Back to Journals » Open Access Rheumatology: Research and Reviews » Volume 13
Pain: A Review of Interleukin-6 and Its Roles in the Pain of Rheumatoid Arthritis
Authors Sebba A
Received 27 November 2020
Accepted for publication 22 January 2021
Published 5 March 2021 Volume 2021:13 Pages 31—43
DOI https://doi.org/10.2147/OARRR.S291388
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Chuan-Ju Liu
Anthony Sebba
Division of Rheumatology, University of South Florida, Tampa, FL, USA
Correspondence: Anthony Sebba
Division of Rheumatology, University of South Florida, 13220 USF Laurel Drive, Tampa, FL, 33612, USA
Tel +1 727 773 9793
Fax +1 727 773 0674
Email [email protected]
Abstract: Pain is a major and common symptom reported as a top priority in patients with rheumatoid arthritis (RA). Intuitively, RA-related pain is often considered to be a natural consequence of peripheral inflammation, so treatment of RA is expected to manage pain concurrently as part of inflammation control. However, pain in patients with RA can be poorly correlated with objective measures of inflammation, for example, in patients who are otherwise in remission. Joint damage appears to account for only a fraction of this residual pain. Emerging evidence suggests that alteration of peripheral and central pain processing contributes to RA-related pain; this is parallel to, but somewhat independent of, joint inflammation. Interleukin (IL)-6 is a proinflammatory cytokine that contributes to the pathogenesis of RA. It exerts systemic effects via signaling through soluble forms of the IL-6 receptor (“trans-signaling”). Evidence from preclinical studies demonstrates that intra-articular IL-6 can produce long-lasting peripheral sensitization to mechanical stimulation and suggests an important role for IL-6 in central pain sensitization. This may be partly explained by its ability to activate neurons through trans-signaling, affecting nociceptive plasticity and nerve fiber regrowth. Local activity at neuron endings may culminate in altered pain processing in the central nervous system because of persistent signaling from sensitized peripheral neurons. Peripheral and central sensitization can promote the development of chronic pain, which can have a significant impact on patients’ health and quality of life. A proportion of pain in RA may be more appropriately managed as an entity separate from inflammation. Both the peripheral and central nervous systems should be recognized as important potential systems targeted by RA. The substantial burden of RA-related chronic pain suggests that pain should be a key focus in RA management and should be assessed and addressed early and separately from the inflammatory component.
Keywords: animal model, non-inflammatory, preclinical
Introduction
Rheumatoid arthritis (RA) is a chronic, autoimmune disease that affects about 1% of the global population. It is characterized by synovial joint tissue inflammation and pain, and by potential direct and indirect involvement of other organ systems, such as the cardiovascular, pulmonary, and skeletal systems, and RA is often associated with a significant psychological impact.1,2 The pathogenesis of RA is driven by a complex combination of inflammatory cells and mediators, which are the targets of disease-modifying agents used in the management of RA.
Pain is the hallmark symptom of RA and can be debilitating. Improvement in pain has consistently been rated as the highest priority among health outcomes for patients with RA.3,4 Many cytokines, including tumor necrosis factor alpha (TNFα), interleukin (IL)-1, IL-6, and IL-10, are known to influence RA pain. However, while pain in RA is presumed to be largely a product of cytokine-driven inflammation in the joint, mounting evidence suggests that other non-inflammatory initiators (eg, neuropeptides) and cytokines may also mediate pain via a direct effect on the nervous system.5 In particular, preclinical evidence suggests a key role for IL-6, which might mediate RA-related pain through a range of mechanisms, in addition to indirectly via inflammation in the joint.6,7
This review presents the concept of non-inflammatory pain in RA and the evidence for the role of IL-6 in non-inflammatory RA-related pain. Preclinical models of RA disease activity and the clinical implications for the management of patients with RA are discussed. Articles cited here were identified via literature reviews or PubMed search. Search terms included, but were not limited to, IL-6, pain, non-inflammatory, rheumatoid arthritis/RA, inflammation, preclinical, and animal model. Articles were included based on peer-reviewed publication and author assessment of relevant scientific content.
Concept of Non-Inflammatory Pain in RA
Most patients with RA report experiencing moderate-to-severe pain. Pain in joints is the most common, and often the only, complaint of patients with RA. The mechanisms of pain are increasingly well understood.
Acute pain is detected by specialized nerve receptors (nociceptors) that detect and respond to strong signals that relay information about danger to the organism, presumably so that the organism can mobilize defenses. In RA, persistent signaling from within inflamed joints can lead to lowering of the threshold for stimulation of nociceptors with resultant hypersensitivity to nociceptive stimulation (peripheral sensitization). It is likely that local factors, such as cytokines, may exert direct, non-inflammatory effects on sensory neurons.8
Central sensitization refers to anomalies in spinal and brain pain processing which result in increased pain sensitivity at diffuse sites and a heightened overall response state throughout the central nervous system (CNS) in response to sensory impulses, which are interpreted in the brain through a complex filter as enhanced pain. This category may include psychosocial interplay with pain perception to a chronic pain state. In central sensitization, new nociceptive pathways are created, for example by recruiting mechanoreceptors to conduct pain. This occurs mainly by producing increased sensitization in the spine and, in time, this state can become self-perpetuating even in the absence of injury, and unrelated to any protective purpose.
Thus, in a patient with underlying inflammatory pain from RA which might intrinsically cause mild pain only, this pain is exaggerated and experienced as a much more intense pain (hyperalgesia). Sensations of pain in this state may also be produced from usually nonpainful stimuli such as movement (eg, making a fist) which are normally interpreted as informational inputs (allodynia).8,9 Clinical observation suggests a disconnect between pain and inflammation in some cases of early RA; for example, bilateral symmetrical small joint arthralgias can occur prior to any objective evidence of joint inflammation.10 Conversely, some patients with RA may present with joint inflammation (and joint damage) only, without pain (the “robust rheumatoid” type).11 There may in fact be a spectrum of RA presentation with phenotypes ranging from patients with mainly identifiable synovitis with accompanying pain to those who present with pain only. Attempts have been made to identify these latter patients with RA who have non-inflammatory pain earlier in the course of disease, for example through the use of the swollen to tender joint count ratio.12
Discordance between patient self-reported pain and objective evaluation of inflammation has been reported, and patient-reported pain appears to correlate poorly with physicians’ assessments and measures of inflammation across studies.13–16 The view that pain is not solely derived from inflammation is also supported by observations that anti-cytokine treatments produce pain relief well before reduction in inflammation,17 and that residual pain persists in some 12–50% of patients in remission, and in 82% of patients with “somewhat to completely controlled” disease following treatment of the inflammatory component of RA.18,19 Rheumatologists often attribute this residual pain to fibromyalgia or to underlying joint damage from RA, but the latter explanation seems unlikely given radiographic damage has been shown to account for only 2.1% of patient-reported pain.20 The proportion of patients with RA fulfilling fibromyalgia classification criteria also increases throughout the course of disease progression, which again suggests central pain augmentation.21
RA is of course a disease involving joints, but pain is often reported as being extra-articular and it may be diffuse, with widespread achiness in remote, nonarticular sites as well as in joints, and can vary in location and time. This pain does not appear to be related to synovitis and is likely due to the alteration of central pain processing.22 To the perceptive clinician, there are sometimes clues hinting that aspects of pain in patients with RA may derive from nervous system involvement, for example, when sensations of pain are reported as tingling, burning, or as sharp. CNS involvement is getting clearer; a substantial proportion of patients with RA have some form of identifiable neuropathic involvement.23 Autonomic neuropathy too is increasingly identified as associated with RA.24 These findings support the concept of the CNS being a significant associated organ system in RA.
The many clinically apparent dissociations between pain and inflammation suggest some RA pain may be an independent problem overlapping with, but not solely due to, inflammation, with multiple mechanisms involved.25 The current working hypothesis is that patient pain in RA originates with some form of inflammation or injury, but, in addition, RA patients feel pain that is also concurrently and independently triggered (with no or minor sensory stimulation), passed on by the peripheral nervous system, amplified in the spinal cord, and experienced in the CNS. In the brain, more distal sensations are interwoven with external psychosocial factors. Indeed, pain has been described as “an opinion.”26 This pain is a distinct entity from the pain of inflammation, and this form of pain is at least partly due to aberrancies related to cytokine dysregulation.
The notion of “unbundling” aspects of the clinical manifestations of the disease that we call RA, ie, looking at components independently of joint inflammation, is not novel and is not unique to pain. Even in those in whom synovitis remains active, radiographic progression is slowed in patients taking TNF or IL-6 inhibitors,27 possibly suggesting that these processes are somewhat unrelated and can perhaps be addressed separately.
In summary, it is understood that cytokines cause joint inflammation (which in turn causes pain), but there appear to be other effects of cytokines on peripheral and also other parts of the nervous system that lead to pain more directly and do not necessarily correlate with inflammation. As detailed below, the cytokine IL-6 seems to have an important role in this.
Overview of IL-6 in RA and Preclinical Models
Role and Activity of IL-6 in RA and Pain
IL-6 clearly has a role in inflammation and thus in pain; it increasingly appears that it also has a role in pain independent from inflammation.
IL-6 exerts its effects through binding to its receptor (IL-6R), which leads to dimerization of glycoprotein 130 (gp130), a component of the transmembrane type 1 cytokine receptor, and initiation of intracellular signaling via the Janus kinase (JAK)–signal transducer and activator of transcription and Ras Raf–mitogen-activated protein kinase (MAPK) pathways. IL-6 is able to activate cells through both membrane-bound and soluble forms of IL-6R, referred to as “classic” (cis) and “trans” signaling, respectively.28 Gp130 is ubiquitous and is expressed in the membrane of all cells. The trans-signaling ability of IL-6 allows for pleiotropic effects of this cytokine.6,29 For example, IL-6 can signal through neuronal cells and has been implicated in many neurologic diseases, including multiple sclerosis and Parkinson’s disease, and also appears likely to be involved in RA-induced CNS-related symptoms and comorbidities.6,30
Animal models of RA demonstrate elevated levels of IL-6 in synovial fluid and serum.31,32 Levels of IL-6 can also be elevated up to hundreds of times above normal values in the synovial fluid of patients with RA, with serum levels increased up to 10-fold.33,34 Although IL-6 can be produced at many sites, in active RA this differential in concentration may imply that IL-6 is produced largely intra-articularly by stromal and immune cells, and subsequently migrates into the systemic circulation. Elevated IL-6 levels can induce further increases in IL-6 levels through positive feedback loops. Peripherally, IL-6 is able to activate primary afferent sensory neurons through trans-signaling, which is an initial step leading to the sensation of pain.35 IL-6 signaling is linked to nociceptive plasticity through enhanced translation in sensory neurons,36 including the upregulation of transient receptor potential vanilloid (TRPV)1 and TRPV2 ion channels which may lead to lowered neuronal response thresholds, potentially resulting in peripheral sensitization.8,37 In addition, IL-6 has a role in central pain perception; persistent signaling from sensitized peripheral neurons can induce prolonged hyperexcitability of CNS neurons (central sensitization) resulting in tactile allodynia and secondary hyperalgesia, as well as primary hyperalgesia, consistent with behavior observed in preclinical models of RA and in patients with RA.38,39
Relation of Preclinical Pain Models to RA Disease Activity in Patients
Animal models of chronic pain and RA contribute to the understanding of pain in RA. While no single animal model reflects all aspects of human RA, features from joint inflammation to destruction of bone and cartilage in at least 1 peripheral joint provide useful similarities.5,40
Traditional, monoarticular models of RA—complete Freund’s adjuvant- and carrageenan-induced arthritis models—involve subcutaneous or intra-articular injection of adjuvants to drive local acute inflammation, mainly as a result of the innate immune response.40 These models are not fully representative of mechanisms underlying pain in human RA, as they do not include aspects of the adaptive immune response, which could impact conditions leading to sensitization of neurons in the joint.
Immunization and transfer models are more commonly used in the study of RA as they provide a longer-lasting alternative, and present with additional features similar to those of human RA. These include the collagen-induced arthritis (CIA), antigen-induced arthritis (AIA), collagen antibody-induced arthritis (CAIA), and K/BxN models.40 Pain in these models can occur in sites distant from the affected joint, suggesting the systemic effects of a mediator. Each of these models also presents with other features of RA, including immune cell infiltration, synovial inflammation, and bone and cartilage destruction, and exhibit pain-like behavior, including thermal and mechanical hypersensitivity and decreased locomotion/altered gait.5 In the CIA and CAIA models, pain-like behavior can be detected prior to the onset of joint inflammation, which resembles some human presentations. In the CAIA and K/BxN models pain-like behavior can outlast evidence of inflammation, consistent with residual pain reported by patients with RA, while signs of nerve injury have been reported in the CIA, CAIA, and K/BxN models, an occurrence observed in human RA.5
Overall, these models suggest involvement of peripheral and central neurological factors in RA-related pain which have similarities to patient-reported pain.
IL-6 and Pain Sensitization
Peripheral Nervous System
Healthy synovial tissue contains sympathetic neurons located around the vascular networks that control articular blood flow, as well as unmyelinated C-fibers (nociceptive fibers) that are responsible for the transmission of pain during potential tissue damage, and also potentially in inflammatory conditions.41 Joints are supplied by nerve branches from muscles acting on the joint which innervate the fibrous capsule, with some neurons reaching the synovial membrane and extending into the intimal layers.
In RA, changes in nerve distribution occur in and around inflamed joints. Proliferation of blood vessels in inflamed synovial tissue from patients with RA is accompanied by a simultaneous proliferation of new sympathetic nerve fibers,42 although the superficial intimal region of the synovium does not seem to be innervated. This could be a result of damage to the peripheral terminals of nerve fibers from exposure to active proinflammatory mediators or rapid proliferation of synovium41,43,44 which outpaces nerve growth.
Several lines of evidence point to an important role for IL-6 in peripherally induced pain. For example, in humans IL-6 is rapidly produced at high concentrations at the sites of surgical wounds. Serum levels appear to correlate with the magnitude of injury, and the degree of IL-6 elevation appears to correlate with the magnitude of pain.45 In a simple animal model, IL-6 injection in rats causes hypersensitivity to stimuli from both mechanical and thermal sources.46–49 After nerve injury in a rat model, a clear relationship was found between mechanical allodynia and IL-6-positive cells in the sciatic nerve. Rats with no allodynia had the fewest IL-6-responsive cells.50 There is evidence that IL-6 has a role in neuron regeneration and nerve survival,51 and that injured nerves that have regenerated, particularly mechanoreceptors, can demonstrate increased sensitivity.52
In the specific settings of inflammation, nerve damage, and/or constant stimulation, adrenergic sprouting may occur, which is linked to increased sensitivity to mechanical stimulation. IL-6 is able to induce such nerve fiber sprouting in response to injury of peripheral neurons in vivo and in vitro, and in hippocampal cultures in vitro.30,53,54 In animal models, this can be demonstrated in IL-6 knock-out (KO) mice by the decrease in adrenergic sprouting observed following sciatic nerve injury, and these mice also display a significant delay in the onset of mechanical allodynia, and a reduction in its severity,51,54 implying a key role for IL-6 in pain induction associated with microanatomic changes.
The role of IL-6 in mediating mechanical hyperalgesia is evidenced by the observation that the magnitude of the hyperalgesia exhibited by IL-6 KO mice was lower than in IL-6 wild-type mice following subcutaneous carrageenan injection.55 Moreover, injection of IL-6 into rat hind paw is sufficient to produce mechanical hyperalgesia,56 induced by the upregulation of C-fibers which results in nonlocalized mechanoreceptor pain.49 Interestingly, although injected into only one hind paw, pain was evoked in both hind paws, mimicking the symmetry of pain usually seen in RA. The onset of contralateral pain was both delayed and dose-related, suggesting systemic absorption of some factor from the joint, or possibly the induction of another pain mechanism that signals to the contralateral joint. Intraplantar co-injection of an IL-6 inhibitor (indomethacin or Lys-
Injection of soluble gp130 (an antagonist of IL-6 activation) reduced pain behavior in the AIA rodent model and did not reduce inflammation.57 Other studies too show that intra-articular injections of gp130 in animal models of RA have led to significant attenuation of pain-related behavior.6,58 This effect is less pronounced when soluble gp130 is administered systemically, which again implies that local intra-articular effects (presumably on nerve conduction) are more important than extra-articular effects. This seems to suggest that IL-6 has an effect on nerve conduction unrelated to inflammation.57 Intra-articular injection of IL-6 into healthy rat knee joints can result in long-lasting sensitization of C-fibers in the joint in response to mechanical stimulation of the knee joint.49,59
Taken together, these data suggest that IL-6 is important in driving sensitization at the neuron level in the peripheral nervous system and, importantly, this seems to occur with or without inflammation in the joint.
CNS
The effects of IL-6 on the nervous system are not limited to the stimulation of peripheral nerves and spinal transmission; there is a key role for IL-6 in central pain processing and enhancement in both the spinal cord and the brain. Of note, pain experienced in higher brain centers is clearly moderated by psychological status60 and psychosocial considerations are intimately interwoven with both peripheral and central pain processing.
IL-6 is present in glial and neuronal cells and, because of its pleiotropism, all cells are susceptible to IL-6 trans-signaling, including the neurons and glial cells of the spinal cord and dorsal root ganglia (DRG).6,28 These cells not only express receptors but neurons, astrocytes, and microglia also produce IL-6 so it seems unsurprising that IL-6 is elevated in the spinal cord and DRG in various models of non-inflammatory pain.7 In addition, circulating IL-6 may also cross the blood–brain barrier to access the CNS, particularly when this is compromised during states of chronic inflammation as is observed in the CIA model of RA.30,61
Of note, many pharmacologic treatments for RA, including IL-6 inhibitors, are too large to cross the blood–brain barrier, and it thus follows that central effects of IL-6 may persist or be only partially remediated, despite biologic disease-modifying anti-rheumatic drug (DMARD) treatment.
IL-6 appears to have an effect on the DRG, which is regarded as an amplification source of nociceptive pain. The cell bodies of peripheral sensory neurons are located within the DRG, from where the peripheral portion of the axon extends to the periphery of the body and the central portion extends into the CNS enabling the transmission of peripheral sensory information, including nociceptive information, to the CNS.62 Mice with gp130 KO specifically in sensory DRG neurons displayed reduced inflammation-induced pain.63 Moreover, co-injection of IL-6 and soluble IL-6R into the knee joint or spinal cord of healthy rats resulted in increased responses of dorsal horn neurons to mechanical stimulation of the knee or ankle, which could be blocked through spinal application of gp130.59 This suggests that the increase of sensitization in noninjected joints reflects increased central pain sensitization, not local pain.
In the absence of IL-6 expression in KO mice, vincristine-induced mechanical allodynia was reduced and spinal nerve lesion-induced mechanical allodynia was delayed.51,64 Furthermore, in a rat model of osteoarthritis, inhibition of IL-6 signaling with tocilizumab reduced the activation of astrocytes and microglia in DRG and mitigated mechanical allodynia,65 while systemic administration of an IL-6 inhibitor to mice with spinal cord injury suppressed allodynia and hyperalgesia.66
Intrathecal administration of IL-6 induced hyperalgesia or allodynia in animal models that had an induced sciatic injury and, interestingly, these animals experienced contralateral hyperalgesia on the noninjured side.48 In the same study, intracerebroventricular injection of IL-6 induced an immediate thermal hyperalgesia. Together these observations imply a substantial role for IL-6 in transmission of sensation via central mechanisms.
IL-6 may also have an orchestrating role in regulating cytokines that modulate pain in and around the spine. Specifically, inhibition of IL-6 suppressed the elevation of TNFα, IL-1, and IL-6 messenger RNA in the spinal cord and inhibited the release of TNFα and IL-1 protein into cerebrospinal fluid.67
These data suggest that, in animal models at least, in the context of IL-6 dysregulation, normal descending inhibitory tracts are less functional and ascending pain conduction may be amplified, resulting in enhanced ascending activation and reduced descending inhibition, promoting pain. Whether this is also the case in human RA-related pain is not yet clear, although there is some evidence of altered central sensory processing in response to pressure pain in patients with RA of >5 years’ duration.68
In the brain, preclinical and in vivo data provide evidence for elevated CNS IL-6 under conditions of neuroinflammation or brain injury. For example, the rate of post-traumatic healing of brain tissue is lowered in IL-6 KO mice and accelerated in transgenic mice overexpressing IL-6 in the CNS, likely due to the role of IL-6 in nerve regrowth discussed above.30
In humans, RA patients have been shown to have an enhanced cortical response to painful stimuli.69 A “damping” in cortical pain response to heat stimulus (evaluated by positron emission tomography scan) was seen in the brains of RA patients when compared with healthy controls, and the authors inferred that the mechanism of pain perception may be different in RA and that there may be a possible thalamocortical control.70 A review of brain changes by imaging concluded that there are significant changes in cortical blood flow in RA.71
IL-6 plays a role in several RA symptoms and comorbidities with CNS involvement, including psychologic disorders (depression, anxiety), fatigue, and pain. Pain and fatigue/depression are well correlated in RA72 and are likely to be bidirectional. Fatigue in RA is multifactorial, although growing evidence indicates disruption of the hypothalamic–pituitary–adrenal axis, which can be regulated by IL-6.6,29 Hyperactivity of this axis has also been connected to severe depression, and levels of IL-6 are significantly increased in depressed individuals, so it is possible that IL-6 may be involved in depression in RA.30,73
Taken together, there appears to be a link between higher CNS processing and IL-6 in the joint, IL-6 in the spinal cord, and central sensitization, suggesting a potential overlap in mechanisms of persistent local joint pain and persistent widespread pain.8,59
IL-6 in Chronic Pain
Chronic pain should not be regarded as acute pain of extended duration. Unlike prolonged acute pain, the mechanism for chronic pain in RA may be related to the duration and/or intensity of prior pain (the pain “area under the curve”), or the source of pain exposure.74
Chronic pain is pain that lasts at least 3–6 months and in this present discussion an apt definition is “pain that extends beyond the expected period of healing”; for example, unanticipated prolonged pain in patients with RA who appear to be in remission.75 Chronic pain is often associated with central sensitization and may be a manifestation of aberrant neuronal activity from peripheral sensitization of primary sensory neurons and subsequent or additional sensitization of neurons in the CNS.76 There may be little or no relationship to prior or current inflammation in this state.
Interestingly, the immune system (obviously important in RA with respect to inflammation) also has a role in modulating pain and may play a role in the development of chronic pain.77 Macrophages and monocytes are attracted to the DRG after adjuvant injection into the paw of healthy rats and may modulate neuronal activity through the secretion of pronociceptive or anti-nociceptive cytokines.78 As discussed above, increased levels of inflammatory cytokines, including IL-6, have been demonstrated in the DRG and serum of rats with chronic pain.37
Elevated levels of IL-6 and IL-6R were evident in a mouse model of chronic back pain (intervertebral disc injury model), and on administration of an IL-6 inhibitor, expression of the pain-related neuropeptide calcitonin gene-related peptide in the DRG was reduced.79 In clinical studies, anti-IL-6R inhibitors have been reported to suppress pain. Tocilizumab has been shown to reduce leg and lower back pain in a small proof-of-concept trial of patients with sciatica.80
It is possible that short episodes of acute inflammation may drive long-lasting sensory neuron plasticity which contributes to chronic pain74 and also to reciprocal signaling between neurons and immune cells in the CNS.81 IL-6 and its receptor are upregulated after nerve injury, and IL-6 is linked to microglia activation; in turn, microglia directly influence the establishment of neuropathic pain.82 Therefore, the processes leading to development of chronic pain have the potential to become self-perpetuating.
Evidence from preclinical and human studies suggests a role for IL-6 in chronic pain conditions; thus, IL-6 has been targeted for the treatment of chronic pain (Figure 1).
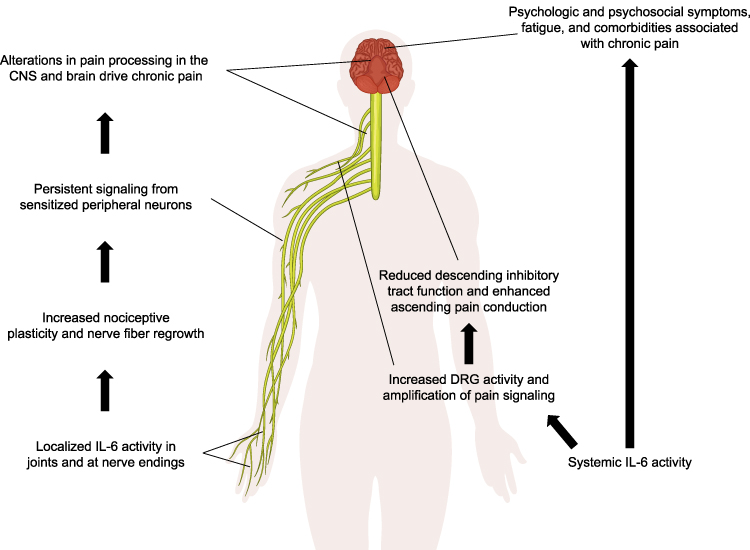 |
Figure 1 Potential mechanisms of IL-6 in RA-related pain, independent of clinical joint inflammation. Abbreviations: CNS, central nervous system; DRG, dorsal root ganglion; IL-6, interleukin-6; RA, rheumatoid arthritis. |
Clinical Implications
Implications of Long-Term Chronic Pain for Patients
The existence of chronic pain in and of itself is not benign. Undertreatment of non-inflammatory pain in RA can lead to a continued, substantial burden on patients’ overall health, with impact on numerous patient-reported outcomes (PROs).83–87 Moreover, patients who have chronic pain have a higher incidence of diabetes, hypertension, depression and fatigue, and are ultimately at increased risk of premature death.72,88 There are also initial hints that pain itself may be proinflammatory; patients with juvenile RA who were exposed to a painful stimulus had enhanced IL-6 production by peripheral blood cells.89,90
In addition, diffuse anatomic changes in the body, including structural changes in the brain, have been associated with chronic pain. A reduction in gray matter has been described across a range of chronic pain conditions, including chronic back pain, fibromyalgia, and osteoarthritis, as well as RA.91 Reductions in gray matter have been linked to pain duration in a study of patients with chronic back pain, and the authors suggested that changes were reflective of neurodegeneration.92 The structural changes in the RA brain may be similarly linked to chronic pain, and it is possible this may contribute to the cognitive impairment observed in patients with RA.93 Structural changes in the brain may also help to explain the mechanism behind the bidirectional relationship of mental health, including depression and anxiety, with pain in RA.94–96 Intriguingly, both functional and anatomic changes may potentially reverse with pain mitigation.97,98
Pain is usually independently treated for reasons of compassion, to help provide the patient with relief from this unpleasant sensory experience while waiting for the improvement of the disease with one or other DMARD. Given that pain duration is a major factor in central sensitization, however, there is a compelling argument to selectively address active pain in RA early and robustly.
Accurately Identifying a Different Target in RA
Accurate identification of the problem of pain as an entity on its own needs to be addressed. Since painful or tender joints contribute toward disease activity scores (eg, the Disease Activity Score with 28-joint count, which increases with the number of tender joints),99,100 high levels of patient-reported pain in early stages of the disease may lead to misidentification of the target in these patients whose treatment can be accelerated by “treat-to-target” approaches, exposing the patient to unnecessary risk.
At the other end of the treatment spectrum, residual RA-related pain in the absence of inflammation suggests that for many currently available therapies the treatment of pain is insufficient compared with the treatment of inflammation. Many rheumatologists would assess these patients for inflammation by examination, or by evaluating laboratory tests of inflammation. If inflammation is apparent, switching biologics would be considered. Perhaps a failure of pain relief (even in a patient in a low disease state) should be considered in a similar way by the treating physician. The issue of residual pain should not necessarily be treated by switching biologics, but (pending a specific treatment for central and peripheral sensitization) a switch in mechanism of action might be considered in cases where current treatment has demonstrated unsatisfactory pain reduction even in the absence of signs of inflammation.
Other therapeutic options may, in due course, provide an alternative for the treatment of peripheral and central pain sensitization, although they are not well trialed in RA.21
Does Mechanism of Action Differentially Affect Selection of Pain Treatment?
Adequate treatment of pain in RA is an unmet need, with most patients reporting dissatisfaction with their RA pain management.19 Guidelines recommend nonsteroidal anti-inflammatory drugs (NSAIDs) for the pharmacologic management of pain in RA,101 yet these and other standard RA treatments frequently do not sufficiently improve pain outcomes.21,102 NSAIDs and inhibition of certain cytokines (including TNFα) seem to decrease pain-like behavior only during inflammatory disease in animal models of RA, suggesting that reversal of central sensitization may depend on specific and potentially different inhibition of involved neuroimmune mechanisms.103–105
The move from preclinical evaluation of pain in rodent models to humans is starting to take place and there are now the beginnings of data in RA patients that support the disconnect between assessed disease activity and pain. Treatment with the JAK inhibitor baricitinib improved pain independently of inflammation in patients with RA,106 which is of interest, given the suppression of IL-6 signaling by JAK inhibitors in such patients.107
With respect to IL-6 inhibitors in patients with RA, sarilumab and tocilizumab have demonstrated positive effects on inflammation and also pain outcomes (MOBILITY and OPTION/LITHE/TOWARD trials, respectively) in patients with inadequate response to methotrexate, as well as in patients with inadequate response to TNF inhibitors (TARGET and RADIATE trials, respectively).108–114 It is difficult to establish whether the improvement in pain in these studies is related to impacts on the mechanisms of pain themselves or mainly to the reduction in inflammation. However, a recently published abstract demonstrated that patients treated with tocilizumab had a numerically similar improvement in pain, irrespective of whether they did or did not achieve a 20% reduction in swollen joint count.115 This again implies that pain can be relieved despite minimal reduction in inflammation.
Looking at traditional metrics of improvements in trials (eg, American College of Rheumatology scores or Clinical Disease Activity Index), cytokine inhibitors seem to result in broadly similar treatment outcomes in RA. It is worth considering whether targeting pain (and other PROs) in RA should be a goal in clinical practice. This would certainly be helpful in terms of patient satisfaction.
It is possible that by looking at specific cytokines involved in peripheral and/or central sensitization and then comparing these in trials with pain as a target, differences might emerge. In this regard, both sarilumab and tocilizumab, in very similarly designed head-to-head studies, with no confounding co-treatment with other DMARDs, demonstrated superiority to the TNF inhibitor adalimumab (MONARCH and ADACTA, respectively) with respect to changes in pain measurements (in MONARCH in a prespecified analysis and in ADACTA in a post hoc analysis).111,116–118 This could be due to a differential favoring IL-6-specific inhibition of mechanisms around pain sensitization with IL-6 inhibitors and/or a differential in the anti-inflammatory effect, as changes in disease activity scores were also superior with tocilizumab and sarilumab vs adalimumab. Regardless of the mechanism of action, there was a consistent difference, favoring IL-6 inhibitors, with respect to pain experienced by patients in both of these comparator studies. It therefore seems reasonable to consider whether this difference in pain may be related to the different cytokine targets.
Ideally, pharmacologic treatment of RA would of course manage inflammatory mechanisms of disease as well as non-inflammatory mechanisms contributing to RA-related pain.
The selection of RA treatment focused on the PRO of pain rather than more traditional metrics is a relatively novel concept, but further evaluation of this seems to be supported by the studies above. Many newer studies do report on pain as a PRO but standard metrics are traditionally the primary endpoints of trials.
Conclusions
As inflammation and joint damage are commonly assumed to be the proximate causes of RA-related pain, treatment of RA directed largely at inflammation is expected to manage pain in conjunction with inflammation.119,120 Recent studies suggest that multiple mechanisms are involved in the generation of RA-related pain, with IL-6 playing a key role. There can be a disconnect between pain and inflammation in RA, and the substantial burden of long-term chronic pain (and sequelae) in patients with RA indicates that pain alleviation should perhaps be considered as seriously as the prevention of joint erosions. These concerns call for thorough pain assessment in addition to and in parallel with measurements of inflammation at baseline and subsequent visits. In view of the fact that central sensitization seems to be related partially to duration of pain exposure at a minimum, pain should be considered as potentially distinct from joint inflammation and should be measured and addressed separately and early.
Rheumatologists are aware of other conditions that may be comorbid with RA (eg, osteoporosis, cardiovascular disease), but they do not typically think of CNS involvement as part of the comorbid process of RA. Evidence seems to suggest this should change and that we should be cognizant of nervous system involvement as a fairly common and major target organ that merits separate consideration when treating patients.
There is a need for additional studies to investigate the persistence of central sensitization in RA and how its development may be prevented early in the disease course. Prospective studies that are specifically designed to study non-inflammatory pain in patients with RA will further our understanding of RA-related pain and help to inform patient management.
Abbreviations
AIA, antigen-induced arthritis; CAIA, collagen antibody-induced arthritis; CIA, collagen-induced arthritis; CNS, central nervous system; DMARD, disease-modifying anti-rheumatic drug; DRG, dorsal root ganglia; gp130, glycoprotein 130; IL, interleukin; IL-6R, interleukin-6 receptor; JAK, Janus kinase; K/BxN, mice expressing both the T cell receptor transgene KRN and the major histocompatibility complex class II molecule Ag7; KO, knock-out; MAPK, mitogen-activated protein kinase; NSAID, nonsteroidal anti-inflammatory drug; PET, positron emission tomography; PRO, patient-reported outcome; RA, rheumatoid arthritis; TNF, tumor necrosis factor; TRPV, transient receptor potential vanilloid.
Data Sharing Statement
Data sharing is not applicable to this article as no datasets were generated or analyzed during the course of drafting this publication.
Acknowledgments
Medical writing support, under the sole direction of the author, was provided by Adelphi Communications, Ltd, Macclesfield, UK (Frances Smith, Gregory Bezkorovainy, and Joseph Hodgson), and was funded by Sanofi Genzyme, Cambridge, MA, USA, in accordance with Good Publications Practices (GPP3).
Funding
Medical writing support, under the sole direction of the author, was provided by Adelphi Communications Ltd, Macclesfield, UK, and was funded by Sanofi, Cambridge, MA, USA. The author received no payment or other compensation for developing this paper. The funder had no role in the preparation, content, or decision to submit this publication.
Disclosure
AS has received consulting fees from or participated in speakers’ bureaus for Genentech, Gilead, Lilly, Regeneron Pharmaceuticals Inc., Roche, and Sanofi.
References
1. Cross M, Smith E, Hoy D, et al. The global burden of rheumatoid arthritis: estimates from the global burden of disease 2010 study. Ann Rheum Dis. 2014;73(7):1316–1322. doi:10.1136/annrheumdis-2013-204627
2. McInnes IB, Schett G. The pathogenesis of rheumatoid arthritis. N Engl J Med. 2011;365(23):2205–2219. doi:10.1056/NEJMra1004965
3. Heiberg T, Finset A, Uhlig T, Kvien TK. Seven year changes in health status and priorities for improvement of health in patients with rheumatoid arthritis. Ann Rheum Dis. 2005;64(2):191–195. doi:10.1136/ard.2004.022699
4. Heiberg T, Kvien TK. Preferences for improved health examined in 1024 patients with rheumatoid arthritis: pain has highest priority. Arthritis Rheum. 2002;47(4):391–397. doi:10.1002/art.10515
5. Bas DB, Su J, Wigerblad G, Svensson CI. Pain in rheumatoid arthritis: models and mechanisms. Pain Manag. 2016;6(3):265–284. doi:10.2217/pmt.16.4
6. Choy EHS, Calabrese LH. Neuroendocrine and neurophysiological effects of interleukin 6 in rheumatoid arthritis. Rheumatology (Oxford). 2018;57(11):1885–1895. doi:10.1093/rheumatology/kex391
7. Zhou YQ, Liu Z, Liu ZH, et al. Interleukin-6: an emerging regulator of pathological pain. J Neuroinflammation. 2016;13(1):141. doi:10.1186/s12974-016-0607-6
8. Schaible H. Nociceptive neurons detect cytokines in arthritis. Arthritis Res Ther. 2014;16(5):470. doi:10.1186/s13075-014-0470-8
9. Cazzola M, Atzeni F, Boccassini L, et al. Physiopathology of pain in rheumatology. Reumatismo. 2014;66(1):4–13. doi:10.4081/reumatismo.2014.758
10. Deane KD, Norris JM, Holers VM. Preclinical rheumatoid arthritis: identification, evaluation, and future directions for investigation. Rheum Dis Clin North Am. 2010;36(2):213–241. doi:10.1016/j.rdc.2010.02.001
11. de Haas WH, de Boer W, Griffioen F, Oosten-Elst P. Rheumatoid arthritis, typus robustus. Ann Rheum Dis. 1973;32(1):91–92. doi:10.1136/ard.32.1.91
12. Kristensen LE, Bliddal H, Christensen R, et al. Is swollen to tender joint count ratio a new and useful clinical marker for biologic drug response in rheumatoid arthritis? Results from a Swedish cohort. Arthritis Care Res (Hoboken). 2014;66(2):173–179. doi:10.1002/acr.22107
13. Challa DN, Kvrgic Z, Cheville AL, et al. Patient-provider discordance between global assessments of disease activity in rheumatoid arthritis: a comprehensive clinical evaluation. Arthritis Res Ther. 2017;19(1):212. doi:10.1186/s13075-017-1419-5
14. Khan NA, Spencer HJ, Abda E, et al. Determinants of discordance in patients’ and physicians’ rating of rheumatoid arthritis disease activity. Arthritis Care Res (Hoboken). 2012;64(2):206–214. doi:10.1002/acr.20685
15. Studenic P, Radner H, Smolen JS, Aletaha D. Discrepancies between patients and physicians in their perceptions of rheumatoid arthritis disease activity. Arthritis Rheum. 2012;64(9):2814–2823. doi:10.1002/art.34543
16. Hammer H, Hansen I, Järvinen P, et al. Major reduction of ultrasound detected synovitis during subcutaneous tocilizumab treatment; results from a multicenter 24 weeks study of patients with rheumatoid arthritis [abstract]. Arthritis Rheumatol. 2018;70(Suppl 10).
17. Maini R, St Clair EW, Breedveld FJ, et al. Infliximab (chimeric anti-tumour necrosis factor alpha monoclonal antibody) versus placebo in rheumatoid arthritis patients receiving concomitant methotrexate: a randomised Phase III trial. ATTRACT study group. Lancet. 1999;354(9194):1932–1939. doi:10.1016/S0140-6736(99)05246-0
18. Lee YC, Cui J, Lu B, et al. Pain persists in DAS28 rheumatoid arthritis remission but not in ACR/EULAR remission: a longitudinal observational study. Arthritis Res Ther. 2011;13(3):R83. doi:10.1186/ar3353
19. Taylor P, Manger B, Alvaro-Gracia J, et al. Patient perceptions concerning pain management in the treatment of rheumatoid arthritis. J Int Med Res. 2010;38(4):1213–1224. doi:10.1177/147323001003800402
20. Sarzi-Puttini P, Fiorini T, Panni B, et al. Correlation of the score for subjective pain with physical disability, clinical and radiographic scores in recent onset rheumatoid arthritis. BMC Musculoskelet Disord. 2002;3:18. doi:10.1186/1471-2474-3-18
21. McWilliams DF, Walsh DA. Pain mechanisms in rheumatoid arthritis. Clin Exp Rheumatol. 2017;35(5):94–101.
22. Flodin P, Martinsen S, Altawil R, et al. Intrinsic brain connectivity in chronic pain: a resting-state fMRI study in patients with rheumatoid arthritis. Front Hum Neurosci. 2016;10:107. doi:10.3389/fnhum.2016.00107
23. Biswas M, Chatterjee A, Ghosh SK, et al. Prevalence, types, clinical associations, and determinants of peripheral neuropathy in rheumatoid patients. Ann Indian Acad Neurol. 2011;14(3):194–197. doi:10.4103/0972-2327.85893
24. Adlan AM, Lip GY, Paton JF, Kitas GD, Fisher JP. Autonomic function and rheumatoid arthritis: a systematic review. Semin Arthritis Rheum. 2014;44(3):283–304. doi:10.1016/j.semarthrit.2014.06.003
25. Walsh DA, McWilliams DF. Mechanisms, impact and management of pain in rheumatoid arthritis. Nat Rev Rheumatol. 2014;10(10):581–592. doi:10.1038/nrrheum.2014.64
26. Ramachandran VS, Blakeslee S. Phantoms in the Brain: Human Nature and the Architecture of the Mind. HarperCollins; 1998.
27. Landewe R, van der Heijde D, Klareskog L, et al. Disconnect between inflammation and joint destruction after treatment with etanercept plus methotrexate: results from the trial of etanercept and methotrexate with radiographic and patient outcomes. Arthritis Rheum. 2006;54(10):3119–3125. doi:10.1002/art.22143
28. Rose-John S. IL-6 trans-signaling via the soluble IL-6 receptor: importance for the pro-inflammatory activities of IL-6. Int J Biol Sci. 2012;8(9):1237–1247. doi:10.7150/ijbs.4989
29. Srirangan S, Choy EH. The role of interleukin 6 in the pathophysiology of rheumatoid arthritis. Ther Adv Musculoskelet Dis. 2010;2(5):247–256. doi:10.1177/1759720X10378372
30. Erta M, Quintana A, Hidalgo J. Interleukin-6, a major cytokine in the central nervous system. Int J Biol Sci. 2012;8(9):1254–1266. doi:10.7150/ijbs.4679
31. Ferraccioli G, Bracci-Laudiero L, Alivernini S, et al. Interleukin-1β and interleukin-6 in arthritis animal models: roles in the early phase of transition from acute to chronic inflammation and relevance for human rheumatoid arthritis. Mol Med. 2010;16(11–12):552–557. doi:10.2119/molmed.2010.00067
32. Palmblad K, Erlandsson-Harris H, Tracey KJ, Andersson U. Dynamics of early synovial cytokine expression in rodent collagen-induced arthritis: a therapeutic study using a macrophage-deactivating compound. Am J Pathol. 2001;158(2):491–500. doi:10.1016/S0002-9440(10)63991-0
33. Arvidson NG, Gudbjornsson B, Elfman L, et al. Circadian rhythm of serum interleukin-6 in rheumatoid arthritis. Ann Rheum Dis. 1994;53(8):521–524. doi:10.1136/ard.53.8.521
34. Desgeorges A, Gabay C, Silacci P, et al. Concentrations and origins of soluble interleukin 6 receptor-alpha in serum and synovial fluid. J Rheumatol. 1997;24(8):1510–1516.
35. Svensson CI. Interleukin-6: a local pain trigger? Arthritis Res Ther. 2010;12(5):145. doi:10.1186/ar3138
36. Melemedjian OK, Asiedu MN, Tillu DV, et al. IL-6- and NGF-induced rapid control of protein synthesis and nociceptive plasticity via convergent signaling to the EIF4F complex. J Neurosci. 2010;30(45):15113–15123. doi:10.1523/JNEUROSCI.3947-10.2010
37. Fang D, Kong LY, Cai J, et al. Interleukin-6-mediated functional upregulation of TRPV1 receptors in dorsal root ganglion neurons through the activation of JAK/PI3K signaling pathway: roles in the development of bone cancer pain in a rat model. Pain. 2015;156(6):1124–1144. doi:10.1097/j.pain.0000000000000158
38. Nieto FR, Clark AK, Grist J, et al. Neuron-immune mechanisms contribute to pain in early stages of arthritis. J Neuroinflammation. 2016;13(1):96. doi:10.1186/s12974-016-0556-0
39. Woolf CJ. Central sensitization: implications for the diagnosis and treatment of pain. Pain. 2011;152(3Suppl):S2–S15. doi:10.1016/j.pain.2010.09.030
40. Krock E, Jurczak A, Svensson CI. Pain pathogenesis in rheumatoid arthritis—what have we learned from animal models? Pain. 2018;159:S98–S109. doi:10.1097/j.pain.0000000000001333
41. Mapp PI. Innervation of the synovium. Ann Rheum Dis. 1995;54(5):398–403. doi:10.1136/ard.54.5.398
42. Widenfalk B. Sympathetic innervation of normal and rheumatoid synovial tissue. Scand J Plast Reconstr Surg Hand Surg. 1991;25(1):31–33. doi:10.3109/02844319109034920
43. Mapp PI, Kidd BL, Gibson SJ, et al. Substance P-, calcitonin gene-related peptide- and C-flanking peptide of neuropeptide Y-immunoreactive fibres are present in normal synovium but depleted in patients with rheumatoid arthritis. Neuroscience. 1990;37(1):143–153. doi:10.1016/0306-4522(90)90199-E
44. Pereira da Silva JA, Carmo-Fonseca M. Peptide containing nerves in human synovium: immunohistochemical evidence for decreased innervation in rheumatoid arthritis. J Rheumatol. 1990;17(12):1592–1599.
45. Holzheimer RG, Steinmetz W. Local and systemic concentrations of pro- and anti-inflammatory cytokines in human wounds. Eur J Med Res. 2000;5(8):347–355.
46. Poole S, Cunha FQ, Selkirk S, Lorenzetti BB, Ferreira SH. Cytokine-mediated inflammatory hyperalgesia limited by interleukin-10. Br J Pharmacol. 1995;115(4):684–688. doi:10.1111/j.1476-5381.1995.tb14987.x
47. Oka T, Oka K, Hosoi M, Hori T. Intracerebroventricular injection of interleukin-6 induces thermal hyperalgesia in rats. Brain Res. 1995;692(1–2):123–128. doi:10.1016/0006-8993(95)00691-I
48. DeLeo JA, Colburn RW, Nichols M, Malhotra A. Interleukin-6-mediated hyperalgesia/allodynia and increased spinal IL-6 expression in a rat mononeuropathy model. J Interferon Cytokine Res. 1996;16(9):695–700. doi:10.1089/jir.1996.16.695
49. Brenn D, Richter F, Schaible HG. Sensitization of unmyelinated sensory fibers of the joint nerve to mechanical stimuli by interleukin-6 in the rat: an inflammatory mechanism of joint pain. Arthritis Rheum. 2007;56(1):351–359. doi:10.1002/art.22282
50. Cui JG, Holmin S, Mathiesen T, et al. Possible role of inflammatory mediators in tactile hypersensitivity in rat models of mononeuropathy. Pain. 2000;88(3):239–248. doi:10.1016/S0304-3959(00)00331-6
51. Ramer MS, Murphy PG, Richardson PM, Bisby MA. Spinal nerve lesion-induced mechanoallodynia and adrenergic sprouting in sensory ganglia are attenuated in interleukin-6 knockout mice. Pain. 1998;78(2):115–121. doi:10.1016/S0304-3959(98)00121-3
52. Krarup C, Rosen B, Boeckstyns M, et al. Sensation, mechanoreceptor, and nerve fiber function after nerve regeneration. Ann Neurol. 2017;82(6):940–950. doi:10.1002/ana.25102
53. Hakkoum D, Stoppini L, Muller D. Interleukin-6 promotes sprouting and functional recovery in lesioned organotypic hippocampal slice cultures. J Neurochem. 2007;100(3):747–757. doi:10.1111/j.1471-4159.2006.04257.x
54. Murphy PG, Ramer MS, Borthwick L, et al. Endogenous interleukin-6 contributes to hypersensitivity to cutaneous stimuli and changes in neuropeptides associated with chronic nerve constriction in mice. Eur J Neurosci. 1999;11(7):2243–2253. doi:10.1046/j.1460-9568.1999.00641.x
55. Xu XJ, Hao JX, Andell-Jonsson S, et al. Nociceptive responses in interleukin-6-deficient mice to peripheral inflammation and peripheral nerve section. Cytokine. 1997;9(12):1028–1033. doi:10.1006/cyto.1997.0243
56. Cunha F, Poole S, Lorenzetti B, Ferreira SH. The pivotal role of tumour necrosis factor alpha in the development of inflammatory hyperalgesia. Br J Pharmacol. 1992;107(3):660–664. doi:10.1111/j.1476-5381.1992.tb14503.x
57. Obreja O, Biasio W, Andratsch M, et al. Fast modulation of heat-activated ionic current by proinflammatory interleukin 6 in rat sensory neurons. Brain. 2005;128:1634–1641. doi:10.1093/brain/awh490
58. Boettger MK, Weber K, Gajda M, et al. Spinally applied ketamine or morphine attenuate peripheral inflammation and hyperalgesia in acute and chronic phases of experimental arthritis. Brain Behav Immun. 2010;24(3):474–485. doi:10.1016/j.bbi.2009.12.002
59. Vazquez E, Kahlenbach J, Segond von Banchet G, et al. Spinal interleukin-6 is an amplifier of arthritic pain in the rat. Arthritis Rheum. 2012;64(7):2233–2242. doi:10.1002/art.34384
60. Schweinhardt P, Kalk N, Wartolowska K, et al. Investigation into the neural correlates of emotional augmentation of clinical pain. Neuroimage. 2008;40(2):759–766. doi:10.1016/j.neuroimage.2007.12.016
61. Nishioku T, Yamauchi A, Takata F, et al. Disruption of the blood-brain barrier in collagen-induced arthritic mice. Neurosci Lett. 2010;482(3):208–211. doi:10.1016/j.neulet.2010.07.035
62. Esposito MF, Malayil R, Hanes M, Deer T. Unique characteristics of the dorsal root ganglion as a target for neuromodulation. Pain Med. 2019;20(2):S23–S30. doi:10.1093/pm/pnz012
63. Andratsch M, Mair N, Constantin CE, et al. A key role for gp130 expressed on peripheral sensory nerves in pathological pain. J Neurosci. 2009;29(43):13473–13483. doi:10.1523/JNEUROSCI.1822-09.2009
64. Kiguchi N, Maeda T, Kobayashi Y, et al. The critical role of invading peripheral macrophage-derived interleukin-6 in vincristine-induced mechanical allodynia in mice. Eur J Pharmacol. 2008;592(1–3):87–92. doi:10.1016/j.ejphar.2008.07.008
65. Lin Y, Liu L, Jiang H, et al. Inhibition of interleukin-6 function attenuates the central sensitization and pain behavior induced by osteoarthritis. Eur J Pharmacol. 2017;811:260–267. doi:10.1016/j.ejphar.2017.06.032
66. Murakami T, Kanchiku T, Suzuki H, et al. Anti-interleukin-6 receptor antibody reduces neuropathic pain following spinal cord injury in mice. Exp Ther Med. 2013;6(5):1194–1198. doi:10.3892/etm.2013.1296
67. Schoeniger-Skinner DK, Ledeboer A, Frank MG, et al. Interleukin-6 mediates low-threshold mechanical allodynia induced by intrathecal HIV-1 envelope glycoprotein gp120. Brain Behav Immun. 2007;21(5):660–667. doi:10.1016/j.bbi.2006.10.010
68. Leffler A, Kosek E, Lerndal T, et al. Somatosensory perception and function of diffuse noxious inhibitory controls (DNIC) in patients suffering from rheumatoid arthritis. Eur J Pain. 2002;6(2):161–167. doi:10.1053/eujp.2001.0313
69. Hummel T, Schiessl C, Wendler J, Kobal G. Peripheral and central nervous changes in patients with rheumatoid arthritis in response to repetitive painful stimulation. Int J Psychophysiol. 2000;37(2):177–183. doi:10.1016/S0167-8760(00)00087-8
70. Jones AK, Derbyshire SW. Reduced cortical responses to noxious heat in patients with rheumatoid arthritis. Ann Rheum Dis. 1997;56(10):601–607. doi:10.1136/ard.56.10.601
71. Peyron R, Laurent B, Garcia-Larrea L. Functional imaging of brain responses to pain. A review and meta-analysis. Neurophysiol Clin. 2000;30(5):263–288. doi:10.1016/S0987-7053(00)00227-6
72. Nikolaus S, Bode C, Taal E, van de Laar MA. Fatigue and factors related to fatigue in rheumatoid arthritis: a systematic review. Arthritis Care Res (Hoboken). 2013;65(7):1128–1146. doi:10.1002/acr.21949
73. Hodes GE, Ménard C, Russo SJ. Integrating interleukin-6 into depression diagnosis and treatment. Neurobiol Stress. 2016;29(4):15–22. doi:10.1016/j.ynstr.2016.03.003
74. Raoof R, Willemen HLDM, Eijkelkamp N. Divergent roles of immune cells and their mediators in pain. Rheumatology (Oxford). 2018;57(3):429–440. doi:10.1093/rheumatology/kex308
75. Turk D, Okifuji A. Pain terms and taxonomies. In: Bonica’s Management of Pain.
76. Yang S, Chang M. Chronic pain: structural and functional changes in brain structures and associated negative affective states. Int J Mol Sci. 2019;20(13):3130. doi:10.3390/ijms20133130
77. Ji R, Chamessian A, Zhang Y. Pain regulation by non-neuronal cells and inflammation. Science. 2016;354(6312):572–577. doi:10.1126/science.aaf8924
78. Jeon S, Lee K, Park E, et al. Monocyte chemoattractant protein-1 immunoreactivity in sensory ganglia and hindpaw after adjuvant injection. Neuroreport. 2008;19(2):183–186. doi:10.1097/WNR.0b013e3282f3c781
79. Sainoh T, Orita S, Miyagi M, et al. Interleukin-6 and interleukin-6 receptor expression, localization, and involvement in pain-sensing neuron activation in a mouse intervertebral disc injury model. J Orthop Res. 2015;33(10):1508–1514. doi:10.1002/jor.22925
80. Ohtori S, Miyagi M, Eguchi Y, et al. Efficacy of epidural administration of anti-interleukin-6 receptor antibody onto spinal nerve for treatment of sciatica. Eur Spine J. 2012;21(10):2079–2084. doi:10.1007/s00586-012-2183-5
81. Vardeh D, Mannion R, Woolf CJ. Toward a mechanism-based approach to pain diagnosis. J Pain. 2016;17(9Suppl):T50–69. doi:10.1016/j.jpain.2016.03.001
82. Brázda V, Klusáková I, Svíženská IH, Dubový P. Dynamic response to peripheral nerve injury detected by in situ hybridization of IL-6 and its receptor mRNAs in the dorsal root ganglia is not strictly correlated with signs of neuropathic pain. Mol Pain. 2013;9:42. doi:10.1186/1744-8069-9-42
83. Courvoisier DS, Agoritsas T, Glauser J, et al. Pain as an important predictor of psychosocial health in patients with rheumatoid arthritis. Arthritis Care Res (Hoboken). 2012;64(2):190–196. doi:10.1002/acr.20652
84. Lee YC. Effect and treatment of chronic pain in inflammatory arthritis. Curr Rheumatol Rep. 2013;15(1):300. doi:10.1007/s11926-012-0300-4
85. Nicassio PM, Ormseth SR, Kay M, et al. The contribution of pain and depression to self-reported sleep disturbance in patients with rheumatoid arthritis. Pain. 2012;153(1):107–112. doi:10.1016/j.pain.2011.09.024
86. Sturgeon JA, Finan PH, Zautra AJ. Affective disturbance in rheumatoid arthritis: psychological and disease-related pathways. Nat Rev Rheumatol. 2016;12(9):532–542. doi:10.1038/nrrheum.2016.112
87. Wolfe F, Michaud K. Severe rheumatoid arthritis (RA), worse outcomes, comorbid illness, and sociodemographic disadvantage characterize RA patients with fibromyalgia. J Rheumatol. 2004;31(4):695–700.
88. Carson HJ. Anatomical changes correlated with chronic pain in forensic medicine. Forensic Sci Res. 2017;2(3):145–151. doi:10.1080/20961790.2017.1341364
89. Griffis CA, Compton P, Doering L. The effect of pain on leukocyte cellular adhesion molecules. Biol Res Nurs. 2006;7(4):297–312. doi:10.1177/1099800405286071
90. Roupe van der Voort C, Heijnen CJ, Wulffraat N, et al. Stress induces increases in IL-6 production by leucocytes of patients with the chronic inflammatory disease juvenile rheumatoid arthritis: a putative role for alpha(1)-adrenergic receptors. J Neuroimmunol. 2000;110(1–2):223–229. doi:10.1016/S0165-5728(00)00328-3
91. Wartolowska K, Hough MG, Jenkinson M, et al. Structural changes of the brain in rheumatoid arthritis. Arthritis Rheum. 2012;64(2):371–379. doi:10.1002/art.33326
92. Apkarian AV, Sosa Y, Sonty S, et al. Chronic back pain is associated with decreased prefrontal and thalamic gray matter density. J Neurosci. 2004;24(46):10410–10415. doi:10.1523/JNEUROSCI.2541-04.2004
93. Vitturi BK, Nascimento BAC, Alves BR, et al. Cognitive impairment in patients with rheumatoid arthritis. J Clin Neurosci. 2019;69:81–87. doi:10.1016/j.jocn.2019.08.027
94. Bondesson E, Larrosa Pardo F, Stigmar K, et al. Comorbidity between pain and mental illness - evidence of a bidirectional relationship. Eur J Pain. 2018;22(7):1304–1311. doi:10.1002/ejp.1218
95. Euesden J, Matcham F, Hotopf M, et al. The relationship between mental health, disease severity, and genetic risk for depression in early rheumatoid arthritis. Psychosom Med. 2017;79(6):638–645. doi:10.1097/PSY.0000000000000462
96. Lu M, Guo H, Lin M, et al. Bidirectional associations between rheumatoid arthritis and depression: a nationwide longitudinal study. Sci Rep. 2016;6:20647. doi:10.1038/srep20647
97. May A. Chronic pain may change the structure of the brain. Pain. 2008;137(1):7–15. doi:10.1016/j.pain.2008.02.034
98. Seminowicz DA, Wideman TH, Naso L, et al. Effective treatment of chronic low back pain in humans reverses abnormal brain anatomy and function. J Neurosci. 2011;31(20):7540–7550. doi:10.1523/JNEUROSCI.5280-10.2011
99. Joharatnam N, McWilliams DF, Wilson D, et al. A cross-sectional study of pain sensitivity, disease-activity assessment, mental health, and fibromyalgia status in rheumatoid arthritis. Arthritis Res Ther. 2015;17(1):11. doi:10.1186/s13075-015-0525-5
100. Ton E, Bakker MF, Verstappen SMM, et al. Look beyond the disease activity score of 28 joints (DAS28): tender points influence the DAS28 in patients with rheumatoid arthritis. J Rheumatol. 2012;39(1):22–27. doi:10.3899/jrheum.110072
101. Geenen R, Overman CL, Christensen R, et al. EULAR recommendations for the health professional’s approach to pain management in inflammatory arthritis and osteoarthritis. Ann Rheum Dis. 2018;77(6):767–807.
102. McWilliams DF, Walsh DA. Factors predicting pain and early discontinuation of tumour necrosis factor-α-inhibitors in people with rheumatoid arthritis: results from the British Society for Rheumatology Biologics Register. BMC Musculoskelet Disord. 2016;17:337. doi:10.1186/s12891-016-1192-7
103. Christianson CA, Corr M, Firestein GS, et al. Characterization of the acute and persistent pain state present in K/BxN serum transfer arthritis. Pain. 2010;151(2):394–403. doi:10.1016/j.pain.2010.07.030
104. Inglis JJ, Notley CA, Essex D, et al. Collagen-induced arthritis as a model of hyperalgesia: functional and cellular analysis of the analgesic actions of tumor necrosis factor blockade. Arthritis Rheum. 2007;56(12):4015–4023. doi:10.1002/art.23063
105. Bas DB, Su J, Sandor K, et al. Collagen antibody-induced arthritis evokes persistent pain with spinal glial involvement and transient prostaglandin dependency. Arthritis Rheum. 2012;64(12):3886–3896. doi:10.1002/art.37686
106. Taylor PC, Lee YC, Fleischmann R, et al. Achieving pain control in rheumatoid arthritis with baricitinib or adalimumab plus methotrexate: results from the RA-BEAM trial. J Clin Med. 2019;8(6):831. doi:10.3390/jcm8060831
107. Migita K, Izumi Y, Jiuchi Y, et al. Effects of Janus kinase inhibitor tofacitinib on circulating serum amyloid A and interleukin-6 during treatment for rheumatoid arthritis. Clin Exp Immunol. 2014;175:208–214. doi:10.1111/cei.12234
108. Emery P, Keystone E, Tony HP, et al. IL-6 receptor inhibition with tocilizumab improves treatment outcomes in patients with rheumatoid arthritis refractory to anti-tumour necrosis factor biologicals: results from a 24-week multicentre randomised placebo-controlled trial. Ann Rheum Dis. 2008;67(11):1516–1523. doi:10.1136/ard.2008.092932
109. Smolen JS, Beaulieu A, Rubbert-Roth A, et al. Effect of interleukin-6 receptor inhibition with tocilizumab in patients with rheumatoid arthritis (OPTION study): a double-blind, placebo-controlled, randomised trial. Lancet. 2008;371(9617):987–997. doi:10.1016/S0140-6736(08)60453-5
110. Strand V, Kosinski M, Chen CI, et al. Sarilumab plus methotrexate improves patient-reported outcomes in patients with active rheumatoid arthritis and inadequate responses to methotrexate: results of a phase III trial. Arthritis Res Ther. 2016;18:198. doi:10.1186/s13075-016-1096-9
111. Strand V, Michalska M, Birchwood C, et al. Impact of tocilizumab monotherapy on patient-reported outcomes in patients with rheumatoid arthritis from two randomised controlled trials. RMD Open. 2017;3(2):e000496. doi:10.1136/rmdopen-2017-000496
112. Kremer JM, Blanco R, Brzosko M, et al. Tocilizumab inhibits structural joint damage in rheumatoid arthritis patients with inadequate responses to methotrexate: results from the double-blind treatment phase of a randomized placebo-controlled trial of tocilizumab safety and prevention of structural joint damage at one year. Arthritis Rheum. 2011;63(3):609–621. doi:10.1002/art.30158
113. Strand V, Reaney M, Chen CI, et al. Sarilumab improves patient-reported outcomes in rheumatoid arthritis patients with inadequate response/intolerance to tumour necrosis factor inhibitors. RMD Open. 2017;3(1):e000416. doi:10.1136/rmdopen-2016-000416
114. Genovese MC, Burmester GR, Gossec L, et al. Associations between rheumatoid arthritis disease activity and patient-reported outcomes in sarilumab clinical trials. Ann Rheum Dis. 2020;79(Suppl1):299. doi:10.1136/annrheumdis-2019-216852
115. Sebba A, Han J, Mohan S. SAT0121 Pain and other patient-reported outcomes in patients with rheumatoid arthritis who did or did not achieve treatment response based on improvement in swollen joints in tocilizumab clinical trials. [abstract]. Ann Rheum Dis. 2020;79:994–995. doi:10.1136/annrheumdis-2020-eular.967
116. Burmester GR, Lin Y, Patel R, et al. Efficacy and safety of sarilumab monotherapy versus adalimumab monotherapy for the treatment of patients with active rheumatoid arthritis (MONARCH): a randomised, double-blind, parallel-group phase III trial. Ann Rheum Dis. 2017;76(5):840–847. doi:10.1136/annrheumdis-2016-210310
117. Gabay C, Emery P, van Vollenhoven R, et al. Tocilizumab monotherapy versus adalimumab monotherapy for treatment of rheumatoid arthritis (ADACTA): a randomised, double-blind, controlled Phase 4 trial. Lancet. 2013;381(9877):1541–1550. doi:10.1016/S0140-6736(13)60250-0
118. Strand V, Gossec L, Proudfoot CWJ, et al. Patient-reported outcomes from a randomized phase III trial of sarilumab monotherapy versus adalimumab monotherapy in patients with rheumatoid arthritis. Arthritis Res Ther. 2018;20(1):129. doi:10.1186/s13075-018-1614-z
119. Crofford L. Use of NSAIDs in treating patients with arthritis. Arthritis Res Ther. 2013;15:S2. doi:10.1186/ar4174
120. Guo Q, Wang Y, Xu D, et al. Rheumatoid arthritis: pathological mechanisms and modern pharmacologic therapies. Bone Res. 2018;5:15. doi:10.1038/s41413-018-0016-9
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.