma-associated antigen. Int J Cancer. 2009;125(3):639–648. | |
Principe M, Ceruti P, Shih NY, et al. Targeting of surface alpha-enolase inhibits the invasiveness of pancreatic cancer cells. Oncotarget. 2015;6(13):11098–11113. | |
Capello M, Ferri-Borgogno S, Cappello P, Novelli F. Alpha-enolase: a promising therapeutic and diagnostic tumor target. FEBS J. 2011;278(7):1064–1074. | |
Cappello P, Rolla S, Chiarle R, et al. Vaccination with ENO1 DNA prolongs survival of genetically engineered mice with pancreatic cancer. Gastroenterology. 2013;144(5):1098–1106. | |
Castino GF, Cortese N, Capretti G, et al. Spatial distribution of B cells predicts prognosis in human pancreatic adenocarcinoma. OncoImunology. Epub 2015 Sep 11. | |
Galluzzi L, Senovilla L, Zitvogel L, Kroemer G. The secret ally: immunostimulation by anticancer drugs. Nat Rev Drug Discov. 2012;11(3):215–233. | |
Beatty GL, Torigian DA, Chiorean EG, et al. A phase I study of an agonist CD40 monoclonal antibody (CP-870,893) in combination with gemcitabine in patients with advanced pancreatic ductal adenocarcinoma. Clin Cancer Res. 2013;19(22):6286–6295. | |
Germano G, Frapolli R, Belgiovine C, et al. Role of macrophage targeting in the antitumor activity of trabectedin. Cancer Cell. 2013; 23(2):249–262. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.