Back to Journals » Neuropsychiatric Disease and Treatment » Volume 17
P53/miR-154 Pathway Regulates the Epithelial-Mesenchymal Transition in Glioblastoma Multiforme Cells by Targeting TCF12
Authors Zhu G, Yang S, Wang R, Lei J, Ji P, Wang J, Tao K, Yang C, Ge S, Wang L ![]()
Received 31 July 2020
Accepted for publication 18 January 2021
Published 26 February 2021 Volume 2021:17 Pages 681—693
DOI https://doi.org/10.2147/NDT.S273578
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Yu-Ping Ning
Gang Zhu,1,* Shirong Yang,2,* Ronglin Wang,3,* Jie Lei,4 Peigang Ji,1 Jiancai Wang,1 Kai Tao,1 Chen Yang,1 Shunnan Ge,1 Liang Wang1
1Department of Neurosurgery, Tangdu Hospital, The Fourth Military Medical University, Xi’an, Shaanxi, People’s Republic of China; 2Department of General Surgery, Tangdu Hospital, The Fourth Military Medical University, Xi’an, Shaanxi, People’s Republic of China; 3Department of Oncology, Tangdu Hospital, The Fourth Military Medical University, Xi’an, Shaanxi, People’s Republic of China; 4Department of Neurosurgery, Wuhan General Hospital of PLA, Wuhan, Hubei, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Liang Wang; Shunnan Ge
Department of Neurosurgery, Tangdu Hospital, The Fourth Military Medical University, Xinsi Road, Xi’an, 710038, People’s Republic of China
Tel +86-29-84717851
; Tel +86-29-84717824
Email [email protected]; [email protected]
Purpose: Glioblastoma multiforme (GBM) is an aggressive brain tumor with a rather short survival time. Mutation of p53 has been observed and reported to play critical roles in the progression of GBM. However, the pathological mechanisms are still unclear. This study was designed to identify the role of miR-154 in mediating the biological functions of p53 in glioblastoma multiforme.
Methods: In the current study, the expression of miR-154 in GBM tissue samples and cell lines with wt-p53 or mutant p53 was evaluated. The functions of miR-154 in tumor migration, invasion and epithelial-mesenchymal transition were analyzed in vitro. A luciferase reporter assay was used to identify the target of miR-154.
Results: We found that expression of miR-154 was much lower in patient tissues with mutant p53. Further study revealed that p53 was a transcription factor of miR-154 and that the R273H mutation led to its inactivation. In addition, overexpression of miR-154 remarkably suppressed cell migration, invasion and EMT in vitro and tumor growth in vivo. Moreover, TCF12 was proven to be a direct target of miR-154, and the tumor suppressive effect of miR-154 was reversed by TCF12.
Conclusion: Overall, miR-154, which was regulated by wt-p53, inhibited migration, invasion and EMT of GBM cells by targeting TCF12, indicating that miR-154 may act as a biomarker and that the p53/miR-154/TCF12 pathway could be a potential therapeutic target for GBM.
Keywords: glioblastoma, p53, microRNA, EMT, TCF12
Introduction
Glioblastoma multiforme (GBM) is the most common and malignant primary brain tumor in adults and is classified as a grade IV glioma by the World Health Organization. Although significant advancements in studies on GBM have been achieved and multiple new therapeutic approaches have been introduced in the clinic, the median survival time of GBM patients is approximately 1 year after diagnosis.1 This is much shorter than the 6–8 years for low-grade (I and II) gliomas.2,3 The poor prognosis of GBM is mainly due to its highly invasive, chemotherapy resistant and genetically unstable cells, especially the epithelial-mesenchymal transition (EMT) of GBM cells, which gives it a more aggressive nature.4 Hence, finding an effective way to prevent EMT is essential to improve the poor prognosis of GBM patients.
MicroRNAs (miRNAs) are a class of 19 to 25 nucleotide noncoding RNAs that regulate gene expression at the posttranscriptional level by interacting with the 3’-untranslated regions (3’-UTRs) of their target messenger (m)RNAs.5,6 Ongoing research on miRNAs in recent years has demonstrated that they are involved in many biological processes, including development, apoptosis, metabolism, immunity and tumor progression.7–10 In particular, miRNAs have been found to contribute to modulating the initiation, progression, invasion and metastasis of cancers, such as GBM.11,12
miRNA-154 (miR-154), which is located in the human imprinted 14q32 domain, has been identified as a tumor suppressor in various types of human cancer, including lung cancer, colorectal cancer, melanoma and GBM.13–16 Previous studies revealed that miR-154 expression is downregulated in human primary GBM tissues, and the overexpression of miR-154 can suppress proliferation and metastasis and induce apoptosis of GBM.17 However, the molecular mechanisms by which it exerts its functions and how it is regulated remain largely unknown.
p53, a tumor suppressor, is well known for preserving genomic stability and preventing tumor formation. Mutations in p53 have been found in approximately 60% of patients with secondary GBMs and 25% of patients with primary GBMs.18,19 For the GBM cell lines, U87 and A172 cells contain wild-type p53, while U251 and U373 are p53 mutants.20 The reactivation of endogenous p53 function represents an important tool in GBM treatment.19,21 The p53 protein is a transcription factor, and several microRNAs are known to be controlled by p53. Previous research has shown that p53 is an activator of several microRNAs, including miR-21, miR-200a, miR-192, miR-215, miR-16-2 and miR-182.22–25 This suggests that miR-154 might also be regulated by p53.
In the present study, we found that the expression of miR-154 was downregulated in GBM tissues and GBM cell lines and that wt-p53 overexpression led to an increase in miR-154 in the U251 cell line. Furthermore, we verified that transcription factor 12 (TCF12, also known as HEB), which is overexpressed in certain tumor types and has been associated with the cancer EMT and stem cell proliferation,26,27 mediated the p53/miR-154-controlled tumorigenesis, invasion and EMT in GBM. Our study is the first to provide evidence that the p53/miR-154 network contributes to suppressing the progression of GBM and that TCF12 plays a role as the target of miR-154.
Materials and Methods
Ethics Statement
This study was performed in compliance with the ethical guidelines of the Declaration of Helsinki. The use of human samples was approved by the research ethics committee of The Fourth Military Medical University. All animal experiments were approved by the Animal Care and Use Committee of The Fourth Military Medical University (approval number: 201903–141) and followed the National Guidelines for the Care and Use of Laboratory Animals. All efforts were made to minimize the number of animals and their suffering.
Cell Culture and Tissue Samples
Tumor cell lines U251, U373, U87, A172 and normal human astrocytes (NHAs) were obtained from the American Type Culture Collection (Manassas, VA, USA). Cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum and 1% antibiotics at 37°C in a humidified atmosphere containing 5% CO2. Human GBM tissues and adjacent normal brain tissues were obtained from patients with GBM without previous radiotherapy or chemotherapy at Tangdu Hospital affiliated with The Fourth Military Medical University. Immediately after surgery, samples were snap-frozen in liquid nitrogen and stored at −80°C. Prior to our research study, all patients gave informed written consent.
Cell Transfection and Transduction
Human wild-type p53 genes or mutant p53 genes (R273H) were inserted into pcDNA3 vectors (Invitrogen, Carlsbad, CA). The TCF12 plasmid was purchased from GeneChem (Shanghai, China). The miR-154 mimic and inhibitor were purchased from RiboBio (Guangzhou, China). The lentiviral lenti-pCMV-GFP-miR-154, lenti-pCMV-GFP-miR-154 inhibitor and empty lentivirus vector (GeneChem, Shanghai, China) were prepared. Cells were infected with lentivirus, and 1 μg/mL puromycin was applied to purify stably expressing cells and control cells. For transfection, the indicated plasmids were transfected into cells with Lipofectamine 2000 (Invitrogen), and the following assays were conducted 24 h later. The sequences used for transfection were as follows: shRNA for p53: 5-AGUGUUUCUGUCAUCCAAAUACUCCAC-3’. miR-154 mimic: sense, 5’-UAGGUUAUCCGUGUUGCCUUCG-3’; antisense, 5’-AAGGCAACACGAUAACCUAUU-3’, miR‐154 inhibitor: 5’-CGAAGGGAACACGGAUAAC CUA-3’.
Western Blot
Total protein samples were separated by SDS-PAGE and transferred to polyvinylidene fluoride (PVDF) membranes (Millipore, Billerica, MA) as previously described.28 Then, membranes were blocked with 5% skim milk in TBST buffer for 2 h and incubated with primary antibodies (p53, 1:1000, 2524, Cell Signaling; TCF12, 1:1000, ab70746, Abcam; Vimentin, 1:800, ab24525, Abcam; E-cadherin, 1:1000, 3195, Cell Signaling; N-cadherin, 1:500, ab18203, Abcam; β-actin, 1:1000, ab8227, Abcam) at 4°C overnight. After being washed three times in TBST for 5 min each, membranes were incubated with HRP-conjugated secondary antibodies (1:5000) and washed three times. Finally, blots were detected with enhanced chemiluminescence reagents by X-ray films. Band densities were quantified using ImageJ software (National Institutes of Health, Bethesda, MD). The blots were performed in three experiments with similar results.
Reverse Transcription and Quantitative Real-Time PCR (RT-qPCR)
For gene expression analysis, TRIzol (Invitrogen; Thermo Fisher Scientific, Inc.) was used to isolate total RNA from tissues and cells according to the manufacturer’s protocol. cDNA was synthesized from 10 ng of total RNA using the TaqMan MicroRNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA, USA). Quantitative real-time reverse transcription–polymerase chain reaction (qRT–PCR) was performed on an ABI 7900 Fast System with a TaqMan MicroRNA Assay kit (both from Applied Biosystems). Small nuclear RNA U6 was used as an endogenous control for miR-154 expression analysis. The thermocycling conditions were as follows: 95°C for 10 min, 40 cycles of denaturation at 95°C for 15 sec, annealing at 60°C for 1 min, and a final elongation step at 72°C for 10 min. Each sample was examined in triplicate. The cycle threshold (CT) was recorded, and the amount of miR-154 relative to RNA U6 was calculated. The primer sequences used were as follows: miR-154, 5’-CGCGAATTCGCATCTAGGACCTCCATCAC-3’ (forward) and 5’ - ACGGGATCCGAACCATCCCTTCACTTACC-3’ (reverse); U6, 5’ - CTCGCTTCGGCAGCACA-3’ (forward) and 5’ - AACGCTTCA CGAATTTGCGT-3’ (reverse).
Transwell Invasion Assay
The Transwell invasion assay was performed using 24-well Transwell permeable inserts containing polycarbonate membranes with 8 μm pore size, which were coated with 25 μg of Matrigel (BD Biosciences). U251 or U87 cells (1 × 105) were added to each well in serum-free medium, and medium containing 5% FBS was added to the bottom chamber as a chemoattractant. The Transwell plates were then incubated at 37°C for 24 h. Then, the membranes were fixed in methanol and stained with crystal violet solution. Noninvasive cells on the top side of the membranes were gently removed using a cotton swab, and invading cells on the underside of the membranes were counted using an inverted microscope.
Wound Healing Assay
U251 and U87 cells were seeded into a 6-well plate at 2 × 104 cells/cm2 in DMEM supplemented with 10% FBS. Each confluent monolayer was scraped by a 200 μL plastic pipette tip to make a cell-free area, and then detached cells were washed off with DMEM. The cells were incubated at 37 °C in DMEM supplemented with 10% FBS for 24 h. The cells were observed at 0 and 24 h using a Nikon Ti2 microscope, and the distance between the edges of the cell-free areas was measured using ImageJ software.
Immunohistochemistry
The fresh-frozen tissue samples were fixed with 4% paraformaldehyde and dehydrated in graded sucrose. Then, the frozen sections were blocked in 1% goat serum in PBS and 0.5% Triton X-100 for 30 min prior to incubation with an anti-TCF12 antibody (1:200, ab70746, Abcam). The sections were then washed with PBS three times and then incubated with system-labeled HRP anti-mouse secondary antibody at room temperature for 2 h. Next, the sections were incubated in DAB, counterstained in Mayer’s hematoxylin, and dehydrated in alcohol and xylene. A Nikon Ti2 microscope was used to observe the positive cells.
Luciferase Reporter Assay
A luciferase reporter assay was performed as previously described.29 Briefly, TargetScan software was applied to predict the potential binding site of miR-154 at the 3’-UTR of TCF12 mRNA. The 3’-UTR sequences of TCF12 containing wild-type (wt) or mutant (mut) miR-154 binding sites were cloned into the pGL-3 luciferase reporter vector. Cells (4 × 104) were seeded in 24-well plates and cultured for 24 h before cotransfection with luciferase reporter vectors and miR-154 (GeneChem, Shanghai, China) or NC vector using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s instructions. Luciferase and Renilla signals were measured 36 h after transfection using a Dual Luciferase Reporter Assay Kit (Promega) according to the manufacturer’s protocol. The activity of firefly luciferase was normalized to that of Renilla luciferase.
P53 Mutation Identification
DNA was extracted from freshly frozen tissue using DNeasy blood and tissue kits (QIAGEN) and quantified using a Qubit 2.0 Fluorometer (Invitrogen). DNA quality was assessed by absorptiometric analyses. Coding sequences of the p53 gene (exons 2–11) were PCR-amplified using primers and conditions as described previously30 and sequenced using an ABI 3730 DNA Analyzer (Applied Biosystems). Mutational analysis was performed using Mutation Surveyor software version 3.97 (Soft Genetics), and sequence data were aligned to the p53 reference sequence NC_000017.10.
CCK-8 Assay
The CCK8 assay was used to measure proliferation of U251 and U87 cells after miR-154 overexpression. 1000 cells per well were cultured in five replicate wells in a 96-well plate in medium containing 10% FBS. CCK-8 reagent (beyotime, Beijing, China) was added to each well according to the manufacturer’s instructions at the indicated time. The absorbance at 450 nm was measured.
Tumor Model in Nude Mice
Five-week-old male nude mice were housed in a temperature- and light-controlled environment with a 14/10 h light/dark cycle. U251 cells were transduced with negative control lentivirus or lenti-miR-154. Cells were harvested and suspended at 1×107 cells per mL before subcutaneous injection into the flanks. The mice were sacrificed, and the tumors were isolated 4 weeks after injection. The size of the tumor was calculated as 0.5×length×width2.
For intracranial transplantation, the mice were anesthetized with 10% chloral hydrate. Then, a small hole was drilled 2 mm forward from bregma and 2 mm lateral from the midline at the right side of the cranium with an electric drill. U251 cells (1x 105) stably expressing miR-154 or control cells suspended in 10 μL PBS were transplanted into the forebrain at a depth of 3 mm with a Hamilton syringe. The mice were carefully observed every day for 60 d to record the survival curves. Brain specimens were harvested from mice 60 d after transplantation. The brain slices were subjected to DYPI staining and observed by Nikon A1 confocal microscopy.
Statistical Analysis
One-way ANOVA or t-test was used to assess significant differences among the experimental groups. All cellular experiments were performed at least three times. Data are presented as the mean ± SEM. Statistical differences were determined by a two-tailed t test. The Prism5 software package was used for data analysis. For all comparisons, a P-value<0.05 was considered significant.
Results
MiR-154 Expression Decreases in Glioblastoma Tissues and Cell Lines, Especially in P53 Mutant Tissues and Cell Lines
Previous studies demonstrated that the expression of miR-154 is downregulated in human GBM tissues.17 We measured the relative levels of miR-154 in 21 paired GBM tissues and adjacent normal tissues by qRT-PCR analysis. The results showed that the expression of miR-154 in tumor tissues was lower than that in adjacent normal tissues in 18 paired tissues (Figure 1A). We then separated the tissues into two groups depending on whether the expression of miR-154 in tumor tissues decreased more than 50% compared to that in normal tissues. Interestingly, DNA sequencing assays revealed that 6 out of 7 tissues in the lower miR-154 group were p53 mutant and 11 out of 14 in the other group were p53 wild-type tissues (Figure 1B). This suggested that the function of p53 might be associated with the regulation of miR-154. Another 48 GBM tissues were taken to assess miR-154 expression after p53 gene sequencing. The miR-154 level was much lower in the p53 mutant group than in the p53 wild-type group (Figure 1C). Furthermore, this finding was confirmed in glioma cell lines. U251 and U373 cells, which are p53 mutant cell lines, expressed less miR-154 than A172 and U87 cells, which are p53 wild-type cell lines. The miR-154 levels were all decreased compared to those in NHAs, except in U87 cells (Figure 1D).
 |
Figure 1 The level of miR-154 was decreased in GBM tissue, especially in p53 mutant tissue. (A) Twenty-one paired GBM and adjacent normal tissues were collected, and a decrease in miR-154 was found in 18 GBM tissues. Eight representative paired tissues are shown. (B) P53 DNA sequencing analysis was performed in the 21 GBM tissues that were divided into groups according to the degree of miR-154 decline. (C) MiR-154 was detected in 48 other GBM tissues with different p53 backgrounds. (D) The miR-154 level is shown in NHA and GBM cell lines. *Compared with NHA, #compared with A172 (*, #p<0.05, **p<0.01, ***, ###p<0.001, ns: no significance). |
To investigate the regulatory role of p53 on miR-154, we searched for transcription factor binding sites of putative promoters upstream of miR-154 using the Footer algorithm. Three p53 binding elements were found within 1.5 kb upstream of miR-154, as we hypothesized (Figure 2A). U251 and U87 cell lines were used to detect the effect of p53 on miR-154. We prepared a wt-p53 DNA plasmid and transfected it into p53 mutant U251 cells. In addition, the sh p53 plasmid was transfected into p53 wild-type U87 cells (Figure 2B). The expression of miR-154 was detected by qRT-PCR 36 h after transfection. Exogenous wt-p53 dramatically upregulated the transcription of miR-154 in U251 cells. Conversely, miR-154 was downregulated after transfection with the sh p53 plasmid in U87 cells (Figure 2C). Then, a mut-p53 (p53-R273H) plasmid was transfected into U251 and U373 cells. We found no significant difference in miR-154 expression between mut-P53 and NC cells in either cell line. (Figure 2D).
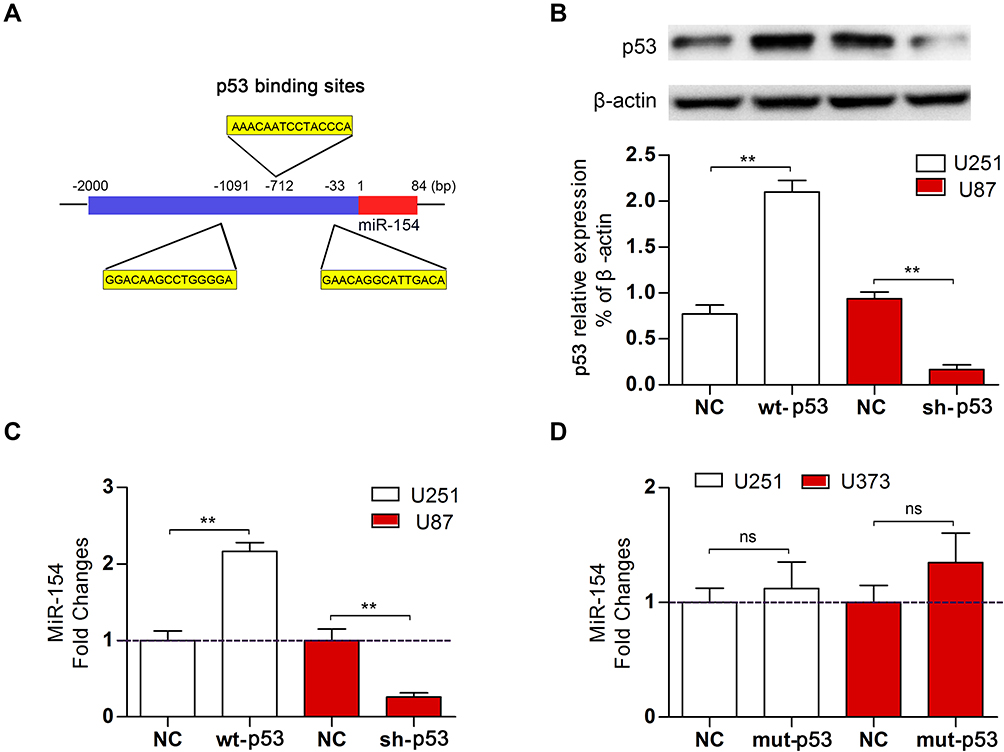 |
Figure 2 Wt-p53 regulated the transcription of miR-154. (A) Three putative p53 binding sites were predicted upstream of the miR-154 gene. (B) We overexpressed wt-p53 in U251 cells and knocked it down in U87 cells, and the p53 level was detected by Western blot. (C) MiR-154 expression in U251 cells and U87 cells after transfection is shown. (D) Mut-p53 (p53-R273H) was transfected into U251 and U373 cells, and miR-154 was not affected (**p<0.01, ns: no significance). |
Overexpressing miR-154 Inhibit the Migration, Invasion and EMT of GBM in the P53 Mutant Cell Line (U251) and P53 Wild-Type Cell Line (U87)
To validate the effect of miR-154 on the progression of GBM, wound healing, Transwell assays and EMT detection were performed on U251 and U87 cell lines. We stably transduced the cell lines using lentivirus that carries a negative control vector or a miR-154 overexpression vector. miR-154 expression was determined by qRT-PCR assay (Figure 3A). In the Transwell assay, increased miR-154 significantly decreased the invading cells (Figure 3B). The wound healing assay showed that miR-154 inhibited cell migration in U251 and U87 cells (Figure 3C). Then, we detected the EMT markers E-cadherin, N-cadherin and vimentin by Western blot. Overexpressed miR-154 increased the level of E-cadherin and decreased the levels of N-cadherin and vimentin, which indicated that the EMT was inhibited by miR-154 (Figure 3D). Interestingly, despite the different p53 backgrounds, the overexpressed miR-154 weakened the malignancy of both the U251 and U87 cell lines. Moreover, we generated tumor xenograft models using nude mice. Before the experiment, a CCK-8 assay was applied to show that miR-154 could not affect the proliferation of U251 and U87 cells (Figure 4A). Our data showed that miR-154-overexpressing U251 cells developed much smaller tumors than NC cells in either subcutaneous models or intracranial models (Figure 4B and C). Furthermore, mice transplanted with miR-154-overexpressing U251 cells showed a much longer life span than the control animals (Figure 4D).
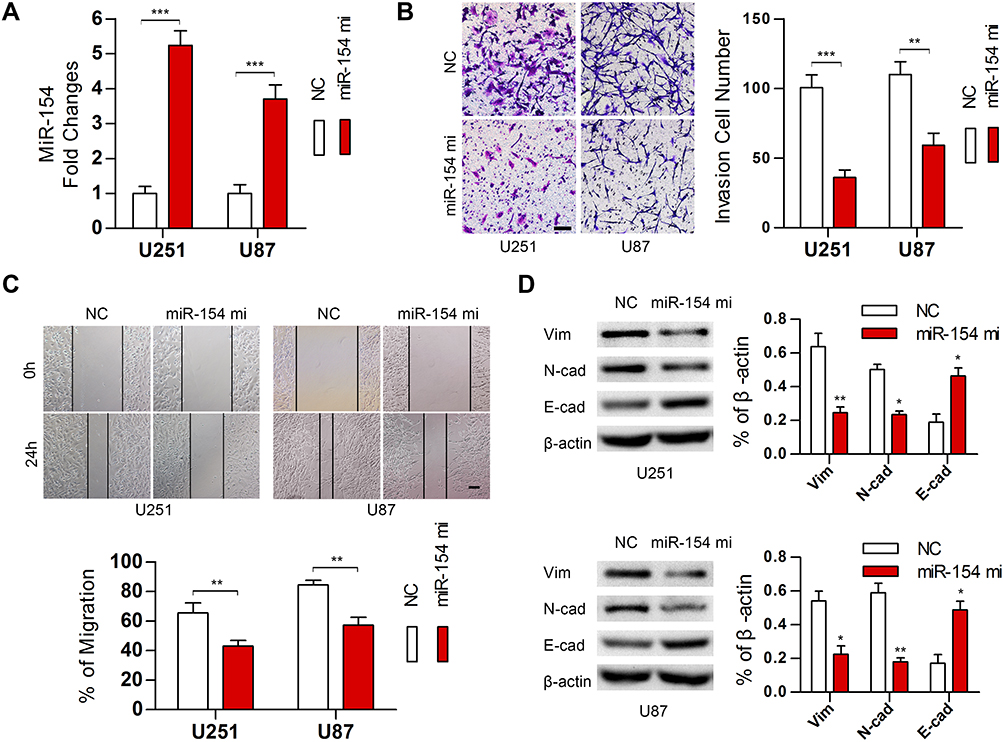 |
Figure 3 MiR-154 suppressed the migration, invasion and EMT in GBM in vitro. (A) MiR-154 was detected in U251 and U87 cells stably expressing miR-154. (B) Transwell assays indicated that miR-154 inhibited the invasion of GBM cells. (C) Wound healing assays showed a decreased migration distance of miR-154-overexpressing cells. (D) EMT markers were detected, and miR-154 reduced the expression of vimentin and N-cadherin and increased E-cadherin (*p<0.05, **p<0.01, ***p<0.001, Bar=100 μm). |
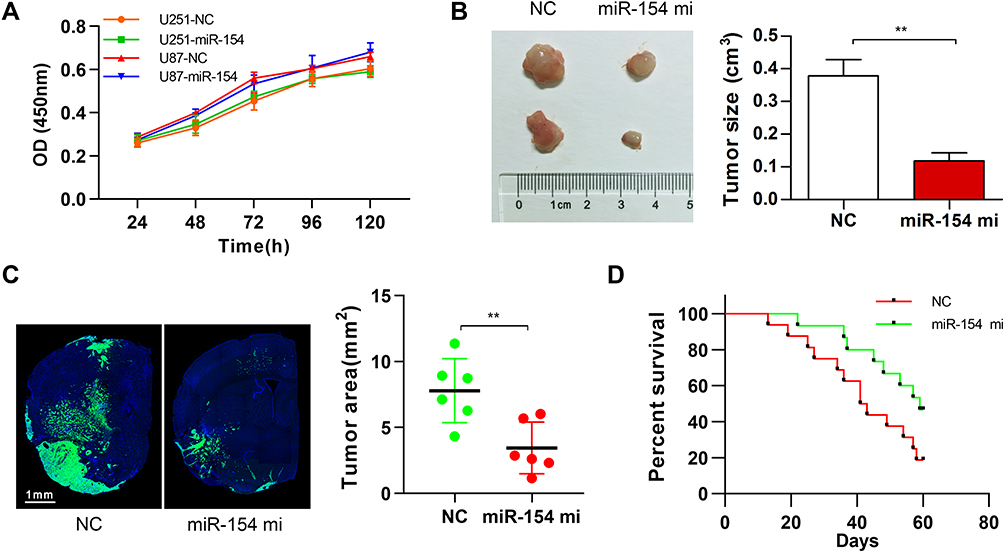 |
Figure 4 MiR-154 promoted the growth of GBM in mouse tumor transplantation models. (A) CCK-8 assay showed no influence on the proliferation of miR-154 in either U251 or U87 cells. (B) U251 cells were transduced with NC vector lentivirus or miR-154 lentivirus and then were subcutaneously injected into nude mice. The tumors were harvested 6 weeks later. Representative tumors are shown, and the size was calculated. MiR-154 significantly suppressed the tumor formation of U251 cells. n=8 per group. (C) U251 cells stably expressing miR-154 and control cells were used as an intracranial tumor model. Representative images of the tumor-transplanted cerebral hemisphere are shown, and the maximum tumor areas were calculated. (D) The survival curve of the intracranial tumor-transplanted mice was recorded (n=16 for NC and n=15 for miR-154 mi) (**p<0.01). |
TCF12 is Expressed at a High Level in GBM Tissues and Can Be Regulated by miR-154
TCF12 has been reported to be overexpressed in several cancers. We detected the expression of TCF12 in GBM tissues using Western blot analysis and observed that the expression of TCF12 protein was greatly upregulated (Figure 5A). Immunohistochemistry staining showed that TCF12 was greatly increased in the nucleus and cytoplasm in GBM tissues compared with adjacent normal tissues (Figure 5B). Moreover, it was indicated that TCF12 might be a binding target of miR-154 by searching TargetScan. Then, we performed a luciferase reporter assay and found that miR-154 mimic significantly suppressed the luciferase activity of the wild-type 3’-UTR of TCF12 in U251 cells, while the luciferase activity of the mutant 3’-UTR of TCF12 was not affected (Figure 5C). Furthermore, we detected the TCF12 protein level in U251 and U373 MG cells stably overexpressing miR-154, and the TCF12 level was much lower than that in negative control (NC) cells (Figure 5D). Taken together, these findings suggest that miR-154 directly targets TCF12 by binding to its 3’UTR in GBM cells and suppresses the protein expression of TCF12.
 |
Figure 5 TCF12 was highly expressed in GBM tissues and could be regulated by miR-154. (A) Western blot analysis showed increased expression of TCF12 in mut-p53 GBM tissues compared with adjacent normal tissues. (B) TCF12-positive cells were increased and highly expressed in both the nucleus and cytoplasm of mut-p53 GBM tissues. (C) The luciferase reporter activity was decreased when cells were cotransfected with miR-154 mi and wt-TCF12 3’-UTR. (D) TCF12 protein levels were detected in U251 and U373 cells after transduction with miR-154 lentivirus (*p<0.05, **p<0.01, ns: no significance). |
P53 Regulates the Expression of TCF12 in an miR-154-Dependent Manner
Our study revealed that TCF12 was a target of miR-154, but whether p53 regulates TCF12 by miR-154 is still unknown. We assessed the expression of TCF12 in U251 cells transfected with wt-p53 or mut-p53 and in U87 cells transfected with p53 knockdown plasmid. Western blot analysis showed that wt-P53, not mut-P53, downregulated the expression of TCF12, and sh p53 led to increased TCF12 (Figure 6A and B). Furthermore, if U251 cells were cotransfected with wt-p53 and miR-154 inhibitor, the decrease in TCF12 was absent (Figure 6C). Correspondingly, TCF12 levels were not increased in U87 cells cotransfected with the sh p53 plasmid and miR-154 mimic (Figure 6D). Together, this result indicates that p53 is negatively correlated with the expression of TCF12. MiR-154 played a critical role in this process due to its inhibitory effect on TCF12. The novel P53/miR-154/TCF12 pathway is important in the progression of GBM.
 |
Figure 6 The regulation of TCF12 by p53 was mediated by miR-154. (A) TCF12 was detected in U251 cells after transfection with wt-p53 or mut-p53. (B) TCF12 was detected in U87 cells after transfection with sh p53. (C) MiR-154 inhibitor blocked the p53-induced decrease in TCF12 in U251 cells. (D) The miR-154 mimic reduced the sh p53-induced increase in TCF12 in U87 cells (*p<0.05, **p<0.01, ns: no significance). |
Overexpressed TCF12 Blocks the miR-154-Induced Inhibition of Migration, Invasion and EMT in GBM Cells
Although TCF12 was suggested to be regulated by miR-154, whether it mediates the effects of miR-154 on GBM still needs further investigation. We transfected TCF12 into Lenti-miR-154 transduced U251 cells. Western blot analysis showed that the protein level of TCF12 was higher in the co-overexpressed cells than in the cells transduced with lenti-miR-154 only, and TCF12 overexpression did not affect the level of miR-154 (Figure 7A and B). We conducted wound healing and Transwell assays in the above cells. We discovered that exogenously introduced TCF12 reversed the tumor suppressive effects of miR-154 on GBM cell lines. The migration distance and the number of invading cells all recovered to the control level (Figure 7C and D). We then detected the expression of EMT markers, including E-cadherin, N-cadherin and vimentin, in the cells. The increase in E-cadherin and the decrease in N-cadherin and vimentin induced by miR-154 were all reversed after TCF12 transfection. TCF12 blocked the inhibitory effect of miR-154 on EMT in GBM (Figure 7E).
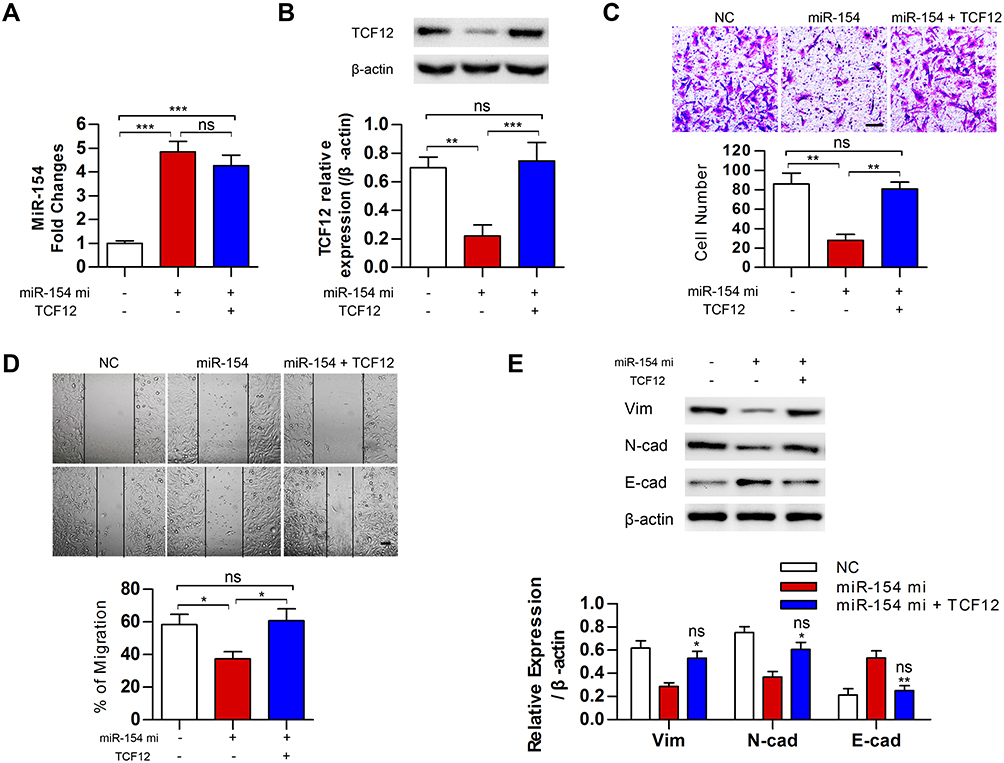 |
Figure 7 MiR-154 inhibited the malignancy of GBM cells by downregulating TCF12. (A) MiR-154 levels were analyzed in miR-154 stably expressed U251 cells after transfected with TCF12. (B) TCF12 protein levels were analyzed by Western blot in miR-154 stably expressed U251 cells after transfection. (C) Transwell assays showed that TCF12 increased the number of invasive cells compared with the number when overexpressing miR-154 alone. The experiment was conducted 24 h after transfection. (D) The migration distance of TCF12 transfected miR-154 stably expressed cells was longer than witch of the miR-154 only overexpressed cells. The experiment was conducted 24 h after transfection. (E) TCF12 prevented the miR-154-induced reduction in vimentin and N-cadherin and the increase in E-cadherin. *: compared with miR-154, ns: compared with NC (*p<0.05, **p<0.01, ***p<0.001, ns: no significance, Bar=100 μm). |
Discussion
In the current study, we identified the critical role of miR-154 in GBM and revealed its regulatory mechanisms. Aberrantly expressed miR-154 has been reported in several diseases and exerts different influences. miR-154 acts as a molecular marker to predict poor prognosis in renal cell carcinoma.31 In a rat model study, miR-154 promoted myocardial apoptosis in acute myocardial infarction.32 However, there is currently no clear understanding of the complex regulatory mechanisms of miR-154 in GBM. The important finding in our work was that wild-type p53 was indicated to be upstream of miR-154, and the mutation of p53 resulted in a decrease in miR-154 in GBM. Furthermore, TCF12 was identified as the direct target of miR-154, and miR-154 suppressed the progression of GBM by downregulating TCF12.
The tumor suppressor role of miR-154 has been reported in several cancers, but why its decrease occurs in GBM is still under investigation. Many miRNAs are reported to be regulated by proteins, especially some transcription factors. For instance, Twist1 regulates miR-148a, TGF-β1 promotes the expression of miR-205 and MYB controls miR-486-3p transcription.33–35 A previous study reported that SMAD3 can bind to the putative promoter of miR-154 in human lung fibroblasts.36 In our study, a p53 binding site-enriched region was detected within 1.5 kD upstream of the miR-154 gene, which was regarded as the promoter region. The deregulation of p53 has been widely described, and its mutations are widely observed in GBM tissues. The reactivation of endogenous p53 function has been explored in the treatment of GBM.37,38 Hence, we hypothesized that p53 acts as a promotive factor of miR-154, and its mutation leads to the loss of this function. Interestingly, miR-154 expression was found to be much lower in p53 mutant GBM tissues than in wt-p53 tissues. The same results were observed in GBM cell lines that contain different p53 gene backgrounds. We transfected p53 mutant U251 cells with the wt-p53 plasmid and found a great increase in miR-154. The expression of miR-154 was significantly decreased in wt-p53 U87 cells after knockdown of wt-p53. It can be assumed that mutant p53 loses its binding capacity or is inactivated to promote the expression of miR-154. As a great oncosuppressive transcription factor, p53 inhibits the progression of various tumors by regulating a set of downstream tumor-associated proteins, including p21, cdc2, survivin, cyclin G, and Bax.39–41 Our data reveal that miR-154 is also a target of wt-p53. Moreover, increased miR-154 inhibited the EMT in GBM and thereby decreased the migration and invasion of GBM cells. The in vivo tumor formation assays also confirm that miR-154 suppresses the growth of GBM. This indicates that the p53/miR-154 pathway plays an important role in the progression of GBM.
By altering the expression level of miR-154 in GBM cells, we proved that overexpressed miR-154 inhibits the growth of tumors and blocks the epithelial-mesenchymal transition of GBM cells. However, miRNAs must bind to the 3’-UTRs of their target mRNAs to come into play. Previous studies have reported some different targets of miR-154. For example, miR-154 functions as a tumor suppressor in non-small cell lung cancer by directly targeting BMI-1.42 Xu et al reported that miR-154 inhibits the growth and invasion of breast cancer cells by targeting E2F5.43 It was also reported that ZEB2 and RUNX2 act as targets of miR-154 in non-small cell lung cancer.13 In the current study, we found that highly expressed miR-154 prevented the EMT in GBM cells by increasing E-cadherin and decreasing N-cadherin and vimentin. By searching on TargetScan, TCT12, which was recently reported to be highly associated with EMT, was assumed to be a novel target of miR-154. TCF12 is a transcription factor because of its ability to recognize the DNA E-box motif as a member of the basic helix-loop-helix family.44,45 This protein is widely expressed in many tissues, and its level is elevated in several kinds of cancers.46,47 TCF12 was found to act as a transcriptional repressor of E-cadherin, and its overexpression was significantly correlated with the occurrence of colorectal cancer metastasis.48 Another study revealed that TCF12 targets HDAC1 to downregulate EMT markers, by which it influences the invasion of gallbladder cancer cells.27 The expression of TCF12 was increased in GBM tissues and negatively related to miR-154 in our study, which suggests that miR-154 suppresses the expression of TCF12. Further investigation revealed that miR-154 significantly suppresses the luciferase activity of the wild-type 3’-UTR of TCF12 in U251 cells but not that of the mutant 3’-UTR of TCF12. Moreover, when TCF12 was overexpressed in GBM cells, the increased miR-154 did not prevent the EMT or decrease the migration and invasion ability of GBM cells. Overall, TCF12 was proven to be the target of miR-154, and the tumor suppression function of miR-154 was shown to depend on the downregulation of TCF12.
The role of miR-154 in the development of glioma has been concerned in the recent years. Several studies have been conducted trying to illustrate the specific function of miR-154 in glioma. Although the tumor suppressive role of miR-154 has been reported in majority of the investigations, how miR-154 is downregulated in GBM is still controversial. Our study is the first to find that classical transcription factor p53 could regulate the expression of miR-154. Importantly, we discovered the relationship between the status of p53 mutation and the expression of miR-154. This novel p53/ miR-154/ TCF12 pathway offers possible therapeutic target for GBM, especially the p53 mutated subgroup of GBM.
Conclusion
In conclusion, microRNAs have emerged as key regulators of tumorigenesis and tumor progression. In this study, we provide evidence for the tumor suppressor role of miR-154 in GBM and provide novel insights into the mechanism by which miR-154 affects the development of GBM. Our results suggest that wt-p53 promotes the expression of miR-154, while miR-154 is downregulated in mutant p53 GBM tissues. Furthermore, TCF12 acts as a direct target of miR-154, and increased miR-154 contributes to the inhibition of EMT, migration, invasion and growth in GBM by downregulating TCF12. The present data highlight the important role of a novel p53/miR-154/TCF12 pathway in GBM. These findings provide potential clinical application of the p53/miR-154/TCF12 pathway in the treatment of GBM.
Abbreviations
GBM, glioblastoma multiforme; miRNAs, microRNAs; EMT, epithelial-mesenchymal transition; NHA, normal human astrocyte; 3’-UTRs, 3’-untranslated regions; TCF12, transcription factor 12; NC, negative control; DMEM, Dulbecco’s modified Eagle’s medium; PVDF, polyvinylidene fluoride; qRT–PCR, quantitative real-time reverse transcription–polymerase chain reaction.
Acknowledgment
We thank the financial support of the National Natural Science Foundation for Young Programs of China (No. 81903072, No. 81701304) and the National Natural Science Foundation for General Programs of China (No. 81772661, No. 81402081). We also thank Meng Xu for clinical tissue preparation.
Disclosure
All authors report no conflicts of interest in this work.
References
1. Mooney J, Bernstock JD, Ilyas A, et al. Current approaches and challenges in the molecular therapeutic targeting of glioblastoma. World Neurosurg. 2019;129:90–100. doi:10.1016/j.wneu.2019.05.205
2. Brown TJ, Brennan MC, Li M, et al. Association of the extent of resection with survival in glioblastoma: a systematic review and meta-analysis. JAMA oncol. 2016;2(11):1460–1469. doi:10.1001/jamaoncol.2016.1373
3. Sanai N, Berger MS. Surgical oncology for gliomas: the state of the art. Nat Rev Clin Oncol. 2017;15(2):112–125. doi:10.1038/nrclinonc.2017.171
4. Iwadate Y. Epithelial-mesenchymal transition in glioblastoma progression. Oncol Lett. 2016;11(3):1615–1620. doi:10.3892/ol.2016.4113
5. Wang J, Zhang KY, Liu SM, Sen S. Tumor-associated circulating microRNAs as biomarkers of cancer. Molecules. 2014;19(2):1912–1938. doi:10.3390/molecules19021912
6. O’Brien J, Hayder H, Zayed Y, Peng C. Overview of MicroRNA Biogenesis, Mechanisms of Actions, and Circulation. Front Endocrinol. 2018;9:402. doi:10.3389/fendo.2018.00402
7. Kumar S, Reddy AP, Yin X, Reddy PH. Novel MicroRNA-455-3p and its protective effects against abnormal APP processing and amyloid beta toxicity in Alzheimer’s disease. Biochimica Et Biophysica Acta Mol Basis Dis. 2019;1865(9):2428–2440. doi:10.1016/j.bbadis.2019.06.006
8. Subramaniam S, Jeet V, Clements JA, Gunter JH, Batra J. Emergence of MicroRNAs as Key Players in Cancer Cell Metabolism. Clin Chem. 2019;65(9):1090–1101. doi:10.1373/clinchem.2018.299651
9. Kumar Kingsley SM, Vishnu Bhat B. Role of MicroRNAs in the development and function of innate immune cells. Int Rev Immunol. 2017;36(3):154–175. doi:10.1080/08830185.2017.1284212
10. Lo Sardo F, Forcato M, Sacconi A, et al. MCM7 and its hosted miR-25, 93 and 106b cluster elicit YAP/TAZ oncogenic activity in lung cancer. Carcinogenesis. 2017;38(1):64–75. doi:10.1093/carcin/bgw110
11. Wu P, Cai J, Chen Q, et al. Lnc-TALC promotes O(6)-methylguanine-DNA methyltransferase expression via regulating the c-Met pathway by competitively binding with miR-20b-3p. Nat Commun. 2019;10(1):2045. doi:10.1038/s41467-019-10025-2
12. Banelli B, Forlani A, Allemanni G, Morabito A, Pistillo MP, Romani M. MicroRNA in Glioblastoma: an Overview. Int J Genomics. 2017;2017:7639084. doi:10.1155/2017/7639084
13. Lingling J, Xiangao J, Guiqing H, Jichan S, Feifei S, Haiyan Z. SNHG20 knockdown suppresses proliferation, migration and invasion, and promotes apoptosis in non-small cell lung cancer through acting as a miR-154 sponge. Biomed Pharmacother. 2019;112:108648. doi:10.1016/j.biopha.2019.108648
14. Xu M, Chen X, Lin K, et al. The long noncoding RNA SNHG1 regulates colorectal cancer cell growth through interactions with EZH2 and miR-154-5p. Mol Cancer. 2018;17(1):141. doi:10.1186/s12943-018-0894-x
15. Wang J, Fang Y, Liu YF, et al. MiR-154 inhibits cells proliferation and metastasis in melanoma by targeting AURKA and serves as a novel prognostic indicator. Eur Rev Med Pharmacol Sci. 2019;23(10):4275–4284. doi:10.26355/eurrev_201905_17932
16. Yang L, Yan Z, Wang Y, Ma W, Li C. Down-expression of miR-154 suppresses tumourigenesis in CD133(+) glioblastoma stem cells. Cell Biochem Funct. 2016;34(6):404–413. doi:10.1002/cbf.3201
17. Wang X, Sun S, Tong X, et al. MiRNA-154-5p inhibits cell proliferation and metastasis by targeting PIWIL1 in glioblastoma. Brain Res. 2017;1676:69–76. doi:10.1016/j.brainres.2017.08.014
18. Parsons DW, Jones S, Zhang X, et al. An integrated genomic analysis of human glioblastoma multiforme. Science. 2008;321(5897):1807–1812. doi:10.1126/science.1164382
19. England B, Huang T, Karsy M. Current understanding of the role and targeting of tumor suppressor p53 in glioblastoma multiforme. Tumour Biol. 2013;34(4):2063–2074. doi:10.1007/s13277-013-0871-3
20. Chen L, Jiang Z, Ma H, et al. Volatile oil of Acori Graminei Rhizoma-Induced apoptosis and Autophagy are dependent on p53 Status in Human Glioma Cells. Sci Rep. 2016;6(1):21148. doi:10.1038/srep21148
21. Khoo KH, Verma CS, Lane DP. Drugging the p53 pathway: understanding the route to clinical efficacy. Nat Rev Drug Discov. 2014;13(3):217–236.
22. Wu CW, Peng ML, Yeh KT, Tsai YY, Chiang CC, Cheng YW. Inactivation of p53 in pterygium influence miR-200a expression resulting in ZEB1/ZEB2 up-regulation and EMT processing. Exp Eye Res. 2016;146:206–211. doi:10.1016/j.exer.2016.03.012
23. Chen J, Wang J, Li H, Wang S, Xiang X, Zhang D. p53 activates miR-192-5p to mediate vancomycin induced AKI. Sci Rep. 2016;6:38868. doi:10.1038/srep38868
24. Moscetti I, Cannistraro S, Bizzarri AR. Probing direct interaction of oncomiR-21-3p with the tumor suppressor p53 by fluorescence, FRET and atomic force spectroscopy. Arch Biochem Biophys. 2019;671:35–41. doi:10.1016/j.abb.2019.05.026
25. Zhang B, Wang X, Deng J, et al. p53-dependent upregulation of miR-16-2 by sanguinarine induces cell cycle arrest and apoptosis in hepatocellular carcinoma. Cancer Lett. 2019;459:50–58. doi:10.1016/j.canlet.2019.05.042
26. Tang X, Hou Y, Yang G, et al. Stromal miR-200s contribute to breast cancer cell invasion through CAF activation and ECM remodeling. Cell Death Differ. 2016;23(1):132–145. doi:10.1038/cdd.2015.78
27. He J, Shen S, Lu W, et al. HDAC1 promoted migration and invasion binding with TCF12 by promoting EMT progress in gallbladder cancer. Oncotarget. 2016;7(22):32754–32764. doi:10.18632/oncotarget.8740
28. Zhu G, Tao L, Wang R, et al. Endoplasmic reticulum stress mediates distinct impacts of sevoflurane on different subfields of immature hippocampus. J Neurochem. 2017;142(2):272–285. doi:10.1111/jnc.14057
29. Wang J, Zhu G, Huang L, et al. Morphine administration induces change in anxiety-related behavior via Wnt/beta-catenin signaling. Neurosci Lett. 2017;639:199–206. doi:10.1016/j.neulet.2017.01.005
30. Crane EK, Kwan SY, Izaguirre DI, et al. Nutlin-3a: a Potential Therapeutic Opportunity for TP53 Wild-Type Ovarian Carcinomas. PLoS One. 2015;10(8):e0135101. doi:10.1371/journal.pone.0135101
31. Lin C, Li Z, Chen P, et al. Oncogene miR-154-5p regulates cellular function and acts as a molecular marker with poor prognosis in renal cell carcinoma. Life Sci. 2018;209:481–489. doi:10.1016/j.lfs.2018.08.044
32. Sun HY, Wang XL, Ma LC, et al. Influence of MiR-154 on myocardial apoptosis in rats with acute myocardial infarction through Wnt/beta-catenin signaling pathway. Eur Rev Med Pharmacol Sci. 2019;23(2):818–825. doi:10.26355/eurrev_201901_16896
33. Haftmann C, Stittrich AB, Zimmermann J, et al. miR-148a is upregulated by Twist1 and T-bet and promotes Th1-cell survival by regulating the proapoptotic gene Bim. Eur J Immunol. 2015;45(4):1192–1205. doi:10.1002/eji.201444633
34. Duan Y, Chen Q. TGF-beta1 regulating miR-205/miR-195 expression affects the TGF-beta signal pathway by respectively targeting SMAD2/SMAD7. Oncol Rep. 2016;36(4):1837–1844. doi:10.3892/or.2016.5023
35. Bianchi E, Bulgarelli J, Ruberti S, et al. MYB controls erythroid versus megakaryocyte lineage fate decision through the miR-486-3p-mediated downregulation of MAF. Cell Death Differ. 2015;22(12):1906–1921. doi:10.1038/cdd.2015.30
36. Milosevic J, Pandit K, Magister M, et al. Profibrotic role of miR-154 in pulmonary fibrosis. Am J Respir Cell Mol Biol. 2012;47(6):879–887. doi:10.1165/rcmb.2011-0377OC
37. Zhang Y, Dube C, Gibert M, et al. The p53 Pathway in Glioblastoma. Cancers. 2018;10(9):9. doi:10.3390/cancers10090297
38. Doan P, Musa A, Candeias NR, Emmert-Streib F, Yli-Harja O, Kandhavelu M. Alkylaminophenol Induces G1/S Phase Cell Cycle Arrest in Glioblastoma Cells Through p53 and Cyclin-Dependent Kinase Signaling Pathway. Front Pharmacol. 2019;10:330. doi:10.3389/fphar.2019.00330
39. Kruiswijk F, Labuschagne CF, Vousden KH. p53 in survival, death and metabolic health: a lifeguard with a licence to kill. Nat Rev Mol Cell Biol. 2015;16(7):393–405. doi:10.1038/nrm4007
40. Guo SL, Ye H, Teng Y, et al. Akt-p53-miR-365-cyclin D1/cdc25A axis contributes to gastric tumorigenesis induced by PTEN deficiency. Nat Commun. 2013;4(1):2544. doi:10.1038/ncomms3544
41. Renner G, Janouskova H, Noulet F, et al. Integrin alpha5beta1 and p53 convergent pathways in the control of anti-apoptotic proteins PEA-15 and survivin in high-grade glioma. Cell Death Differ. 2016;23(4):640–653. doi:10.1038/cdd.2015.131
42. Liu S, Yang Y, Chen L, Liu D, Dong H. MicroRNA-154 functions as a tumor suppressor in non-small cell lung cancer through directly targeting B-cell-specific Moloney murine leukemia virus insertion site 1. Oncol Lett. 2018;15(6):10098–10104.
43. Xu H, Fei D, Zong S, Fan Z. MicroRNA-154 inhibits growth and invasion of breast cancer cells through targeting E2F5. Am J Transl Res. 2016;8(6):2620–2630.
44. Yi S, Yu M, Yang S, Miron RJ, Zhang Y. Tcf12, A member of basic helix-loop-helix transcription factors, mediates bone marrow mesenchymal stem cell osteogenic differentiation in vitro and in vivo. Stem Cells. 2017;35(2):386–397. doi:10.1002/stem.2491
45. Sjogren H, Wedell B, Meis-Kindblom JM, Kindblom LG, Stenman G, Kindblom JM. Fusion of the NH2-terminal domain of the basic helix-loop-helix protein TCF12 to TEC in extraskeletal myxoid chondrosarcoma with translocation t(9;15)(q22;q21). Cancer Res. 2000;60(24):6832–6835.
46. Wang X, Gao S, Xie F, et al. High expression of TCF12 contributes to gastric cancer development via being target regulated by miR-183 and activating PI3K/AKT pathway. J Cell Biochem. 2019;120:13903.
47. Chen Y, Wang Q, Wang Q, et al. DEAD-box helicase 5 interacts with transcription factor 12 and promotes the progression of osteosarcoma by stimulating cell cycle progression. Front Pharmacol. 2018;9:1558. doi:10.3389/fphar.2018.01558
48. Lee CC, Chen WS, Chen CC, et al. TCF12 protein functions as transcriptional repressor of E-cadherin, and its overexpression is correlated with metastasis of colorectal cancer. J Biol Chem. 2012;287(4):2798–2809. doi:10.1074/jbc.M111.258947
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.