Back to Journals » Journal of Inflammation Research » Volume 19
P-Selectin and Platelet–Monocyte Interaction in Inflammation: Mechanisms, Hypothesis, and Open Questions
Authors Fernandes MKC ![]() , De Paula MML, Oliveira RTR, Dos Santos JPRS, Mancini MCB
, De Paula MML, Oliveira RTR, Dos Santos JPRS, Mancini MCB ![]() , Hottz ED
, Hottz ED ![]()
Received 22 December 2025
Accepted for publication 23 April 2026
Published 9 June 2026 Volume 2026:19 556871
DOI https://doi.org/10.2147/JIR.S556871
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Qing Lin
Mayara Karen Carvalho Fernandes, Marcelo Miranda Lima De Paula, Renata Tôrres Rêgo Oliveira, João Pedro Ribeiro Soares Dos Santos, Maria Clara Bertorelli Mancini, Eugenio D Hottz
Department of Biochemistry, Federal University of Juiz de Fora, Juiz de Fora, Minas Gerais, Brazil
Correspondence: Eugenio D Hottz, Email [email protected]
Abstract: This review provides an integrated overview of how P-selectin-mediated platelet–monocyte interactions orchestrate key inflammatory processes in sterile and non-sterile pathological contexts. Inflammation presents a paradox: although it is a fundamental physiological process, uncontrolled activation can progress to a chronic state, contributing to the development of various inflammatory diseases. Platelets, previously considered to play a role exclusively in hemostasis, are now recognized as important immunomodulatory effectors capable of releasing inflammatory mediators and interact with leukocytes upon activation. The interaction between platelet P-selectin and monocyte PSGL-1 promotes the formation of platelet–monocyte aggregates, that together exert broad and profound inflammatory effects. These aggregates have been implicated in multiple pathological contexts, both sterile, such as atherosclerosis, obesity, autoimmune diseases, and hypertension, and non-sterile, such as sepsis and viral infections, highlighting their role as a common axis of immune and inflammatory regulation. This review integrates shared but contextualized mechanistic pathways and outlines current hypotheses regarding how platelet–monocyte interactions may operate in sterile and pathogen-induced inflammation. We further discuss emerging evidence connecting P-selectin signaling to metabolic reprogramming, vascular dysfunction, and immunothrombosis, as well as its possible influence on monocyte activation and phenotype in diverse inflammatory settings, discussing open questions, perspectives, and challenges in the field. By integrating interdisciplinary findings, this review highlights P-selectin-dependent platelet–leukocyte interactions as central axis in inflammation and promising therapeutic targets.
Keywords: P-selectin, platelet activation, platelet–monocyte aggregates, thromboinflammation, coagulation
Introduction
Inflammation is a fundamental physiological process and one of the body’s most effective defense mechanisms against infection, injury, and exposure to foreign particles or stress.1 Inflammation can be triggered by infectious agents (non-sterile inflammation)2 or by disruptions in cellular and tissue homeostasis, such as injury or metabolic stress (sterile inflammation).3,4 Traditionally viewed as a crucial protective response, acute inflammation mobilizes specialized cell types, such as neutrophils and monocytes, from the circulation to the site of injury. This response is vital for host defense, orchestrating a cascade of events that leads to the eradication of invasive pathogens and facilitates tissue repair.5 Therefore, inflammation is a vital mechanism of homeostasis; however, uncontrolled inflammation can become chronic, contributing to a variety of diseases. This inflammation paradox highlights the complexity of the immune system and the need to differentiate between pathological and protective inflammatory responses.6,7 In both physiological and pathological contexts, inflammation can intersect with coagulation in a process known as thromboinflammation, which describes the interplay between coagulation and immune activation particularly within the vasculature. Thromboinflammation is driven by both humoral and cellular elements of the coagulation and immune continuum, including leukocytes, endothelial cells, and platelets, potentially leading to microvascular thrombosis and organ dysfunction.8–10
For a long time, platelets were recognized solely by their roles in hemostasis and pathological thrombosis. However, mounting evidence now positions them as active immunomodulatory and “sentinel” cells within the innate immune system.11–15 Upon activation by various inflammatory or procoagulant stimuli, platelets release a wide range of stored and newly-synthesized factors, interacting in complex ways with other cells, especially leukocytes.16–19 Monocytes, along with their macrophage derivatives, are central components of the inflammatory response, acting in both host defense and pathogenesis. The interaction between platelets and monocytes has been reported to regulate monocyte responses, being indispensable in both hemostasis and inflammation.20,21 Circulating human monocytes subdivide into three main subsets according to the expression of CD14 and CD16.21,22 Classical monocytes (CD14highCD16−) represent more than 90% of circulating monocytes and are characterized by strong phagocytic capacity, high expression of scavenger and pattern-recognition receptors, and robust inflammatory responses.23,24 Intermediate monocytes (CD14highCD16⁺) display enhanced antigen-presenting capacity and are potent inducers of T cells.24,25 Non-classical monocytes (CD14lowCD16⁺), in contrast, are specialized in endothelial patrolling and surveillance, exhibit increased migratory potential and regulation of vascular inflammation.23,26,27 Such functional heterogeneity is particularly relevant in the context of platelet–monocyte aggregate (PMA) formation, as the composition and downstream consequences of PMAs may differ depending on the monocyte subset engaged, thereby influencing inflammatory amplification, endothelial dysfunction, and thromboinflammatory outcomes.
Selectins are calcium-dependent vascular adhesion molecules composed of three known members, namely E-selectin (CD62E), P-selectin (CD62P), and L-selectin (CD62L). CD62E is mainly expressed on endothelial cells, while CD62L is expressed in leukocytes and CD62P in both endothelial cells and platelets.28 The main role of selectins is to regulate leukocyte recruitment to sites of inflammation. The initial tethering and rolling of monocytes over the vascular endothelium are dependent on interactions between selectins and their corresponding glycosylated ligands and are imperative for extravasation into inflammatory lesions.29,30 Importantly, selectin binding not only tethers the cells together but signals gene expression, regulating inflammation. Selectins, particularly CD62P, have been shown to contribute to many inflammatory diseases.31,32
Like many platelet granular contents, CD62P is stored in α-granules after being synthesized by parent megakaryocytes and upon procoagulant, inflammatory or pathogen-derived stimuli is rapidly translocated to platelet surface alongside α-granule releasate.28 Its surface and/or soluble (sCD62P) forms serve as markers of platelet activation. Platelet CD62P binding to its ligand P-selectin glycoprotein ligand-1 (PSGL-1) on leukocytes is the dominant molecular event in platelet–leukocyte interactions, with significant inflammatory consequences across a wide range of diseases. Patients with inflammatory disorders typically have higher levels of activated platelets and endothelial cells displaying CD62P, which subsequently increases platelet–leukocyte aggregation and leukocyte-endothelium adhesion, rolling, and infiltration.33–35
In this review, we discuss the central role of P-selectin in mediating platelet–monocyte interactions through a spectrum of inflammatory diseases, addressing common mechanisms and specific characteristics in different pathological contexts, from infectious to sterile inflammation. Our focus is to discuss the effects of PMA on disease mechanisms and outcomes. The review is structured into two major sections: sterile inflammatory conditions and infectious diseases. Within each section, we highlight shared and disease-specific mechanisms of P-selectin-mediated platelet–monocyte responses and their roles in diseases’ pathogenesis. Finally, therapeutic implications and future research directions are addressed in the concluding section.
P-Selectin-Mediated Interaction in Sterile Inflammatory Diseases
Obesity
Abdominal obesity, one of the most prevalent metabolic disorders of the 21st century, has reached epidemic proportions worldwide and is now recognized as a chronic disease by the World Health Organization (WHO).36,37 Obesity is associated with reduced life expectancy and higher risk of several comorbidities, many of which are components of metabolic syndrome, a cluster of interrelated cardiometabolic risk factors that significantly increase susceptibility to cardiovascular disease (CVD) and type 2 diabetes (T2DM).38–40 Obesity independently increases the risk for adverse cardiovascular outcomes, which remains the main cause of obesity-related morbidity and mortality.41,42 Chronic low-grade inflammation and a prothrombotic state are hallmarks of obesity, further amplifying the risk for thrombotic events even after procedures like percutaneous coronary intervention.43,44 This condition stems from enhanced platelet response, a state of hypercoagulability and hypofibrinolysis,45 and enhanced platelet–monocyte interactions.46–48
Dysfunctional adipose tissue, combined with systemic immune activation, promotes chronic inflammation, monocyte, and macrophage activation, and increased tissue factor (TF) expression, enhancing thrombin generation, and coagulopathy in obesity.44,49,50 Clinical evidence shows elevated sCD62P levels in obesity, likely reflecting platelet and endothelial activation.51 PMA formation is also increased in obesity,49,52 a phenomenon that correlates with elevated TF expression in monocytes.49 Bariatric surgery induces significant weight loss and reverses many of these thromboinflammatory alterations, improving insulin sensitivity, systemic inflammation, and metabolic syndrome characteristics.53 Platelet transcriptomic analyses reveal normalization of inflammatory signaling and reduced platelet hyperreactivity one year after surgery.53 Likewise, a significant decrease in PMA formation has been reported after surgery,54 highlighting the reversibility of obesity-associated platelet and monocyte dysregulation.
Studies in experimental models of obesity (ob/ob mice and diet-induced obesity) demonstrate that visceral adipose tissue is an active focus of inflammation, marked by increased leukocyte rolling and adhesion, platelet aggregation, and elevated expression of CD62P, CD62E, and ICAM-1 on the endothelium. Platelet activation occurs primarily within the adipose tissue itself, where platelets exhibit high surface CD62P and form PMA via PSGL-1, favoring macrophage recruitment and activation. These findings show that visceral obesity promotes intensified interactions between endothelium, platelets, and leukocytes, sustaining local inflammation.55
Recently, we showed increased platelet activation and PMA formation in obesity, which formed especially among CD16- and TF-expressing monocytes. Ex vivo mechanistic experiments demonstrated that platelet adhesion mainly through CD62P promotes reciprocal platelet and monocyte activation and secretion of inflammatory mediators.52 Platelets from obese patients induced TF and IL-1β expression in monocytes partially depending on CD62P, CD40L, and integrin αIIb/β3 signaling, while secreted IL-1 amplified inflammation by activating endothelial cells.52 These findings indicate a platelet-driven amplification of inflammation in obesity.
Metabolic syndrome represents the convergence of obesity with comorbidities including insulin resistance, dyslipidemia, and hypertension. Patients with metabolic syndrome exhibit increased platelet activation, evidenced by greater fibrinogen binding (activated integrin αIIb/β3), CD62P translocation,56 and greater aggregation under high shear.57 Notably, platelet contact plays a key role in shaping monocyte phenotypes in metabolic syndrome. Patients with metabolic syndrome exhibit greater monocyte activation, with elevated CD11b (Mac-1) expression across all monocyte subsets.56 PMA formation is higher in obese patients with metabolic syndrome and hypertension, accompanied by elevated TF expression on monocytes, while the shift from classical to intermediate and non-classical subsets associate with metabolic syndrome independently of diabetes or hypertension.52 Importantly, both platelet and monocyte activation correlate positively with the blood glucose levels.56
Together, these findings indicate enhanced platelet activation and platelet–monocyte interactions in obesity, contributing to a prothrombotic state. These features are consistent with a broader thromboinflammatory environment that may extend beyond adipose tissue and influence vascular function. Whether targeted modulation of platelet–monocyte interactions can interrupt this thromboinflammatory loop and reduce cardiometabolic risk in obesity remains an open question.
Atherosclerosis and Coronary Arterial Disease
Atherosclerosis is a chronic inflammatory disease characterized by the progressive accumulation of lipid-rich and fibrous plaques in medium and large-sized arteries, driven by risk factors such as hyperlipidemia, hypertension, smoking, diabetes, and chronic inflammation.58,59 Atherosclerotic plaques are complex and dynamic structures characterized by the accumulation of lipids, particularly oxidized low-density lipoprotein (oxLDL), within the intima, in association with endothelial activation and an inflammatory microenvironment.60–64 These lesions contain multiple interacting cell types, including macrophages derived from recruited monocytes, lipid-laden foam cells, and vascular smooth muscle cells, which contribute to plaque organization and progression.65–69 Structurally, plaques typically exhibit a lipid-rich necrotic core covered by a fibrous cap formed by smooth muscle cells and extracellular matrix components.68,69 In addition, they display persistent inflammatory activity and extracellular matrix remodeling, which can compromise structural integrity and promote plaque instability, increasing susceptibility to rupture, thrombus formation, and vascular occlusion.70–75 Consequently, plaque progression leads to varying degrees of arterial stenosis, which can impair blood flow and result in tissue hypoxia.70,76–78 Metabolic disturbances associated with obesity including insulin resistance, endothelial dysfunction, and chronic low-grade inflammation exacerbate these processes by promoting inflammatory signaling and monocyte recruitment within the vascular wall.79–83 Consistently, obesity is a major modifiable risk factor for atherosclerosis and cardiovascular diseases.84,85
Platelet activation is a consistent feature of atherosclerosis, playing major pathological roles. During atherogenesis, high platelet CD62P surface expression supports PMA formation, preparing the environment for lipid accumulation and monocyte transformation into foam cells, which are strong predictors of cardiovascular risk (Figure 1).86,87 Activated platelets adhere to the inflamed endothelium enhancing leukocyte recruitment and extravasation through CD62P-dependent mechanisms (Figure 1a and b).88–91 Research with animal models demonstrates that CD62P-PSGL-1 interaction is crucial for monocyte accumulation. Activated platelets, when infused into mice, rapidly adhere to monocytes in a CD62P-dependent manner and transfer chemokines such as RANTES/CCL5 and PF4/CXCL4, amplifying leukocyte recruitment and accelerating the formation of atherosclerotic lesions (Figure 1b).89 Ly6C-expressing monocytes depend on PSGL-1 for selective rolling and accumulation in the initial atherosclerotic endothelium (Figure 1c). Consistently, ApoE knockout mice (a model of atherosclerosis) with PSGL-1 deficiency exhibit reduced leukocyte infiltration and atherosclerotic progression.92 In vivo chimeric models (WT platelet transfusion into CD62P knockout),93 together with ex vivo studies,94 reinforce the predominance of platelet-derived CD62P over endothelial CD62P in mediating leukocyte adhesion.
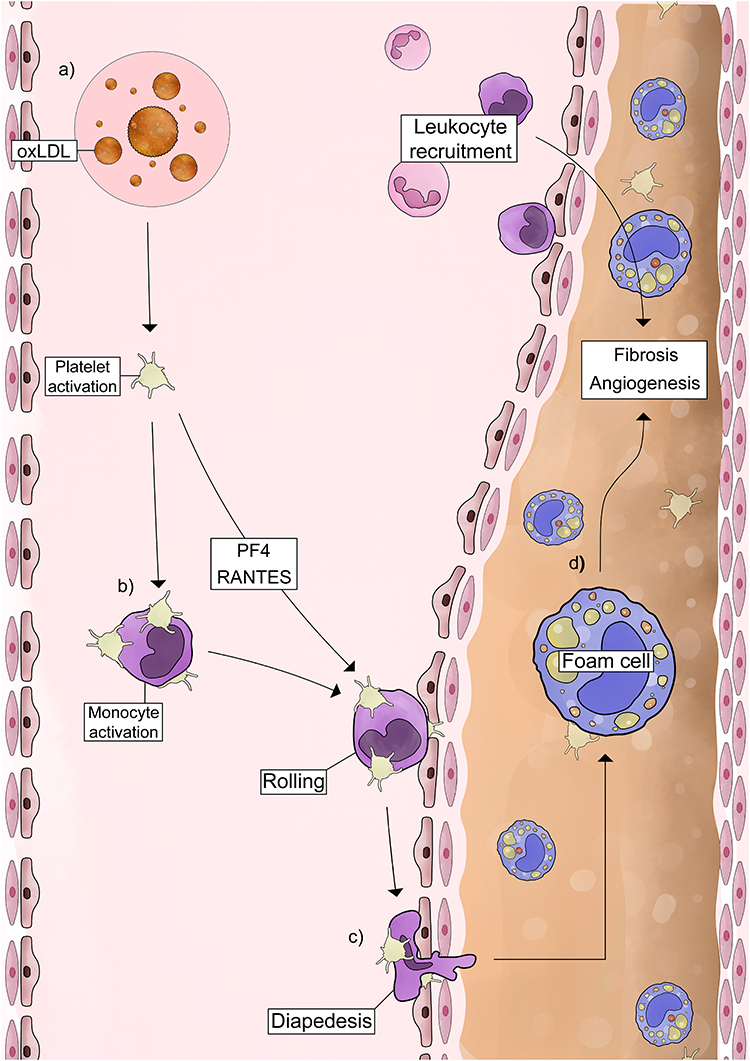 |
Figure 1 Mechanisms of platelet activation and platelet-mediated monocyte responses in atherosclerosis. Schematic representation of (a) OxLDL-driven platelet activation and CD62P exposure, which promote (b) chemokine secretion and PMA formation, (c) increase monocyte adhesion to the endothelium, and (d) support their differentiation into foam cells. See the text for details and references. Abbreviations: OxLDL, oxidized LDL; CD62P, P-selectin; PMA, platelet-monocyte aggregates; ROS, reactive oxygen species; PSGL-1, selectin glycoprotein ligand-1. |
Dyslipidemia intensifies these mechanisms by promoting chronic low-grade inflammation, platelet hyperactivity,87 and PMA formation.95 OxLDL, but not native LDL, induces platelet activation and CD62P exposure (Figure 1a),96 which promotes PSGL-1 dependent PMA formation (Figure 1b), enhancing monocyte adhesion to the endothelium (Figure 1c) and differentiation into foam cells (Figure 1c and d).89 Platelet adhesion increases OxLDL uptake by monocytes in a dose-dependent manner through mechanisms depending on CD62P. In murine models of atherosclerosis, platelet depletion reduces monocytes/macrophage accumulation within lesions, demonstrating that platelets accelerate monocyte infiltration and foam cell formation in vivo.89 OxLDL-activated platelets also increase CD11b activation in monocytes, reinforcing their inflammatory and pro-migratory phenotype.97
Pharmacological interventions further support the role of lipid-driven platelet activation in atherosclerosis pathogenesis. Lipid-lowering therapies, including statins and ezetimibe, have been shown to reduce platelet activation and CD62P expression.98,99 Similarly, monoclonal antibodies targeting the reduction of LDL by PCSK9 inhibition, such as evolocumab and alirocumab, also attenuate oxidative stress and platelet reactivity.100–102 Together, these findings indicate that modulation of lipid metabolism may indirectly limit platelet activation and its pathophysiological consequences. However, their relevance in reducing PMA formation remains to be fully established.
The cellular mechanisms underlying atherosclerosis translate clinically into coronary artery disease (CAD), characterized by narrowing or occlusion of the coronary arteries.103 Increased levels of PMA have been consistently reported in patients with cardiovascular diseases, including myocardial infarction and other vascular complications.104–107 PMA formation has been suggested as reliable markers of early-stage cardiovascular disease.105 Functionally, platelet–monocyte interactions are associated with changes in both cell types, including increased secretion of pro-coagulant mediators, which may contribute to angiopathy and thrombotic events.107 Studies in human coronary arteries using intracoronary blood sampling show increased platelet CD62P and PMA in stenosed regions, with local monocyte and platelet activation specifically in the damaged area.108 Animal models confirm that CD62P promotes inflammation and cardiac remodeling, since genetic ablation of CD62P reduces fibrosis and tissue inflammation.93 Besides, transferring of wild-type platelets restores inflammatory and fibrotic responses.93 These findings highlight the central role of platelet CD62P in aggregate formation,103,109 localized inflammation,108 and tissue remodeling,93 reinforcing its prognostic significance and its potential as a therapeutic target in cardiovascular diseases.110
Different clinical contexts rely on distinct cellular components of the CD62P-PSGL-1 axis to amplify thromboinflammation. In stable CAD, heightened platelet reactivity is reflected by circulating degranulated platelets and high levels of PMA,106,111,112 even without increased CD62P expression.109 In acute coronary syndromes (ACS), however, plaque rupture triggers local activation of monocytes with upregulated PSGL-1 at the lesion site, promoting intense PMA formation and fueling local inflammatory and thrombotic activation.113 Rather than representing a direct comparison between CAD and ACS, these observations illustrate how platelet-driven or monocyte-driven mechanisms may predominate depending on the pathological setting. Strategies that inhibit platelet activation or disrupt the CD62P-PSGL-1 interaction, such as the analog peptide IELLQAR, reduce monocyte recruitment and attenuate key steps in foam cell formation, offering potential to slow disease progression.88,114 These processes reinforce the central role of P-selectin-mediated platelet–monocyte interactions in coronary atherosclerosis and position them as promising therapeutic targets.115,116
Importantly, P-selectin-mediated thromboinflammation is not confined to the coronary arteries. Structural alterations associated with arteriolosclerosis and nephrosclerosis, including progressive intimal fibroelastosis, subendothelial hyalinosis, and intimal fibrosis in afferent arterioles, demonstrate that these mechanisms extend to multiple organs potentially contributing to hypertension and its associated vascular and renal complications (reviewed in117), as discussed in the next chapter.
Hypertension
Hypertension is a multifactorial disease influenced by genetic, endocrine, dietary, lifestyle, immune, and environmental factors.118,119 Vascular remodeling, including smooth muscle hypertrophy, endothelial injury, and immune cell infiltration, leads to extracellular matrix deposition and increased vascular stiffness.120,121 These structural and inflammatory alterations create a prothrombotic environment, with CD62P-mediated platelet–monocyte interactions amplifying vascular inflammation, endothelial dysfunction, and cardiac remodeling. Chronic platelet activation in hypertensive patients is reflected by elevated PMA formation in circulation, which correlates with tissue inflammation and early fibrosis in both experimental models and clinical studies.122
Experimental93 and clinical123 evidence indicate that CD62P is a key mediator of vascular and organ damage associated with hypertension. In angiotensin II–induced hypertension models, wild-type mice develop cardiac inflammation, macrophage infiltration, and early fibrosis, while CD62P deficiency markedly attenuates these responses despite similar elevation in blood pressure.93 CD62P-PSGL-1-facilitated PMA promote the recruitment of monocytes to cardiac tissue, where they differentiate into macrophages that secrete pro-inflammatory cytokines and induce fibrosis.93 Consistently, clinical studies demonstrate that hypertensive patients exhibit increased platelet and endothelial activation, including elevated plasma and myocardial levels of CD62P.123 Increased platelet activation, paralleled by low-grade inflammation, is more pronounced in individuals with signs of renal injury.123 Furthermore, antiplatelet therapy with aspirin reduces surface and soluble CD62P levels, reinforcing the role of platelet and endothelial activation in cardiovascular and renal complications during hypertension.124
These findings demonstrate that platelet activation and CD62P-mediated platelet–monocyte interaction represent key mechanisms of inflammation and tissue remodeling associated with hypertension.
Type 2 Diabetes Mellitus
Type 2 diabetes mellitus (T2DM) represents one of the most prevalent and clinically relevant consequences of overweight and obesity, as progressive weight gain is a major driver of metabolic dysfunction.125 T2DM is characterized by insulin resistance, hyperglycemia, inflammation, and progressive β-cell dysfunction.126–129 Environmental factors and sustained nutrient excess induce epigenetic modifications that impair insulin signaling, reduce peripheral glucose uptake, increase hepatic glucose production, and elevate circulating free fatty acids.130 Beyond metabolic dysregulation, T2DM is associated with chronic low-grade inflammation, which accelerates β-cell failure and disease progression.129,131
T2DM patients exhibit a prothrombotic state associated with heightened platelet activation132 and PMA formation,95 which directly correlate with poor glycemic control133,134 and to the presence of carotid plaques in patients.135 The coexistence of T2DM and obesity further exacerbates platelet activation and hyperreactivity.136 Increased PMA formation in T2DM is particularly higher among patients with higher adiposity.54,137–139 A positive correlation between PMA and the body mass index supports a role for excess adipose tissue in amplifying platelet–monocyte interactions. Similar associations are observed with other metabolic disorders, such as dyslipidemia.54,140 Mechanistically, initial CD62P-mediated platelet–monocyte interactions are subsequently stabilized by monocyte Mac-1 (CD11b/CD18), enhancing firm adhesion to the endothelium and promoting local inflammation.141,142 Notably, Mac-1 expression in monocytes increases rapidly in response to acute glucose excursions, further reinforcing the formation and stability of PMA under hyperglycemic conditions.142 This process initiated by increased CD62P on platelets, facilitates their interaction with leukocytes and rapid PMA formation,133,134 representing a central mechanism linking hyperglycemia, vascular inflammation, and atherothrombotic risk in T2DM.
Pharmacological strategies that reduce postprandial hyperglycemia, such as acarbose,143 decrease platelet activation144 and PMA formation145 by mitigating acute glycemic fluctuations in patients with T2DM. These findings suggest that short-term glycemic excursions rather than long-term glycemic control are key drivers of thromboinflammation in T2DM. Together, these observations suggest that different metabolic, inflammatory, and coagulation disturbances may converge to sustain platelet–monocyte interactions and thromboinflammation.
Fatty Liver Disease (Hepatic Steatosis)
Non-alcoholic Fatty Liver Disease (NAFLD) represents the hepatic manifestation of metabolic dysfunction, closely associated with obesity, insulin resistance, hypertension, and type 2 diabetes.146–149 During obesity, dietary energy surplus becomes a central driver of NAFLD development.150–152 When energy intake exceeds the adipose tissue expenditure capacity, excess lipids accumulate ectopically in organs such as the liver.153–155 Insulin normally exerts an antilipolytic effect and promotes triglyceride storage in adipose tissue, thus, insulin resistance plays a central pathogenic role by increasing free fatty acid flow to the liver and enhancing hepatic lipid accumulation.156–158 Within this spectrum, non-alcoholic steatohepatitis (NASH) represents a more severe form, characterized by steatosis accompanied by liver necro-inflammation and varying degrees of fibrosis, potentially progressing to cirrhosis, liver failure, and, in some cases, hepatocellular carcinoma.159 The transition from metabolic dysfunction-associated steatotic liver disease to metabolic dysfunction-associated steatohepatitis (MASH) and advanced fibrosis involves a cascade of inflammatory and fibrogenic events.160
NASH is associated with a chronic proinflammatory and prothrombotic state.161 Higher hepatic fat content positively correlates with increased coagulation activation,162,163 while the platelet and endothelial activation markers sICAM-1 and sCD62P are associated with markers of liver injury, such as aminotransferases enzymes.164,165 Similarly, patients with metabolic dysfunction-associated steatotic liver disease (MASLD) exhibit elevated serum thromboxane B2 (TxB2) and sCD62-P, which progressively increase from steatosis to steatohepatitis and cirrhosis, positively correlating with the stage of fibrosis.166,167 PMA formation also increases during NASH and correlates with platelet activation and plasma aminotransferases, while increased monocyte activation was associated with end-stage liver cirrhosis.165 Within this context, platelet activation and PMA formation may link inflammation, coagulation, and fibrogenesis, contributing to both pro-inflammatory and pro-fibrotic responses that sustain hepatic injury.160 Preclinical studies with genetic or pharmacological interventions targeting platelet activation or CD62P are still needed to better understand their role in NASH.
Ulcerative Colitis
Ulcerative colitis (UC) and inflammatory bowel disease are characterized by inflammation of the colonic mucosa, in which genetic susceptibility, alterations in gut microbial composition and environmental factors collectively contribute to its development.168 This persistent inflammatory environment leads to platelet activation, contributing to thromboembolic complications.169 Patients in UC flare exhibit increased platelet activation and elevated CD62P expression,170 which supports PMA formation primarily relying on CD62P-PSGL-1 interactions.171 In patients with severe endoscopic inflammation, PMAs were markedly increased and more abundant in regions of extensive mucosal damage. Moreover, PMA displayed higher CD16 expression compared to platelet-free monocytes, indicating preferential interaction with inflammatory monocytes.172 However, opposing data have been reported on whether PMA formation is associated with disease severity in UC by inducing or inhibiting inflammatory response.171–174 Zamora et al (2018) found reduced PMA formation in flare patients with endotoxemia, linked to decreased monocyte PSGL-1 expression.171 This finding was consistent with studies that lipopolysaccharide (LPS) downregulates PSGL-1, thereby impairing PMA formation.173,174 In a murine model of DSS-induced colitis, however, blocking either PF4/CXCL4 or CXCR3 expression alleviated inflammation.175 Although the study did not specifically evaluate circulating PMA, it provides mechanistic insight into how platelet activation influence monocyte and macrophage response within the intestine, thereby amplifying local inflammation and contributing to disease progression. Despite ongoing debate regarding PMA dynamics, their association with severe mucosal damage supports the notion that they may contribute to local thromboinflammatory events in the colon.175
Notably, therapeutic strategies targeting these interactions provide indirect clinical support for their pathogenic relevance. Adsorptive cytapheresis has been shown to significantly reduce circulating PMAs, with clinical response correlating with the magnitude of platelet depletion, reinforcing the concept that disrupting platelet–monocyte crosstalk may attenuate intestinal inflammation.176 However, further studies are needed to determine whether PMA serve as reliable biomarkers of disease activity or prognosis and potential therapeutic targets in UC.
Autoimmune Diseases
Autoimmune diseases (ADs) are characterized by the presence of autoreactive immune cells, in which overactivation and recruitment of T and B lymphocytes result in immune-mediated damage.177 Cardiovascular complications frequently contribute to increased mortality in ADs, with systemic lupus erythematosus (SLE) and type 1 diabetes mellitus (T1DM) ranking among those with the highest cardiovascular risk.178 The increased risk of thrombotic events in patients with ADs is primarily driven by systemic inflammation, which promotes a hypercoagulable state associated with endothelial dysfunction and enhanced platelet activation.179
In SLE, the immune system inappropriately targets nucleic acids-containing cellular components.180 Among its various immunopathological processes, enhanced platelet activation and PMA formation have been identified as major players to vascular inflammation and thrombotic complications.181 PMAs consistently exhibit higher monocyte activation markers when compared to monocytes alone,182 and PMA formation is positively correlated to C-reactive protein levels,183 indicating that PMA formation is associated with enhanced inflammatory activity in SLE. Elevated platelet activation and platelet–leukocyte interactions have been associated with enhanced TF-dependent coagulation activation in SLE.184 Platelet–monocyte interactions are often described favoring inflammatory amplification, but PSGL-1/CD62P-mediated interactions can also contribute to tolerogenic responses.185 Notably, PSGL-1 deficient mice develop a scleroderma-like syndrome, whereas the absence of its main ligand, CD62P, disrupts immune homeostasis and leads to a progressive lupus-like autoimmune disorder with many features observed in lupus-prone mice. Consistently, patients with cutaneous lupus exhibit reduced endothelial CD62P expression in dermal vessels, implying that decreased CD62P expression could play a role in disease development.186,187 Monocyte PSGL-1 expression is reduced in SLE patients despite increased PMA formation.188 Monocyte CD40 and platelet CD40L may act as an alternative pathway mediating platelet–monocyte binding in SLE. Altogether, these findings emphasize the complexity and multifaceted nature of platelet–monocyte interactions in SLE. Understanding how these pathways cooperate, compensate, or become dysregulated may provide valuable insights for developing targeted therapies to mitigate loss of tolerance and thromboinflammatory complications in SLE.188
T1DM is a chronic disorder characterized by autoimmune-mediated destruction of pancreatic islet β-cells leading to insulin deficiency and hyperglycemia.189 Patients with T1DM often exhibit a hypercoagulable state, characterized by platelet hyperreactivity, elevated plasma coagulation factors, impaired fibrinolysis,134 and increased PMA formation.95 In individuals with T1DM who also develop insulin resistance, platelets shift toward a state of heightened reactivity while becoming less responsive to the inhibitory actions of prostacyclin (PGI2). This combination of increased activation and reduced inhibitory sensitivity creates a platelet phenotype that is inherently harder to restrain, offering a clearer explanation for why these patients face an amplified risk of cardiovascular complications.190 CD62P-positive platelets and PMA formation are associated with the degree of hyperglycemia and dyslipidemia in T1DM.191 Overall, this evidence indicates that persistent metabolic imbalance in T1DM enhances platelet activation and promotes the formation of PMA, amplifying thromboinflammation and contributing to a higher cardiovascular risk.
Sickle Cell Disease
Sickle cell disease (SCD) is a hereditary hemoglobinopathy caused by the substitution of glutamic acid for valine in the β-globin chain, characterized by chronic hemolysis, recurrent episodes of painful vaso-occlusive crises, multi-organ dysfunction, and premature death.192 These crises result from vaso-occlusive aggregates formed of sickle-shaped erythrocytes, activated platelets, and leukocytes to the endothelium, leading to tissue ischemia.193 Platelet bind to erythrocytes, monocytes, and neutrophils, form multicellular aggregates that contribute to blood flow abnormalities and vascular inflammation.194–197 Monocytes from patients with SCD exhibit a highly activated phenotype, with increased expression of pro-inflammatory cytokines such as IL-1β and TNF-α, promoting greater endothelial adhesion and further amplification of the inflammatory response.198,199
In experimental SCD models, CD62P blocking or genetic deficiency significantly reduces leukocyte adhesion and protects mice from vaso-occlusion.196,200,201 Mice deficient in CD62P and CD62E exhibit markedly reduced leukocyte rolling and adhesion, a marked reduction in interactions between sickle cells and adherent leukocytes, preservation of microvascular flow, and increased survival during the intravital experimental procedure, highlighting that selectin-dependent leukocyte recruitment is crucial for vaso-occlusion.196,201,202
In humans, PMA are more abundant in patients with SCD than in healthy controls203 contributing to increased CD11b expression, inflammatory amplification, greater endothelial adhesion, and tissue damage.199,204 Circulating platelets remain chronically activated, showing higher levels of CD40L205,206 and CD62P,207,208 and TNFSF14,209 a cytokine associated with endothelial activation and inflammation. CD40L promotes the release of α and dense granules, intensifying platelet activation with increased CD62P exposure favoring the interaction with monocytes.206 In addition to the classic CD62P/PSGL-1 pathway, monocyte podoplanin binding to platelet CLEC-2 also contributes to platelet activation and the formation of PMA in patients with SCD, which was correlated with CD62P expression and disease severity, including hemolysis and coagulation activation markers, and history of vaso-occlusive crises.203
Those preclinical and translational findings provided the basis for therapeutic interventions targeting CD62P. Crizanlizumab, a humanized monoclonal antibody that blocks the CD62P-PSGL-1 interaction, significantly reduces the annual frequency of vaso-occlusive crises, prolongs the time between crises, and decreases hospital stays, without a relevant increase in adverse events.210 These results directly demonstrate that CD62P inhibition can effectively modify disease outcomes in humans, showing that platelet–leukocyte interactions are not only essential mechanistic contributors but also actionable therapeutic targets that translate into significant clinical benefits. Building on this success, it will be valuable to explore whether CD62P blockade could provide similar benefits to other thromboinflammatory conditions where CD62P contributes to PMAs formation and poorer disease outcomes, representing a promising avenue for future investigation.
P-Selectin-Mediated Interaction in Infectious Diseases
Dengue Virus
Dengue viruses (DENV) are the most epidemiologically relevant arboviruses worldwide, responsible for approximately 400 million infections per year on the planet.211 Transmission occurs through the bite of blood-feeding mosquitoes of the genus Aedes, especially Aedes aegypti.212 Clinical manifestations range from self-limiting febrile illness to severe syndromes characterized by endothelial dysfunction and hemorrhage.213 Among the most striking laboratory findings in severe forms are thrombocytopenia and cytokine storm, resulting in exacerbated systemic inflammation.214 Platelet activation is evidenced by increased expression of classical markers such as CD62P, PAC-1, and phosphatidylserine,215–217 accompanied by secretion of inflammatory mediators including PF4/CXCL4, CCL5/RANTES218 and nitric oxide.219 This activation drives the formation of PMA, which are elevated in dengue patients220,221 and correlate with thrombocytopenia and increased vascular permeability in patients.215,216,219,221 Proteomic studies reinforce this profile, demonstrating higher expression of proteins associated with platelet activation and immunoregulation in platelets from infected patients.222
In vitro infection models demonstrate increased platelet activation215,216 and aggregation with monocytes223 resulting in the induction of inflammatory cytokines, chemokines and COX-2 expression, alongside monocyte metabolic reprogramming leading to lipid droplet-dependent PGE2 production.223 Ex vivo studies corroborate these findings, demonstrating that platelets from dengue patients stimulates monocytes to produce inflammatory cytokines such as IL-8, IL-1β, and IL-10 in a process depending on CD62P-mediated adhesion and phosphatidylserine recognition by the monocyte.221 In addition to P-selectin-mediated adhesion, secreted mediators such as MIF also participate in monocyte reprogramming.223 Likewise, PF4-CXCR3 signaling has shown immunomodulatory proviral effects enhancing DENV replication in monocytes by reducing type I interferon while promoting proinflammatory cytokine production.224 Together, these in vitro and ex vivo data indicate that platelet activation drives PMA formation and modulates monocyte metabolic and inflammatory responses.
Experimental DENV infection of interferon-deficient (A129) mice also shows a robust increase in platelet activation and platelet–leukocyte aggregate formation alongside bone marrow hypocellularity and thrombocytopenia.225 P-selectin neutralization in vivo significantly recovered marrow cellularity and peripheral platelet counts. In addition, blocking P-selectin reduced platelet–monocyte and platelet-neutrophil aggregates formation, thereby mitigating chemokine levels and neutrophil infiltration to the lungs, reducing tissue damage. Together, these clinical and experimental evidence highlight the central role of CD62P in mediating thromboinflammation in dengue. Targeting these pathways may attenuate pathophysiological processes linked to vascular dysfunction, providing potential therapeutic insights.
COVID-19 and Post Acute Sequelae of COVID-19 (PASC)
Severe COVID-19 is a complication of SARS-CoV-2 pneumonia characterized by systemic thromboinflammation.226–230 Thromboembolic events are a major cause of mortality in severe COVID-19, with a growing body of evidence showing coagulation activation and platelet activation markers associated with poor outcomes.35,226,228,231–238 COVID-19 patients platelets exhibit a hyperactivated phenotype with increased CD62P surface expression, TXB2 secretion, EV release, and heightened responsiveness to stimuli such as thrombin, collagen, and fibrinogen, besides platelet–leukocyte aggregates formation among neutrophils, monocytes, and T-cells.226,228,232,235,239–242 Notably, platelet activation and platelet-induced monocyte reprogramming correlate with disease severity, thrombosis, and mortality.226,243,244
Although ACE2 and TMPRSS2 are central receptors for SARS-CoV-2 entry in multiple cell types,245,246 platelets internalize the virus via ACE2-independent mechanisms, as most studies did not detect ACE2 on platelets.239,245,246 Yet, the viral RNA is found within platelets from patients or in vitro infection models.239,247–251 Many alternative receptors have been proposed for SARS-CoV-2 binding on platelets, including CD42b, CD147, and CD26, although consensus on whether they are involved in viral entry was not reached.229,252–256 Recently, P-selectin was proposed as a target for SARS-CoV-2 binding on platelets under flow.229,256,257 Regardless of viral entry, SARS-CoV-2 or Spike protein binding to most of these receptors triggers signal transduction inducing platelet activation and PMA formation.229,252–256 New studies are still necessary to determine whether SARS-CoV-2 binding to Platelet P-selectin may play a role in infection and/or platelet-mediated responses during COVID-19.
Severe COVID-19 is marked by systemic inflammation with elevated circulating cytokines such as TNF-α, IL-1β, and IL-6.226,228–230,258,259 Plasma from COVID-19 patients is sufficient to induce platelet activation, PMA formation, and TF expression independently of viral presence and depending on IL-6 receptor.228 The PMA phenotype induced in this model is similar to the one reported during severe COVID-19, involving majorly CD16+ monocyte subsets with higher TF and lower HLA-DR expression.12,35,229,241 These monocyte characteristics were related to disease severity and mortality in COVID-19 patients.226,260
Mechanistic experiments with platelet–monocyte co-cultures using platelets from COVID-19 patients ex vivo, or SARS-CoV-2 infection in vitro, showed that platelets are determinant and sufficient to induce TF expression in monocytes depending on CD62P and integrin αIIbβ3.12,226 Neutralization of CD62P, but not integrin αIIb/β3, disrupted PMA formation, confirming P-selectin as the dominant adhesion molecule mediating platelet–monocyte binding, even though both adhesion molecules signal monocyte activation and TF expression.12,226 While platelets from COVID-19 patients induced monocyte inflammatory responses, monocytes from patients were also hyperresponsive to platelets and to immobilized CD62P or fibrinogen, producing higher levels of TNF-α and IL-1β.12 Consistently, platelets from COVID-19 patients trigger monocyte TNF-α and IL-1β secretion.12,35 Similar results have been reported with in vitro models using SARS-CoV-2 Spike protein-induced platelet activation and PMA formation, promoting monocyte IL-1β and IL-6 secretion through CD62P and CD40L signaling.229 Parallel neutralization of P-selectin, integrin αIIb/β3 and TF in both, ex vivo and in vitro experimental models, revealed a sequential mechanism of platelet-driven monocyte inflammatory amplification in COVID-19. P-selectin and integrin αIIb/β3 surface signaling induce the secretion of inflammatory cytokines and chemokines plus TF expression in the monocyte (Figure 2a). TF then amplifies the inflammatory signaling by triggering PAR-1 engagement in platelets and PAR-1 and PAR-2 in monocytes (Figure 2b). This reciprocal activation loop amplifies platelet degranulation and is responsible for monocyte CD16 expression and TNF-α and IL-1β secretion (Figure 2c).12,35 This P-selectin-induced and TF-amplified signaling mechanism contributes to hyperinflammation and hypercoagulability in severe COVID-19 (Figure 2d).
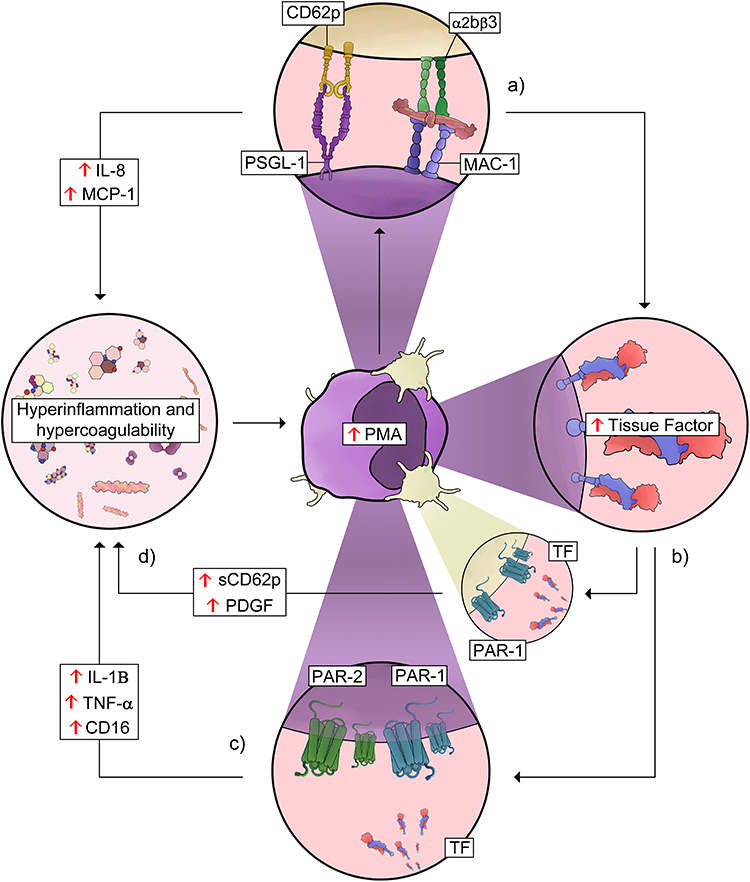 |
Figure 2 Platelet–monocyte interaction-driven thromboinflammatory loop in COVID-19. Schematic representation of thromboinflammatory amplification mediated by platelet–monocyte aggregates (PMA). (a) Activated platelets interact with monocytes mainly through P-selectin (CD62P)–PSGL-1 binding and additional integrin-mediated interactions, such as αIIbβ3 and MAC-1, promoting PMA formation. This interaction enhances the release of inflammatory mediators, including IL-8 and MCP-1, contributing to a hyperinflammatory and hypercoagulable state. (b) Platelet–monocyte interactions promote tissue factor (TF) expression in monocytes, increasing the procoagulant potential and contributing to thrombus formation. (c) TF-driven activation of the coagulation cascade leads to signaling through protease-activated receptors (PAR-1 and PAR-2) on platelets and monocytes, amplifying inflammatory responses and sustaining the thromboinflammatory loop, with increased levels of mediators such as IL-1β, TNF-α, CD16, soluble P-selectin (sCD62P), and platelet-derived growth factor (PDGF). (d) Mediators from activated plateets and monocytes amplify inflammation and hypercoagulability in COVID-19. See the text for details and references. Abbreviations: PMA, platelet–monocyte aggregates; PSGL-1, P-selectin glycoprotein ligand-1; MAC-1, macrophage-1 antigen (CD11b/CD18); CD62P, P-selectin; αIIbβ3, integrin alpha-IIb/beta-3 (glycoprotein IIb/IIIa); sCD62P, soluble P-selectin; PDGF, platelet-derived growth factor; MCP-1, monocyte chemoattractant protein-1; PAR-1, protease-activated receptor-1; PAR-2, protease-activated receptor-2. |
Even though pre-clinical data described above may suggest a major role for P-selectin in inflammatory amplification during COVID-19, clinical evidence remains limited. A randomized, double-blind, placebo-controlled pilot trial investigating hospitalized COVID-19 patients treated with the anti-P-selectin monoclonal antibody crizanlizumab found no significant effects on inflammatory mediator levels or clinical outcomes.261 However, given the small sample size, these findings should be interpreted with caution, and larger studies are required to determine whether targeting P-selectin or its downstream pathways, including TF, could provide clinical benefit.
Beyond the immune alterations observed during acute COVID-19, a subset of individuals develops a syndrome known as PASC or long COVID, defined by the persistence of symptoms for more than 12 weeks after acute SARS-CoV-2 infection. This post-acute condition involves immunological and inflammatory mechanisms that, although distinct, arise from the dysregulated response triggered during the initial infection. Clinical presentations include fatigue, post-exertional malaise, cognitive impairment, dyspnea, and arthralgia.262,263 The most relevant manifestations result from pulmonary involvement with dyspnea, chest pain, and fatigue associated with tissue remodeling and infiltration observed in computed tomography scans.264,265
Chronic low-grade inflammation in PASC sustains a persistently activated platelet phenotype, marked by increased expression of P-selectin, αIIbβ3 activation, and platelet aggregation in individuals who have had severe COVID-19.266,267 Persistent platelet activation is observed months after infection, accompanied by elevated platelet–leukocyte aggregates and coagulation markers such as D-dimer.268–270 Platelet–monocyte and platelet–granulocyte aggregates formed in PASC express higher TF levels and correlate to the extent of lung parenchymal damage, linking platelet hyperactivation to common PASC symptoms, including fatigue, breathlessness, and persistent pulmonary inflammation.265,270 The PASC inflammatory milieu is sufficient to drive platelet activation, as plasma from PASC patients induces platelet activation and platelet–leukocyte aggregates formation through mechanisms depending on Fcγ-R and IL-6R, but independent of PAR-1 signaling.268,270 Future studies should further examine the CD62P/PSGL-1 axis to elucidate how CD62P-mediated signaling shapes monocyte function in PASC. The enrichment of PMA and the heightened procoagulant phenotype of monocytes in PASC raise the possibility that sustained platelet-driven reprogramming may represent a key mechanism underlying chronic thromboinflammation in long COVID.
Influenza Virus
Influenza is one of the most prevalent respiratory infections in humans, occurring both as occasional pandemic outbreaks and recurrent seasonal epidemics, resulting in substantial morbidity and a significant number of deaths. Its pathogenesis is multifaceted, involving virological and host immune system determinants.271–273 In severe cases, dysregulated inflammation contributes to lung injury,274–276 a process largely orchestrated by the vascular endothelium, whose activation and dysfunction promote platelet adhesion and leukocyte recruitment.277 Clinical research confirms in vivo platelet activation in critically ill H1N1 patients, including enhanced PMA formation.278
In vitro studies have confirmed the relationship between Influenza A Virus (IAV) strains and platelet/endothelial activation, possibly impacting strain-dependent lung pathology. The highly pathogenic IAV strain H5N1 induces pulmonary microvascular endothelial cells to express selectins, including CD62P and CD62E, whereas seasonal strains such as H1N1 and H3N2 did not.279–281 Even though, seasonal H3N2 promotes platelet-endothelium adhesion in vitro through mechanisms independent of CD62P but dependent on αV/β1 integrin.281 These findings highlight the capacity for platelets and endothelial cells to recognize viral subtypes, influencing adhesion-mediated responses.
In murine models of lethal influenza infection, increased sCD62P in bronchoalveolar lavage fluid indicates platelet activation in the pulmonary microenvironment. Additional approaches, including direct platelet counting and immunohistochemistry, demonstrated platelet migration into the airways and lung tissue, associated with congestion, infiltration of monocytes and neutrophils, interstitial and alveolar hemorrhages, and thrombosis.282 Murine models infected with H5N1 show increased macrophage and neutrophil infiltration in the lungs.283 Antiplatelet pharmacological interventions, including aspirin or the GPIIb/IIIa antagonist eptifibatide, reduced CD62P activation and prevented lung injury, highlighting the functional role of platelets in pulmonary pathogenesis during influenza.281,282
Influenza vaccination is widely used as a controlled model of systemic inflammation to investigate platelet–monocyte interactions. Following vaccination, increased PSGL-1 expression and PMA formation were observed, particularly within the intermediate monocyte subset. This subset was more sensitive to interactions with activated platelets, correlating positively with platelet activation and systemic inflammatory markers.284–286 Co-culture studies demonstrated that PMA formation and increased CD16 expression in monocytes depended directly on CD62P/PSGL-1-mediated interactions exhibiting enhanced cytokine production and adhesion with platelets and endothelial cells. In contrast, the addition of platelet releasate or recombinant sCD62P does not fully reproduce this phenotype, indicating that physical contact with activated platelets is essential for functional modulation. COX-2 inhibition abolished CD16 upregulation but did not affect PMA formation, suggesting that specific intracellular pathways, such as COX-2/PGE2, modulate distinct aspects of the monocytic phenotype during PMA formation.223,287
Further studies are needed to understand how platelet CD62P-mediated leukocyte recruitment contributes to pulmonary pathogenesis during severe influenza infection, and whether PMA formation amplifies inflammation and exacerbates tissue injury. Investigating these mechanisms may reveal novel strategies to modulate inflammatory responses without compromising antiviral immunity.
HIV Infection
Human immunodeficiency virus 1 (HIV-1) is the most prevalent retrovirus in the world, being highly transmissible and widely distributed.288 Acute infection progresses with high viremia and CD4+ T-cell depletion, followed by chronic viral replication and progressive immune deterioration. Acquired immunodeficiency syndrome (AIDS) is characterized by severe immunosuppression and occurrence of opportunistic infections.289,290 However, with the introduction of antiretroviral therapy (ART), the epidemiology of HIV infection underwent an important transition, from high mortality due to opportunistic infections in AIDS to the predominance of long-term non-infectious comorbidities among chronically infected individuals.291,292 With sustained virological control by ART, people living with HIV (PLHIV) now live longer, but remain susceptible to long-term comorbidities including hypertension, metabolic syndrome, dyslipidemia, cardiovascular disease, neurocognitive disorders, and non-AIDS-related cancers.290,293–303
In untreated HIV, or AIDS patients, viral replication and systemic inflammation promote intense platelet activation with CD62P and CD40L surface expression,304 which correlates with increased TF in monocytes.304 Untreated HIV patients exhibit increased PMA formation, especially within CD16+ monocytes, and show elevated levels of sCD163 and sCD14, indicating increased monocyte activation.305 Both, platelet activation and PMA formation correlate positively with the viral load and negatively with CD4+ T-cell count, supporting a role in AIDS pathophysiology (Figure 3a). 305–307 In addition, platelets from AIDS patients become exhausted, secreting lower levels of chemokines when stimulated ex vivo. Complementing these observations, studies in non-human primate models infected with SIV show increased platelet activation, with CD62P and CD40L favoring PMA formation primarily with CD16⁺ monocytes and correlating with thrombocytopenia.308 These data suggest that platelet activation and PMA formation may be related to viral replication in AIDS. However, recent studies have shown residual platelet activation in PLHIV under ART, with increased CD62P and sCD40L persisting for months to years after virological suppression.306,309–316 Additional analyses of PLHIV on stable ART show persistent platelet exhaustion evidencing chronic platelet activation,309 and elevated levels of sCD163,317 indicating that platelet and monocyte activation involves other mechanisms besides viremia. Like HIV, SIV-infected primate models receiving ART exhibit persistent monocyte and macrophage activation.318 These findings indicate that, despite viral suppression achieved through ART, several mechanisms continue to contribute to a persistent inflammatory state in PLHIV (Figure 3b). Among these mechanisms, viral persistence and latency, as well as microbial translocation, with continuous activation of Pattern Recognition Receptors in the innate immunity are probably involved.318
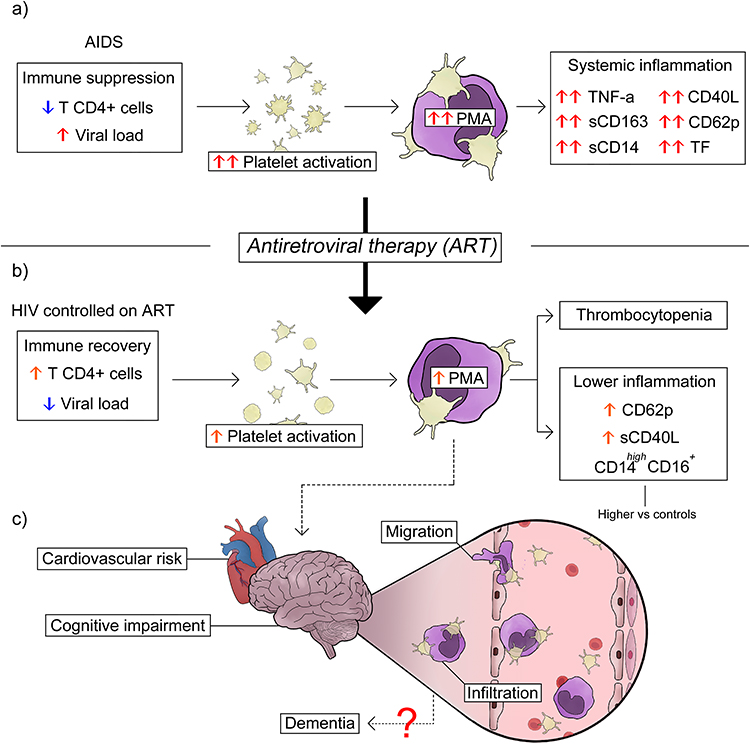 |
Figure 3 PMAs in HIV-associated inflammation. (a) HIV-infected untreated AIDS patients exhibit increased platelet activation and PMA formation in association with viral load and immunosuppression. (b) Platelet activation and PMA formation in individuals living with HIV under ART-induced virological control. ART partially reduces platelet activation and PMA formation, resulting in decreased inflammatory markers and improved immune recovery. (c) Despite this improvement, residual activation persists contributing to PMA infiltration into the parenchyma of target organs, such as the brain, which is associated with increased neuroinflammation. See the text for details and references. Abbreviations: AIDS, immunodeficiency syndrome; PMA, platelet-monocyte aggregates; CD62P, P-selectin; CD40L, CD40 ligand; TF, tissue factor; sCD163, soluble CD163; sCD14, soluble CD14; ART, antiretroviral therapy; HIV, human immunodeficiency virus. |
In addition to the peripheral blood, platelet activation and PMA formation have functional effects in target tissues, including the brain, potentially amplifying local and systemic inflammatory responses, with implications for long-term comorbidities in PLHIV.319 Particularly relevant for neurological pathogenesis, studies in animal models and human tissues indicate PMA adhesion and infiltration to the brain.319,320 In vitro experiments using brain microvascular endothelial cell monolayers showed that monocytes within PMAs exhibit increased CCR2, PSGL-1, and CD40 expression, along with enhanced adhesion and trans-endothelial migration in a CD62P-dependent manner.320 Studies showed that platelet-derived sCD40L increases blood–brain barrier permeability and promotes monocyte adhesion and migration into the brain.319 Clinical studies have shown that the expansion of non-classical monocytes was correlated with poor cognitive performance in PLHIV with neurological disorder, highlighting an association between monocyte activation and HIV-associated cognitive impairment.314 Corroborating these findings, post-mortem brain tissues from patients with HIV-associated encephalitis exhibited higher numbers of PMAs marginated along post-capillary venule walls compared with HIV-negative individuals, suggesting that platelet activation and PMA formation directly contribute to HIV-associated neuroinflammation (Figure 3c).320 These data indicate that, even under ART, HIV-1 infection induces residual platelet activation and PMA formation, contributing to monocyte infiltration into the brain and neuroinflammation, but potentially contributing to other long-term comorbidities involving other target tissues.
Bacterial Infection/Sepsis
Sepsis is characterized by a dysregulated immune response to infection that can progress to multiple organ dysfunction, septic shock, and death.321 Although it may arise from bacterial, viral, or fungal pathogens, Gram-positive bacteria such as Staphylococcus aureus and Streptococcus pneumoniae are among the most frequently associated with sepsis.322 Platelets recognize bacterial structures through many receptors such as Toll-like receptors, GPIb, and GPIIb/IIIa, among others.323 In addition, bacteria release soluble products capable of stimulating platelets,324 or proteins that associate with plasma proteins, such as vWF and fibrinogen, forming molecular bridges that indirectly activate platelets.325 Once activated, platelets promote immune cell activation and drive the formation of PMAs, playing central roles in sepsis by contributing to microvascular thrombosis and organ dysfunction.326–328
Transcriptomic analysis of Peripheral Blood Mononuclear Cells from patients with sepsis revealed that platelets and monocytes are the cell types exhibiting the most pronounced transcriptional disturbances associated with inflammation and coagulation during the transition from high risk to clinical sepsis.329 This pattern indicates a coordinated immune response between platelets and monocytes that potentially contributes to disease progression. Clinical studies further support the importance of platelet–monocyte interactions in sepsis. Gram-positive sepsis is associated with higher platelet activation, platelet hyperreactivity, and increased PMA formation compared with sepsis caused by common Gram-negative pathogens.311 In elderly septic patients, PMA formation was associated with mortality, a relationship not observed in younger individuals.330 Another recent study examined PMAs in septic patients who developed respiratory distress syndrome (ARDS), showing that PMA levels were significantly elevated in ARDS group compared with sepsis alone, and positively correlated with APACHE II severity scores.331
Experimental sepsis reinforces the central role of platelet–monocyte interactions. In a murine model of peritoneal sepsis, early and robust upregulation of CD62P was accompanied by a marked increase in PMA formation within hours after abdominal sepsis.332 The functional importance of this pathway is demonstrated in CD62P-deficient mice, which show reduced PMA formation, impaired bacterial clearance, dysregulated inflammatory responses, and significantly worse clinical outcomes, including increased mortality, during pneumosepsis.333,334 These findings support that CD62P-mediated platelet–monocyte interactions are not only biomarkers of disease severity but actively contribute to host defense mechanisms in sepsis.
The importance of platelet–monocyte interactions for bacterial clearance has also been demonstrated in in vitro models of Klebsiella pneumoniae and Brucella abortus infection, where platelets enhance phagocytosis, activation, and cytokine production (Figure 4a).335,336 However, certain pathogens have evolved mechanisms to counteract this host defense strategy. Staphylococcus aureus, for instance, secretes the extracellular fibrinogen-binding protein (Efb), which blocks interaction between CD62P and PSGL-1, thereby inhibiting the formation of platelet–leukocyte complexes (Figure 4b).337 This evasion mechanism highlights the importance of PMA formation in antibacterial immunity, as bacteria capable of disrupting these interactions can effectively dampen inflammatory responses and improve their survival within the host.
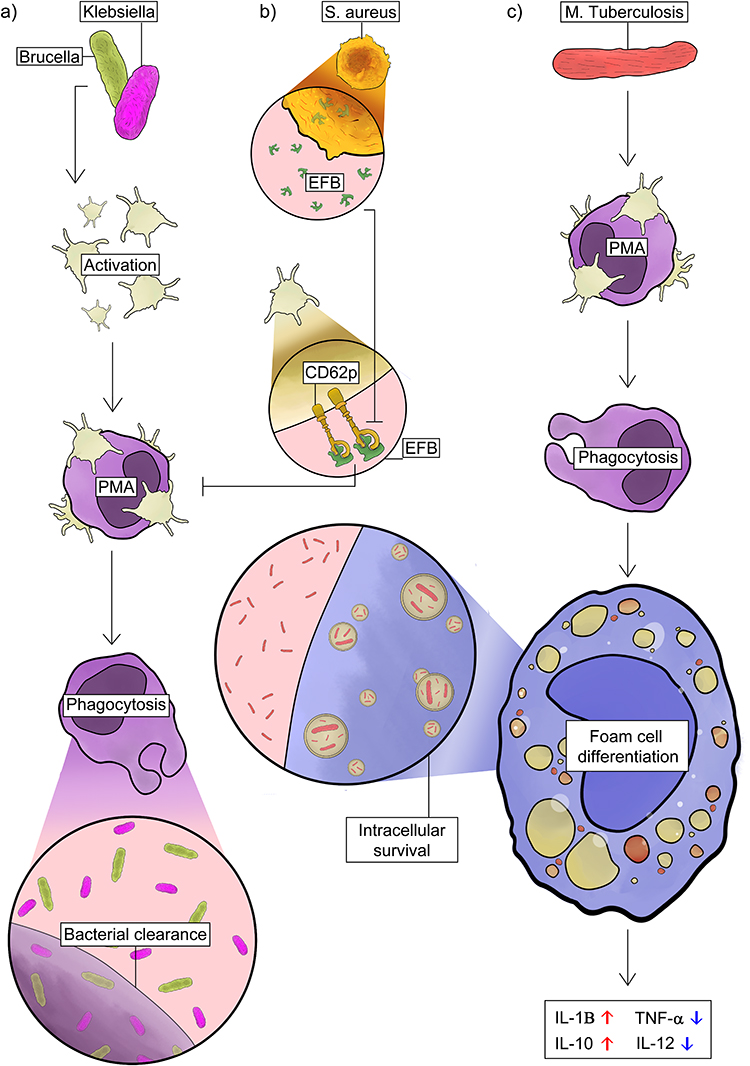 |
Figure 4 Platelet–monocyte interactions in bacterial infections. (a) During infection with Klebsiella pneumoniae and Brucella abortus, platelets interact with monocytes promoting their activation, enhancing phagocytosis and cytokine production, which contributes to bacterial clearance. (b) In Staphylococcus aureus infection, the bacterial protein Efb interferes with CD62P-PSGL-1 interactions, impairing platelet–leukocyte aggregate formation and favoring bacterial persistence and survival. (c) In active pulmonary tuberculosis caused by Mycobacterium tuberculosis, increased levels of platelet–monocyte aggregates (PMAs) promote monocyte differentiation into foam cells, a phenotype associated with high lipid load, increased IL-1β and IL-10 production, and reduced TNF-α and IL-12, which favors pathogen intracellular survival. See the text for details and references. Abbreviations: PMAs, platelet-monocyte aggregates; Efb, fibrinogen-binding protein; CD62P, P-selectin. |
Increasing evidence indicates that platelets play an active role during Mycobacterium tuberculosis (M.tb) infection, contributing to inflammatory and tissue-damage processes that exacerbate tuberculosis pathogenesis. Tuberculosis primarily affects the lungs and is characterized by an intense immune response, extensive tissue remodeling, and frequent pulmonary cavitation.338 Patients with active pulmonary TB show significantly elevated levels of circulating PMAs.339 In the presence of platelets, human monocytes differentiate into large epithelioid-like multinucleated foam cells resembling those found in tuberculous granulomas.340 This process requires platelet phagocytosis by monocyte and results in the formation of foam cells with a distinctly immunoregulatory phenotype, characterized by increased IL-10 and IL-1β alongside reduced TNF-α and IL-12 release. Together, these changes indicate platelet-driven monocyte differentiation towards permissive macrophage subsets during mycobacterial infection which in turn contributes to increased intracellular bacterial viability (Figure 4c).341,342 Furthermore, monocytes co-stimulated with platelets and M.tb showed higher expression of matrix metalloproteinases MMP-1, enzyme strongly linked to collagen degradation and pulmonary tissue destruction in TB.342 These studies demonstrate that platelets can modulate monocyte responses and may contribute to granuloma organization and potentially to the balance between pathogen control and immune regulation in tuberculosis.
Thus, platelet–monocyte interactions in bacterial infections occupy a central and paradoxical position, acting as essential components of host defense while also driving pathological inflammation, promoting tissue remodeling, and even facilitating pathogen survival within monocytes.
Conclusion
CD62P-mediated platelet–monocyte interactions represent a central axis of inflammation across diverse pathological conditions. These interactions contribute to multiple pathophysiological processes including leukocyte recruitment, chemokine transfer, monocyte activation, and foam cell formation, contributing to vascular remodeling, fibrosis, and tissue injury. PMA formation is primarily driven by platelet activation and P-selectin-PSGL-1 interactions, in both sterile and non-sterile inflammatory conditions, leading to monocyte activation and amplification of thromboinflammatory responses that contribute to inflammation and organ dysfunction. However, the upstream triggers and functional outcomes differ significantly. Sterile inflammation is driven by damage-associated molecular patterns, such as oxidized lipids and metabolic stress signals, leading to sustained formation of PMAs. These aggregates are often associated with endothelial dysfunction and proatherogenic processes. In non-sterile inflammation, pathogen-associated molecular patterns directly activate innate immune and platelet receptors, resulting in an acute and robust inflammatory response with enhanced PMA formation. In bacterial infections, PMAs may also contribute to host defense by enhancing pathogen recognition, promoting monocyte activation, and facilitating bacterial clearance. Thus, while the core mechanisms of PMA formation are shared, their regulation and pathological consequences are highly context dependent.
Likewise, therapeutic strategies targeting CD62P, including PSGL-1 analogs and anti-P-selectin antibodies, have also shown context-dependent efficacy. Clinical benefit has been demonstrated primarily in SCD,210 in which crizanlizumab supports the mechanistic relevance of CD62P-mediated platelet–leukocyte interactions. However, similar benefits have not been consistently observed in other thromboinflammatory diseases, many of which CD62P-targeted therapies have not yet been clinically evaluated. In hospitalized COVID-19 patients, a randomized, placebo-controlled pilot trial of the crizanlizumab showed no significant effects on inflammatory markers or clinical outcomes,261 suggesting that CD62P blockade alone may be insufficient to modulate inflammation in certain contexts. Larger, adequately powered trials are needed to determine whether targeting P-selectin or its downstream pathways, such as TF, may provide clinical benefit. Therefore, while effective in SCD, CD62P-targeted strategies likely depend on disease-specific inflammatory drivers and the relative contribution of CD62P-mediated pathways, requiring further evaluation across diverse conditions with consideration for combination therapies and patient stratification.
Pharmacological approaches targeting platelet–monocyte interactions through anticoagulation also exhibit context-dependent effects. While anticoagulant treatment has been associated with a reduction in PMA formation in conditions, such as dyslipidemia, this effect appears limited or insignificant in type 1 and type 2 diabetes mellitus.95 These observations suggest that in metabolic diseases characterized by chronic inflammation and hyperglycemia, additional mechanisms beyond coagulation may sustain platelet–monocyte interactions. Antidiabetic155 and lipid lowering agents99 have been shown to reduce platelet activation and PMA formation. Therefore, even though isolated P-selectin-based or anti-platelet therapeutic approaches may be insufficient to effectively modulate PMA formation, combined strategies, including glucose-lowering or hypolipidemic agents emerge as promising alternatives to reduce PMA formation and its pathological consequences. Thus, an integrated view of these mechanisms not only broadens the understanding of the pathophysiology involved but also points towards the development of more targeted and effective therapeutic interventions.
Despite these advances, important gaps remain in the understanding of CD62P-mediated platelet–leukocyte interactions. Most studies have focused on platelet-neutrophil aggregates,204,343–351 and the specific contributions of PMA to human pathophysiology remains incompletely defined, particularly across heterogeneous inflammatory and infectious diseases. Future research should address these gaps at multiple levels. At the mechanistic level, further studies are needed to elucidate the signaling pathways and cell-specific contributions underlying PMA formation and function. At the clinical level, improved characterization of patient heterogeneity and disease-specific inflammatory profiles will be essential to better define the contexts in which PMA are most relevant. At the therapeutic level, the development of combination strategies and targeted interventions should be prioritized to overcome the limited efficacy observed with CD62P blockade alone.
In summary, CD62P-dependent platelet–monocyte interactions are key mechanisms in sterile and non-sterile inflammation. Integrating mechanistic, translational, and clinical research will be essential for developing novel therapeutic strategies aimed at mitigating thromboinflammatory complications and improving patient outcomes. This holistic perspective reinforces research and clinical significance of PMAs and highlights critical avenues for future investigation. Understanding these pathways will be essential for developing new therapeutical approaches and improving clinical outcomes.
Abbreviations
CD62E, E-selectin; CD62P, P-selectin; CD62L, L-selectin; PSGL-1, P-selectin glycoprotein ligand-1; PMA, platelet–monocyte aggregates; sCD62P, soluble P-selectin; OxLDL, oxidized LDL; CAD, coronary artery disease; ACS, acute coronary syndromes; CVD, cardiovascular disease; T2DM, type 2 diabetes mellitus; TF, tissue factor; NASH, Nonalcoholic Fatty Liver Disease; MASH, metabolic dysfunction-associated steatohepatitis; vWF, von Willebrand factor; SCD, sickle cell disease; PDPN, Podoplanin; ADs, autoimmune diseases; SLE, systemic lupus erythematosus; T1DM, type 1 diabetes mellitus; UC, ulcerative colitis; LPS, lipopolysaccharide; DENV, dengue viruses; PS, phosphatidylserine; LD, lipid droplet; TxB2, thromboxane B2; PASC, Post-Acute Sequelae of COVID-19; hPMEC, human pulmonary microvascular endothelial cells; HIV, human immunodeficiency virus; AIDS, immunodeficiency syndrome; ART, antiretroviral therapy; PLHIV, people living with HIV; SIV, simian immunodeficiency virus; ARDS, respiratory distress syndrome; Efb, fibrinogen-binding protein; M.tb, Mycobacterium tuberculosis; TB, tuberculosis.
Data Sharing Statement
Data sharing is not applicable to this article as no data were created or analyzed in this study.
Acknowledgments
The authors thank the Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq), the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES) and the Instituto Serrapilheira for the funding support.
Author Contributions
MKCF, MMLdP, RTRO, JPRSdS, and MCBM: Writing – original draft. MKCF and EDH: Writing – review and editing. EDH: Conceptualization, funding acquisition, project administration, resources, and supervision. All authors discussed the concepts.
All authors made a significant contribution to the work reported, whether that is in the conception, study design, literature search, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq); Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES); and Instituto Serrapilheira.
Disclosure
The authors report no conflicts of interest in this article.
References
1. Chovatiya R, Medzhitov R. Stress, inflammation, and defense of homeostasis. Mol Cell. 2014;54(2):281–29. doi:10.1016/j.molcel.2014.03.030
2. Casanova JL, Abel L. Mechanisms of viral inflammation and disease in humans. Science. 2021;374(6571):1080–1086. doi:10.1126/science.abj7965
3. Gupta L, Thomas J, Ravichandran R, Singh M, Nag A, Panjiyar BK. Inflammation in cardiovascular disease: a comprehensive review of biomarkers and therapeutic targets. Cureus. 2023;15(9):e45483. doi:10.7759/cureus.45483
4. Soták M, Clark M, Suur BE, Börgeson E. Inflammation and resolution in obesity. Nat Rev Endocrinol. 2024;21(1):45–61. doi:10.1038/s41574-024-01047-y
5. Chen GY, Nuñez G. Sterile inflammation: sensing and reacting to damage. Nat Rev Immunol. 2010;10(12):826–837. doi:10.1038/nri2873
6. Nathan C, Ding A. Nonresolving Inflammation. Cell. 2010;140(6):871–882. doi:10.1016/j.cell.2010.02.029
7. Zhou Y, Hong Y, Huang H. Triptolide attenuates inflammatory response in membranous glomerulo-nephritis rat via downregulation of NF-κB signaling pathway. Kidney Blood Press Res. 2016;41(6):901–910. doi:10.1159/000452591
8. Ekdahl KN, Teramura Y, Asif S, Jonsson N, Magnusson PU, Nilsson B. Thromboinflammation in therapeutic medicine. Adv Exp Med Biol. 2015;865:3–17. doi:10.1007/978-3-319-18603-0_1
9. Wagner DD, Heger LA. Thromboinflammation: from atherosclerosis to COVID-19. Arterioscler Thromb Vasc Biol. 2022;42(9):1103. doi:10.1161/ATVBAHA.122.317162
10. Iba T, Helms J, Levi M, Levy JH. Thromboinflammation in acute injury: infections, heatstroke, and trauma. J Thromb Haemost. 2024;22(1):7–22. doi:10.1016/j.jtha.2023.07.020
11. Semple JW, Italiano JE, Freedman J. Platelets and the immune continuum. Nat Rev Immunol. 2011;11(4):264–274. doi:10.1038/nri2956
12. Hottz ED, Martins-Gonçalves R, Palhinha L, et al. Platelet-monocyte interaction amplifies thromboinflammation through tissue factor signaling in COVID-19. Blood Adv. 2022;6(17):5085–5099. doi:10.1182/BLOODADVANCES.2021006680
13. Scherlinger M, Richez C, Tsokos GC, Boilard E, Blanco P. The role of platelets in immune-mediated inflammatory diseases. Nat Rev Immunol. 2023;23(8):495–510. doi:10.1038/s41577-023-00834-4
14. de Paula MML, Oliveira RTR, Hottz ED. Platelets and platelet–leukocyte interactions in infectious diseases. Curr Opin Hematol. 2025;32(5):261–269. doi:10.1097/MOH.0000000000000878
15. Zheng L, Feng D, Wu Y, et al. Platelets as immune sensors: monitoring immune dynamics and diagnosing disease states across multiple disorders. EBioMedicine. 2026;125:106174. doi:10.1016/j.ebiom.2026.106174
16. Larsen E, Palabrica T, Sajer S, et al. PADGEM-dependent adhesion of platelets to monocytes and neutrophils is mediated by a lineage-specific carbohydrate, LNF III (CD15). Cell. 1990;63(3):467–474. doi:10.1016/0092-8674(90)90443-I
17. Palabrica T, Lobb R, Furie BC, et al. Leukocyte accumulation promoting fibrin deposition is mediated in vivo by P-selectin on adherent platelets. Nature. 1992;359(6398):848–851. doi:10.1038/359848A0
18. Greinacher A, Warkentin TE. Platelet factor 4 triggers thrombo-inflammation by bridging innate and adaptive immunity. Int J Lab Hematol. 2023;45(S2):11–22. doi:10.1111/ijlh.14075
19. Liu Z, Li L, Zhang H, et al. Platelet factor 4(PF4) and its multiple roles in diseases. Blood Rev. 2024;64:101155. doi:10.1016/j.blre.2023.101155
20. Hrachovinová I, Cambien B, Hafezi-Moghadam A, et al. Interaction of P-selectin and PSGL-1 generates microparticles that correct hemostasis in a mouse model of hemophilia A. Nat Med. 2003;9(8):1020–1025. doi:10.1038/NM899
21. Williams H, Mack C, Baraz R, et al. Monocyte Differentiation and Heterogeneity: inter-Subset and Interindividual Differences. Int J Mol Sci. 2023;24(10):8757. doi:10.3390/ijms24108757
22. Ziegler-Heitbrock L, Ancuta P, Crowe S, et al. Nomenclature of monocytes and dendritic cells in blood. Blood. 2010;116(16):e74–e80. doi:10.1182/blood-2010-02-258558
23. Wong KL, Yeap WH, Tai JJY, Ong SM, Dang TM, Wong SC. The three human monocyte subsets: implications for health and disease. Immunol Res. 2012;53(1–3):41–57. doi:10.1007/s12026-012-8297-3
24. Wong KL, Tai JJY, Wong WC, et al. Gene expression profiling reveals the defining features of the classical, intermediate, and nonclassical human monocyte subsets. Blood. 2011;118(5):e16–e31. doi:10.1182/blood-2010-12-326355
25. Marcu-Malina V, Heijhuurs S, Van Buuren M, et al. Redirecting αβ T cells against cancer cells by transfer of a broadly tumor-reactive γδT-cell receptor. Blood. 2011;118(1):50–59. doi:10.1182/blood-2010-12-325993
26. Cros J, Cagnard N, Woollard K, et al. Human CD14dim monocytes patrol and sense nucleic acids and viruses via TLR7 and TLR8 receptors. Immunity. 2010;33(3):375–386. doi:10.1016/j.immuni.2010.08.012
27. Vas J, Grnwal C, Silverman GJ. Fundamental roles of the innate-like repertoire of natural antibodies in immune homeostasis. Front Immunol. 2013;4(FRB). doi:10.3389/fimmu.2013.00004
28. Smith BAH, Bertozzi CR. The clinical impact of glycobiology: targeting selectins, Siglecs and mammalian glycans. Nat Rev Drug Discov. 2021;20(3):217. doi:10.1038/S41573-020-00093-1
29. Liu Z, Miner JJ, Yago T, et al. Differential regulation of human and murine P-selectin expression and function in vivo. J Exp Med. 2010;207(13):2975–2987. doi:10.1084/JEM.20101545
30. Zarbock A, Ley K, McEver RP, Hidalgo A. Leukocyte ligands for endothelial selectins: specialized glycoconjugates that mediate rolling and signaling under flow. Blood. 2011;118(26):6743–6751. doi:10.1182/BLOOD-2011-07-343566
31. Tardif JC, Tanguay JF, Wright SS, et al. Effects of the P-selectin antagonist inclacumab on myocardial damage after percutaneous coronary intervention for non-st-segment elevation myocardial infarction: results of the SELECT-ACS trial. J Am Coll Cardiol. 2013;61(20):2048–2055. doi:10.1016/j.jacc.2013.03.003
32. Tokarz-Deptuła B, Baraniecki Ł, Palma J, Stosik M, Deptuła W. Characterization of platelet receptors and their involvement in immune activation of these cells. Int J Mol Sci. 2024;25(23):12611. doi:10.3390/ijms252312611
33. Blann AD, Nadar SK, Lip GYH. The adhesion molecule P-selectin and cardiovascular disease. Eur Heart J. 2003;24(24):2166–2179. doi:10.1016/J.EHJ.2003.08.021
34. McEver RP. Selectins: initiators of leucocyte adhesion and signalling at the vascular wall. Cardiovasc Res. 2015;107(3):331. doi:10.1093/CVR/CVV154
35. Hottz ED, Bozza PT. Platelet-leukocyte interactions in COVID-19: contributions to hypercoagulability, inflammation, and disease severity. Res Pract Thromb Haemost. 2022;6(3):e12709. doi:10.1002/RTH2.12709
36. World Health Organization. Guidelines for drinking-water quality. 1997. Available from: https://iris.who.int/server/api/core/bitstreams/5430ae89-5967-49e7-bd64-7764d8a1cba9/content.
37. Burki T. European Commission classifies obesity as a chronic disease. Lancet Diabetes Endocrinol. 2021;9(7):418. doi:10.1016/S2213-8587(21)00145-5
38. Williams EP, Mesidor M, Winters K, Dubbert PM, Wyatt SB. Overweight and obesity: prevalence, consequences, and causes of a growing public health problem. Curr Obes Rep. 2015;4(3):363–370. doi:10.1007/S13679-015-0169-4
39. Alberti KGMM, Eckel RH, Grundy SM, et al. Harmonizing the metabolic syndrome: a joint interim statement of the International Diabetes Federation Task Force on Epidemiology and Prevention; National Heart, Lung, and Blood Institute; American Heart Association; World Heart Federation; International. Circulation. 2009;120(16):1640–1645. doi:10.1161/CIRCULATIONAHA.109.192644
40. González-Muniesa P, Mártinez-González MA, Hu FB, et al. Obesity. Nat Rev Dis Primers. 2017;3(1):1–18. doi:10.1038/nrdp.2017.34
41. MacMahon S, Baigent C, Duffy S, et al. Body-mass index and cause-specific mortality in 900 000 adults: collaborative analyses of 57 prospective studies. Lancet. 2009;373(9669):1083–1096. doi:10.1016/S0140-6736(09)60318-4
42. Wang H, Naghavi M, Allen C, et al. Global, regional, and national life expectancy, all-cause mortality, and cause-specific mortality for 249 causes of death, 1980-2015: a systematic analysis for the Global Burden of Disease Study 2015. Lancet. 2016;388(10053):1459–1544. doi:10.1016/S0140-6736(16)31012-1
43. GBD 2015 Obesity Collaborators. Health effects of overweight and obesity in 195 countries over 25 years. N Engl J Med. 2017;377(1):13–27. doi:10.1056/NEJMOA1614362.
44. Piché ME, Tchernof A, Després JP. Obesity Phenotypes, diabetes, and cardiovascular diseases. Circ Res. 2020;126(11):1477–1500. doi:10.1161/CIRCRESAHA.120.316101
45. Blokhin IO, Lentz SR. Mechanisms of thrombosis in obesity. Curr Opin Hematol. 2013;20(5):437. doi:10.1097/MOH.0B013E3283634443
46. De Pergola G, Pannacciulli N. Coagulation and fibrinolysis abnormalities in obesity. J Endocrinol Invest. 2002;25(10):899–904. doi:10.1007/BF03344054
47. Stein PD, Beemath A, Olson RE. Obesity as a risk factor in venous thromboembolism. Am J Med. 2005;118(9):978–980. doi:10.1016/j.amjmed.2005.03.012
48. Borch KH, Nyegaard C, Hansen JB, et al. Joint effects of obesity and body height on the risk of venous thromboembolism: the Tromsø Study. Arterioscler Thromb Vasc Biol. 2011;31(6):1439–1444. doi:10.1161/ATVBAHA.110.218925
49. Minervino D, Gumiero D, Nicolazzi MA, et al. Leukocyte activation in obese patients: effect of bariatric surgery. Medicine. 2015;94(40):e1382. doi:10.1097/MD.0000000000001382
50. Hotamisligil GS. Inflammation, metaflammation and immunometabolic disorders. Nature. 2017;542(7640):177–185. doi:10.1038/NATURE21363
51. De Pergola G, Pannacciulli N, Coviello M, et al. sP-selectin plasma levels in obesity: association with insulin resistance and related metabolic and prothrombotic factors. Nutr Metab Cardiovasc Dis. 2008;18(3):227–232. doi:10.1016/j.numecd.2006.09.010
52. Dib P, Fernandes M, Venerando L, et al. Platelet activation and platelet-monocyte interaction amplify thromboinflammation in obesity through adhesion molecules and endothelial IL-1R signaling. J Thromb Haemost. 2026 in press. doi:10.1016/j.jtha.2026.02.002
53. Ezzaty Mirhashemi M, Shah RV, Kitchen RR, et al. The dynamic platelet transcriptome in obesity and weight loss. Arterioscler Thromb Vasc Biol. 2020;41(2):854. doi:10.1161/ATVBAHA.120.315186
54. Periasamy M, Lieb DC, Butcher MJ, et al. Bariatric surgery decreases monocyte-platelet aggregates in blood: a pilot study. Obes Surg. 2014;24(8):1410–1414. doi:10.1007/s11695-014-1278-y
55. Nishimura S, Manabe I, Nagasaki M, et al. In vivo imaging in mice reveals local cell dynamics and inflammation in obese adipose tissue. J Clin Invest. 2008;118(2):710–721. doi:10.1172/JCI33328
56. Marques P, Collado A, Martinez-Hervás S, et al. Systemic inflammation in metabolic syndrome: increased platelet and leukocyte activation, and key role of CX3CL1/CX3CR1 and CCL2/CCR2 axes in arterial platelet-proinflammatory monocyte adhesion. J Clin Med. 2019;8(5):708. doi:10.3390/JCM8050708
57. Serebruany VL, Malinin A, Ong S, Atar D. Patients with metabolic syndrome exhibit higher platelet activity than those with conventional risk factors for vascular disease. J Thromb Thrombolysis. 2008;25(2):207–213. doi:10.1007/S11239-007-0047-3
58. Melaku L, Dabi A. The cellular biology of atherosclerosis with atherosclerotic lesion classification and biomarkers. Bull Natl Res Cent. 2021;45(1):225. doi:10.1186/s42269-021-00685-w
59. Blankenberg S, Barbaux S, Tiret L. Adhesion molecules and atherosclerosis. Atherosclerosis. 2003;170(2):191–203. doi:10.1016/S0021-9150(03)00097-2
60. Rangaswamy S, Penn MS, Saidel GM, Chisolm GM. Exogenous oxidized low-density lipoprotein injures and alters the barrier function of endothelium in rats in vivo. Circ Res. 1997;80(1):37–44. doi:10.1161/01.RES.80.1.37
61. Steffen Y, Jung T, Klotz LO, Schewe T, Grune T, Sies H. Protein modification elicited by oxidized low-density lipoprotein (LDL) in endothelial cells: protection by (–)-epicatechin. Free Radic Biol Med. 2007;42(7):955–970. doi:10.1016/j.freeradbiomed.2006.12.024
62. Klouche M, May AE, Hemmes M, et al. Enzymatically modified, nonoxidized LDL induces selective adhesion and transmigration of monocytes and T-lymphocytes through human endothelial cell monolayers. Arterioscler Thromb Vasc Biol. 1999;19(3):784–793. doi:10.1161/01.ATV.19.3.784
63. Shaw SK, Bamba PS, Perkins BN, Luscinskas FW, Kajiya F. Oxidized LDL specifically promotes the initiation of monocyte invasion during transendothelial migration with upregulated PECAM-1 and downregulated VE-cadherin on endothelial junctions. Atherosclerosis. 2007;194(2):e9–e17. doi:10.4049/jimmunol.167.4.2323
64. Keiper T, Al-Fakhri N, Chavakis E, et al. The role of junctional adhesion molecule-C (JAM-C) in oxidized LDL-mediated leukocyte recruitment. FASEB J. 2005;19(14):2078–2080. doi:10.1096/fj.05-4196fje
65. Hurt-Camejo E, Camejo G, Rosengren B, et al. Effect of arterial proteoglycans and glycosaminoglycans on low density lipoprotein oxidation and its uptake by human macrophages and arterial smooth muscle cells. Arterioscler Thromb. 1992;12(5):569–583. doi:10.1161/01.ATV.12.5.569
66. Østerud B, Bjørklid E. Role of monocytes in atherogenesis. Physiol Rev. 2003;83(4):1069–1112. doi:10.1152/physrev.00005.2003
67. Territo M, Berliner JA, Fogelman AM. Effect of monocyte migration on low density lipoprotein transport across aortic endothelial cell monolayers. J Clin Invest. 1984;74(6):2279–2284. doi:10.1172/JCI111655
68. Zernecke A, Schober A, Bot I, et al. SDF-1alpha/CXCR4 axis is instrumental in neointimal hyperplasia and recruitment of smooth muscle progenitor cells. Circ Res. 2005;96(7):784–791. doi:10.1161/01.RES.0000162100.52009.38
69. Bennett MR, Sinha S, Owens GK. Vascular smooth muscle cells in atherosclerosis. Circ Res. 2016;118(4):692. doi:10.1161/CIRCRESAHA.115.306361
70. Bentzon JF, Otsuka F, Virmani R, Falk E. Mechanisms of plaque formation and rupture. Circ Res. 2014;114(12):1852–1866. doi:10.1161/CIRCRESAHA.114.302721
71. Van Der Wal AC, Becker AE. Atherosclerotic plaque rupture--pathologic basis of plaque stability and instability. Cardiovasc Res. 1999;41(2):334–344. doi:10.1016/S0008-6363(98)00276-4
72. Holvoet P, Collen D. Thrombosis and atherosclerosis. Curr Opin Lipidol. 1997;8(5):320–328. doi:10.1097/00041433-199710000-00012
73. Rosendaal FR. Venous thrombosis: a multicausal disease. Lancet. 1999;353(9159):1167–1173. doi:10.1016/S0140-6736(98)10266-0
74. Cheng GC, Loree HM, Kamm RD, Fishbein MC, Lee RT. Distribution of circumferential stress in ruptured and stable atherosclerotic lesions. A structural analysis with histopathological correlation. Circulation. 1993;87(4):1179–1187. doi:10.1161/01.cir.87.4.1179
75. Noonan J, Cardoso L, Bobik A, Peter K. Atherosclerotic plaque instability and rupture: recommended mouse models to empower clinically relevant discoveries, diagnostics and therapeutics. Arterioscler Thromb Vasc Biol. 2025;45(10):1707. doi:10.1161/ATVBAHA.125.321011
76. Holmstedt CA, Turan TN, Chimowitz MI. Atherosclerotic intracranial arterial stenosis: risk factors, diagnosis, and treatment. Lancet Neurol. 2013;12(11):1106–1114. doi:10.1016/S1474-4422(13)70195-9
77. Falk E. Pathogenesis of Atherosclerosis. J Am Coll Cardiol. 2006;47(8 Suppl):C7–C12. doi:10.1016/j.jacc.2005.09.068
78. Sima AV, Stancu CS, Simionescu M. Vascular endothelium in atherosclerosis. Cell Tissue Res. 2009;335(1):191–203. doi:10.1007/s00441-008-0678-5
79. Ding X, Wang X, Wu J, Zhang M, Cui M. Triglyceride-glucose index and the incidence of atherosclerotic cardiovascular diseases: a meta-analysis of cohort studies. Cardiovasc Diabetol. 2021;20(1). doi:10.1186/s12933-021-01268-9
80. Renovato-Martins M, Moreira-Nunes C, Atella GC, Barja-Fidalgo C, de Moraes JA. Obese adipose tissue secretion induces inflammation in preadipocytes: role of toll-like receptor-4. Nutrients. 2020;12(9):1–16. doi:10.3390/nu12092828
81. Kwaifa IK, Bahari H, Yong YK, Md Noor S. Endothelial dysfunction in obesity-induced inflammation: molecular mechanisms and clinical implications. Biomolecules. 2020;10(2):291. doi:10.3390/biom10020291
82. Steven S, Dib M, Hausding M, et al. CD40L controls obesity-associated vascular inflammation, oxidative stress, and endothelial dysfunction in high fat diet-treated and db/db mice. Cardiovasc Res. 2018;114(2):312–323. doi:10.1093/cvr/cvx197
83. Li M, Cui M, Li G, et al. The pathophysiological associations between obesity,NAFLD,and atherosclerotic cardiovascular diseases. Hormone Metab Res. 2024;56(10):683–696. doi:10.1055/a-2266-1503
84. World Health Organization. Cardiovascular diseases (CVDs). 2025. Available from: https://www.who.int/news-room/fact-sheets/detail/cardiovascular-diseases-cvds.
85. Powell-Wiley TM, Poirier P, Burke LE, et al. Obesity and cardiovascular disease: a scientific statement from the American heart association. Circulation. 2021;143(21):e984. doi:10.1161/CIR.0000000000000973
86. Hilgendorf I, Swirski FK, Robbins CS. Monocyte fate in atherosclerosis. Arterioscler Thromb Vasc Biol. 2015;35(2):272–279. doi:10.1161/ATVBAHA.114.303565
87. Alfhili MA, Alotaibi GA, Alfaifi M, Almoghrabi Y, Alsughayyir J. Association of platelet-monocyte ratio with dyslipidemia in Saudi Arabia: a large, population-based study. Life. 2023;13(8):1685–1699. doi:10.3390/LIFE13081685
88. Burger PC, Wagner DD. Platelet P-selectin facilitates atherosclerotic lesion development. Blood. 2003;101(7):2661–2666. doi:10.1182/BLOOD-2002-07-2209
89. Huo Y, Schober A, Forlow SB, et al. Circulating activated platelets exacerbate atherosclerosis in mice deficient in apolipoprotein E. Nat Med. 2003;9(1):61–67. doi:10.1038/NM810
90. Von Hundelshausen P, Weber KSC, Huo Y, et al. RANTES deposition by platelets triggers monocyte arrest on inflamed and atherosclerotic endothelium. Circulation. 2001;103(13):1772–1777. doi:10.1161/01.CIR.103.13.1772
91. Schober A, Manka D, Von Hundelshausen P, et al. Deposition of platelet RANTES triggering monocyte recruitment requires P-selectin and is involved in neointima formation after arterial injury. Circulation. 2002;106(12):1523–1529. doi:10.1161/01.CIR.0000028590.02477.6F
92. An G, Wang H, Tang R, et al. PSGL-1 is highly expressed on Ly-6Chi monocytes and a major determinant for Ly-6Chi monocyte recruitment to sites of atherosclerosis in mice. Circulation. 2008;117(25):3227–3237. doi:10.1161/CIRCULATIONAHA.108.771048
93. Liu G, Liang B, Song X, et al. P-selectin increases angiotensin II-induced cardiac inflammation and fibrosis via platelet activation. Mol Med Rep. 2016;13(6):5021–5028. doi:10.3892/mmr.2016.5186
94. Ramos CL, Huo Y, Jung U, et al. Direct demonstration of P-selectin- and VCAM-1-dependent mononuclear cell rolling in early atherosclerotic lesions of apolipoprotein E-deficient mice. Circ Res. 1999;84(11):1237–1244. doi:10.1161/01.RES.84.11.1237
95. Gunaseelan C, Lomozová Z, Hrubša M, et al. Occurrence of platelet-monocyte aggregates in patients with metabolic disorders and effect of direct anticoagulants. Thromb Haemost. 2025. doi:10.1055/a-2747-8963
96. Coleman LG, Polanowska-Grabowska RK, Marcinkiewicz M, Gear ARL. LDL oxidized by hypochlorous acid causes irreversible platelet aggregation when combined with low levels of ADP, thrombin, epinephrine, or macrophage-derived chemokine (CCL22). Blood. 2004;104(2):380–389. doi:10.1182/BLOOD-2003-08-2961
97. Badrnya S, Schrottmaier WC, Kral JB, et al. Platelets mediate oxidized low-density lipoprotein-induced monocyte extravasation and foam cell formation. Arterioscler Thromb Vasc Biol. 2014;34(3):571–580. doi:10.1161/ATVBAHA.113.302919
98. Di Costanzo A, Indolfi C, Sorrentino S, Esposito G, Spaccarotella CAM. The effects of statins, ezetimibe, PCSK9-inhibitors, inclisiran, and icosapent ethyl on platelet function. Int J Mol Sci. 2023;24(14):11739. doi:10.3390/ijms241411739
99. Puccetti L, Pasqui AL, Pastorelli M, et al. Platelet hyperactivity after statin treatment discontinuation. Thromb Haemost. 2003;90(3):476–482. doi:10.1160/th03-02-0111
100. Navarese EP, Kolodziejczak M, Winter MP, et al. Association of PCSK9 with platelet reactivity in patients with acute coronary syndrome treated with prasugrel or ticagrelor: the PCSK9-REACT study. Int J Cardiol. 2017;227:644–649. doi:10.1016/j.ijcard.2016.10.084
101. Wang S, Fu D, Liu H, Peng D. Independent association of PCSK9 with platelet reactivity in subjects without statin or antiplatelet agents. Front Cardiovasc Med. 2022;9. doi:10.3389/fcvm.2022.934914.
102. Barale C, Bonomo K, Frascaroli C, et al. Platelet function and activation markers in primary hypercholesterolemia treated with anti-PCSK9 monoclonal antibody: a 12-month follow-up. Nutr Metab Cardiovasc Dis. 2020;30(2):282–291. doi:10.1016/j.numecd.2019.09.012
103. Linden MD, Furman MI. Monocyte-platelet aggregates in patients with ischemic heart disease. Cardiovasc Biomarkers. 2006;487–493. doi:10.1007/978-1-59745-051-5_28
104. Ahn KC, Jun AJ, Pawar P, et al. Preferential binding of platelets to monocytes over neutrophils under flow. Biochem Biophys Res Commun. 2005;329(1):345–355. doi:10.1016/j.bbrc.2005.01.146
105. Allen N, Barrett TJ, Guo Y, et al. Circulating monocyte-platelet aggregates are a robust marker of platelet activity in cardiovascular disease. Atherosclerosis. 2019;282:11–18. doi:10.1016/j.atherosclerosis.2018.12.029
106. Kossmann H, Rischpler C, Hanus F, et al. Monocyte-platelet aggregates affect local inflammation in patients with acute myocardial infarction. Int J Cardiol. 2019;287:7–12. doi:10.1016/j.ijcard.2019.04.009
107. Maugeri N, Evangelista V, Celardo A, et al. Polymorphonuclear leukocyte-platelet interaction: role of P-selectin in thromboxane B2 and leukotriene C4 cooperative synthesis. Thromb Haemost. 1994;72(3):450–456. doi:10.1055/s-0038-1648888
108. Yong ASC, Pennings GJ, Chang M, et al. Intracoronary shear-related up-regulation of platelet P-selectin and platelet-monocyte aggregation despite the use of aspirin and clopidogrel. Blood. 2011;117(1):11–20. doi:10.1182/BLOOD-2010-04-278812
109. Furman MI, Benoit SE, Barnard MR, et al. Increased platelet reactivity and circulating monocyte-platelet aggregates in patients with stable coronary artery disease. J Am Coll Cardiol. 1998;31(2):352–358. doi:10.1016/S0735-1097(97)00510-X
110. Han J, Bloxham CJ, Kirmes K, et al. Platelet-leukocyte aggregates in cardiovascular disease: prognostic significance and therapeutic potential. Cardiovasc Res. 2025;121(11):1679–1696. doi:10.1093/cvr/cvaf105
111. Stojkovic S, Wadowski PP, Haider P, et al. Circulating MicroRNAs and monocyte-platelet aggregate formation in acute coronary syndrome. Thromb Haemost. 2021;121(7):913–922. doi:10.1055/s-0040-1722226
112. Ghorbani M, Bashash D, Gheydari ME, et al. Platelet–leukocyte aggregate and interleukin-6: an emerging perspective on a new diagnostic and therapeutic clue for acute coronary syndrome, a case–control study. Health Sci Rep. 2024;7(12):e70209. doi:10.1002/hsr2.70209
113. Ozaki Y, Imanishi T, Teraguchi I, et al. Association between P-selectin glycoprotein ligand-1 and pathogenesis in acute coronary syndrome assessed by optical coherence tomography. Atherosclerosis. 2014;233(2):697–703. doi:10.1016/j.atherosclerosis.2013.12.052
114. Ye Z, Zhong L, Zhu S, et al. The P-selectin and PSGL-1 axis accelerates atherosclerosis via activation of dendritic cells by the TLR4 signaling pathway. Cell Death Dis. 2019;10(7):1–15. doi:10.1038/s41419-019-1736-5
115. Merten M, Chow T, Hellums JD, Thiagarajan P. A new role for P-selectin in shear-induced platelet aggregation. Circulation. 2000;102(17):2045–2050. doi:10.1161/01.CIR.102.17.2045
116. Chelliah RK, Lucking AJ, Tattersall L, et al. P-selectin antagonism reduces thrombus formation in humans. J Thromb Haemost. 2009;7(11):1915–1919. doi:10.1111/j.1538-7836.2009.03587.x
117. Chen L, Fukuda N, Matsumoto T, Abe M. Role of complement 3 in the pathogenesis of hypertension. Hypertens Res. 2020;43(4):255–262. doi:10.1038/s41440-019-0371-y
118. Sarafidis PA, Georgianos P, Bakris GL. Resistant hypertension--its identification and epidemiology. Nat Rev Nephrol. 2013;9(1):51–58. doi:10.1038/nrneph.2012.260
119. Hall JE, Granger JP, Do Carmo JM, et al. Hypertension: physiology and pathophysiology. Compr Physiol. 2012;2(4):2393–2442. doi:10.1002/cphy.c110058
120. Harrison DG, Gongora MC. Oxidative stress and hypertension. Med Clin North Am. 2009;93(3):621–635. doi:10.1016/j.mcna.2009.02.015
121. Cuhlmann S, Van Der Heiden K, Saliba D, et al. Disturbed blood flow induces RelA expression via c-Jun N-terminal kinase 1: a novel mode of NF-κB regulation that promotes arterial inflammation. Circ Res. 2011;108(8):950–959. doi:10.1161/CIRCRESAHA.110.233841
122. Gkaliagkousi E, Corrigall V, Becker S, et al. Decreased platelet nitric oxide contributes to increased circulating monocyte-platelet aggregates in hypertension. Eur Heart J. 2009;30(24):3048–3054. doi:10.1093/EURHEARTJ/EHP330
123. Ferroni P, Guagnano MT, Falco A, et al. Association of low-grade inflammation and platelet activation in patients with hypertension with microalbuminuria. Clin Sci. 2008;114(6):449–455. doi:10.1042/CS20070307
124. Ferroni P, Martini F, D’Alessandro R, et al. In vivo platelet activation is responsible for enhanced vascular endothelial growth factor levels in hypertensive patients. Clin Chim Acta. 2023;388(1–2):33–37. doi:10.1016/J.CCA.2007.09.026
125. Ruze R, Liu T, Zou X, et al. Obesity and type 2 diabetes mellitus: connections in epidemiology, pathogenesis, and treatments. Front Endocrinol. 2023;14:1161521. doi:10.3389/fendo.2023.1161521
126. Schneider DJ. Factors contributing to increased platelet reactivity in people with diabetes. Diab Care. 2009;32(4):525. doi:10.2337/DC08-1865
127. American Diabetes Association. Classification and diagnosis of diabetes: standards of medical care in diabetes-2019. Diab Care. 2019;42(Suppl 1):S13–S28. doi:10.2337/dc19-S002
128. Mandrup-Poulsen T. Type 2 Diabetes Mellitus. A metabolic autoinflammatory disease. Dermatol Clin. 2013;31(3):495–506. doi:10.1016/j.det.2013.04.006
129. Eizirik DL, Pasquali L, Cnop M. Pancreatic β-cells in type 1 and type 2 diabetes mellitus: different pathways to failure. Nat Rev Endocrinol. 2020;16(7):349–362. doi:10.1038/s41574-020-0355-7
130. Keating S, El-Osta A. Epigenetic changes in diabetes. Clin Genet. 2013;84(1):1–10. doi:10.1111/cge.12121
131. Versini M, Jeandel PY, Rosenthal E, Shoenfeld Y. Obesity in autoimmune diseases: not a passive bystander. Autoimmun Rev. 2014;13(9):981–1000. doi:10.1016/j.autrev.2014.07.001
132. Agarwal S, Saha S, Ghosh R, et al. Elevated glycosylation of CD36 in platelets is a risk factor for oxLDL-mediated platelet activation in type 2 diabetes. FEBS J. 2024;291(2):376–391. doi:10.1111/febs.16976
133. Fontana P, Gaussem P, Aiach M, Fiessinger JN, Emmerich J, Reny JL. P2Y12 H2 haplotype is associated with peripheral arterial disease: a case-control study. Circulation. 2003;108(24):2971–2973. doi:10.1161/01.CIR.0000106904.80795.35
134. Stratmann B, Tschoepe D. Pathobiology and cell interactions of platelets in diabetes. Diab Vasc Dis Res. 2005;2(1):16–23. doi:10.3132/DVDR.2005.001
135. Shoji T, Koyama H, Fukumoto S, et al. Platelet-monocyte aggregates are independently associated with occurrence of carotid plaques in type 2 diabetic patients. J Atheroscler Thromb. 2005;12(6):344–352. doi:10.5551/jat.12.344
136. Guglielmini G, Falcinelli E, De Fano M, et al. Obesity and type 2 diabetes mellitus add up to induce platelet hyperreactivity and platelet activation. Thromb Res. 2026;259:109626. doi:10.1016/j.thromres.2026.109626
137. Malle E, Sattler W. Platelets and the lipoproteins: native, modified and platelet modified lipoproteins. Platelets. 1994;5(2):70–83. doi:10.3109/09537109409005516
138. Martín-Timón I, Sevillano-Collantes C, Segura-Galindo A, Del Cañizo-Gómez FJ. Type 2 diabetes and cardiovascular disease: have all risk factors the same strength? World J Diab. 2014;5(4):444. doi:10.4239/wjd.v5.i4.444
139. Beckman JA, Creager MA. Vascular complications of diabetes. Circ Res. 2016;118(11):1771–1785. doi:10.1161/CIRCRESAHA.115.306884
140. Sener A, Ozsavci D, Oba R, Demirel GY, Uras F, Yardimci KT. Do platelet apoptosis, activation, aggregation, lipid peroxidation and platelet-leukocyte aggregate formation occur simultaneously in hyperlipidemia? Clin Biochem. 2005;38(12):1081–1087. doi:10.1016/j.clinbiochem.2005.09.005
141. Rolling CC, Barrett TJ, Berger JS. Platelet-monocyte aggregates: molecular mediators of thromboinflammation. Front Cardiovasc Med Frontiers Media S A. 2023;10. doi:10.3389/fcvm.2023.960398.
142. Sampson MJ, Davies IR, Brown JC, Ivory K, Hughes DA. Monocyte and neutrophil adhesion molecule expression during acute hyperglycemia and after antioxidant treatment in type 2 diabetes and control patients. Arterioscler Thromb Vasc Biol. 2002;22(7):1187–1193. doi:10.1161/01.ATV.0000021759.08060.63
143. Nusca A, Tuccinardi D, Pieralice S, et al. Platelet effects of anti-diabetic therapies: new perspectives in the management of patients with diabetes and cardiovascular disease. Front Pharmacol. 2021;12:670155. doi:10.3389/fphar.2021.670155
144. Santilli F, Formoso G, Sbraccia P, et al. Postprandial hyperglycemia is a determinant of platelet activation in early type 2 diabetes mellitus. J Thromb Haemost. 2010;8(4):828–837. doi:10.1111/j.1538-7836.2010.03742.x
145. Kaplar M, Kappelmayer J, Veszpremi A, Szabo K, Udvardy M. The possible association of in vivo leukocyte-platelet heterophilic aggregate formation and the development of diabetic angiopathy. Platelets. 2001;12(7):419–422. doi:10.1080/09537100120078368
146. Lonardo A, Bellentani S, Argo CK, et al. Epidemiological modifiers of non-alcoholic fatty liver disease: focus on high-risk groups. Digestive Liver Dis. 2015;47(12):997–1006. doi:10.1016/j.dld.2015.08.004
147. Lomonaco R, Leiva EG, Bril F, et al. Advanced liver fibrosis is common in patients with type 2 diabetes followed in the outpatient setting: the need for systematic screening. Diab Care. 2021;44(2):399–406. doi:10.2337/dc20-1997
148. Dixon JB, Bhathal PS, O’Brien PE. Nonalcoholic fatty liver disease: predictors of nonalcoholic steatohepatitis and liver fibrosis in the severely obese. Gastroenterology. 2001;121(1):91–100. doi:10.1053/gast.2001.25540
149. Jinjuvadia R, Antaki F, Lohia P, Liangpunsakul S. The association between nonalcoholic fatty liver disease and metabolic abnormalities in United States population. J Clin Gastroenterol. 2017;51(2):160. doi:10.1097/MCG.0000000000000666
150. Fan JG, Kim SU, Wong VWS. New trends on obesity and NAFLD in Asia. J Hepatol. 2017;67(4):862–873. doi:10.1016/j.jhep.2017.06.003
151. Yki-Järvinen H. Nutritional modulation of nonalcoholic fatty liver disease and insulin resistance: human data. Curr Opin Clin Nutr Metab Care. 2010;13(6):709–714. doi:10.1097/MCO.0b013e32833f4b34
152. Sakurai Y, Kubota N, Yamauchi T, Kadowaki T. Role of insulin resistance in MAFLD. Int J Mol Sci. 2021;22(8):4156. doi:10.3390/ijms22084156
153. Byrne CD, Targher G. NAFLD: a multisystem disease. J Hepatol. 2015;62(S1):S47–S64. doi:10.1016/j.jhep.2014.12.012
154. Byrne CD. Ectopic fat, insulin resistance and non-alcoholic fatty liver disease. Proc Nutr Soc. 2013;72(4):412–419. doi:10.1017/S0029665113001249
155. Takamura T, Misu H, Ota T, Kaneko S. Fatty liver as a consequence and cause of insulin resistance: lessons from type 2 diabetic liver. Endocr J. 2012;59(9):745–763. doi:10.1507/endocrj.EJ12-0228
156. Byrne CD, Targher G. Ectopic fat, insulin resistance, and nonalcoholic fatty liver disease: implications for cardiovascular disease. Arterioscler Thromb Vasc Biol. 2014;34(6):1155–1161. doi:10.1161/ATVBAHA.114.303034
157. Tanase DM, Gosav EM, Costea CF, et al. The intricate relationship between type 2 diabetes mellitus (T2DM), insulin resistance (IR), and nonalcoholic fatty liver disease (NAFLD). J Diabetes Res. 2020;2020:1–16. doi:10.1155/2020/3920196
158. Zechner R, Kienesberger PC, Haemmerle G, Zimmermann R, Lass A. Adipose triglyceride lipase and the lipolytic catabolism of cellular fat stores. J Lipid Res. 2009;50(1):3–21. doi:10.1194/jlr.R800031-JLR200
159. Lazarus JV, Mark HE, Villota-Rivas M, et al. The global NAFLD policy review and preparedness index: are countries ready to address this silent public health challenge? J Hepatol. 2022;76(4):771–780. doi:10.1016/j.jhep.2021.10.025
160. Dalbeni A, Castelli M, Zoncapè M, Minuz P, Sacerdoti D. Platelets in Non-alcoholic Fatty Liver Disease. Front Pharmacol. 2022;13:842636. doi:10.3389/FPHAR.2022.842636/FULL
161. Targher G, Chonchol M, Miele L, Zoppini G, Pichiri I, Muggeo M. Nonalcoholic fatty liver disease as a contributor to hypercoagulation and thrombophilia in the metabolic syndrome. Semin Thromb Hemost. 2009;35(3):277–287. doi:10.1055/S-0029-1222606
162. Kotronen A, Joutsi-Korhonen L, Sevastianova K, et al. Increased coagulation factor VIII, IX, XI and XII activities in non-alcoholic fatty liver disease. Liver Int. 2011;31(2):176–183. doi:10.1111/J.1478-3231.2010.02375.X
163. Verrijken A, Francque S, Mertens I, et al. Prothrombotic factors in histologically proven nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Hepatology. 2014;59(1):121–129. doi:10.1002/HEP.26510
164. Fricker ZP, Pedley A, Massaro JM, et al. Liver fat is associated with markers of inflammation and oxidative stress in analysis of data from the framingham heart study. Clin Gastroenterol Hepatol. 2019;17(6):1157–1164.e4. doi:10.1016/j.cgh.2018.11.037
165. Panasiuk A, Zak J, Kasprzycka E, Janicka K, Prokopowicz D. Blood platelet and monocyte activations and relation to stages of liver cirrhosis. World J Gastroenterol. 2005;11(18):2754–2758. doi:10.3748/wjg.v11.i18.2754
166. Castelli M, Zoncapè M, Meneguzzi A, et al. Platelet functional profile is altered in metabolic dysfunction-associated steatotic liver disease. Liver Int. 2025;45(8):e70231. doi:10.1111/liv.70231
167. Baratta F, Cocomello N, Carpino G, et al. Platelet Thromboxane B2 overproduction associated with liver fibrosis severity in patients with MASLD. Thromb Res. 2026;257:109540. doi:10.1016/j.thromres.2025.109540
168. Kobayashi T, Siegmund B, Le Berre C, et al. Ulcerative colitis. Nat Rev Dis Primers. 2020;6(1). doi:10.1038/S41572-020-0205-X
169. Tufano A, Testa A, Patturelli M, et al. Venous and arterial thromboembolism in out-patients with inflammatory bowel disease: a single center study. Thrombosis Update. 2025;20:100212. doi:10.1016/J.TRU.2025.100212
170. Tekelioglu Y, Uzun H, Sisman G. Activated platelets in patients suffering from inflammatory bowel disease. Bratisl Lek Listy. 2014;115(2):83–85. doi:10.4149/BLL_2014_018
171. Zamora C, Canto E, Nieto JC, et al. Inverse Association between circulating monocyte-platelet complexes and inflammation in ulcerative colitis patients. Inflamm Bowel Dis. 2018;24(4):818–828. doi:10.1093/IBD/IZX106
172. Sano Y, Tomiyama T, Yagi N, et al. Platelet activation through CD62P and the formation of platelet–monocyte complexes are associated with the exacerbation of mucosal inflammation in patients with ulcerative colitis. Sci Rep. 2024;14(1):28055. doi:10.1038/s41598-024-78462-8
173. Marsik C, Mayr F, Cardona F, Schaller G, Wagner OF, Jilma B. Endotoxin down-modulates P-selectin glycoprotein ligand-1 (PSGL-1, CD162) on neutrophils in humans. J Clin Immunol. 2004;24(1):62–65. doi:10.1023/B:JOCI.0000018064.13793.83
174. Pamuk GE, Vural Ö, Turgut B, Demir M, Ümit H, Tezel A. Increased circulating platelet-neutrophil, platelet-monocyte complexes, and platelet activation in patients with ulcerative colitis: a comparative study. Am J Hematol. 2006;81(10):753–759. doi:10.1002/AJH.20655
175. Niu Y, Li A, Xu W, et al. Platelet activation stimulates macrophages to enhance ulcerative colitis through PF4/CXCR3 signaling. Int J Mol Med. 2025;55(5):1–20. doi:10.3892/IJMM.2025.5519
176. Schneider M, Waitz G, Prophet H, Schober HC, Ramlow W. Adsorptive cytapheresis for ulcerative colitis with focus on removing platelets and platelet-aggregates. Therapeut Apheresis Dial. 2023;27(3):452–463. doi:10.1111/1744-9987.13942
177. Pisetsky DS. Pathogenesis of autoimmune disease. Nat Rev Nephrol. 2023;19(8):509–524. doi:10.1038/S41581-023-00720-1
178. Conrad N, Verbeke G, Molenberghs G, et al. Autoimmune diseases and cardiovascular risk: a population-based study on 19 autoimmune diseases and 12 cardiovascular diseases in 22 million individuals in the UK. Lancet. 2022;400(10354):733–743. doi:10.1016/S0140-6736(22)01349-6
179. Menichelli D, Cormaci VM, Marucci S, et al. Risk of venous thromboembolism in autoimmune diseases: a comprehensive review. Autoimmun Rev. 2023;22(11):103447. doi:10.1016/J.AUTREV.2023.103447
180. Kaul A, Gordon C, Crow MK, et al. Systemic lupus erythematosus. Nat Rev Dis Primers. 2016;2(1):16039. doi:10.1038/nrdp.2016.39
181. Joseph JE, Harrison P, Mackie IJ, Isenberg DA, Machin SJ. Increased circulating platelet-leucocyte complexes and platelet activation in patients with antiphospholipid syndrome, systemic lupus erythematosus and rheumatoid arthritis. Br J Haematol. 2001;115(2):451–459. doi:10.1046/J.1365-2141.2001.03101.X
182. Peshkova AD, Saliakhutdinova SM, Sounbuli K, Andrianova IA, Litvinov RI, Weisel JW. Blood levels and composition of leukocyte–platelet aggregates in inflammatory diseases of various etiologies. Thromb Haemost. 2025. doi:10.1055/a-2742-3449
183. Baroni Pietto MC, Glembotsky AC, Lev PR, et al. Toll-like receptor expression and functional behavior in platelets from patients with systemic lupus erythematosus. Immunobiology. 2024;229(1):152782. doi:10.1016/j.imbio.2023.152782
184. Manzano EM, Fernández-Bello I, Sanz RJ, et al. Insights into the procoagulant profile of patients with systemic lupus erythematosus without antiphospholipid antibodies. J Clin Med. 2020;9(10). doi:10.3390/JCM9103297
185. Urzainqui A, Martínez Del Hoyo G, Lamana A, et al. Functional role of P-selectin glycoprotein ligand 1/P-selectin interaction in the generation of tolerogenic dendritic cells. J Immunol. 2007;179(11):7457–7465. doi:10.4049/JIMMUNOL.179.11.7457
186. Pérez-Frías A, González-Tajuelo R, Núñez-Andrade N, et al. Development of an autoimmune syndrome affecting the skin and internal organs in P-selectin glycoprotein ligand 1 leukocyte receptor-deficient mice. Arthritis Rheumatol. 2014;66(11):3178–3189. doi:10.1002/ART.38808
187. González-Tajuelo R, Silván J, Pérez-Frías A, et al. P-Selectin preserves immune tolerance in mice and is reduced in human cutaneous lupus. Sci Rep. 2017;7(1):41841. doi:10.1038/srep41841
188. Mariscal A, Zamora C, Magallares B, et al. Phenotypic and functional consequences of PLT binding to monocytes and its association with clinical features in SLE. Int J Mol Sci. 2021;22(9):4719. doi:10.3390/IJMS22094719
189. Katsarou A, Gudbjörnsdottir S, Rawshani A, et al. Type 1 diabetes mellitus. Nat Rev Dis Primers. 2017;3(1). doi:10.1038/NRDP.2017.16
190. Sagar RC, Yates DM, Pearson SM, et al. Insulin resistance in type 1 diabetes is a key modulator of platelet hyperreactivity. Diabetologia. 2025;68(7):1544–1558. doi:10.1007/S00125-025-06429-Z
191. Zahran AM, El-Badawy O, Mohamad IL, Tamer DM, Abdel-Aziz SM, Elsayh KI. Platelet activation and platelet-leukocyte aggregates in type i diabetes mellitus. Clin Appl Thromb Hemost. 2018;24(9_suppl):230S–239S. doi:10.1177/1076029618805861
192. Hebbel RP, Eaton JW, Steinberg MH, White JG. Erythrocyte/endothelial interactions and the vasocclusive severity of sickle cell disease. Prog Clin Biol Res. 1981;55:145–162.
193. Rees DC, Williams TN, Gladwin MT. Sickle-cell disease. Lancet. 2010;376(9757):2018–2031. doi:10.1016/S0140-6736(10)61029-X
194. Wun T, Paglieroni T, Rangaswami A, et al. Platelet activation in patients with sickle cell disease. Br J Haematol. 1998;100(4):741–749. doi:10.1046/J.1365-2141.1998.00627.X
195. Frenette PS, Johnson RC, Hynes RO, Wagner DD. Platelets roll on stimulated endothelium in vivo: an interaction mediated by endothelial P-selectin. Proc Natl Acad Sci U S A. 1995;92(16):7450–7454. doi:10.1073/pnas.92.16.7450
196. Frenette PS. Sickle cell vasoocclusion: heterotypic, multicellular aggregations driven by leukocyte adhesion. Microcirculation. 2004;11(2):167–177. doi:10.1080/10739680490278556
197. Bennewitz MF, Watkins SC, Sundda P. Quantitative intravital two-photon excitation microscopy reveals absence of pulmonary vaso-occlusion in unchallenged sickle cell disease mice. Intravital. 2014;3(2):e29748. doi:10.4161/INTV.29748
198. Belcher JD, Marker PH, Weber JP, Hebbel RP, Vercellotti GM. Activated monocytes in sickle cell disease: potential role in the activation of vascular endothelium and vaso-occlusion. Blood. 2000;96(7):2451–2459. doi:10.1182/BLOOD.V96.7.2451
199. Wun T, Cordoba M, Rangaswami A, Cheung AW, Paglieroni T. Activated monocytes and platelet-monocyte aggregates in patients with sickle cell disease*. Clin Lab Haematol. 2002;24(2):81–88. doi:10.1046/J.1365-2257.2002.00433.X
200. Kaul DK, Hebbel RP. Hypoxia/reoxygenation causes inflammatory response in transgenic sickle mice but not in normal mice. J Clin Invest. 2000;106(3):411–420. doi:10.1172/JCI9225
201. Turhan A, Weiss LA, Mohandas N, Coller BS, Frenette PS. Primary role for adherent leukocytes in sickle cell vascular occlusion: a new paradigm. Proc Natl Acad Sci U S A. 2002;99(5):3047. doi:10.1073/PNAS.052522799
202. Bennewitz MF, Tutuncuoglu E, Gudapati S, et al. P-selectin-deficient mice to study pathophysiology of sickle cell disease. Blood Adv. 2020;4(2):266–273. doi:10.1182/BLOODADVANCES.2019000603
203. Borba-Junior IT, da S Barbosa M, Moraes CRP, et al. Evaluation of the podoplanin/C-type lectin-like receptor-2 (CLEC-2) pathway as a mediator of platelet and coagulation activation in sickle cell disease. Res Pract Thromb Haemost. 2025;9(6):103168. doi:10.1016/j.rpth.2025.103168
204. Polanowska-Grabowska R, Wallace K, Field JJ, et al. P-selectin mediated platelet-neutrophil aggregate formation activates neutrophils in mouse and human sickle cell disease. Arterioscler Thromb Vasc Biol. 2010;30(12):2392. doi:10.1161/ATVBAHA.110.211615
205. Lee SP, Ataga KI, Orringer EP, Phillips DR, Parise LV. Biologically active CD40 ligand is elevated in sickle cell anemia: potential role for platelet-mediated inflammation. Arterioscler Thromb Vasc Biol. 2006;26(7):1626–1631. doi:10.1161/01.ATV.0000220374.00602.a2
206. Inwald DP, McDowall A, Peters MJ, Callard RE, Klein NJ. CD40 is constitutively expressed on platelets and provides a novel mechanism for platelet activation. Circ Res. 2003;92(9):1041–1048. doi:10.1161/01.RES.0000070111.98158.6C
207. Inwald DP, Kirkham FJ, Peters MJ, et al. Platelet and leucocyte activation in childhood sickle cell disease: association with nocturnal hypoxaemia. Br J Haematol. 2000;111(2):474–481. doi:10.1046/J.1365-2141.2000.02353.X
208. Tomer A, Harker LA, Kasey S, Eckman JR. Thrombogenesis in sickle cell disease. J Lab Clin Med. 2001;137(6):398–407. doi:10.1067/MLC.2001.115450
209. Garrido VT, Proença-Ferreira R, Dominical VM, et al. Elevated plasma levels and platelet-associated expression of the pro-thrombotic and pro-inflammatory protein, TNFSF14 (LIGHT), in sickle cell disease. Br J Haematol. 2012;158(6):788–797. doi:10.1111/j.1365-2141.2012.09218.x
210. Ataga KI, Kutlar A, Kanter J, et al. Crizanlizumab for the prevention of pain crises in sickle cell disease. N Engl J Med. 2016;376(5):429. doi:10.1056/NEJMOA1611770
211. Roy SK, Bhattacharjee S. Dengue virus: epidemiology, biology, and disease aetiology. Can J Microbiol. 2021;67(10):687–702. doi:10.1139/cjm-2020-0572
212. Pourzangiabadi M, Najafi H, Fallah A, Goudarzi A, Pouladi I. Dengue virus: etiology, epidemiology, pathobiology, and developments in diagnosis and control – a comprehensive review. Infection Genet Evol Elsevier B V. 2025;127. doi:10.1016/j.meegid.2024.105710.
213. Kularatne SA, Dalugama C. Dengue infection: global importance, immunopathology and management. Clin Med J Royal College Phys London. 2022;22(1):9–13. doi:10.7861/clinmed.2021-0791
214. Khazali AS, Hadrawi WH, Ibrahim F, Othman S, Nor Rashid N. Thrombocytopenia in dengue infection: mechanisms and a potential application. Expert Rev Mol Med. 2024;26. doi:10.1017/erm.2024.18.
215. Ojha A, Nandi D, Batra H, et al. Platelet activation determines the severity of thrombocytopenia in dengue infection. Sci Rep. 2017;7(1). doi:10.1038/srep41697
216. Hottz ED, Oliveira MF, Nunes PCG, et al. Dengue induces platelet activation, mitochondrial dysfunction and cell death through mechanisms that involve DC-SIGN and caspases. J Thromb Haemost. 2013;11(5):951–962. doi:10.1111/jth.12178
217. Vedpathak S, Sharma A, Palkar S, et al. Platelet derived exosomes disrupt endothelial cell monolayer integrity and enhance vascular inflammation in dengue patients. Front Immunol. 2023;14:1285162. doi:10.3389/fimmu.2023.1285162
218. Quirino-Teixeira AC, Rozini SV, Barbosa-Lima G, et al. Inflammatory signaling in dengue-infected platelets requires translation and secretion of nonstructural protein 1. Blood Adv. 2020;4(9):2018–2031. doi:10.1182/bloodadvances.2019001169
219. Pinheiro MBM, Rozini SV, Quirino-Teixeira AC, et al. Dengue induces iNOS expression and nitric oxide synthesis in platelets through IL-1R. Front Immunol. 2022;13:1029213. doi:10.3389/fimmu.2022.1029213
220. Tsai JJ, Jen YH, Chang JS, Hsiao HM, Noisakran S, Perng GC. Frequency alterations in key innate immune cell components in the peripheral blood of dengue patients detected by FACS analysis. J Innate Immun. 2011;3(5):530–540. doi:10.1159/000322904
221. Hottz ED, Medeiros-de-moraes IM, Vieira-de-Abreu A, et al. Platelet activation and apoptosis modulate monocyte inflammatory responses in dengue. J Immunol. 2014;193(4):1864–1872. doi:10.4049/JIMMUNOL.1400091
222. de O Trugilho MR, Hottz ED, Brunoro GVF, et al. Platelet proteome reveals novel pathways of platelet activation and platelet-mediated immunoregulation in dengue. PLoS Pathog. 2017;13(5). doi:10.1371/journal.ppat.1006385
223. Barbosa-Lima G, Hottz ED, de Assis EF, et al. Dengue virus-activated platelets modulate monocyte immunometabolic response through lipid droplet biogenesis and cytokine signaling. J Leukoc Biol. 2020;108(4):1293–1306. doi:10.1002/JLB.4MA0620-658R
224. Singh A, Ghosh R, Asuru TR, et al. Inhibition of cellular activation induced by platelet factor 4 via the CXCR3 pathway ameliorates Japanese encephalitis and dengue viral infections. J Thromb Haemost. 2024;22(3):818–833. doi:10.1016/j.jtha.2023.11.015
225. Batista VL, Martins JR, Dias ASL, et al. P2Y12–P-selectin mediated platelet activation drives dengue-associated thrombocytopenia. bioRxiv. 2025. doi:10.1101/2025.09.08.674864
226. Hottz ED, Azevedo-Quintanilha IG, Palhinha L, et al. Platelet activation and platelet-monocyte aggregate formation trigger tissue factor expression in patients with severe COVID-19. Blood. 2020;136(11):1330–1341. doi:10.1182/BLOOD.2020007252
227. Hu B, Huang S, Yin L. The cytokine storm and COVID-19. J Med Virol. 2021;93(1):250–256. doi:10.1002/JMV.26232
228. Canzano P, Brambilla M, Porro B, et al. Platelet and endothelial activation as potential mechanisms behind the thrombotic complications of COVID-19 patients. JACC Basic Transl Sci. 2021;6(3):202–218. doi:10.1016/j.jacbts.2020.12.009
229. Li T, Yang Y, Li Y, et al. Platelets mediate inflammatory monocyte activation by SARS-CoV-2 spike protein. J Clin Invest. 2022;132(4). doi:10.1172/JCI150101
230. Hiti L, Markovič T, Lainscak M, Farkaš Lainščak J, Pal E, Mlinarič-Raščan I. The immunopathogenesis of a cytokine storm: the key mechanisms underlying severe COVID-19. Cytokine Growth Factor Rev. 2025;82:1–17. doi:10.1016/J.CYTOGFR.2024.12.003
231. Yu HH, Qin C, Chen M, Wang W, Tian DS. D-dimer level is associated with the severity of COVID-19. Thromb Res. 2020;195:219–225. doi:10.1016/J.THROMRES.2020.07.047
232. Agrati C, Sacchi A, Tartaglia E, et al. The role of P-selectin in COVID-19 coagulopathy: an updated review. Int J Mol Sci. 2021;22(15):7942. doi:10.3390/IJMS22157942
233. Sui J, Noubouossie DF, Gandotra S, Cao L. Elevated plasma fibrinogen is associated with excessive inflammation and disease severity in COVID-19 patients. Front Cell Infect Microbiol. 2021;11:734005. doi:10.3389/FCIMB.2021.734005/BIBTEX
234. Mizurini DM, Hottz ED, Bozza PT, Monteiro RQ. Fundamentals in Covid-19-associated thrombosis: molecular and cellular aspects. Front Cardiovasc Med. 2021;8:785738. doi:10.3389/FCVM.2021.785738/FULL
235. Srihirun S, Sriwantana T, Srichatrapimuk S, et al. Increased platelet activation and lower platelet-monocyte aggregates in COVID-19 patients with severe pneumonia. PLoS One. 2023;18(3):e0282785. doi:10.1371/JOURNAL.PONE.0282785
236. Lippi G, Mullier F, Favaloro EJ. D-dimer: old dogmas, new (COVID-19) tricks. Clin Chem Lab Med. 2023;61(5):841–850. doi:10.1515/CCLM-2022-0633/XML
237. Othman HY, Zaki IAH, Isa MR, Ming LC, Zulkifly HH. A systematic review of thromboembolic complications and outcomes in hospitalised COVID-19 patients. BMC Infect Dis. 2024;24(1):484. doi:10.1186/S12879-024-09374-1
238. Valencia I, Lumpuy-Castillo J, Magalhaes G, Sánchez-Ferrer CF, Lorenzo Ó, Peiró C. Mechanisms of endothelial activation, hypercoagulation and thrombosis in COVID-19: a link with diabetes mellitus. Cardiovasc Diabetol. 2024;23(1):75. doi:10.1186/S12933-023-02097-8
239. Kanth Manne B, Denorme F, Middleton EA, et al. Platelet gene expression and function in patients with COVID-19. Blood. 2020;136(11):1317–1329. doi:10.1182/BLOOD.2020007214
240. Eichhorn T, Weiss R, Huber S, et al. Expression of tissue factor and platelet/leukocyte markers on extracellular vesicles reflect platelet–leukocyte interaction in severe COVID-19. Int J Mol Sci. 2023;24(23):16886. doi:10.3390/ijms242316886
241. Rolling CC, Sowa MA, Wang TT, et al. P2Y12 inhibition suppresses proinflammatory platelet-monocyte interactions. Thromb Haemost. 2023;123(2):231–244. doi:10.1055/s-0042-1758655
242. Scavone M, Ghali C, Calogiuri M, et al. Impairment of platelet function in both mild and severe COVID-19 patients. Br J Haematol. 2023;203(4):656–667. doi:10.1111/bjh.19062
243. Barrett TJ, Lee AH, Xia Y, et al. Platelet and vascular biomarkers associate with thrombosis and death in coronavirus disease. Circ Res. 2020;127(7):945–947. doi:10.1161/CIRCRESAHA.120.317803
244. Vassiliou AG, Keskinidou C, Jahaj E, et al. ICU admission levels of endothelial biomarkers as predictors of mortality in critically ill COVID-19 patients. Cells. 2021;10(1):186. doi:10.3390/CELLS10010186
245. Beyerstedt S, Casaro EB, Rangel ÉB. COVID-19: angiotensin-converting enzyme 2 (ACE2) expression and tissue susceptibility to SARS-CoV-2 infection. Eur J Clin Microbiol Infect Dis. 2021;40(5):905–919. doi:10.1007/S10096-020-04138-6
246. Hoffmann M, Kleine-Weber H, Schroeder S, et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell. 2020;181(2):271–280.e8. doi:10.1016/j.cell.2020.02.052
247. Campbell RA, Boilard E, Rondina MT. Is there a role for the ACE2 receptor in SARS-CoV-2 interactions with platelets? J Thromb Haemost. 2021;19(1):46–50. doi:10.1111/JTH.15156
248. Zaid Y, Puhm F, Allaeys I, et al. Platelets can associate with SARS-CoV-2 RNA and are hyperactivated in COVID-19. Circ Res. 2020;127(11):1404–1418. doi:10.1161/CIRCRESAHA.120.317703;SUBPAGE:STRING:FULL
249. Barrett TJ, Bilaloglu S, Cornwell M, et al. Platelets contribute to disease severity in COVID-19. J Thromb Haemost. 2021;19(12):3139–3153. doi:10.1111/jth.15534
250. Koupenova M, Corkrey HA, Vitseva O, et al. SARS-CoV-2 initiates programmed cell death in platelets. Circ Res. 2021;129(6):631–646. doi:10.1161/CIRCRESAHA.121.319117;WGROUP:STRING:PUBLICATION
251. Trugilho MRO, Azevedo-Quintanilha IG, Gesto JSM, et al. Platelet proteome reveals features of cell death, antiviral response and viral replication in covid-19. Cell Death Discovery. 2022;8(1):324. doi:10.1038/s41420-022-01122-1
252. Shen S, Zhang J, Fang Y, et al. SARS-CoV-2 interacts with platelets and megakaryocytes via ACE2-independent mechanism. J hematol oncol. 2021;14(1):72. doi:10.1186/S13045-021-01082-6
253. Jackson CB, Farzan M, Chen B, Choe H. Mechanisms of SARS-CoV-2 entry into cells. Nat Rev Mol Cell Biol. 2021;23(1):3–20. doi:10.1038/s41580-021-00418-x
254. Toral A, Martinez-Sobrido L, Lim S, Zhang M, Chang TL. ACE2-independent alternative receptors for SARS-CoV-2. Viruses. 2022;14(11):2535. doi:10.3390/V14112535
255. Maugeri N, De Lorenzo R, Clementi N, et al. Unconventional CD147-dependent platelet activation elicited by SARS-CoV-2 in COVID-19. J Thromb Haemost. 2022;20(2):434–448. doi:10.1111/jth.15575
256. Moreno CL, Castanheira FVS, Stella AO, et al. P selectin promotes SARS-CoV-2 interactions with platelets and the endothelium. J Clin Investig. 2025;135(22). doi:10.1172/JCI184514
257. Wang C, Wang S, Ma X, et al. P-selectin facilitates SARS-CoV-2 Spike 1 subunit attachment to vesicular endothelium and platelets. ACS Infect Dis. 2024;10(8):2656–2667. doi:10.1021/acsinfecdis.3c00728
258. Granai M, Warm V, Vogelsberg A, et al. Impact of P-selectin–PSGL-1 axis on platelet-endothelium-leukocyte interactions in fatal COVID-19. Lab Invest. 2023;103(8):100179. doi:10.1016/j.labinv.2023.100179
259. Goonewardena SN, Chen Q, Tate AM, et al. Monocyte-mediated thrombosis linked to circulating tissue factor and immune paralysis in COVID-19. Arterioscler Thromb Vasc Biol. 2024;44(5):1124–1134. doi:10.1161/ATVBAHA.122.318721
260. Haschka D, Petzer V, Burkert FR, et al. Alterations of blood monocyte subset distribution and surface phenotype are linked to infection severity in COVID-19 inpatients. Eur J Immunol. 2022;52(8):1285–1296. doi:10.1002/EJI.202149680;CTYPE:STRING:JOURNAL
261. Leucker TM, Osburn WO, Reventun P, et al. Effect of crizanlizumab, a P-selectin inhibitor, in COVID-19: a placebo-controlled, randomized trial. JACC Basic Transl Sci. 2021;6(12):935–945. doi:10.1016/J.JACBTS.2021.09.013
262. Thaweethai T, Jolley SE, Karlson EW, et al. Development of a definition of postacute sequelae of SARS-CoV-2 infection. JAMA. 2023;329(22):1934–1946. doi:10.1001/JAMA.2023.8823
263. Low RN, Low RJ, Akrami A. A review of cytokine-based pathophysiology of Long COVID symptoms. Front Med Lausanne. 2023;10. doi:10.3389/FMED.2023.1011936.
264. Martini K, Larici AR, Revel MP, et al. COVID-19 pneumonia imaging follow-up: when and how? A proposition from ESTI and ESR. Eur Radiol. 2021;32(4):2639–2649. doi:10.1007/S00330-021-08317-7
265. Rodrigues RS, Motta Ribeiro G, Barreto MM, et al. Increased lung immune metabolic activity in COVID-19 survivors. Clin Nucl Med. 2022;47(12):1019. doi:10.1097/RLU.0000000000004376
266. Nara N, Shimizu M, Yamamoto M, Nakamizo T, Hayakawa A, Johkura K. Prolonged platelet hyperactivity after COVID-19 infection. Br J Haematol. 2024;204(2):492–496. doi:10.1111/BJH.19125;PAGEGROUP:STRING:PUBLICATION
267. Whitcomb LA, Berry K, LaVergne SM, et al. Blood Pro-thrombotic analytes and platelet activation predict post-acute sequelae of COVID-19. BMC Infect Dis. 2025. doi:10.21203/RS.3.RS-7132372/V1
268. Martins-Gonçalves R, Campos MM, Palhinha L, et al. Persisting platelet activation and hyperactivity in COVID-19 survivors. Circ Res Lippincott Williams Wilkins. 2022;131(11):944–947. doi:10.1161/CIRCRESAHA.122.321659
269. Young Wang SS, Chee K, Wong SW, et al. Increased platelet activation demonstrated by elevated CD36 and P-selectin expression in 1-year post-recovered COVID-19 patients. Semin Thromb Hemost. 2023;49(5):561–564. doi:10.1055/S-0043-1762578/ID/JR03102-15/BIB
270. Brambilla M, Fumoso F, Conti M, et al. Low-grade inflammation in long COVID syndrome sustains a persistent platelet activation associated with lung impairment. JACC Basic Transl Sci. 2024;10(1):20–39. doi:10.1016/j.jacbts.2024.09.007
271. Kuiken T, Riteau B, Fouchier RAM, Rimmelzwaan GF. Pathogenesis of influenza virus infections: the good, the bad and the ugly. Curr Opin Virol. 2012;2(3):276–286. doi:10.1016/j.coviro.2012.02.013
272. Fukuyama S, Kawaoka Y. The pathogenesis of influenza virus infections: the contributions of virus and host factors. Curr Opin Immunol. 2011;23(4):481–486. doi:10.1016/j.coi.2011.07.016
273. Foucault ML, Moules V, Rosa-Calatrava M, Riteau B. Role for proteases and HLA-G in the pathogenicity of influenza A viruses. J Clin Virol. 2011;51(3):155–159. doi:10.1016/j.jcv.2011.04.013
274. Cheung CY, Poon LLM, Lau AS, et al. Induction of proinflammatory cytokines in human macrophages by influenza A (H5N1) viruses: a mechanism for the unusual severity of human disease? Lancet. 2002;360(9348):1831–1837. doi:10.1016/S0140-6736(02)11772-7
275. De Jong MD, Simmons CP, Thanh TT, et al. Fatal outcome of human influenza A (H5N1) is associated with high viral load and hypercytokinemia. Nature Med. 2006;12(10):1203–1207. doi:10.1038/nm1477
276. Kobasa D, Jones SM, Shinya K, et al. Aberrant innate immune response in lethal infection of macaques with the 1918 influenza virus. Nature. 2007;445(7125):319–323. doi:10.1038/nature05495
277. Rumbaut RE, Thiagarajan P. Platelet-Vessel Wall Interactions in Hemostasis and Thrombosis. Available from. https://www.ncbi.nlm.nih.gov/books/NBK53450/.
278. Rondina MT, Brewster BA, Grissom CK, et al. In vivo platelet activation in critically ill patients with primary 2009 influenza A(H1N1). Chest. 2012;141(6):1490–1495. doi:10.1378/chest.11-2860
279. Zeng H, Pappas C, Belser JA, et al. Human pulmonary microvascular endothelial cells support productive replication of highly pathogenic avian influenza viruses: possible involvement in the pathogenesis of human H5N1 virus infection. J Virol. 2012;86(2):667–678. doi:10.1128/JVI.06348-11
280. Ocaña-Macchi M, Bel M, Guzylack-Piriou L, et al. Hemagglutinin-dependent tropism of H5N1 avian influenza virus for human endothelial cells. J Virol. 2009;83(24):12947–12955. doi:10.1128/JVI.00468-09/FORMAT/EPUB
281. Sugiyama MG, Gamage A, Zyla R, et al. Influenza virus infection induces platelet-endothelial adhesion which contributes to lung injury. J Virol. 2015;90(4):1812–1823. doi:10.1128/JVI.02599-15
282. Lê VB, Schneider JG, Boergeling Y, et al. Platelet activation and aggregation promote lung inflammation and influenza virus pathogenesis. Am J Respir Crit Care Med. 2015;191(7):804–819. doi:10.1164/RCCM.201406-1031OC
283. Perrone LA, Plowden JK, García-Sastre A, Katz JM, Tumpey TM. H5N1 and 1918 Pandemic influenza virus infection results in early and excessive infiltration of macrophages and neutrophils in the lungs of mice. PLoS Pathog. 2008;4(8):e1000115. doi:10.1371/JOURNAL.PPAT.1000115
284. Lanza GA, Barone L, Scalone G, et al. Inflammation-related effects of adjuvant influenza A vaccination on platelet activation and cardiac autonomic function. J Intern Med. 2011;269(1):118–125. doi:10.1111/J.1365-2796.2010.02285.X
285. Passacquale G, Vamadevan P, Pereira L, Hamid C, Corrigall V, Ferro A. Monocyte-platelet interaction induces a pro-inflammatory phenotype in circulating monocytes. PLoS One. 2011;6(10):e25595. doi:10.1371/JOURNAL.PONE.0025595
286. Layne K, Di Giosia P, Ferro A, Passacquale G. Anti-platelet drugs attenuate the expansion of circulating CD14highCD16+ monocytes under pro-inflammatory conditions. Cardiovasc Res. 2016;111(1):26–33. doi:10.1093/CVR/CVW089
287. Dixon DA, Tolley ND, Bemis-Standoli K, et al. Expression of COX-2 in platelet-monocyte interactions occurs via combinatorial regulation involving adhesion and cytokine signaling. J Clin Invest. 2006;116(10):2727–2738. doi:10.1172/JCI27209
288. Williams A, Menon S, Crowe M, et al. Geographic and population distributions of human immunodeficiency virus (HIV)-1 and HIV-2 circulating subtypes: a systematic literature review and meta-analysis (2010-2021). J Infect Dis. 2023;228(11):1583–1591. doi:10.1093/infdis/jiad327
289. Bekker LG, Beyrer C, Mgodi N, et al. HIV infection. Nat Rev Dis Primers Nat Res. 2023;9(1). doi:10.1038/s41572-023-00452-3
290. Landovitz RJ, Scott H, Deeks SG. Prevention, treatment and cure of HIV infection. Nat Rev Microbiol Nat Res. 2023;21(10):657–670. doi:10.1038/s41579-023-00914-1
291. Palella FJ, Baker RK, Moorman AC, et al. Mortality in the highly active antiretroviral therapy era: changing causes of death and disease in the HIV outpatient study. J Acquir Immune Defic Syndr. 2006;43(1):27–34. doi:10.1097/01.QAI.0000233310.90484.16
292. Lewden C, Chêne G, Morlat P, et al. HIV-infected adults with a CD4 cell count greater than 500 cells/mm3 on long-term combination antiretroviral therapy reach same mortality rates as the general population. J Acquir Immune Defic Syndr. 2007;46(1):72–77. doi:10.1097/QAI.0B013E318134257A
293. Lazar R, Kersanske L, Xia Q, Daskalakis D, Braunstein SL. Hospitalization rates among people with HIV/AIDS in New York City, 2013. Clin Infect Dis. 2017;65(3):469–476. doi:10.1093/CID/CIX343
294. Marin B, Thiébaut R, Bucher HC, et al. Non-AIDS-defining deaths and immunodeficiency in the era of combination antiretroviral therapy. AIDS. 2009;23(13):1743. doi:10.1097/QAD.0B013E32832E9B78
295. Grulich AE, van Leeuwen MT, Falster MO, Vajdic CM. Incidence of cancers in people with HIV/AIDS compared with immunosuppressed transplant recipients: a meta-analysis. Lancet. 2007;370(9581):59–67. doi:10.1016/S0140-6736(07)61050-2
296. Goehringer F, Bonnet F, Salmon D, et al. Causes of death in HIV-infected individuals with immunovirologic success in a national prospective survey. AIDS Res Hum Retroviruses. 2017;33(2):187–193. doi:10.1089/AID.2016.0222
297. Hasse B, Ledergerber B, Furrer H, et al. Morbidity and aging in HIV-infected persons: the Swiss HIV cohort study. Clin Infect Dis. 2011;53(11):1130–1139. doi:10.1093/CID/CIR626
298. Lang S, Mary-Krause M, Simon A, et al. HIV replication and immune status are independent predictors of the risk of myocardial infarction in HIV-infected individuals. Clin Infect Dis. 2012;55(4):600–607. doi:10.1093/CID/CIS489
299. Lichtenstein KA, Armon C, Buchacz K, et al. Low CD4+ T cell count is a risk factor for cardiovascular disease events in the HIV outpatient study. Clin Infect Dis. 2010;51(4):435–447. doi:10.1086/655144
300. Kuller LH, Tracy R, Belloso W, et al. Inflammatory and coagulation biomarkers and mortality in patients with HIV infection. PLoS Med. 2008;5(10):1496–1508. doi:10.1371/JOURNAL.PMED.0050203
301. McDonald B, Moyo S, Gabaitiri L, et al. Persistently elevated serum interleukin-6 predicts mortality among adults receiving combination antiretroviral therapy in Botswana: results from a clinical trial. AIDS Res Hum Retroviruses. 2013;29(7):993–999. doi:10.1089/AID.2012.0309
302. Tien PC, Choi AI, Zolopa AR, et al. Inflammation and mortality in HIV-infected adults: analysis of the FRAM study cohort. J Acquir Immune Defic Syndr. 2010;55(3):316–322. doi:10.1097/QAI.0B013E3181E66216
303. Heaton RK, Clifford DB, Franklin DR, et al. HIV-associated neurocognitive disorders persist in the era of potent antiretroviral therapy: CHARTER Study. Neurology. 2010;75(23):2087–2096. doi:10.1212/WNL.0b013e318200d727
304. Mayne E, Funderburg NT, Sieg SF, et al. Increased platelet and microparticle activation in HIV infection: upregulation of P-selectin and tissue factor expression. J Acquir Immune Defic Syndr. 2012;59(4):340–346. doi:10.1097/QAI.0b013e3182439355
305. Liang H, Duan Z, Li D, et al. Higher levels of circulating monocyte-platelet aggregates are correlated with viremia and increased sCD163 levels in HIV-1 infection. Cell Mol Immunol. 2015;12(4):435–443. doi:10.1038/cmi.2014.66
306. Nkambule BB, Davison G, Ipp H. Platelet leukocyte aggregates and markers of platelet aggregation, immune activation and disease progression in HIV infected treatment naive asymptomatic individuals. J Thromb Thrombolysis. 2015;40(4):458–467. doi:10.1007/s11239-015-1212-8
307. Holme PA, Müller F, Solum NO, Brosstad F, Frøland SS, Aukrust P. Enhanced activation of platelets with abnormal release of RANTES in human immunodeficiency virus type 1 infection. FASEB J. 1998;12(1):79–90. doi:10.1096/FASEBJ.12.1.79
308. Metcalf Pate KA, Lyons CE, Dorsey JL, et al. Platelet activation and platelet-monocyte aggregate formation contribute to decreased platelet count during acute simian immunodeficiency virus infection in pig-tailed macaques. J Infect Dis. 2013;208(6):874–883. doi:10.1093/infdis/jit278
309. Mesquita EC, Hottz ED, Amancio RT, et al. Persistent platelet activation and apoptosis in virologically suppressed HIV-infected individuals. Sci Rep. 2018;8(1). doi:10.1038/s41598-018-33403-0
310. Wolf K, Tsakiris DA, Weber R, Erb P, Battegay M. Antiretroviral therapy reduces markers of endothelial and coagulation activation in patients infected with human immunodeficiency virus type 1. J Infect Dis. 2002;185(4):456–462. doi:10.1086/338572
311. Tunjungputri RN, van de Heijden W, Urbanus RT, de Groot PG, van der Ven A, de Mast Q. Higher platelet reactivity and platelet-monocyte complex formation in Gram-positive sepsis compared to Gram-negative sepsis. Platelets. 2017;28(6):595–601. doi:10.1080/09537104.2016.1252837
312. O’Bryan TA, Okulicz JF, Bradley WP, Ganesan A, Wang X, Agan BK. Impact of the highly active antiretroviral therapy era on the epidemiology of primary HIV-associated thrombocytopenia. BMC Res Notes. 2015;8(1). doi:10.1186/s13104-015-1548-3
313. Corrales-Medina VF, Simkins J, Chirinos JA, et al. Increased levels of platelet microparticles in HIV-infected patients with good response to highly active antiretroviral therapy. J Acquir Immune Defic Syndr. 2010;54(2):217–219. doi:10.1097/QAI.0B013E3181C8F4C9
314. Singh MV, Uddin N, Covacevich Vidalle M, et al. Role of non-classical monocytes in HIV-associated vascular cognitive impairment. medRxiv. 2023:
315. Deng M, Biao R, Jiang M, Fu J, Zhao H, Du J. Over-activation and dysfunction of platelet-NK cell aggregates in HIV-infected individuals. J Transl Med. 2025;23(1):584. doi:10.1186/s12967-025-06591-3
316. Subia NT, Awamura TK, Dean LS, et al. Dysregulation of complement components associated with inflammation and coagulation in virally suppressed people living with HIV. J Immunol. 2025;214(11):2871–2880. doi:10.1093/jimmun/vkaf227
317. Illanes-álvarez F, Márquez-Ruiz D, Cuesta-Sancho S, et al. Persistent inflammatory activation in people living with HIV. Involvement in atherosclerosis. Front Med Lausanne. 2025;12:1621765. doi:10.3389/fmed.2025.1621765
318. Chen Y, Ding X, Ray S, et al. Persistent activation of monocytes/macrophages and cell senescence in SIV-infected macaques on ART. bioRxiv. 2025:
319. Davidson DC, Hirschman MP, Sun A, Singh MV, Kasischke K, Maggirwar SB. Excess soluble CD40L contributes to blood brain barrier permeability in vivo: implications for HIV-associated neurocognitive disorders. PLoS One. 2012;7(12):e51793. doi:10.1371/journal.pone.0051793
320. Singh MV, Davidson DC, Jackson JW, et al. Characterization of platelet–monocyte complexes in HIV-1–infected individuals: possible role in HIV-associated neuroinflammation. J Immunol. 2014;192(10):4674–4684. doi:10.4049/jimmunol.1302318
321. Demir O. Sepsis: an overview of current therapies and future research. J Clin Pract Res. 2025;47(2):99–110. doi:10.14744/cpr.2025.01903
322. Assinger A, Schrottmaier WC, Salzmann M, Rayes J. Platelets in sepsis: an update on experimental models and clinical data. Front Immunol. 2019;10(JULY). doi:10.3389/fimmu.2019.01687
323. Keane C, Tilley D, Cunningham A, et al. Invasive Streptococcus pneumoniae trigger platelet activation via Toll-like receptor 2. J Thromb Haemost. 2010;8(12):2757–2765. doi:10.1111/j.1538-7836.2010.04093.x
324. Cox D, Kerrigan SW, Watson SP. Platelets and the innate immune system: mechanisms of bacterial-induced platelet activation. J Thromb Haemost. 2011;9(6):1097–1107. doi:10.1111/j.1538-7836.2011.04264.x
325. Jahn K, Kohler TP, Wiebe S, Swiatek LS, Wiebe S. Platelets, bacterial adhesins and the Pneumococcus. Cells. 2022;11(7):1121. doi:10.3390/cells11071121
326. Dewitte A, Lepreux S, Villeneuve J, et al. Blood platelets and sepsis pathophysiology: a new therapeutic prospect in critical ill patients? Ann Intensive Care. Springer Verlag. 2017;7(1). doi:10.1186/s13613-017-0337-7
327. Yang X, Song J, Ma H, et al. The crucial roles of platelets as immune mediators in sepsis. J Inflamm Res. 2025;18:12825–12845. doi:10.2147/JIR.S535701
328. Xu X, Wang Y, Tao Y, Dang W, Yang B, Li Y. The role of platelets in sepsis: a review. Biomol Biomed. 2024;24(4):741. doi:10.17305/bb.2023.10135
329. Sun L, Zhang P, Zhang H, et al. Single-Cell transcriptomic profiles of peripheral blood immune cells reveal early monocyte and platelet activation in the transition from high-risk states to clinical sepsis. Sci Rep. 2025;15(1). doi:10.1038/s41598-025-17078-y
330. Rondina MT, Carlisle M, Fraughton T, et al. Platelet-monocyte aggregate formation and mortality risk in older patients with severe sepsis and septic shock. J Gerontol Series a Biol Sci Med Sci. 2015;70(2):225–231. doi:10.1093/gerona/glu082
331. Huang CM, Li JJ, Wei WK. Clinical significance of platelet mononuclear cell aggregates in patients with sepsis and acute respiratory distress syndrome. World J Clin Cases. 2024;12(5):966. doi:10.12998/WJCC.V12.I5.966
332. Vardon Bounes F, Mémier V, Marcaud M, et al. Platelet activation and prothrombotic properties in a mouse model of peritoneal sepsis. Sci Rep. 2018;8(1):13536. doi:10.1038/S41598-018-31910-8
333. Munoz FM, Hawkins EP, Bullard DC, Beaudet AL, Kaplan SL. Host defense against systemic infection with Streptococcus pneumoniae is impaired in E-, P-, and E-/P-selectin-deficient mice. J Clin Investig. 1997;100(8):2099–2106. doi:10.1172/JCI119744
334. de Stoppelaar SF, Van’t Veer C, Roelofs JJTH, et al. Platelet and endothelial cell P-selectin are required for host defense against Klebsiella pneumoniae-induced pneumosepsis. J Thromb Haemost. 2015;13(6):1128–1138. doi:10.1111/jth.12893
335. Trotta A, Velásquez LN, Milillo MA, et al. Platelets promote Brucella abortus monocyte invasion by establishing complexes with monocytes. Front Immunol. 2018;9(MAY). doi:10.3389/fimmu.2018.01000
336. Gautam I, Huss CW, Storad ZA, et al. Activated platelets mediate monocyte killing of Klebsiella pneumoniae. Infect Immun. 2023;91(3). doi:10.1128/iai.00556-22
337. Posner MG, Upadhyay A, Abubaker AA, et al. Extracellular fibrinogen-binding protein (Efb) from staphylococcus aureus inhibits the formation of platelet-leukocyte complexes. J Biol Chem. 2016;291(6):2764–2776. doi:10.1074/jbc.M115.678359
338. Kirwan DE, Chong DLW, Friedland JS. Platelet activation and the immune response to tuberculosis. Front Immunol. 2021;12. doi:10.3389/fimmu.2021.631696.
339. Kullaya V, van der Ven A, Mpagama S, et al. Platelet-monocyte interaction in Mycobacterium tuberculosis infection. Tuberculosis. 2018;111:86–93. doi:10.1016/j.tube.2018.05.002
340. Feng Y, Dorhoi A, Mollenkopf HJ, et al. Platelets direct monocyte differentiation into epithelioid-like multinucleated giant foam cells with suppressive capacity upon mycobacterial stimulation. J Infect Dis. 2014;210(11):1700–1710. doi:10.1093/infdis/jiu355
341. Feng C, Chen Q, Fan M, et al. Platelet-derived microparticles promote phagocytosis of oxidized low-density lipoprotein by macrophages, potentially enhancing foam cell formation. Ann Transl Med. 2019;7(18):477. doi:10.21037/ATM.2019.08.06
342. Fox KA, Kirwan DE, Whittington AM, et al. Platelets regulate pulmonary inflammation and tissue destruction in tuberculosis. Am J Respir Crit Care Med. 2018;198(2):245–255. doi:10.1164/rccm.201710-2102OC
343. Ramirez GA, Manfredi AA, Maugeri N. Misunderstandings between platelets and neutrophils build in chronic inflammation. Front Immunol. 2019;10(OCT):2491. doi:10.3389/FIMMU.2019.02491
344. Pircher J, Engelmann B, Massberg S, Schulz C, Maximilian L. Platelet-neutrophil crosstalk in atherothrombosis. Thrombosis Haemostasis. 2019;119(08):1274–1282. doi:10.1055/s-0039-1692983
345. Liu Q, Zhu W, Wen X, Da Y. The role of platelet–neutrophil interactions in driving autoimmune diseases. Immunology. 2025;175(1):1–15. doi:10.1111/IMM.13901
346. Patalakh I, Drobotko T, Tykhomyrov A. Platelet-neutrophil aggregates: perspectives for the treatment of postsurgical hemostatic disorders. J Clin Med Surg. 2025;5:1–5
347. Popp SK, Vecchio F, Brown DJ, et al. Circulating platelet-neutrophil aggregates characterize the development of type 1 diabetes in humans and NOD mice. JCI Insight. 2022;7(2):e153993. doi:10.1172/JCI.INSIGHT.153993
348. Kaiser R, Escaig R, Erber J, Nicolai L. Neutrophil-platelet interactions as novel treatment targets in cardiovascular disease. Front Cardiovasc Med. 2021;8:824112. doi:10.3389/FCVM.2021.824112
349. Pitchford S, Pan D, Welch HCE. Platelets in neutrophil recruitment to sites of inflammation. Curr Opin Hematol. 2017;24(1):23. doi:10.1097/MOH.0000000000000297
350. Ma AC, Kubes P. Platelets, neutrophils, and neutrophil extracellular traps (NETs) in sepsis. J Thromb Haemost. 2008;6(3):415–420. doi:10.1111/j.1538-7836.2007.02865.x
351. Li J, Kim K, Barazia A, Tseng A, Cho J. Platelet-neutrophil interactions under thromboinflammatory conditions. Cell Mol Life Sci. 2015;72(14):2627–2643. doi:10.1007/S00018-015-1845-Y/FIGURES/3
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.