Back to Journals » OncoTargets and Therapy » Volume 13
P53 Mutant p53N236S Regulates Cancer-Associated Fibroblasts Properties Through Stat3 Pathway
Authors Liu Q, Yu B, Tian Y, Dan J, Luo Y, Wu X
Received 28 August 2019
Accepted for publication 10 January 2020
Published 14 February 2020 Volume 2020:13 Pages 1355—1363
DOI https://doi.org/10.2147/OTT.S229065
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Geoffrey Pietersz
Qing Liu, Biao Yu, Yingbin Tian, Juhua Dan, Ying Luo, Xiaoming Wu
Laboratory of Molecular Genetics of Aging & Tumor, Medical School, Kunming University of Science and Technology, Kunming, Yunnan 650500, People’s Republic of China
Correspondence: Ying Luo; Xiaoming Wu
Email [email protected]; [email protected]
Background: Cancer-associated fibroblasts (CAFs) play important roles in cancer development and progression. Recent studies show that p53 plays a cell non-autonomous tumor-suppressive role to restrict tumor growth in CAFs. However, the role of p53 mutant in CAFs remains obscure.
Methods: In this study, the contribution of p53 mutant p53N236S (p53S) to CAFs activation was examined using mouse embryonic fibroblasts (MEFs) from wild-type (WT), p53 deficient (p53-/-) and p53S/S mice. The role of p53S in MEFs in inducing prostate cancer cell growth and metastasis was studied by utilizing xenograft models and transwell assays. The effects of p53S on the properties of CAFs were assessed by measuring CAFs-specific factors expression and functional collagen contraction assay. Moreover, Microarray data were analyzed by GSEA and Stat3 signaling was inhibited to further determine p53S’s role in the CAFs activation.
Results: We found that p53S/S MEF accelerated cancer cells growth and metastasis compared with WT and p53-/- MEF. We also found that p53S induced significantly increasing collagen contraction in fibroblasts and overexpression of CAFs-specific factors, such as α-smooth muscle actin (α-SMA), FGF10 and CXCL12. p53S regulated these CAF-specific properties through Stat3 activation.
Conclusion: Our results illustrate that p53S plays an important role in CAFs activation by the Stat3 pathway. The study indicates that cancer cells and fibroblasts interaction promotes prostate cancer cell growth, migration and invasion due to p53S expression in fibroblasts.
Keywords: p53N236S, cancer-associated fibroblasts, Stat3 pathway
Introduction
Although carcinomas, which account for approximately 90% of human cancers, are derived from the epithelia, the tumor stroma exerts a powerful influence on cancer behavior, such as tumor cell growth, invasion, metastasis, and evading immune responses. Cancer-associated fibroblasts (CAFs) are a major cell component of the tumor stroma.1,2 Previous studies have shown that CAFs are differentiated from quiescent fibroblasts and highly express α-smooth muscle actin (α-SMA), fibroblast-specific protein-1 (FSP1) and fibroblast activation protein (FAP) compared with normal fibroblasts (NFs), which have been established as a hallmark of CAFs.3 It is widely accepted that CAFs promote tumor growth and their aggressive features largely through the production and secretion of a variety of soluble pro-carcinogenic factors.
The p53 tumor suppressor is known in the prevention of tumor development by maintaining genome integrity and cellular homeostasis. p53 binds DNA in a sequence-specific manner and controls the expression of target genes involved in the regulation of cell cycle, apoptosis, stem cell differentiation, senescence, DNA repair, and metabolism.4,5 However, recent studies suggested that stromal p53 also possessed non–cell-autonomous functions in tumor biology. Interestingly, p53 expression was significantly lower in CAFs than in NFs.6 Additionally, the suppression of p53 expression in NFs promotes the acquisition of CAFs characteristics.7 p53 accumulation in stromal fibroblasts can reduce tumor growth and enhance the death of adjacent tumor cells.8 Moreover, decreased stromal p53 activity in tumors is more resistant to chemotherapy.9 p53 also regulates macrophage functions in a cell non-cell-autonomous manner, thereby promoting an anti-tumorigenic microenvironment.10
p53 is mutated in more than 50% of human cancers. Cancer-associated mutations in the p53 gene can lead not only to loss of its tumor-suppressive functions (LOF) but often also to gain of tumor-promoting activities (GOF).11 A high frequency of p53 mutations in the tumor surrounding stroma has reported, and p53 mutations in breast cancer stromal cells have been reported to be closely associated with nodal metastasis.12 Mechanistically, altered p53 functionality in CAFs might modify the CAF secretome and exert a cancer-promoting effect of CAFs.13 Based on the above findings, p53 mutation of tumor stromal fibroblasts may play a very important role in carcinoma progression.
The p53N236Smutation (p53N239S in humans, referred to here as p53S) has been reported as one of the less common but recurrent mutations in human cancers and has been labeled as hot-spot mutations in the TCGA database.14 In our previous study, we generated the p53S knock-in mouse. The p53S/S mice manifested highly invasive lymphomas and metastatic sarcomas.15 Nevertheless, it is unknown whether p53S mutation in fibroblasts could play any role in cancer growth and metastasis.
The present study aimed to elucidate the mechanism by which p53S mutation in fibroblast actively contributes to CAFs characteristics to stimulate tumor progression, using three genotypes of mouse embryonic fibroblasts (MEFs) from wild-type (WT), p53-/- and p53S/S. The p53S mutation in MEF led to accelerated prostate cancer cell growth, migration and invasion. Moreover, p53S can induce the expression of CAFs marker α-SMA and collagen contraction in fibroblasts. Strikingly, we found that the repress Stat3 pathway could rescue p53S-mediated biological features of the CAFs. We thus propose that p53S impact CAFs characteristics by the Stat3 pathway.
Materials and Methods
Cell and Mice
As described previously, we harvested MEFs with different genotypes in E13.5 days and cultured in Dulbecco’s Modified Eagle Medium (DMEM) with 10% heat-inactivated fetal bovine serum (FBS) at 37°C with 5% CO2 and 3% O2.
The genotypes of MEFs were WT, p53-/-, p53S/S and p53R172H. All cell lines were authenticated at the beginning of the planned experiments by genotype analysis.
Human PC-3 cell and H1299 cell were purchased from China Infrastructure of Cell Line Resources and cultured in RPMI-1640 supplemented with 10% (v/v) heat-inactivated FBS.
All the mice involved in procedures were in line with the Guide for the Care and Use of Laboratory Animals. Animal experiments and protocols were approved by the Animal Care and Use Committee of Kunming University of Science & Technology.
Xenograft Experiments
Xenograft experiments were performed using male BALB/c nude mice aged 6–8 weeks (n=2 per group). PC-3 cells (2× 106) were suspended in 50 μL PBS and injected subcutaneously into the left and right flank of BALB/c nude mice, respectively, in the presence or absence of 1 × 106 WT, p53-/- or p53S/S MEFs. Tumor size was measured every 3 days to calculate tumor volume using the following formula: (tumor length × [tumor width]2)/2. *P=0.0186; **P=0.0002; P values relate to the last time point. Thirty days after the injections, the animals were sacrificed and tumors were excised and weighted. **P=0.008; **P=0.0002; P values relate to the last time point.
Quantitative Real-Time PCR (qRT-PCR) Analysis
Total RNA was isolated from MEFs using Trizol (Invitrogen) according to the manufacturer’s protocol. RNA quantity was determined by agarose gel electrophoresis and by spectrophotometry. Single-stranded complementary DNA (cDNA) was obtained from reverse transcription of 600 ng of RNA using RT-PCR kit (BD Biosciences). qRT-PCR was performed in triplicate using SYBR (Takara Bio, Japan). β-actin primers: 5ʹ-AGAGGGAAATCGTGCGTGAC −3ʹ, 5ʹ-CAATAGTGATGACCTGGCCGT −3ʹ; α-SMA primers: 5'-ATGCAGAAGGAGATCACAGC-3', 5'-CAGCTTCGTCGTATTCCTGT-3'; FAP primers: 5'-TTCTGCCTCCTCAGTTTGAC-3', 5'-CTGTGAGCTGGTCCTCAACT-3'; FSP1 primers: 5'-AGGGTGACAAGTTCAAGCTG-3', 5'-GCAGGACAGGAAGACACAGT-3'; Vimentin primers: 5'-TCCTCGAGCAGCAGAACAAAATCC-3', 5'-CAGGGCAGCAGTGAGGTC-3'; IL6 primers: 5'-TAGTCCTTCCTACCCCAATTTCC-3', 5'-TTGGTCCTTAGCCACTCCTTC-3'; CXCL12 primers: 5'-TGCATCAGTGACGGTAAACCA-3', 5'-CACAGTTTGGAGTGTTGAGGAT-3'; CXCL10 primers: 5'-CCAAGTGCTGCCGTCATTTTC-3', 5'-GGCTCGCAGGGATGATTTCAA-3'; CXCL1 primers: 5'-GCTTGAAGGTGTTGCCCTCA -3', 5'-CTATGACTTCGGTTTGGGTGC-3'; FGF10 primers: 5ʹ- CACATTGTGCCTCAGCCTTTC-3ʹ, 5ʹ- CCTCTATTCTCTCTTTCAGCTTAC-3ʹ; The relative expression level of each mRNA was analyzed by the 2−ΔΔCt method. All experiments were repeated three times.
Microarray Assay
Mouse embryonic fibroblast cell samples were harvested from E13.5 WT (p53+/+) or p53S/S mouse embryos. To reduce individual heterogeneity, three MEF cell samples were collected from embryos of three individual pregnant mice.
For the microarray assay, the details are described in another paper.33 Briefly, total RNA was extracted from samples using the Trizol reagent (Invitrogen, Carlsbad, CA, USA) and was purified with the mirVana miRNA Isolation Kit (Ambion, Austin, TX, USA), according to manufacturer’s protocol. The RNA samples analyzed by Agilent mouse mRNA Array chips (8960K format, Agilent, Carpinteria, CA, USA). The array data were analyzed for data summarization, normalization, and quality control using the GENESPRING software V12 (Agilent). The expression signals of the microarray were used for GSEA analysis, the Hallmark gene set was used as the reference.
Western Blotting
Cells were lysed in RIPA buffer containing Protease Inhibitor Cocktail (Roche). Sample proteins (20 μg) were separated by SDS-PAGE and then transferred to PVDF membranes. After blocking in 10% nonfat milk for 1 hr at room temperature, membranes were incubated with primary antibodies overnight at 4°C or 2 hrs at room temperature. The membranes were then incubated with horseradish peroxidase-labeled secondary antibodies and visualized with ECL. The following primary antibodies were used: anti-p-Stat3(1:1000, CST), anti-Stat3(1:1000, CST), anti-α-SMA (1:4000, abcam), anti-p-p53 (1:1000,CST), anti-p53 (1:800, santa) and anti-γ-tubulin (1:8000, Millipore).
ELISA
Supernatants from 24-hrs MEFs cultures were harvested, and CXCL12 protein expression was quantified by ELISA according to the manufacturer’s instructions (Abcam). The OD was used at 450 nm on a standard ELISA plate reader. These experiments were performed in triplicate. *P=0.0141; **P<0.001
Fibroblast Contraction Assay
The contraction assay was performed to evaluate the contractility of MEF cells. MEFs were embedded in 400 µL of type I collagen (Corning, Germany). The final cell density was 3 × 105 cells/mL with 1 mg/mL collagen. This mixture was seeded in a 12-well plate, incubated for 24 or 48 hrs in a humidified incubator at 37°C and imaged. To calculate the relative collagen area (%), scanned images were quantified with Image J software.
Conditional Media
MEFs were seeded in DMEM supplemented with 10% FBS on a 10-cm2 dish and allowed to reach confluence. The medium was then replaced with serum-reduced DMEM (0.1% serum). MEFs (WT, p53-/-, p53S/S, p53S/S+Stattic) were incubated at 37°C for 24 hrs. The medium was then collected, centrifuged, and stored at −20°C for later use.
Tumor Cell Migration and Invasion Assays
Transwell chambers without Matrigel were used to examine tumor cell ability of migration and transwell chambers were pre-coated with 50-µl Matrigel to measure cell invasion (BD Biosciences). In brief, on the top of inserts: tumor cells in serum-free medium, 5×103 PC-3 or 8 ×103 H1299, were added to 24-well transwell plates with 8-mm pores. On the bottom chamber: MEFs (WT, p53-/- or p53S/S MEFs, 1×104 each), or conditional media from MEFs (WT, p53-/-, p53S/S MEFs or p53S/S MEF +Stattic), in 10% FBS were added. The control group was added DMEM with 10% FBS in the bottom chamber. After incubation for 18 hrs, the invaded cells were fixed with 70% ethanol, stained with crystal violet, and 5 random fields were counted under a light microscope. Each experiment was repeated twice. Quantification of migration/invasion cells was calculated as the percentage of migration/invasion cells relative to the total number of tumor cells, which were seeded on the top of inserts.
Statistical Analysis
Student′s t-tests were used to assess the significance of the differences between the two groups of data (p < 0.05 is deemed significant).
Results
High Expression of CAFs-Specific Genes in Sarcoma Derived from p53S/S Mice
Our previous study showed that p53S/S mice manifested to an early onset of sarcomas that metastasized to various organs, such as liver and lung.15 We attempted to understand the phenotypes resulting from the p53S mutant that were described above, we performed qRT-PCR analyses on total RNA extracted from sarcoma derived from p53-/- and p53S/S mice. The sarcoma, derived from p53-/- and p53S/S mice, are both primary and not any differences according to their histological staining (not shown in this paper). Surprisingly, p53S/S tumor tissues had higher expression of CAFs markers; α-SMA (p=0.004), FAP (p=0.001), FSP1 (p=0.001) and vimentin (p<0.001) compared with p53-/- tumor tissues (Figure 1A). We further focused on the mRNA expression of CAFs-specific secreted factors, SDF-1, CXCL1, CXCL10, and IL6 in p53S/S tumors. We found that SDF-1 (p=0.012), CXCL1 (p=0.002), CXCL10 (p<0.001), and IL6 (p<0.001) mRNA was significantly up-regulated in p53S/S tumor tissues (Figure 1B).
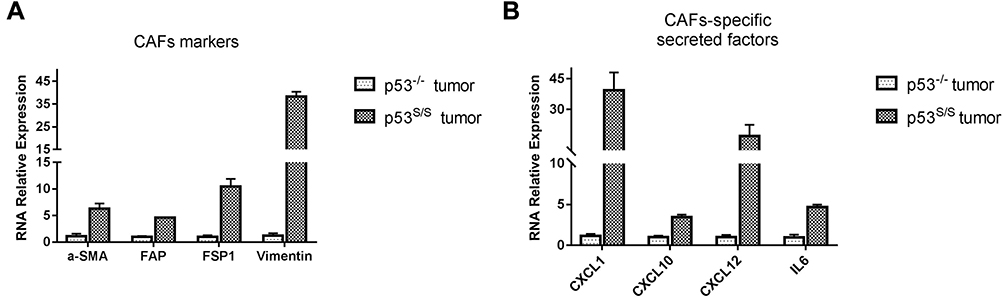 |
Figure 1 High expression of CAFs-specific genes in sarcoma derived from p53S/S mice. (A) High expression of CAFs biomarker genes in p53S/S tumor (n=2 per group). (B) High expression of CAFs-secreted factors in p53S/S tumor (n=2 per group). |
Altogether, these data imply that the presence of p53S in the host would provide a more tumor-promoting microenvironment, contributing to the promotion of tumor growth and metastasis.
p53S in Stromal Fibroblasts Exerts a Gain of Function Effect on Tumor Growth in vivo
CAFs is one major type of component in the tumor microenvironment. It is well established that CAFs promote the proliferation, migration and invasion of cancer cells. To elucidate whether p53S would promote CAFs properties, we prepared MEFs from the wild type (WT), p53 null (p53-/-) or p53 N236S mutant (p53S/S) mouse.
The human prostate epithelial cancer cell line PC-3 were employed subcutaneous injection into male nude mice, either alone or together with WT, p53-/- and p53S/S MEFs. Tumor size was metered with calipers every 3 days. Analysis of tumour growth revealed that co-injection with WT MEFs failed to induce greater tumor growth in this group, while those mice co-injected with p53-/- MEFs showed greater tumor size compared to those injected with PC-3 alone (Figure 2A and 2B). Most notably, co-injection with p53S/S MEF significantly accelerated the growth of PC-3 tumors more than p53-/- MEFs (Figure 2A and 2B), suggesting that expression of p53S in MEF supports tumour growth.
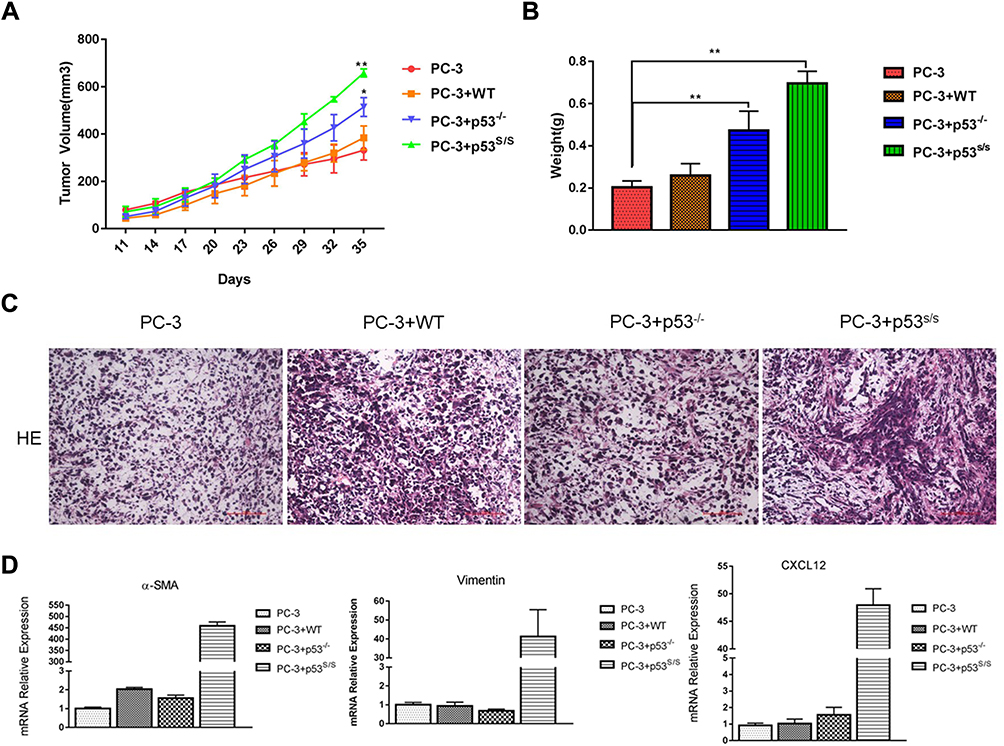 |
Figure 2 Effect of p53S on PC3 tumor growth in nude mice. (A) PC-3 cell was injected, either alone or together, WT, p53-/-, p53S/S MEFs, into the right flank of male nude mice (n = 3 per group; total inoculation volume = 50 μL). *p<0.05; **p<0.005. (B) Tumors were excised and weighted 35 days later. **p<0.005. (C) H&E staining of histologic sections from a representative tumor of each group. (D) qRT-PCR analysis of α-SMA, Vimentin and CXCL12 expression in the tumor tissue. |
Histopathologic examination revealed that tumors generated by PC-3 alone exhibited a modest stromal response. Tumors generated by PC3 together with p53S/S MEFs displayed the highest density of stroma (Figure 2C). Moreover, using qRT-PCR, we observed CAFs markers and CAFs-specific secreted factor overexpression in PC-3 co-injection with p53S/S MEF, such as α-SMA, vimentin, CXCL12 (Figure 2D). Taken together, p53S in the MEF has a new gain of function effect on acceleration of the growth of PC-3 epithelial tumor.
p53S in Stromal Fibroblasts Promotes Cancer Cell Migration and Invasion
CAFs can promote tumor cell migration and invasion. To determine whether p53S in MEFs can promote tumor cell migration and invasion, we cultured PC-3 cells either alone or co-cultured with the WT, p53-/-, or p53S/S MEFs. The transwell assay demonstrated that p53S/S MEF markedly enhanced the migration of PC-3 cells, while WT, p53-/- MEFs only slightly promoted the migration of PC-3 cells compared with untreated cells (Figure 3A and 3B). Moreover, p53S/S MEF also significantly increased the invasion of PC-3 cells (Figure 3B and 3C). To confirm these findings, we also performed transwell assays using lung cancer cell line H1299. Similar to PC-3, H1299 migrated more toward control p53S/S MEF than toward control, WT and p53-/- MEFs (Figure 4D). Therefore, these results demonstrated that p53S enhances the migration and invasion of tumor cell.
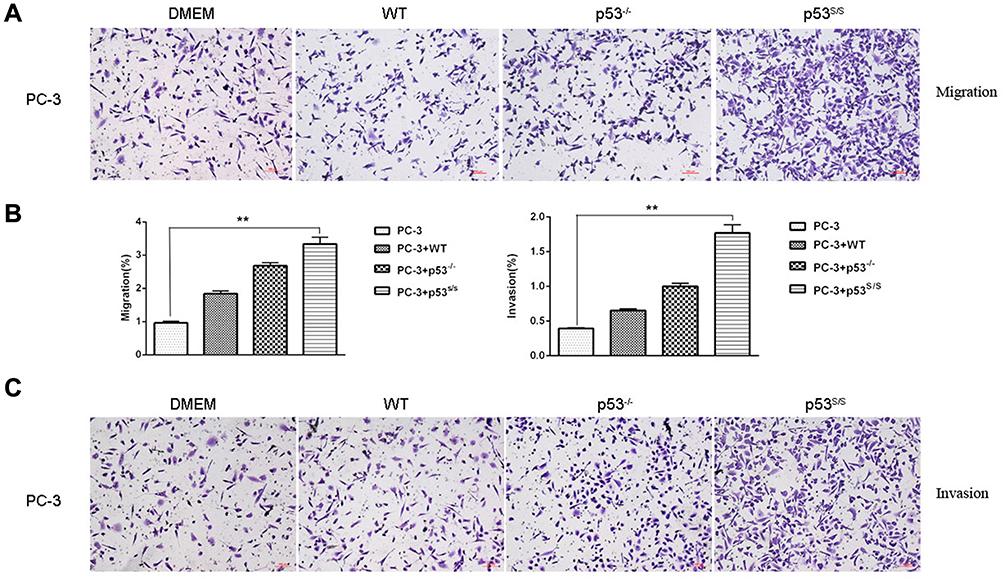 |
Figure 3 Effect of p53S on PC3 tumor migration and invasion. (A). Transwell migration assay of PC-3 cells following alone or co-culture with WT, p53-/-and p53S/S MEFs after 24 hrs. (B) Average migration ±SEM from three independent experiments performed as in A; Average invasion ±SEM from three independent experiments performed as in C. **p<0.005. (C) Transwell invasion assay of PC-3 cells following alone or co-culture with WT, p53-/-and p53S/S MEFs after 24 hrs. |
p53S in Stromal Fibroblasts Play a Stimulatory Role in Contractile Activity and Regulate the Expression of CAF-Specific Genes
CAFs also exhibit an increased ability to induce collagen gel contraction upon growth in such gels, enhanced contractile activity is an important pathological feature of CAFs, we examined whether p53S/S MEF could enhance the cell contractility of fibroblast and cause mechanical stress similar as that of CAFs. Fibroblast contraction assays showed the area of collagen gel with WT MEFs was almost similar with p53-/- MEFs. As expected, p53S/S MEF displayed markedly greater contractile activity than WT and p53-/- MEFs (Figure 4C). Thus, p53S indeed promote the contractile activity of fibroblast.
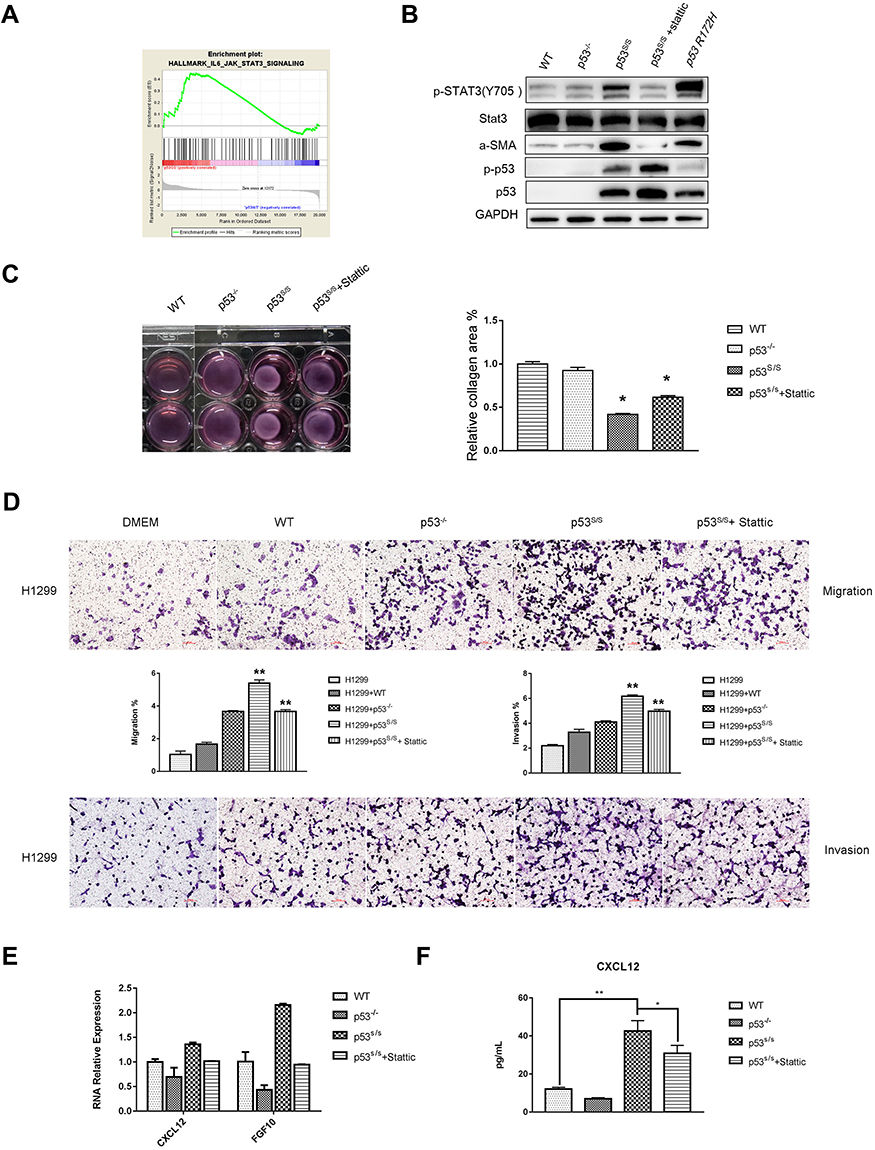 |
Figure 4 p53S Regulates CAFs Properties through Stat3 pathway. (A) GSEA plot indicating significant IL6-JAK-Stat3-activated signatures in p53S/S MEFs. (B) Western blotting analysis of α-SMA, total-Stat3, p-Stat3, total-p53, p-p53 expression in WT, p53-/-, p53S/S MEFs p53S/S MEFs treated with Stattic and p53R172H MEFs. (C) WT, p53-/-, p53S/S MEFs and p53S/S MEFs treated with Stattic were subjected to a collagen gel contraction assay in duplicate and imaged after 24 hrs. Error bars represent SD (n=3), *p<0.05. (D) Transwell migration assay of H1299 cells following alone or co-culture with conditional media from WT, p53-/-and p53S/S MEFs and p53S/S MEF treated with Stattic after 24 hrs. Average migration ±SEM from three independent experiments. Transwell invasion assay of H1299 cells following alone or co-culture with conditional media from WT, p53-/-and p53S/S MEFs and p53S/S MEF treated with Stattic after 24 hrs. Average invasion ±SEM from three independent experiments. **p<0.005. (E) qRT-PCR analysis of α-SMA and FGF10 expression in WT, p53-/-, p53S/S MEFs and p53S/S MEFs treated with Stattic. (F) ELISA analysis of CXCL12 expression in WT, p53-/-, p53S/S MEFs and p53S/S MEFs treated with Stattic. *p<0.05; **p<0.005. |
To explore the factors that are responsible for inducing tumor cell proliferation, migration, invasion and collagen gel contraction in p53S/S MEF, we found that α-SMA, CXCL12 and FGF10 were upregulated in p53S/S MEF (Figure 4B, 4E and 4F). Together, these results indicate that p53S may exert a gain of function led to overexpression of α-SMA, CXCL12 and FGF10, resulting in CAFs activation.
p53S Regulates CAFs Properties Through Stat3 Pathway
We next explore the role of p53S in regulating CAFs properties, we performed microarrays to compare the expression profile of p53S/S MEF with wild-type p53 MEF. We then group the microarray data and analyzed the Gene Set Enrichment Analysis (GSEA). The Hallmark gene set was used for enrichment analysis.
GSEA confirmed that the Stat3 pathway was significantly upregulated in p53S/S MEF (Figure 4A). The Stat3 protein is a key factor in Stat3 pathway activation, and phospho-Stat3 (Tyr705) is the activated status of Stat3. Western blot analysis showed a highly increased p-Stat3Tyr705 in p53S/S MEF. To further confirm whether the effects of p53S on CAFs Properties were Stat3 pathway dependent, we treated p53S/S MEF with 7.5 nM Stattic, an inhibitor of Stat3, and found that Stattic could reverse p53S-mediated CAF marker α-SMA (Figure 4B). Fibroblast contraction assays also showed that Stattic strongly compromised contractile activity in the p53S/S MEF (Figure 4C). Moreover, p53S/S MEF-induced H1299 migration and invasion were clearly inhibited by Stattic (Figure 4D). Consistent with these, we found that p53S-induced CAFs-specific secreted factor CXCL12 and FGF10 augmentation were dependent on the Stat3 pathway (Figure 4E and 4F). Collectively, these data demonstrated that p53S can regulate CAF function through Stat3 pathway.
We evaluated whether other mutant p53 may also exert cell non-autonomous gain of function effect on CAFs activation when expressed in stromal cells. To that end, MEFs were prepared from the p53R172H mice. p53R172H were the mouse equivalent of the human cancer-associated hotspot mutant p53R175H. As seen in Figure 4B, α-SMA and p-Stat3Tyr705 were upregulated in p53R175H, suggesting that p53S has a like hot-spot gain of function effect on CAFs activation.
Discussion
Tumor development depends on a complex interaction between malignant cells and their microenvironment.16 Cancer-associated fibroblasts (CAFs), as a major cellular component of the tumor microenvironment, modulate tumor growth and metastasis.17 CAFs, marked by smooth muscle actin (SMA), acquire a phenotype similar to that of myofibroblasts, which are activated in wound healing and fibrosis, with a morphology and function that differ from those of normal fibroblasts (NFs).3 However, the molecular mechanisms underlying CAFs activation remain largely unclear. Further clarification of the mechanism underlying CAFs activation is important for cancer diagnosis and therapy.
There is increasing evidence that mutant p53 regulates CAFs Characteristics, although conflicting results have been reported. Whereas several studies have shown that p53 mutations in the stromal fibroblasts,12,18–20 other studies have reported that p53 mutations plays no role in CAFs.21,22
Surprisingly, our evidences showed that p53S is actually a significant contributor to the CAFs properties. The properties of CAFs is their supportive role in tumor progression. Indeed, in a xenograft tumor model, p53S in MEFs significantly promoted PC-3 tumor growth. On the other hand, transwell assays demonstrated that p53S in MEFs could promote PC-3 tumor cell migration and invasion. Last, we confirmed p53S could enhance the cell contractility of fibroblast. Together, these data identify a novel function of p53S in CAFs properties activation.
Next, we identified the mechanisms p53S regulated CAFs properties. Our results show that that p53S led to the activation of CAFs, resulting in augmented expression of α-SMA, which was regulated by the Stat3 pathway. Moreover, we used the Stattic to block Stat3 pathway, found their CAF phenotype were deprived. It demonstrated that p53S regulate CAFs properties by Stat3. Of note, Low-level Stat3 activation already occurs by the mere absence of WT p53 (Figure 4B).23,24 Since WT p53 suppresses Stat3, its loss activates Stat3. Importantly, p53S strongly increases Stat3 activation over and above the level of p53 loss. Thus, loss of function of WT p53 and the GOF by p53S each contribute to the robust constitutive hyperactivation of the Stat3 pathway, albeit at different magnitudes.
It was well recognized that the Stat3 pathway played a key role in various malignancies.25 Previous evidences suggest that the Stat3 pathway can promote the proliferation26 and activation of fibroblasts.27 Stat3 pathway induced the differentiation of MSCs into CAFs.28 CAFs induced bladder cancer EMT by the secretion of IL6.29 However, further evidence is needed regarding Stat3-mediated fibroblast activation. Our study revealed that, in the abovementioned p53S-mediated CAFs activation, the Stat3 pathway is a significantly activated pathway. p53S could activate fibroblasts in a Stat3-dependent way.
One possible mechanism might be: p53S bound to Stat3 and enhanced activating Stat3 phosphorylation by competitively displacing its phosphatase SHP2, a negative regulator of Stat3, and upregulated the expression of α-SMA, FGF10 and CXCL12, promoted CAFs activation. SHP2 is known to inhibit Stat3 phosphorylation, and high SHP2 levels in tumors correlate with longer cancer patient survival.30 The coordinated action of the Stat3 pathway may help to fine-tune p53S in CAFs activation.
In recent years, CAFs have become a novel target in cancer treatment.31,32 The identification of p53S as a pivotal participant in the CAFs activation highlights critical pathways that could be targeted for novel therapeutic interventions.
In conclusion, the findings of the present study verified the biological functions of p53S in CAFs activation, and provided evidence that the p53S in fibroblast promoted the proliferation, migration and invasion of PC-3 cells. p53S could enhance the cell contractility of fibroblast. p53S regulate CAFs properties through the activation of the Stat3 pathway.
Acknowledgments
This work was supported by NSFC (81960529, 81760262) fundings and Basic Research Program of Science and Technology Department of Yunnan Province (2019FB110).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Kalluri R, Zeisberg M. Fibroblasts in cancer. Nat Rev Cancer. 2006;6(5):392–401. doi:10.1038/nrc1877
2. Kuzet SE, Gaggioli C. Fibroblast activation in cancer: when seed fertilizes soil. Cell Tissue Res. 2016;365(3):607–619. doi:10.1007/s00441-016-2467-x
3. Madar S, Goldstein I, Rotter V. ‘Cancer associated fibroblasts’–more than meets the eye. Trends Mol Med. 2013;19(8):447–453. doi:10.1016/j.molmed.2013.05.004
4. Levine AJ, Oren M. The first 30 years of p53: growing ever more complex. Nat Rev Cancer. 2009;9(10):749–758. doi:10.1038/nrc2723
5. Kastenhuber ER, Lowe SW. Putting p53 in Context. Cell. 2017;170(6):1062–1078. doi:10.1016/j.cell.2017.08.028
6. Hawsawi NM, Ghebeh H, Hendrayani SF,et al. Breast carcinoma-associated fibroblasts and their counterparts display neoplastic-specific changes. Cancer Res. 2008;68(8):2717–2725. doi:10.1158/0008-5472.CAN-08-0192
7. Procopio MG, Laszlo C, Al Labban D, et al. Combined CSL and p53 downregulation promotes cancer-associated fibroblast activation. Nat Cell Biol. 2015;17(9):1193–1204. doi:10.1038/ncb3228
8. Schmid JO, Dong M, Haubeiss S, et al. Cancer cells cue the p53 response of cancer-associated fibroblasts to cisplatin. Cancer Res. 2012;72(22):5824–5832. doi:10.1158/0008-5472.CAN-12-1201
9. Dudley AC, Shih SC, Cliffe AR, et al. Attenuated p53 activation in tumour-associated stromal cells accompanies decreased sensitivity to etoposide and vincristine. Br J Cancer. 2008;99(1):118–125. doi:10.1038/sj.bjc.6604465
10. Lujambio A, Akkari L, Simon J, et al. Non-cell-autonomous tumor suppression by p53. Cell. 2013;153(2):449–460. doi:10.1016/j.cell.2013.03.020
11. Muller PA, Vousden KH. p53 mutations in cancer. Nat Cell Biol. 2013;15(1):2–8. doi:10.1038/ncb2641
12. Patocs A, Zhang L, Xu Y, et al. Breast-cancer stromal cells with TP53 mutations and nodal metastases. N Engl J Med. 2007;357(25):2543–2551. doi:10.1056/NEJMoa071825
13. Arandkar S, Furth N, Elisha Y, et al. Altered p53 functionality in cancer-associated fibroblasts contributes to their cancer-supporting features. Proc Natl Acad Sci U S A. 2018;115(25):6410–6415. doi:10.1073/pnas.1719076115
14. Chang MT, Asthana S, Gao SP, et al. Identifying recurrent mutations in cancer reveals widespread lineage diversity and mutational specificity. Nat Biotechnol. 2016;34(2):155–163. doi:10.1038/nbt.3391
15. Zhao L, Wang B, Zhao X, et al. Gain of function in the mouse model of a recurrent mutation p53(N236S) promotes the formation of double minute chromosomes and the oncogenic potential of p19(ARF). Mol Carcinog. 2018;57(2):147–158. doi:10.1002/mc.v57.2
16. Mueller MM, Fusenig NE. Friends or foes - bipolar effects of the tumour stroma in cancer. Nat Rev Cancer. 2004;4(11):839–849. doi:10.1038/nrc1477
17. Kalluri R. The biology and function of fibroblasts in cancer. Nat Rev Cancer. 2016;16(9):582–598. doi:10.1038/nrc.2016.73
18. Fukino K, Shen L, Patocs A, et al. Genomic instability within tumor stroma and clinicopathological characteristics of sporadic primary invasive breast carcinoma. JAMA. 2007;297(19):2103–2111. doi:10.1001/jama.297.19.2103
19. Kiaris H, Chatzistamou I, Trimis G, et al. Evidence for nonautonomous effect of p53 tumor suppressor in carcinogenesis. Cancer Res. 2005;65(5):1627–1630. doi:10.1158/0008-5472.CAN-04-3791
20. Addadi Y, Moskovits N, Granot D, et al. p53 status in stromal fibroblasts modulates tumor growth in an SDF1-dependent manner. Cancer Res. 2010;70(23):9650–9658. doi:10.1158/0008-5472.CAN-10-1146
21. Hosein AN, Wu M, Arcand SL, et al. Breast carcinoma-associated fibroblasts rarely contain p53 mutations or chromosomal aberrations. Cancer Res. 2010;70(14):5770–5777. doi:10.1158/0008-5472.CAN-10-0673
22. Qiu W, Hu M, Sridhar A, et al. No evidence of clonal somatic genetic alterations in cancer-associated fibroblasts from human breast and ovarian carcinomas. Nat Genet. 2008;40(5):650–655. doi:10.1038/ng.117
23. Wormann SM, Song L, Ai J, et al. Loss of P53 function activates JAK2-STAT3 signaling to promote pancreatic tumor growth, stroma modification, and gemcitabine resistance in mice and is associated with patient survival. Gastroenterology. 2016;151(1):180–193 e12. doi:10.1053/j.gastro.2016.03.010
24. Lin J, Tang H, Jin X, et al. p53 regulates Stat3 phosphorylation and DNA binding activity in human prostate cancer cells expressing constitutively active Stat3. Oncogene. 2002;21(19):3082–3088. doi:10.1038/sj.onc.1205426
25. Sansone P, Bromberg J. Targeting the interleukin-6/Jak/stat pathway in human malignancies. J Clin Oncol. 2012;30(9):1005–1014. doi:10.1200/JCO.2010.31.8907
26. Liu LD, Dong C-H, Shi H-J, et al. A novel type II membrane receptor up-regulated by IFN-alpha in fibroblasts functions in cell proliferation through the JAK-STAT signalling pathway. Cell Prolif. 2006;39(2):93–103. doi:10.1111/cpr.2006.39.issue-2
27. Hendrayani SF, Al-Khalaf HH, Aboussekhra A. The cytokine IL-6 reactivates breast stromal fibroblasts through transcription factor STAT3-dependent up-regulation of the RNA-binding protein AUF1. J Biol Chem. 2014;289(45):30962–30976. doi:10.1074/jbc.M114.594044
28. Tan HX, Cao Z-B, He -T-T, et al. TGFbeta1 is essential for MSCs-CAFs differentiation and promotes HCT116 cells migration and invasion via JAK/STAT3 signaling. Onco Targets Ther. 2019;12:5323–5334. doi:10.2147/OTT
29. Goulet CR, Champagne A, Bernard G, et al. Cancer-associated fibroblasts induce epithelial-mesenchymal transition of bladder cancer cells through paracrine IL-6 signalling. BMC Cancer. 2019;19(1):137. doi:10.1186/s12885-019-5353-6
30. Huang Y, Wang J, Cao F, et al. SHP2 associates with nuclear localization of STAT3: significance in progression and prognosis of colorectal cancer. Sci Rep. 2017;7(1):17597. doi:10.1038/s41598-017-17604-7
31. Mertens JC, Fingas CD, Christensen JD, et al. Therapeutic effects of deleting cancer-associated fibroblasts in cholangiocarcinoma. Cancer Res. 2013;73(2):897–907. doi:10.1158/0008-5472.CAN-12-2130
32. Chan JSK, Sng MK, Teo ZQ, et al. Targeting nuclear receptors in cancer-associated fibroblasts as concurrent therapy to inhibit development of chemoresistant tumors. Oncogene. 2018;37(2):160–173. doi:10.1038/onc.2017.319
33. Wang B, Dan J, Li H, et al. The transcription and expression profile of p53(N236S) mutant reveals new aspects of gain of function for mutant p53. FEBS Lett. 2018;592(18):3183–3197. doi:10.1002/feb2.2018.592.issue-18
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.