Back to Journals » International Journal of Nanomedicine » Volume 13
Paclitaxel-loaded redox-sensitive nanoparticles based on hyaluronic acid-vitamin E succinate conjugates for improved lung cancer treatment
Authors Song Y ![]() , Cai H
, Cai H ![]() , Yin T, Huo M, Ma P, Zhou J, Lai W
, Yin T, Huo M, Ma P, Zhou J, Lai W
Received 28 October 2017
Accepted for publication 26 January 2018
Published 15 March 2018 Volume 2018:13 Pages 1585—1600
DOI https://doi.org/10.2147/IJN.S155383
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Lei Yang
Yu Song,1,2 Han Cai,1 Tingjie Yin,1 Meirong Huo,1 Ping Ma,3 Jianping Zhou,1 Wenfang Lai2
1Department of Pharmaceutics, China Pharmaceutical University, Nanjing, People’s Republic of China; 2College of Pharmacy, Fujian University of Traditional Chinese Medicine, Fuzhou, People’s Republic of China; 3Formulation Development, Tolmar Inc, Fort Collins, CO, USA
Background: Lung cancer is the primary cause of cancer-related death worldwide. A redox-sensitive nanocarrier system was developed for tumor-targeted drug delivery and sufficient drug release of the chemotherapeutic agent paclitaxel (PTX) for improved lung cancer treatment.
Methods: The redox-sensitive nanocarrier system constructed from a hyaluronic acid-disulfide-vitamin E succinate (HA-SS-VES, HSV) conjugate was synthesized and PTX was loaded in the delivery system. The physicochemical properties of the HSV nanoparticles were characterized. The redox-sensitivity, tumor-targeting and intracellular drug release capability of the HSV nanoparticles were evaluated. Furthermore, in vitro and in vivo antitumor activity of the PTX-loaded HSV nanoparticles was investigated in a CD44 over-expressed A549 tumor model.
Results: This HSV conjugate was successfully synthesized and self-assembled to form nanoparticles in aqueous condition with a low critical micelle concentration of 36.3 µg mL-1. Free PTX was successfully entrapped into the HSV nanoparticles with a high drug loading of 33.5% (w/w) and an entrapment efficiency of 90.6%. Moreover, the redox-sensitivity of the HSV nanoparticles was confirmed by particle size change of the nanoparticles along with in vitro release profiles in different reducing environment. In addition, the HA-receptor mediated endocytosis and the potency of redox-sensitivity for intracellular drug delivery were further verified by flow cytometry and confocal laser scanning microscopic analysis. The antitumor activity results showed that compared to redox-insensitive nanoparticles and Taxol®, PTX-loaded redox-sensitive nanoparticles exhibited much greater in vitro cytotoxicity and apoptosis-inducing ability against CD44 over-expressed A549 tumor cells. In vivo, the PTX-loaded HSV nanoparticles possessed much higher antitumor efficacy in an A549 mouse xenograft model and demonstrated improved safety profile. In summary, our PTX-loaded redox-sensitive HSV nanoparticles demonstrated enhanced antitumor efficacy and improved safety of PTX.
Conclusion: The results of our study indicated the redox-sensitive HSV nanoparticle was a promising nanocarrier for lung cancer therapy.
Keywords: tumor targeting, nanoparticles, redox-sensitive
Introduction
Lung cancer is one of the most common cancers and the leading cause of cancer death in the world.1,2 Due to poor diagnosis in the early stage, when symptoms are present, most lung cancer patients become aware of their malignancy only in the late stages, when surgical treatment is not preferable.3,4 As a result, in order to improve the quality of life and extend survival, systemic chemotherapy is usually the final and main treatment for advanced lung cancer. However, the major problem of current chemotherapies is their low efficacy due to nonselective cytotoxicity to normal tissues and their poor water solubility, which limits their administration at high doses.5 Numerous efforts have been applied to overcome these obstacles and, among them, nanoparticle approach is considered as one of the most promising strategies for lung cancer treatment. In general, nanoparticle vehicles possess the ability to prolong drug circulation time, load more hydrophobic drugs, improve drug accumulation to tumor tissues through enhanced permeability and retention effect, reduce side effects, etc.6 Therefore, different nanoparticle formulations such as liposomes, micelles, and magnetic nanoparticles were designed and these have been reported to have improved therapeutic outcomes of various anticancer drugs.7,8
Recently, a lot of attention has been paid to vitamin E succinate (VES)-based nanomedicines. VES, one of tocopherol derivatives, inhibits tumor cellular proliferation and has the potential to be utilized as an antineoplastic agent in clinic.9,10 Notably, it is nontoxic to normal cells unlike other anticancer agents. In addition, the derivation of the VES structure enhanced its effectiveness,11 and it is reported that nanocarriers that attached to VES chains demonstrated synergistic therapeutic effects.12,13 Furthermore, it is biocompatible and easy to manufacture. Therefore, due to its hydrophobicity and synergistic therapeutic effects, VES is considered to be a promising agent in drug delivery systems14 and thereby is frequently modified as nanocarriers to deliver anticancer drugs. For instance, chitosan-vitamin E succinate copolymer (CS-VES),15 polyethylene glycol-derivatized vitamin E copolymer,16 and vitamin E succinate-conjugated F68 micelles17 have all been synthesized and exhibited a high drug loading (DL) capacity. Unfortunately, the hydrophobic interactions between the encapsulated drug and hydrophobic VES of these nanoparticles led to a problematic slow release. For example, the cumulative release of paclitaxel (PTX) from PTX-loaded CS-VES micelles was only about 44.35% within 168 hours,15 and PTX released <60% from VES-modified pluronic micelles within 72 hours.18 The unfavorably delayed release of drugs may result in slow degradation of nanoparticles in vivo and hinder their effectiveness. Therefore, VES-based nanoparticles possessing bio-response to microenvironment are in great demand.
It is well known that the concentration of glutathione (GSH, a tripeptide containing cysteine) in tumor intracellular microenvironments (~20 mM) is significantly higher than that of blood circulation (2–20 μM).19 This reducing gradient provides the basis of the tumor-specific release potential of reduction-sensitive nanoparticles, where the incorporated redox-responsive conjugate of disulfide bonds is either in the backbone matrix or in auxiliary chains.20,21 Based on the above facts, VES-based nanocarriers with redox-sensitive bridge in their structure respond to redox potential and cleave to trigger release of molecular payload in target tumor cells.
Meanwhile, there was a general consensus that simple passive targeting is insufficient but active targeting is more desirable due to its more preferred accumulation on tumor cells as well as less side effects on normal tissues. A lot of efforts on active targeting have been reported and one of them is hyaluronic acid (HA), a natural anionic polysaccharide. It has a high binding affinity to CD44 receptors, which are overexpressed on the cell surface of numerous tumor cells.22,23 Furthermore, HA possesses many advantages such as being nontoxic, noninflammatory, biocompatible, and biodegradable when it is used as a moiety for anticancer drug delivery.24,25
In summary, an ideal delivery system would be the combination of HA active targeting and VES-based redox-sensitive nanocarriers in that a multifunctional carrier is capable of binding HA receptor-mediated active targeting, followed by stimuli-triggered release and VES-based nanotechnology as shown in Figure 1A, which will further enhance their effectiveness. Here, we report a novel redox-sensitive nanocarrier consisting of HA as a hydrophilic segment, a redox-sensitive connecting bridge containing one disulfide group, and VES as a hydrophobic segment (HA-SS-VES, HSV). The designated HSV nanocarrier was developed as a platform for the intracellular-targeted delivery of PTX, an important anticancer drug for lung cancer treatment. As a control, insensitive analog HA-VES (HV) conjugates were synthesized, which were structurally similar to HSV but the redox-sensitive disulfide bonds were omitted. Herein, reduction responsiveness of the nanocarrier was investigated by evaluation of the disassembly and drug release behavior under different concentrations of GSH in vitro. Next, cellular uptake via HA receptor-mediated endocytosis and triggered intracellular release through reducible linker dissociation were investigated in non-small-cell lung cancer A549 cells. Furthermore, in vitro cytotoxicity and in vitro apoptosis were investigated in A549 cells. Finally, an in vivo antitumor efficacy study of PTX-loaded nanoparticles on A549 tumor xenograft mouse model was conducted.
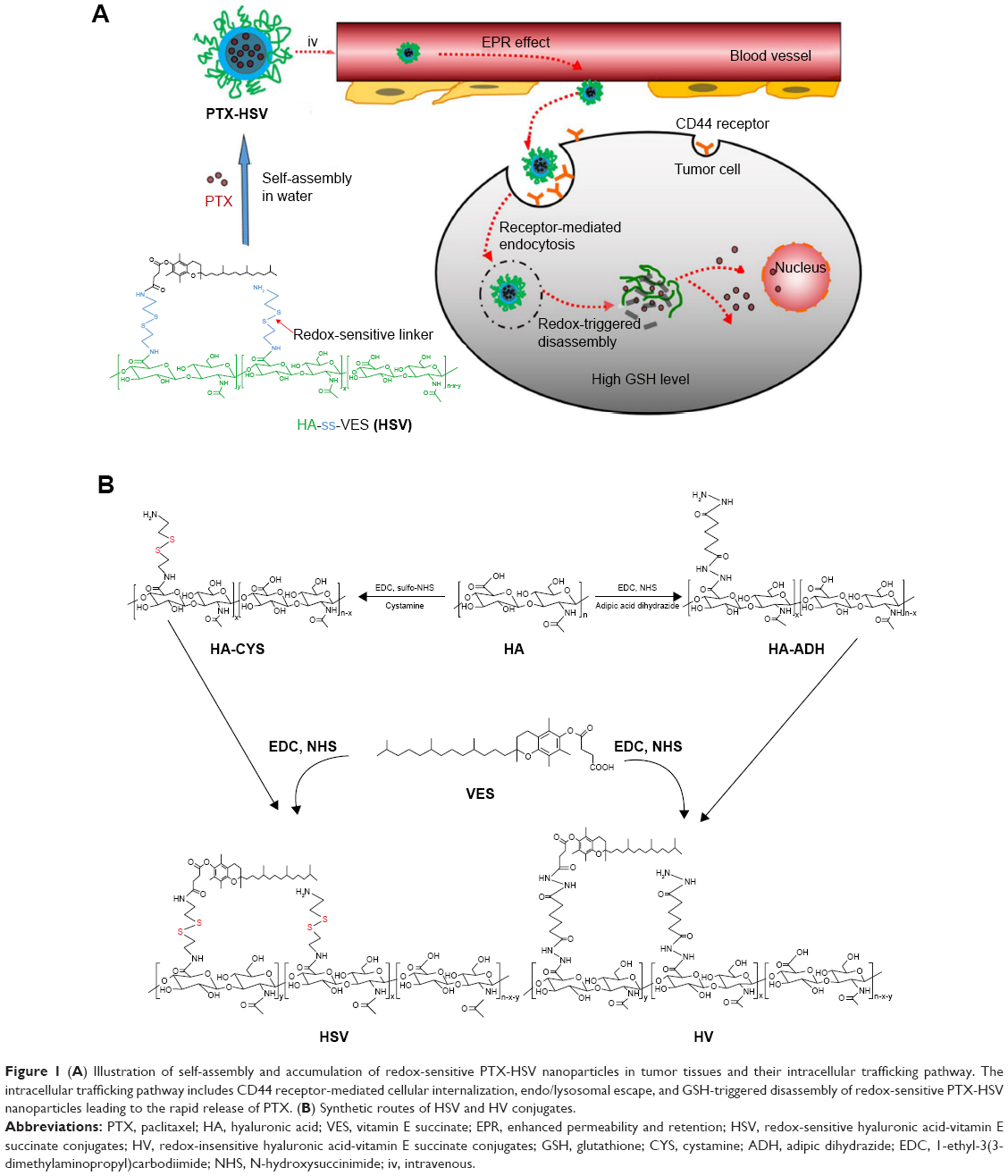 | Figure 1 (A) Illustration of self-assembly and accumulation of redox-sensitive PTX-HSV nanoparticles in tumor tissues and their intracellular trafficking pathway. The intracellular trafficking pathway includes CD44 receptor-mediated cellular internalization, endo/lysosomal escape, and GSH-triggered disassembly of redox-sensitive PTX-HSV nanoparticles leading to the rapid release of PTX. (B) Synthetic routes of HSV and HV conjugates. |
Materials and methods
Materials
Sodium HA (molecular weight 9.9 kDa) was purchased from Zhenjiang Dongyuan Biochem Co, Ltd (Jiangsu, People’s Republic of China). Vitamin E succinate (purity ≥98%) was bought from Zhejiang Medicine Co, Ltd (Zhejiang, People’s Republic of China). PTX was obtained from Shanghai Sanwei Pharmaceuticals Co, Ltd (Shanghai, People’s Republic of China). 1-Ethyl-3(3-dimethylaminopropyl)carbodiimide (EDC), N-hydroxysuccinimide (NHS), adipic acid dihydrazide, and GSH were procured from Aladdin Reagent Database Inc (Shanghai, People’s Republic of China). Cystamine dihydrochloride was purchased from TCI Development Co, Ltd (Shanghai, People’s Republic of China). MTT, Hoechst 33342, Pyrene, and Nile red (NR) were obtained from Sigma-Aldrich (St Louis, MO, USA). RPMI-1640 medium, penicillin–streptomycin, fetal bovine serum (FBS), and trypsin were provided by Thermo Fisher Scientific (Waltham, MA, USA). All other chemicals were of analytical grade.
Synthesis of HSV and HV conjugates
Synthesis of cystamine-modified HA (HA-CYS) and adipic dihydrazide-modified HA (HA-ADH)
Following the synthetic steps shown in Figure 1B, HSV and HV conjugates were synthesized through a two-step process. HA-CYS and HA-ADH were prepared as previously described with modification.26 First, EDC (1.6 mmol) and sulfo-NHS (1.6 mmol) were added to 4 mg mL−1 of HA (0.8 mmol) in phosphate-buffered saline solution (PBS; 0.01 M, pH 7.4) and stirred for 30 minutes to activate the carboxyl groups of HA. After that, 1.8 g of cystamine dihydrochloride (8 mmol) was added and the mixture was reacted at room temperature under stirring for 24 hours. The resulting HA-CYS solution was then extensively dialyzed (molecular weight cutoff [MWCO] 3.5 kDa) in order to remove any reactive impurity. Finally, the HA-CYS powder was obtained by lyophilization, and then stored at 4°C for further use.
To prepare HA-ADH, 0.32 g of HA (0.8 mmol) was dissolved in 80 mL of deionized water to achieve a concentration of 4 mg mL−1; 1.4 g of adipic dihydrazide (8 mmol) and 0.3 g of EDC (1.6 mmol) were then added to the solution. The reaction mixture was maintained at pH 4.75 by the addition of 0.1 M HCl for 2 hours. In order to quench the reaction, the pH of the reaction mixture was adjusted to 7.0 by the addition of 0.1 M NaOH. The resulting solution was then extensively dialyzed (MWCO 3.5 kDa) for 48 hours. Finally, the polymer solutions were lyophilized and stored at 4°C for further use.
Synthesis of HSV and HV conjugates
Amphiphilic HSV conjugates and HV conjugates were prepared by conjugating vitamin E succinate to HA-CYS and HA-ADH through amide formation, respectively. Briefly, the carboxyl groups on vitamin E succinate (0.9 mmol) were activated by EDC (1.8 mmol) and NHS (1.8 mmol) in 10 mL of dimethylformamide at 0°C for 1 hour. Next, HA-CYS (0.45 mmol) or HA-ADH (0.45 mmol) dissolved in 20 mL of formamide was added dropwise to the above solution. When the reaction proceeded at room temperature for 24 hours under gentle stirring, the mixture was precipitated into 250 mL of frozen acetone. After that, the flocculent precipitate was collected by filtration, followed by being dissolved in 20 mL of distilled water. The product solution was then filtered through a 0.45 μm membrane and dialyzed (MWCO 3.5 kDa) against the excess amount of water/ethanol (1:1, v/v) for 24 hours and then deionized water for 48 hours. Finally, after being freeze-dried, the resulting product was obtained and stored at 4°C until further use.
Characterization of HSV conjugates and HV conjugates
The chemical structures of HSV and HV conjugates were confirmed by Fourier transform infrared spectroscopy (FT-IR; Nicolet iS5; Thermo Fisher) and 1H nuclear magnetic resonance (NMR) spectroscopy (Avance 400; Bruker Optik GmbH, Ettlingen, Germany) using D2O as the solvent. Degree of VES substitution (DS) was determined by elemental analysis using a Vario EL III analyzer (Elementar, Langenselbold, Germany).
Preparation and characterization of nanoparticles
Preparation of blank nanoparticles
Reduction-sensitive HSV nanoparticles were prepared by dissolution of HSV-lyophilized polymer (18 mg) in 3 mL of deionized water under stirring for 15 minutes. Next, the solution was sonicated by a probe-type ultrasonicator (JY92-2D; Ningbo Scientz Biotechnology Co, Ltd, Nanjing, People’s Republic of China) for 10 minutes at 100 W in an ice bath to avoid heat buildup, followed by filtration through a 0.45 μm membrane. Non-redox-sensitive HV nanoparticles were prepared in the same manner as HV conjugates.
Determination of critical micelle concentration (CMC)
The CMC of amphiphilic HSV conjugates and HV conjugates was measured by fluorescence spectroscopy method using pyrene as a probe.25 Briefly, 1 mL of pyrene solution (6.0×10−6 M) in acetone was transferred into a series of 10 mL volumetric flasks and then acetone was evaporated under nitrogen flow. Ten milliters of various polymer aqueous solutions at a concentration ranging from 1×10−4 to 1.0 mg mL−1 were added to the volumetric flasks followed by sonication for 30 minutes. The samples were incubated at 50°C for 1 hour and then kept at room temperature overnight. Subsequently, pyrene excitation spectra from 333 to 338 nm were obtained by using a fluorescence spectrophotometer (RF-5301 PC; Shimadzu Corporation, Kyoto, Japan) with an emission of 390 nm. In order to calculate the CMC value, the intensity ratio of I338/I333 against the logarithm of nanoparticle concentration was plotted. The CMC was determined by the point of intersection when the plots were extrapolated.
Reduction-triggered disassembly of nanoparticles
To demonstrate the redox responsiveness, the size change of nanoparticles in response to different concentrations of GSH in PBS (10 mM, pH 7.4) was measured by dynamic light scattering (DLS) using a Nicomp™ 380ZLS Particle Sizer (Particle Sizing Systems Company, Santa Barbara, CA, USA). Briefly, GSH was added to 3 mL of PBS solution with HSV nanoparticles to desired concentrations of 0 μM, 10 μM, 10 mM, and 20 mM. The solution was placed in a shaker with a rotation speed of 100 rpm at 37°C. After 24 hours, the particle size as well as size distribution of the nanoparticles were measured by DLS. The HV nanoparticles were also measured as the control.
Preparation of PTX-loaded nanoparticles
PTX-loaded HSV nanoparticles and PTX-loaded HV nanoparticles were prepared by a simple dialysis technique. Briefly, 10 mg of PTX at a concentration of 25 mg mL−1 was dissolved in ethanol; this PTX solution was then added dropwise to the 6 mg mL−1 aqueous solutions of blank nanoparticles while stirring. The resulting solution was sonicated for 30 minutes at 100 W by a probe-type ultrasonicator in an ice bath. Next, the solution was dialyzed (MWCO 3.5 kDa) against an excess amount of distilled water for 12 hours. Thereafter, the dialyzed solution was centrifuged to remove the unentrapped PTX at a speed of 3,000 rpm for 10 minutes. The supernatant was then filtered through a 0.45 μm filter and then freeze-dried to obtain the PTX-loaded nanoparticles. The entrapment efficiency (EE) and DL were calculated by the following formulae:
|
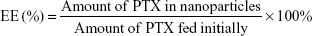
|
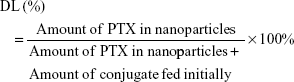
For DL determination, PTX-loaded nanoparticles were dissolved in methanol and measured by high performance liquid chromatography (HPLC; Shimadzu LC-2010 system) using a Diamonsil® Platisil C18 column (5 μm, 250×4.6 mm), and the mobile phase was a mixture consisting of methanol and water (75:25, v/v) at a flow rate of 1.0 mL min−1. The detection wavelength was 227 nm and the injection volume was 20 μL for HPLC analysis.
Physicochemical characterization of nanoparticles
Particle size, polydispersity index (PDI), and zeta potential of blank and PTX-loaded nanoparticles were determined by DLS using a Nicomp™ 380ZLS Zeta Potential/Particle Sizer.
The morphology and particle size distribution of PTX-loaded nanoparticles were observed by transmission electron microscopy (TEM; H-7650; Hitachi Ltd, Tokyo, Japan) and atomic force microscopy (AFM; 5500AFM; Agilent Technologies, Santa Clara, CA, USA). Negative staining of samples was performed by placing one drop of sample solution (0.01 mg mL−1) onto a copper grid coated with carbon and the sample droplet was then tapped with a filter paper to remove surface water, followed by air drying for 5 minutes. Next, the samples were negatively stained with 2% phosphor tungstic acid for 30 seconds and air-dried at room temperature before observation. The interaction of HSV conjugate with PTX was evaluated by differential scanning calorimetry (DSC) and wide angle X-ray diffraction (WAXD) analysis. DSC analysis was carried out using NETZSCH DSC 204 equipment (NETZSCH-Gerätebau GmbH, Selb, Germany), and scanning was performed from 25°C to 400°C at a heating rate of 10°C min−1. WAXD spectrometry was performed using X’Pert3 powder diffraction (PANalytical, Almelo, the Netherlands) with Cu Kα radiation. Samples were scanned from 5° to 40° (2θ) at a scanning speed of 1° min−1 with a step size of 0.05° (2θ). The X-ray tube was operated at a potential of 40 kV and a current of 40 mA.
In vitro reduction-triggered release of PTX from PTX-loaded nanoparticles
The in vitro PTX release behavior of PTX-loaded HSV and HV nanoparticles triggered by different reducing conditions was studied by using a simple dialysis method. Lyophilized PTX-loaded nanoparticles (0.5 mg of PTX) were suspended in 5% glucose solutions and placed in a dialysis bag (MWCO 14 kDa). The entire bag was then immersed into 150 mL of 0.1 M PBS (pH 5.8, pH 7.4) containing 0.1% (w/v) Tween 80 and various amounts of GSH (0 μM, 10 μM, 10 mM, and 20 mM). The experiment was performed in an incubation shaker at 37°C with a speed of 100 rpm. At different time intervals, 2 mL of release medium was withdrawn for HPLC analysis, and an equivalent volume of fresh release medium was added. The release experiments were conducted in triplicate and the average data were calculated.
PTX-loaded HSV nanoparticle stability in plasma
To access the stability of nanoparticles in plasma, the PTX-HSV nanoparticles were mixed with rat plasma and the mixture was incubated in a water bath at 37°C. At different time points, the plasma sample was taken and centrifuged at a speed of 3,000 rpm for 5 minutes. One hundred microliters of the supernatant was then dissolved in 10 mL of methanol and centrifuged at a high speed of 10,000 rpm for 10 minutes. PTX contents in nanoparticles at different time points (Mt) were measured and calculated by HPLC. The nanoparticle stability analysis in plasma was evaluated by the ratio of Mt and M0, where M0 and Mt represent PTX contents in the nanoparticles at initial time (T=0) and sampling time t, respectively.
Cell culture
The human non-small lung cancer cell line A549 was purchased from the Cell Bank of the Chinese Academy of Sciences (Shanghai, People’s Republic of China) and cultured at 37°C under a 5% CO2 atmosphere in RPMI-1640 medium supplemented with 10% FBS and 1% antibiotics (100 U/mL penicillin and 100 μg/mL streptomycin).
Flow cytometry and confocal laser scanning microscopy (CLSM) analysis
To evaluate the ability of selective delivery to tumor cells and intracellular release of nanoparticles in tumor cells, flow cytometry and CLSM were used. NR-loaded nanoparticles were prepared by encapsulating fluorescent probe NR (0.2% loading by weight) into HSV nanoparticles and HV nanoparticles using the same preparation method described in “Preparation of PTX-loaded nanoparticles” section. In order to investigate whether the nanoparticles were specifically taken up through HA receptor-mediated endocytosis, human non-small lung cancer A549 cells (CD44 overexpression) were chosen and the cells were incubated with 100-fold excess of free-HA polymer for 2 hours prior to the addition of NR-loaded nanoparticles. In flow cytometry analysis, all the samples were washed with PBS three times, then harvested by trypsinization, and collected in PBS to measure the fluorescence intensity at 560/620 nm using a flow cytometer (BD FACS Calibur; BD Biosciences, San Jose, CA, USA). In CLSM analysis, after incubation with NR-loaded nanoparticles, the culture media were subsequently removed and the cells were rinsed with PBS three times, followed by the addition of Hoechst 33342 (10 μg mL−1) to stain the cell nuclei. The samples were then observed using a confocal microscope (Olympus FV1000; Olympus Corporation, Tokyo, Japan) at the excitation and emission wavelengths of 352 and 455 nm for Hoechst 33342 and 488 and 530 nm for NR, respectively.
MTT assay
In vitro cytotoxicity of PTX-loaded nanoparticles was evaluated by MTT assay using A549 cells. All samples including Taxol® (Bristol-Myers Squibb, New York, NY, USA), PTX-loaded HV nanoparticles, and PTX-loaded HSV nanoparticles were further diluted with culture medium to a wide range of PTX concentrations ranging from 0.001 to 100 μg mL−1. A549 cells were seeded into 96-well plates at a density of 5×103 cells/well in 100 μL of RPMI-1640 medium and incubated at 37°C in 5% CO2 atmosphere for 24 hours. After that, the samples with different PTX concentrations were added to the wells and cultured for another 48 hours. Next, 20 μL MTT solution (5 mg mL−1 in PBS) was added into each well and the cells were further incubated for 4 hours at 37°C. The supernatant was then carefully removed and 150 mL of dimethyl sulfoxide was added to each well to dissolve the formazan crystals. The OD at 570 nm was measured with a microplate reader (Tecan Infinite®M200 PRO; AmGen Technology Ltd, Thousand Oaks, CA, USA). The cell viability was calculated by the formula: cell viability (%) = (ODtreated/ODcontrol) ×100%. The IC50 values were calculated by GraphPad Prism 6.0 software (GraphPad Software, Inc, La Jolla, CA, USA).
In vitro cell apoptosis
The cell apoptosis of PTX-loaded HSV nanoparticles was further analyzed by flow cytometry using PTX-HV nanoparticles and Taxol® as controls. Briefly, A549 cells were cultured in 24-well plates at a concentration of 5×104 cells/well and incubated at 37°C in 5% CO2 atmosphere for 24 hours. Afterwards, the cells were treated with different samples at a PTX concentration of 1 μg mL−1 for 48 hours. At the end of the treatment period, the cells were harvested, washed, and resuspended in 400 μL of PBS. Five microliters of Annexin V-fluorescein isothiocyanate (FITC) and 5 μL of propidium iodide were then added and incubated for 15 minutes at 37°C in the dark. Then, the cell suspension was immediately analyzed by flow cytometry.
In vivo antitumor efficacy of nanoparticles
All animal experiments were approved by the Ethical Committee of China Pharmaceutical University and were conducted in compliance with the institutional guidelines for animal care and use of China Pharmaceutical University.
In vivo antitumor efficacy of the PTX-loaded nanoparticles was evaluated in a BALB/c nude mice model. Approximately 2×106 A549 cells were inoculated subcutaneously in the armpit region of athymic nude mice. The mice were randomly divided into four groups (n=5) when tumor size reached ~90–100 mm3, and were intravenously administered with saline (control), Taxol®, PTX-loaded HSV nanoparticles, and PTX-loaded HV nanoparticles every 3 days for a total of 18 days at a dose of 10 mg PTX/kg. After treatment, the body weight of each mouse was recorded, and tumor volumes were calculated as (a2×b)/2, where a represents the smallest diameter and b the largest of the tumor. At the end of the experiment (day 18), all mice were sacrificed by cervical dislocation, and the tumors were extracted and weighed to calculate the inhibition ratio using the following formula:
|
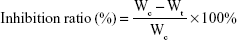
where Wc and Wt denote the average tumor weight for the control group and treatment group, respectively. Subsequently, the tumor and major organs (heart, lung, spleen, kidney, and liver) were separated and embedded in paraffin blocks for histological evaluation by hematoxylin and eosin (H&E) staining. In addition, blood samples of each mouse were collected to evaluate the hepatic and renal damage by measuring various biological markers, that is, alanine transaminase (ALT), aspartate transaminase (AST), blood urea nitrogen (BUN), and serum creatinine (Scr).
Statistical analysis
All quantitative data were expressed as mean ± SD. Statistical analysis was performed by Student’s t-test for two groups, and one-way analysis of variance for multiple groups. A value of p<0.05 was considered statistically significant.
Results and discussion
Synthesis and characterization of HSV and HV
In this paper, a novel redox-sensitive amphiphilic conjugate of HSV, based on HA and VES using cystamine as a linker, has been successfully synthesized. The detailed synthesis steps are described in Figure 1B. As a control, another non-redox-sensitive HV conjugate with a similar structure was synthesized using adipic dihydrazide as the connecting bridge. The chemical structures of both conjugates were characterized by 1H NMR and FT-IR. In the 1H NMR spectra shown in Figure 2, the characteristic peaks of HA were identified at 2.01 ppm (methyl proton of N-acetyl group) and 3.30–4.79 ppm (methylene and hydroxyl groups), whereas the peaks assigned to adipic dihydrazide and cystamine were identified in the range of 1.63–1.79 ppm, 2.19–2.60 ppm (methylene groups in adipic dihydrazide, Figure 2D and E), and 2.82–3.02 ppm (methylene groups of cystamine neighboring disulfide bonds, Figure 2B and C). The signals in the range of 0.60–1.31 ppm attributed to the methyl and methylene protons of the long-chain alkyl group of VES suggested the successful introduction of VES into HA polymers (Figure 2C and F). The amount of cystamine in the conjugates was quantitatively characterized by 1H NMR (22.1% on a molar basis), and the amount of adipic dihydrazide was also determined by 1H NMR (31.5% on a molar basis). The calculated DS of VES in HV and HSV conjugate was 2.78 and 3.04 mol%, respectively.
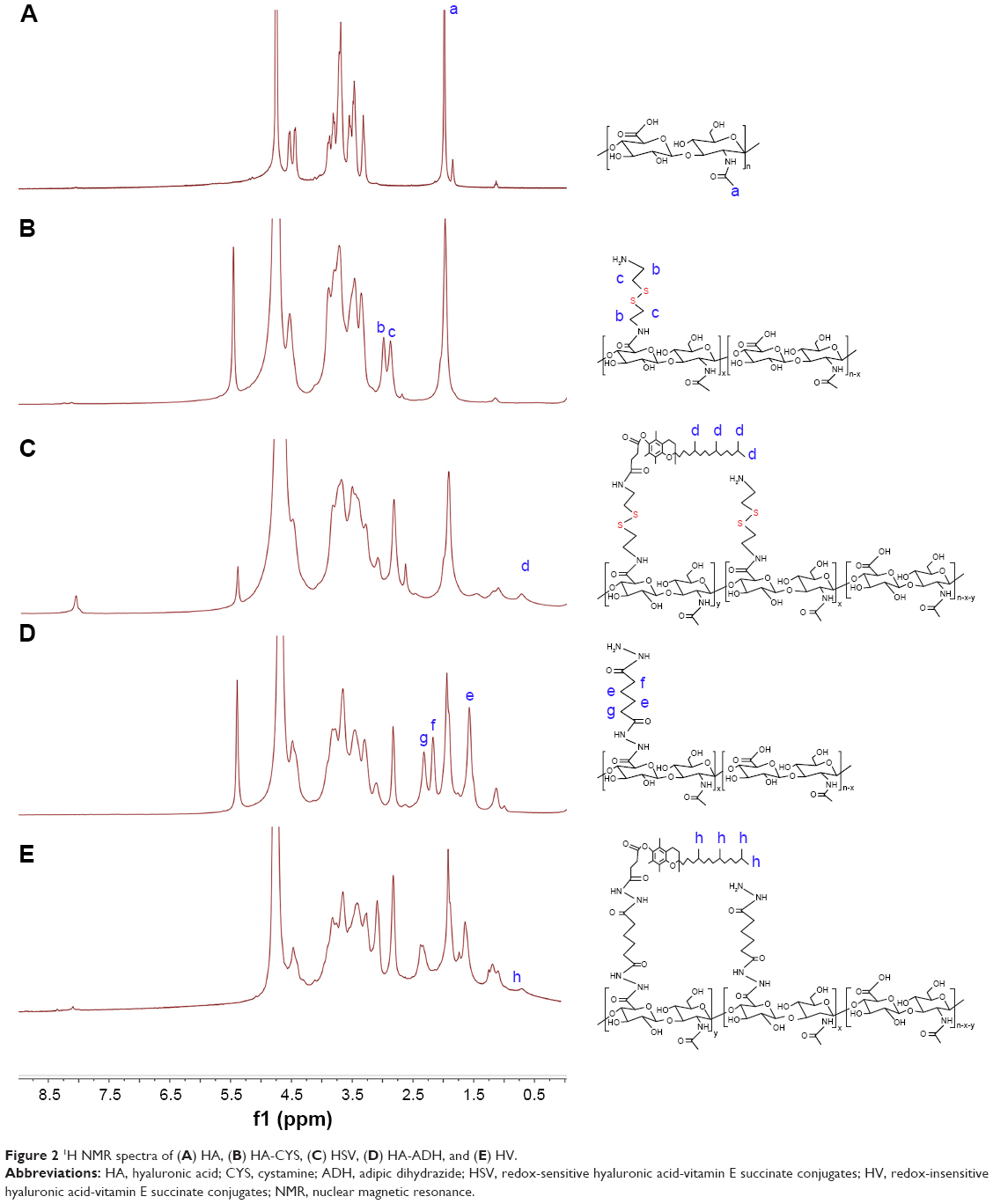 | Figure 2 1H NMR spectra of (A) HA, (B) HA-CYS, (C) HSV, (D) HA-ADH, and (E) HV. |
The structure of the conjugates was further confirmed by FT-IR as shown in Figure 3. Compared to the HA spectrum (Figure 3A), new signals of the conjugates at 1,642, 1,646 (amide I band), and 1,556 cm−1 (amide II band) suggested the formation of a new amide link in HA-CYS (Figure 3B) and HA-ADH (Figure 3D). Similarly, new absorption bands were observed at 1,658 and 1,659 cm−1 (amide I band) as well as at 1,552 and 1,550 cm−1 (amide II band) in HV (Figure 3E) and HSV (Figure 3C) spectrum, respectively. Meanwhile, new bands at 1,734 and 1,714 cm−1 of HSV and HV were associated to the ester bond vibration of VES, which were also present in VES (Figure 3F). All these implied that VES was successfully grafted to the HA chain.
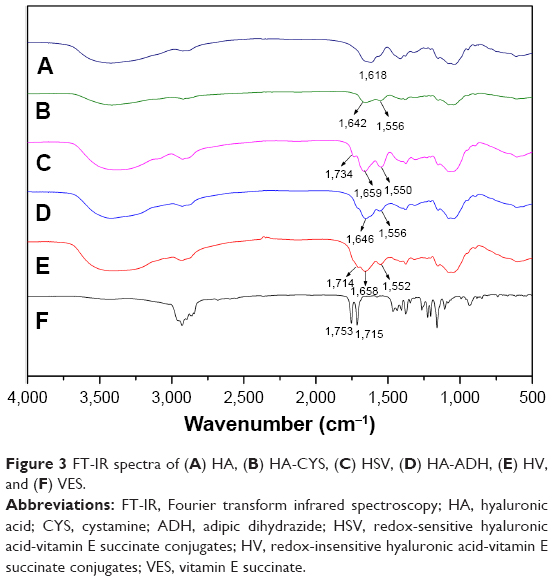 | Figure 3 FT-IR spectra of (A) HA, (B) HA-CYS, (C) HSV, (D) HA-ADH, (E) HV, and (F) VES. |
Physicochemical characterization of HSV, HV, PTX-HSV, and PTX-HV
Characterization of blank nanoparticles
Amphiphilic copolymers of HSV and HV spontaneously formed core–shell structural nanoparticles and the particle size and zeta potential were measured by DLS. As listed in Table 1, the effective diameters of HSV and HV were 200.1 and 207.2 nm with PDI of 0.13 and 0.10, respectively. The low PDI (<0.2) indicated a narrow size distribution of the nanoparticles. The negative charge as measured by zeta potential (<−20 mV) confirmed the shielding effect of HA at the surface of the nanoparticles, which provided sufficient repelling force among particles for improved physical stability. The CMC in aqueous milieu was determined by UV/Vis spectra using pyrene as a fluorescence probe. The CMC values of HSV and HV conjugate nanoparticles were determined to be 36.3 and 43.8 mg L−1, respectively, which indicated that nanoparticles would keep their original morphology in vivo under strong diluted conditions prior to arriving at the target site.27
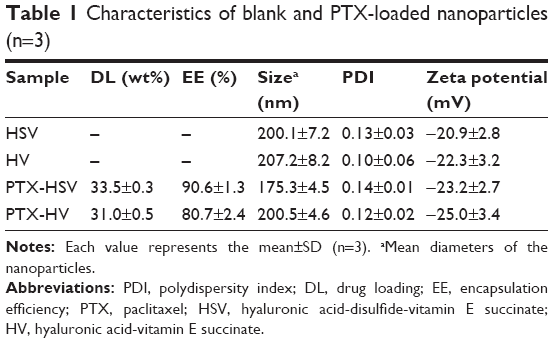 | Table 1 Characteristics of blank and PTX-loaded nanoparticles (n=3) |
It should be noted that the disulfide bridge linkage renders HSV nanoparticles reductively breakable in response to a reductive environment. To demonstrate their responsiveness to different reductive environments, the reduction-sensitivity of nanoparticles was studied by treating nanoparticles under different concentrations of GSH to observe their size distribution change. As shown in Figure 4G, the mean particle size of HSV nanoparticles increased from 200.0 to 410.8 nm in 10 mM GSH and 571.0 nm in 20 mM GSH (representing a reducing environment in tumor cells) at 24 hours following the addition of GSH, indicating that a higher GSH concentration resulted in increased mean particle size. This suggested that the aggregates were formed mostly due to the reductive cleavage of the intermediate disulfide bond. In sharp contrast, very little change in particle size was found when the HSV nanoparticles were treated with 10 μm GSH (simulating normal cells and blood milieus). The same phenomenon was also observed for the nanoparticles without GSH treatment after 24 hours. In the HV control experiment, the particle size showed no significant change in response to different GSH concentrations of 0 and 20 mM GSH, which further confirmed that the disulfide bridge linkage between HA and VES made HSV nanoparticles reductively breakable in specific redox environment and resulted in rapid release of their payloads.
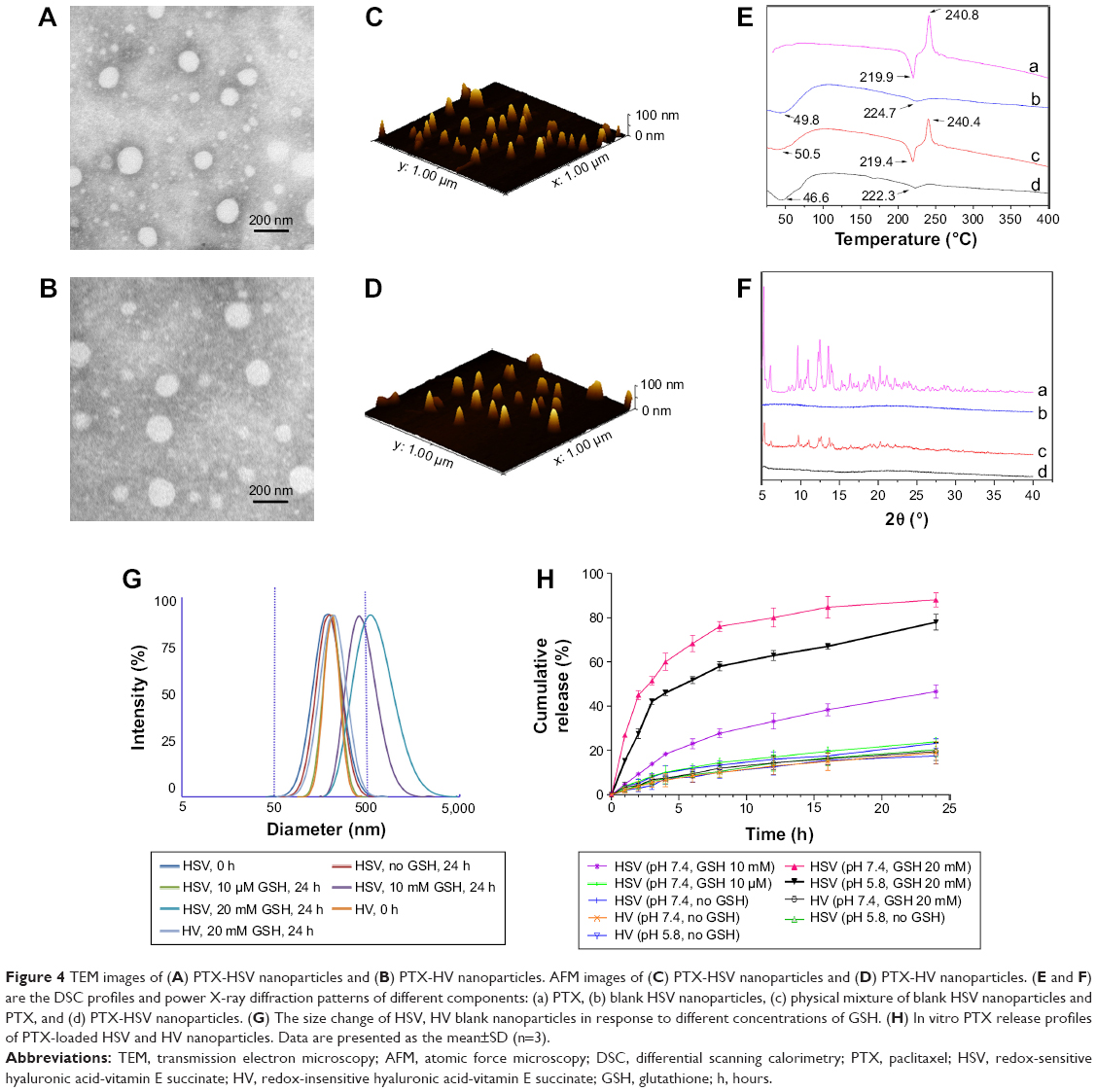 | Figure 4 TEM images of (A) PTX-HSV nanoparticles and (B) PTX-HV nanoparticles. AFM images of (C) PTX-HSV nanoparticles and (D) PTX-HV nanoparticles. (E and F) are the DSC profiles and power X-ray diffraction patterns of different components: (a) PTX, (b) blank HSV nanoparticles, (c) physical mixture of blank HSV nanoparticles and PTX, and (d) PTX-HSV nanoparticles. (G) The size change of HSV, HV blank nanoparticles in response to different concentrations of GSH. (H) In vitro PTX release profiles of PTX-loaded HSV and HV nanoparticles. Data are presented as the mean±SD (n=3). |
Preparation and characterization of PTX-loaded nanoparticles
In order to further effectively solubilize free hydrophobic PTX molecules, PTX-loaded nanoparticles were prepared by probe-type ultrasonic and dialysis methods. The extremely high PTX-loading of HSV and HV nanoparticles was expected and the drug contents of the nanoparticles are listed in Table 1. The hydrophobic PTX content in PTX-HSV and PTX-HV reached up to 33.5% and 31.0%, respectively, with high entrapment efficiencies of 90.6% for PTX-HSV and 80.7% for PTX-HV. Compared to blank nanoparticles (200.1 nm for HSV and 207.2 nm for HV), PTX-HSV and PTX-HV displayed a smaller particle size (175.3 nm for PTX-HSV and 200.5 nm for PTX-HV, respectively), which may be due to the increased cohesive force to form smaller cores resulting from the interactions between PTX and VES segments.28 In addition, TEM and AFM were used to confirm the size and morphology of PTX-HSV (Figure 4A and C) and PTX-HV nanoparticles (Figure 4B and D). The amphiphilic copolymer assembled into a nearly spherical morphology with a narrow size distribution and clear boundary, which confirmed their self-assembly behavior. Compared to the DLS results, the smaller size measured by TEM and AFM was probably explained by the shrinkage of nanoparticles upon drying.28
To confirm the existent form of PTX in the polymeric nanoparticles, DSC and WAXD studies were carried out for PTX, blank HSV nanoparticles, their physical mixture, and PTX-loaded HSV nanoparticles. The DSC thermograms of the above four samples are shown in Figure 4E. Figure 4E(a) demonstrated that pure PTX existed in a crystalline state with an endothermic melting peak at 219.9°C followed by an exothermic decomposition peak at 240.8°C. Meanwhile, Figure 4E(b) represented an endothermal peak at 224.7°C of HSV conjugate. Figure 4E(c) showed that the intense peaks of PTX in the physical mixture of PTX and HSV conjugate were slightly shifted, where the melting and decomposition peaks were at 219.4°C and 240.4°C, respectively. However, lyophilized PTX-HSV nanoparticles (Figure 4E(d)) retained the HSV pattern and only showed a weak single peak at 222.3°C but without characteristic peaks of pure PTX. This implied that PTX reduced its crystallinity and mainly existed in an amorphous or a molecular dispersion state upon encapsulation into the nanoparticles. In addition, as shown in Figure 4F, both PTX and the physical mixture of PTX and drug-free HSV nanoparticles had some intense peaks from 5° to 13° and small peaks at 15°–30°. However, no characteristic peaks of pure PTX were observed in WAXD diagrams of PTX-loaded nanoparticles (Figure 4F(d)) and this was very similar to drug-free HSV nanoparticles (Figure 4F(b)), which further confirmed that the PTX crystalline state was inhibited during nanoparticle encapsulation.
In vitro release of PTX triggered by reducing environments
The drug release behaviors of PTX-loaded nanoparticles in pH 7.4 and pH 5.8 PBS at 37°C with different redox conditions were investigated. In Figure 4H, a slow release of PTX from redox-sensitive nanoparticles was observed when incubated in the pH 7.4 buffer solution with no GSH or at 10 μM GSH (mimicking the extracellular redox conditions), where only ~18.9% and 23.9% of PTX was released after 24 hours, respectively. Similarly, 20.3% and 17.5% of PTX was released in the pH 5.8 buffer solution without GSH after 24 hours for PTX-HSV and PTX-HV, respectively. This indicated that the redox carriers were stable during blood circulation and independent of pH. Notably, the release rate was dramatically increased when PTX-HSV nanoparticles were in the medium containing 20 mM GSH (mimicking reducing environment of tumor cells),19,29,30 which reached up to 68.3% of PTX released in 6 hours and 88.0% in 24 hours. In contrast, only ~46.6% of PTX was released in 10 mM GSH medium, which was due to a reducing reagent concentration-dependent release profile. Additionally, the release profile of PTX from non-redox-sensitive PTX-HV nanoparticles in the medium of 20 mM GSH was the slowest and only 19.5% of PTX was released after 24 hours, which was similar to that without GSH. Together, the in vitro release results clearly demonstrated that the faster release of PTX was attributed to the disassembly of disulfide bond by reducing agents, which was critical to promote breakage of redox-sensitive nanoparticles. Figure 4H also indicated that the PTX release rate in pH 5.8 was slightly slower than that of PTX in pH 7.4 at the same GSH concentration of 20 mM. This might be attributed to the redox-sensitive activity of GSH from its thiol group, which has a pKa of 8.8 and its thiolate form is more reactive in GSH than its sulfhydryl form.26 The above results indicated that the PTX-HSV nanoparticles would achieve rapid drug release when exposed to the intracellular tumor environment, which would enhance the anticancer efficacy and reduce systemic cytotoxicity because of the slow release behavior in blood circulation.
PTX-loaded HSV nanoparticle stability in plasma
It is well known that the in vivo nanocarrier stability will be affected by the existence of a variety of enzymes and proteins in plasma.27 Therefore, the stability of PTX-HSV nanoparticles in rat plasma was investigated. The results (Table 2) showed that >97% PTX was still encapsulated in nanoparticles even incubated in rat plasma at 37°C for 24 hours. This was consistent with its release profile in 10 μM GSH (analogous to blood circulation), which indicated that the redox-sensitive HSV nanoparticles were stable with structural integrity in blood before it reached the tumor target site.
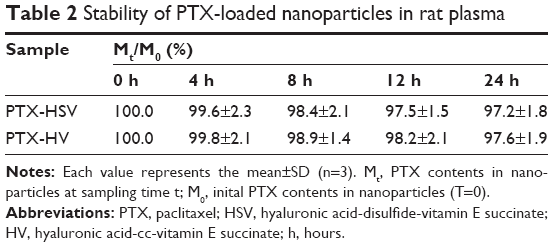 | Table 2 Stability of PTX-loaded nanoparticles in rat plasma |
In vitro cell experiments for redox-sensitive PTX-HSV nanoparticles
Flow cytometry and CLSM analysis
Flow cytometry studies were performed to investigate the cellular uptake behavior of nanoparticles using NR as a fluorescence marker. As seen in Figure 5A, the mean NR fluorescence intensity of the redox-sensitive HSV nanoparticles reached nearly 1.52- and 1.35-fold at 1 and 4 hours, respectively, when compared to redox-insensitive HV nanoparticles in A549 cells. HSV nanoparticles exhibited higher intracellular uptake compared to redox-insensitive HV nanoparticles, which may be due to the lower DS of cystamine in sensitive micelles compared to that of hydrazines in nonsensitive micelles. This was supported by the fact that carboxyl groups of HA were considered as the recognition sites for HA receptors.31,32 Therefore, any chemical modification of carboxyl groups in HA would reduce its targeting ability. To further confirm whether HSV nanoparticles were internalized via CD44 receptor-mediated endocytosis, a competitive study of HSV nanoparticles treated with free HA was performed. After saturation of CD44 receptors, the fluorescence intensity reduced to 40.7% in 1 hour and 32.8% in 4 hours compared to the untreated group, indicating a lower cellular uptake. This indicated that the cellular uptake of nanoparticles was initiated by CD44 receptor-mediated endocytosis. The chemical modification of HA with VES did not affect receptor-mediated intracellular signaling pathways of HA.
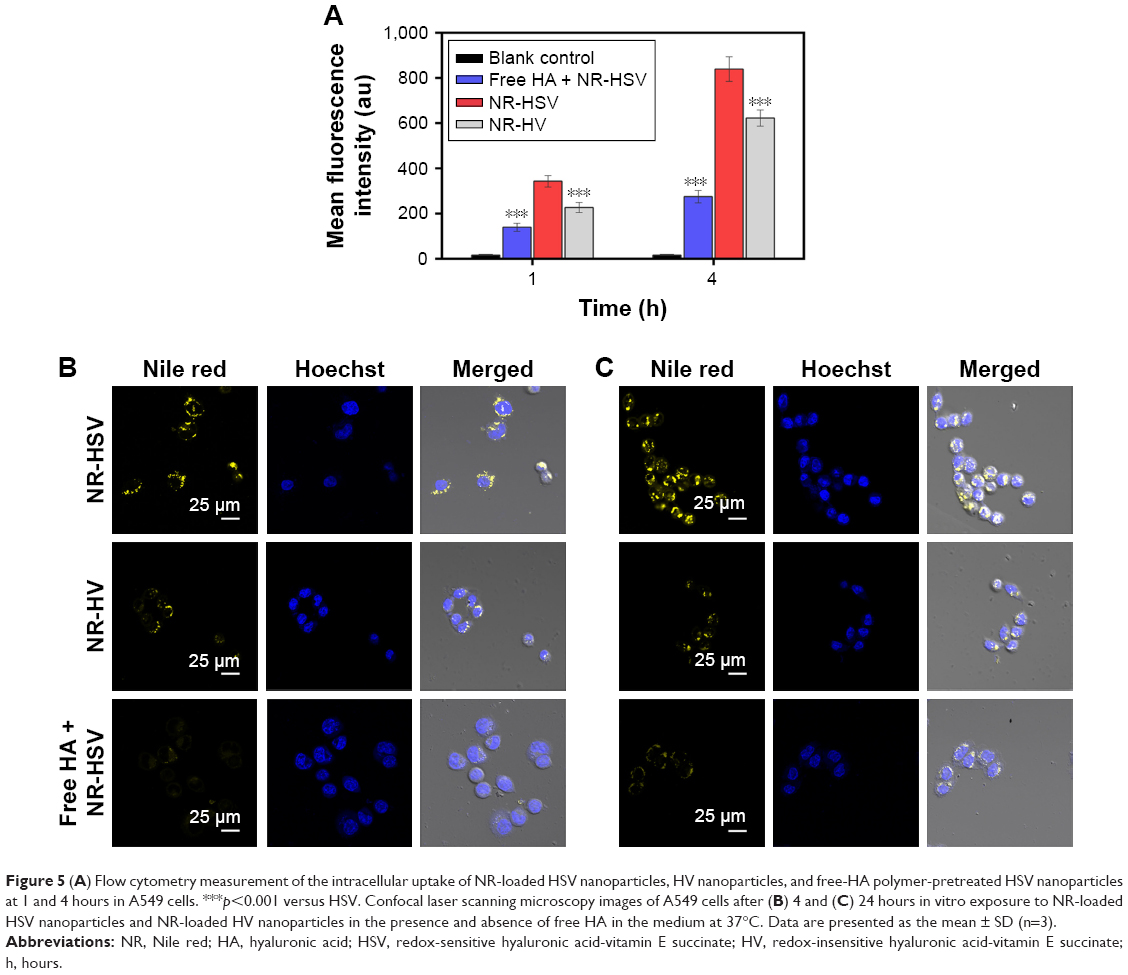 | Figure 5 (A) Flow cytometry measurement of the intracellular uptake of NR-loaded HSV nanoparticles, HV nanoparticles, and free-HA polymer-pretreated HSV nanoparticles at 1 and 4 hours in A549 cells. ***p<0.001 versus HSV. Confocal laser scanning microscopy images of A549 cells after (B) 4 and (C) 24 hours in vitro exposure to NR-loaded HSV nanoparticles and NR-loaded HV nanoparticles in the presence and absence of free HA in the medium at 37°C. Data are presented as the mean ± SD (n=3). |
In addition, CLSM experiments were applied to further investigate the intracellular release of NR-loaded redox-sensitive nanoparticles in A549 cells. As previously reported, NR was used to stain intracellular lipid droplets to be visualized as clearly distinct spherical fluorescent bodies with yellow-gold fluorescence rather than red fluorescence.28 Because intracellular lipid droplets are distributed in the cytoplasm and on the cell membrane, only free NR released from nanoparticles allows detection of lipid droplets. Therefore, more stained intracellular lipid droplets confirmed greater release of NR molecules into the cytosol from the nanoparticles. Figure 5B and C showed the number of stained lipid droplets with yellow-gold fluorescence after NR-loaded HSV and HV nanoparticles had been incubated in A549 cells after 4 and 24 hours, respectively. After 4 hours incubation with NR-loaded HSV nanoparticles, some yellow-gold fluorescence in cytosol was observed in cells. The fluorescence increased over time where the quantity of spherical fluorescence bodies significantly increased after 24 hours incubation. In contrast, redox-insensitive nanoparticles only produced a very weak NR signal even after 24 hours. The results of the CLSM experiment demonstrated that HSV nanoparticles were able to rapidly disintegrate to effectively release cargos into cytosol, where they exerted their cytotoxicity. Notably, the fluorescence intensity of HSV nanoparticles decreased rapidly when cells were preincubated with a high dose of free HA to block CD44 receptors, indicating the CD44 receptor-mediated endocytosis process of HSV nanoparticles.
In vitro cytotoxicity and apoptosis assay
To explore the cell inhibitory effect of drug-loaded nanoparticles, MTT cytotoxicity studies of PTX-loaded HSV and HV nanoparticles against A549 cells were performed. The PTX-loaded nanoparticles exhibited greater cytotoxicity than Taxol® following 48 hours incubation with the designated concentration ranges of PTX (Figure 6A). The half maximal inhibitory concentration (IC50) of Taxol® was extremely high than that of PTX-loaded HSV nanoparticles and HV nanoparticles (Figure 6B). The CD44 receptor-mediated endocytosis of HA-based nanoparticles accounted for the enhanced cytotoxicity to A549 cells. The IC50 of PTX-HSV and PTX-HV nanoparticles was 0.030 and 0.125 μg mL−1, respectively. The redox-sensitive nanoparticles exhibited stronger cytotoxic activity to A549 cells with a significantly lower IC50 than nonsensitive nanoparticles, which could be due to the rapid disassembly of nanoparticles in response to the cytoplasmic reducing environment.
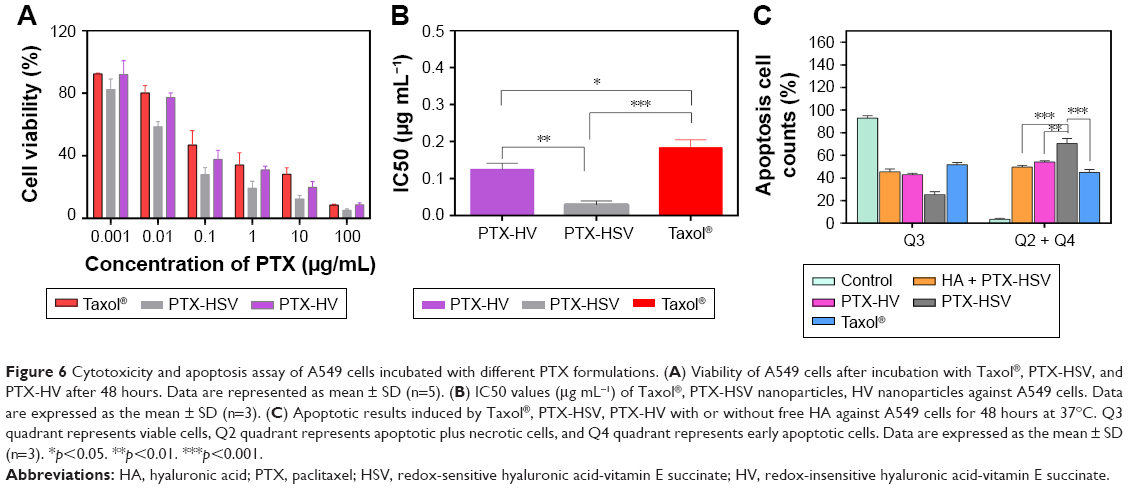 | Figure 6 Cytotoxicity and apoptosis assay of A549 cells incubated with different PTX formulations. (A) Viability of A549 cells after incubation with Taxol®, PTX-HSV, and PTX-HV after 48 hours. Data are represented as mean ± SD (n=5). (B) IC50 values (μg mL−1) of Taxol®, PTX-HSV nanoparticles, HV nanoparticles against A549 cells. Data are expressed as the mean ± SD (n=3). (C) Apoptotic results induced by Taxol®, PTX-HSV, PTX-HV with or without free HA against A549 cells for 48 hours at 37°C. Q3 quadrant represents viable cells, Q2 quadrant represents apoptotic plus necrotic cells, and Q4 quadrant represents early apoptotic cells. Data are expressed as the mean ± SD (n=3). *p<0.05. **p<0.01. ***p<0.001. |
On the other hand, the significant inhibitory effect of PTX-HSV on A549 cells was also investigated by an Annexin V-FITC/PI double labeling assay and cell apoptosis studies by using flow cytometry. The quantitative analysis in Figure 6C exhibited similar results obtained from the MTT assay; the total percentage of apoptosis in A549 cells was 70.5%, 54.0%, and 44.9% for PTX-HSV, PTX-HV, and Taxol®, respectively, which confirmed that PTX-loaded redox-sensitive nanoparticles significantly induced apoptosis and necrotic rate than other treatment groups. Furthermore, competitive inhibition studies indicated that the cell apoptosis induced by PTX-HSV nanoparticles was significantly reduced when A549 cells were pretreated with free HA. The percentage of apoptotic cells was downregulated to 49.6%, which was consistent with the results of flow cytometry and CLSM studies. This was mainly attributed to HA receptor-mediated endocytosis of PTX-loaded HSV nanoparticles which inhibited cell growth and resulted in increased cell apoptosis.
In vivo antitumor efficacy
To further evaluate the in vivo antitumor efficacy of PTX-HSV nanoparticles, mice bearing A549 tumors were intravenously injected with saline, PTX-HSV, PTX-HV, or Taxol®, respectively, when the tumor volume reached ~100 mm3. The comparison of tumor volume change and tumor weight of mice treated with different formulations over 18 days is depicted in Figure 7A and B. The results of tumor growth and tumor weight measurement showed that all PTX formulations were effective in tumor inhibition compared to the saline group. To compare the inhibition ratios of all PTX formulations, the tumor weight at the end of the experiment was used. Results (Figure 7B) demonstrated that the PTX-loaded HSV nanoparticles had the best antitumor efficacy (75.4%), which was significantly higher than that of PTX-HV nanoparticles (55.2%) and Taxol® (39.2%), which probably was attributed to the targeting of the HA backbone and the redox-selective burst release of PTX from sensitive nanoparticles upon internalization into tumor cells. Furthermore, H&E evaluation was adopted to assess the antitumor efficacy after different treatments. As shown in Figure 7C, cell apoptosis patterns in tumor tissues agreed to the results of tumor inhibitory study (Figure 7B). As expected, tumors in the saline control group had some histologic characteristic indications of rapid tumor growth, including hyperchromatic nuclei, scant cytoplasm, large nucleus, and closely arranged tumor cells.22,32 In contrast, the tumors of mice treated with PTX-HSV, PTX-HV, or Taxol® all exhibited shrinkage of tumor cells, a large homogeneous red staining of necrotic tissue, and cell separation. This cell apoptotic phenomenon indicated that the PTX-HSV treatment achieved the highest level of tumor necrosis.
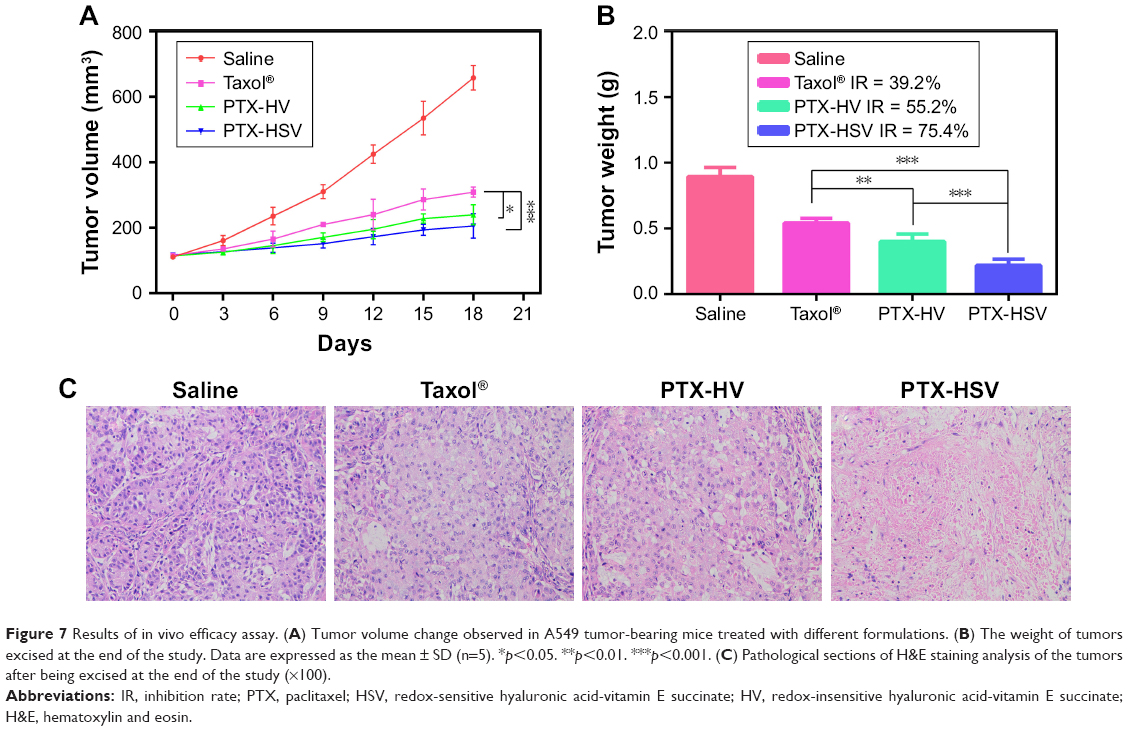 | Figure 7 Results of in vivo efficacy assay. (A) Tumor volume change observed in A549 tumor-bearing mice treated with different formulations. (B) The weight of tumors excised at the end of the study. Data are expressed as the mean ± SD (n=5). *p<0.05. **p<0.01. ***p<0.001. (C) Pathological sections of H&E staining analysis of the tumors after being excised at the end of the study (×100). |
There is a broad consensus that in vivo toxicity evaluation is important for systemic drug delivery systems from a safety perspective. Therefore, we assessed body weight analysis and histological analysis of organs through H&E staining after formulation treatment. As indicated in Figure 8A, compared to the initial body weight of tumor-bearing mice, animals in nanoparticle groups (both redox-sensitive and -insensitive) did not show any weight loss over the duration of the study, while those treated with Taxol® exhibited a significant decrease in body weight (p<0.001). In addition, the results of the tissue section analysis (Figure 8D) indicated that PTX-HSV and PTX-HV nanoparticle treatment groups had no obvious pathological symptoms compared to the saline control group, which further confirmed the safety of PTX-loaded nanoparticles. Furthermore, the hepatotoxicity and nephrotoxicity were determined by plasma samples and the results showed that mice treated with Taxol® had significantly higher serum levels of the liver enzyme (AST and ALT) and renal function marker (BUN and Scr) than those treated with the control group (Figure 8B and C). In contrast, the mice treated with PTX-HSV and PTX-HV nanoparticles induced a negligible increase of those enzymes and markers over the control group. All the results indicated that PTX-HSV and PTX-HV nanoparticles were well tolerated at the tested dose while Taxol® had severe side effects and toxicity.
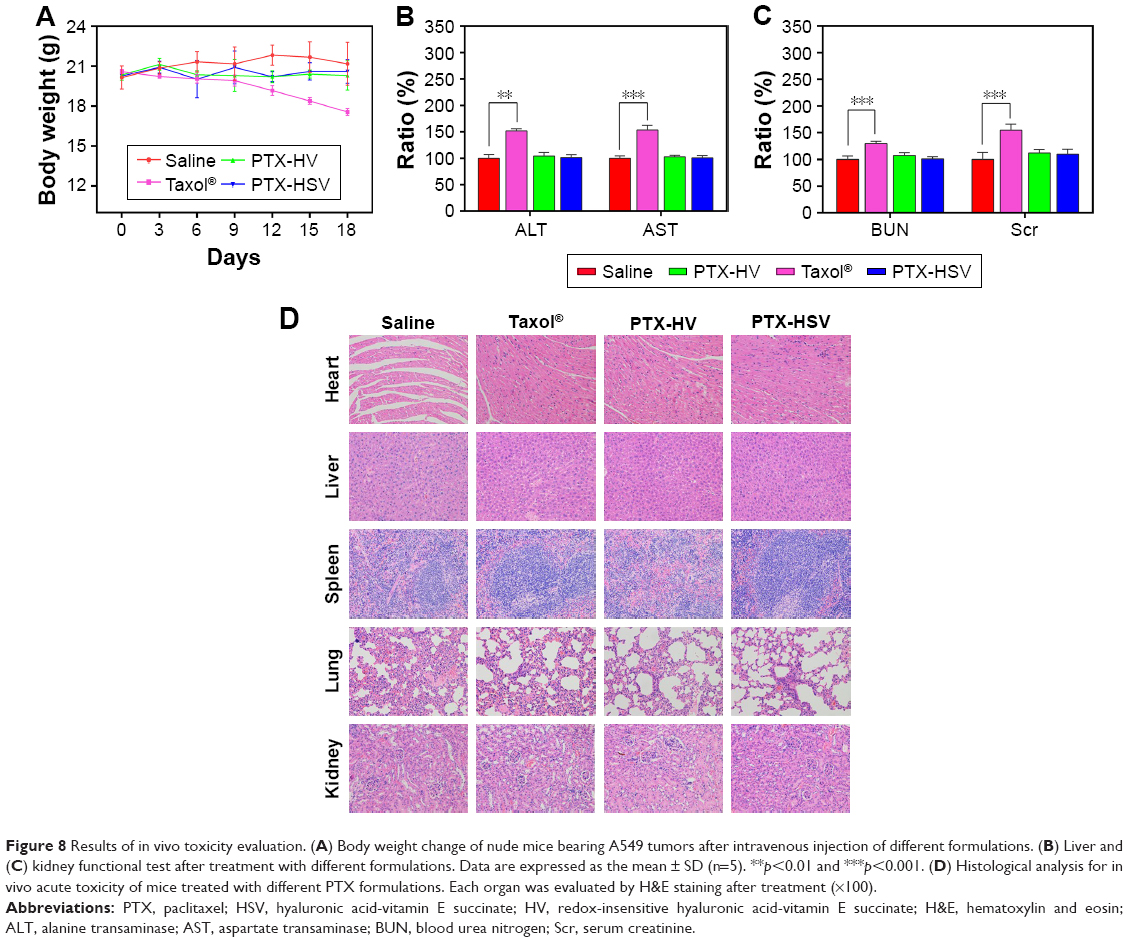 | Figure 8 Results of in vivo toxicity evaluation. (A) Body weight change of nude mice bearing A549 tumors after intravenous injection of different formulations. (B) Liver and (C) kidney functional test after treatment with different formulations. Data are expressed as the mean ± SD (n=5). **p<0.01 and ***p<0.001. (D) Histological analysis for in vivo acute toxicity of mice treated with different PTX formulations. Each organ was evaluated by H&E staining after treatment (×100). |
Conclusion
In this study, PTX-loaded HSV nanoparticles constructed from redox-sensitive HSV conjugates were successfully engineered to treat A549 non-small-cell lung carcinoma. The low CMC value and stability of PTX-loaded HSV nanoparticles in rat plasma indicated that the nanoparticles had strong resistance to the dilution and were stable during blood circulation upon intravenous injection. Moreover, the reduction-sensitivity assessment and in vitro release studies of PTX-loaded HSV nanoparticles demonstrated their stimuli-triggered drug release into highly reducing cytoplasm upon tumor cell arrival. In addition, the redox-sensitive nature of the nanoparticles further enhanced both cytotoxicity and apoptosis rate in A549 cells. In vivo, PTX-loaded HSV nanoparticles demonstrated a greater inhibition rate of tumor growth than that of redox-insensitive nanoparticles and Taxol®. In summary, HSV nanoparticles can be considered as a promising vehicle for PTX-targeted delivery for lung cancer treatment.
Acknowledgment
This work was financially supported by the project of Fujian Science and Technology Department (No 2015Y0065), the key disciplines research program of Chinese Traditional Medicine Analysis, and State Administration of Traditional Chinese Medicine (No X2014101).
Disclosure
The authors report no conflicts of interest in this work.
References
Hoffman RM, Sanchez R. Lung cancer screening. Med Clin North Am. 2017;101(4):769–785. | ||
Gao Q, Jiang X, Huang C. [Clinical advanced in early-stage ALK-positive non-small cell lung cancer patients]. Zhongguo Fei Ai Za Zhi. 2017;20(2):124–129. Chinese [with English abstract]. | ||
Li K, Xiu CL, Gao LM, et al. Screening of specific nucleic acid aptamers binding tumor markers in the serum of the lung cancer patients and identification of their activities. Tumour Biol. 2017;39(7):1010428317717123. | ||
Vicary GW, Roman J. Targeting the mammalian target of rapamycin in lung cancer. Am J Med Sci. 2016;352(5):507–516. | ||
Hussain S. Nanomedicine for treatment of lung cancer. Adv Exp Med Biol. 2016;890:137–147. | ||
Wang Y, Lv SX, Deng MX, Tang ZH, Chen XS. A charge-conversional intracellular-activated polymeric prodrug for tumor therapy. Polym Chem. 2016;7(12):2253–2263. | ||
Wu H, Zhong Q, Zhong R, et al. Preparation and antitumor evaluation of self-assembling oleanolic acid-loaded Pluronic P105/d-alpha-tocopheryl polyethylene glycol succinate mixed micelles for non-small-cell lung cancer treatment. Int J Nanomedicine. 2016;11:6337–6352. | ||
Mandal B, Mittal NK, Balabathula P, Thoma LA, Wood GC. Development and in vitro evaluation of core-shell type lipid-polymer hybrid nanoparticles for the delivery of erlotinib in non-small cell lung cancer. Eur J Pharm Sci. 2016;81:162–171. | ||
Quin J, Engle D, Litwiller A, et al. Vitamin E succinate decreases lung cancer tumor growth in mice. J Surg Res. 2005;127(2):139–143. | ||
Neuzil J, Weber T, Schroder A, et al. Induction of cancer cell apoptosis by alpha-tocopheryl succinate: molecular pathways and structural requirements. FASEB J. 2001;15(2):403–415. | ||
Tomic-Vatic A, Eytina J, Chapman J, Mahdavian E, Neuzil J, Salvatore BA. Vitamin E amides, a new class of vitamin E analogues with enhanced proapoptotic activity. Int J Cancer. 2005;117(2):188–193. | ||
Mi Y, Liu Y, Feng SS. Formulation of docetaxel by folic acid-conjugated d-alpha-tocopheryl polyethylene glycol succinate 2000 (Vitamin E TPGS(2k)) micelles for targeted and synergistic chemotherapy. Biomaterials. 2011;32(16):4058–4066. | ||
Liang D, Wang AT, Yang ZZ, Liu YJ, Qi XR. Enhance cancer cell recognition and overcome drug resistance using hyaluronic acid and alpha-tocopheryl succinate based multifunctional nanoparticles. Mol Pharm. 2015;12(6):2189–2202. | ||
Duhem N, Danhier F, Preat V. Vitamin E-based nanomedicines for anti-cancer drug delivery. J Control Release. 2014;182:33–44. | ||
Lian H, Sun J, Yu YP, et al. Supramolecular micellar nanoaggregates based on a novel chitosan/vitamin E succinate copolymer for paclitaxel selective delivery. Int J Nanomedicine. 2011;6:3323–3334. | ||
Li Y, Liu Q, Li W, et al. Design and validation of PEG-derivatized vitamin E copolymer for drug delivery into breast cancer. Bioconjug Chem. 2016;27(8):1889–1899. | ||
Liu Y, Xu Y, Wu M, et al. Vitamin E succinate-conjugated F68 micelles for mitoxantrone delivery in enhancing anticancer activity. Int J Nanomedicine. 2016;11:3167–3178. | ||
Tao Y, Han J, Wang X, Dou H. Nano-formulation of paclitaxel by vitamin E succinate functionalized pluronic micelles for enhanced encapsulation, stability and cytotoxicity. Colloids Surf B Biointerfaces. 2013;102:604–610. | ||
Lin CJ, Kuan CH, Wang LW, et al. Integrated self-assembling drug delivery system possessing dual responsive and active targeting for orthotopic ovarian cancer theranostics. Biomaterials. 2016;90:12–26. | ||
Hu W, He CY, Tan LJ, et al. Synthesis and micellization of redox-responsive dynamic covalent multi-block copolymers. Polym Chem. 2016;7(18):3145–3155. | ||
Yin T, Wu Q, Wang L, Yin L, Zhou J, Huo M. Well-defined redox-sensitive polyethene glycol-paclitaxel prodrug conjugate for tumor-specific delivery of paclitaxel using octreotide for tumor targeting. Mol Pharm. 2015;12(8):3020–3031. | ||
Lin L, Cai M, Deng S, et al. Amelioration of cirrhotic portal hypertension by targeted cyclooxygenase-1 siRNA delivery to liver sinusoidal endothelium with polyethylenimine grafted hyaluronic acid. Nanomedicine. 2017;13(7):2329–2339. | ||
Zerbinati N, Haddad RG, Bader A, et al. A new hyaluronic acid polymer in the augmentation and restoration of labia majora. J Biol Regul Homeost Agents. 2017;31(2 Suppl 2):153–161. | ||
Saneja A, Nayak D, Srinivas M, et al. Development and mechanistic insight into enhanced cytotoxic potential of hyaluronic acid conjugated nanoparticles in CD44 overexpressing cancer cells. Eur J Pharm Sci. 2017;97:79–91. | ||
Sgorla D, Almeida A, Azevedo C, Bunhak EJ, Sarmento B, Cavalcanti OA. Development and characterization of crosslinked hyaluronic acid polymeric films for use in coating processes. Int J Pharm. 2016;511(1):380–389. | ||
Winterbourn CC, Metodiewa D. Reactivity of biologically important thiol compounds with superoxide and hydrogen peroxide. Free Radic Biol Med. 1999;27(3–4):322–328. | ||
Miller T, Rachel R, Besheer A, Uezguen S, Weigandt M, Goepferich A. Comparative investigations on in vitro serum stability of polymeric micelle formulations. Pharm Res. 2012;29(2):448–459. | ||
Huo M, Liu Y, Wang L, et al. Redox-sensitive micelles based on O,N-hydroxyethyl chitosan-octylamine conjugates for triggered intracellular delivery of paclitaxel. Mol Pharm. 2016;13(6):1750–1762. | ||
Cheng R, Feng F, Meng F, Deng C, Feijen J, Zhong Z. Glutathione-responsive nano-vehicles as a promising platform for targeted intracellular drug and gene delivery. J Control Release. 2011;152(1):2–12. | ||
Deng B, Ma P, Xie Y. Reduction-sensitive polymeric nanocarriers in cancer therapy: a comprehensive review. Nanoscale. 2015;7(30):12773–12795. | ||
He Y, Cheng G, Xie L, Nie Y, He B, Gu Z. Polyethyleneimine/DNA polyplexes with reduction-sensitive hyaluronic acid derivatives shielding for targeted gene delivery. Biomaterials. 2013;34(4):1235–1245. | ||
Banerji S, Wright AJ, Noble M, et al. Structures of the CD44-hyaluronan complex provide insight into a fundamental carbohydrate–protein interaction. Nat Struct Mol Biol. 2007;14(3):234–239. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.