Back to Journals » Drug Design, Development and Therapy » Volume 8
P-glycoprotein- and organic anion-transporting polypeptide-mediated transport of periplocin may lead to drug–herb/drug–drug interactions
Authors Liang S, Deng F, Xing H ![]() , Wen H, Shi X, Martey O, Koomson E, He X
, Wen H, Shi X, Martey O, Koomson E, He X
Received 19 January 2014
Accepted for publication 21 March 2014
Published 9 May 2014 Volume 2014:8 Pages 475—483
DOI https://doi.org/10.2147/DDDT.S61024
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Sheng Liang,* Fengchun Deng,* Haiyan Xing, He Wen, Xiaoyan Shi, Orleans Nii Martey, Emmanuel Koomson, Xin He
School of Chinese Materia Medica, Tianjin University of Traditional Chinese Medicine, Tianjin, People's Republic of China
*These authors contributed equally to this work
Abstract: Periplocin, an active and toxic component of the traditional Chinese herbal medicine Periploca sepium Bge, is a cardiac glycoside compound that has been implicated in various clinical accidents. This study investigated the role of transporters in the intestinal absorption and biliary excretion of periplocin, as well as the possible metabolic mechanism of periplocin in liver S9. In a bidirectional transport assay using Madin–Darby canine kidney (MDCK) and MDCK multidrug-resistance protein (MRP)-1 cell monolayers, both in situ intestinal and liver-perfusion models were used to evaluate the role of efflux and uptake transporters on the absorption and biliary excretion of periplocin. In addition, in vitro metabolism of periplocin was investigated by incubating with human/rat liver S9 homogenate fractions to evaluate its metabolic mechanisms in liver metabolic enzymes. The results showed that P-glycoprotein (P-gp) was involved in the intestinal absorption of periplocin, whereas MRP2 and breast cancer-resistance protein were not. The efflux function of P-gp may be partly responsible for the low permeability and bioavailability of periplocin. Moreover, both inhibitors of P-gp and organic anion-transporting polypeptides (OATPs) increased periplocin biliary excretion. No obvious indications of metabolism were observed in the in vitro incubation system, which suggests that periplocin did not interact with the hepatic drug metabolic enzymes. The results of this study showed that the efflux and uptake transporters P-gp and OATPs were involved in the absorption and biliary excretion of periplocin, which may partially account for its low permeability and bioavailability. As a toxic compound, potential drug–herb/herb–herb interactions based on OATPs and P-gp should be taken into account when using P. sepium Bge in the clinic.
Keywords: periplocin, P-gp, OATPs, toxicity, interactions
Introduction
Cortex periplocae, also known as xiangjiapi or beiwujiapi in traditional Chinese medicine (TCM), is the root of Periploca sepium Bge and has been historically used in the People’s Republic of China. P. sepium Bge has been used for relieving rheumatic conditions and strengthening the bone and the musculature for thousands of years. In recent years, an increasing number of bioactivities of cortex periplocin have been recognized, including tumor suppression and anti-chronic heart failure function.1,2 Clinical observation on treatment of 147 cases of chronic congestive heart failure by a xiangjiapi mixture showed that the effective rate of xiangjiapi mixture is higher than the digoxin group, while the side effects showed no difference.3 Periplocin (also named periplocoside) is a cardiotonic glycoside compound in xiangjiapi that may be the main ingredient responsible for the improvement of chronic congestive heart failure.4 However, arrhythmia is a common side effect that occurs during high plasma concentrations of periplocin. Therefore, periplocin is not only a main active component but also a potential toxic compound in P. sepium Bge. To avoid the occurrence of periplocin side effects and even potential clinical accidents, it is essential to elucidate the mechanism of its actions.
Drug transporters are recognized as key players in the processes of drug absorption, distribution, metabolism, and elimination. These transporters mainly belong to the adenosine triphosphate-binding cassette (ABC) superfamily or the solute-carrier group of membrane transport proteins. The ABC superfamily includes P-gp, breast cancer-resistant protein (BCRP), and multidrug resistance (MDR)-associated protein (MRP), whereas organic anion transporters, organic anion-transporting polypeptides (OATPs), and organic cation transporters are members of the solute-carrier superfamily. The expression of transporters varies among different tissues and organs.5 The major transporters involved in the intestinal absorption and biliary excretion of drugs across the canaliculus membrane include P-gp, MRP2, BCRP, and OATPs.6 Various in vitro models have been developed to investigate the relationship between transporters and specific drugs using various cell lines, such as Caco-2, MDR1-Madin–Darby canine kidney (MDCK), BCRP-MDCK, and MRP2-MDCK.7 In addition to in vitro models, in situ models, such as single-pass intestine perfusion, have been used to investigate drug absorption and transport. On the other hand, drug-metabolism enzymes pertain to another group of factors that are involved in in vivo drug disposition. In the past few years, a growing number of drug–drug interactions (DDIs) and drug–herb interactions (DHIs) associated with metabolic enzymes and transporters have been the focus of extensive research studies, especially in terms of evaluating drug toxicities.8
It has been previously reported that the bioavailability of periplocin is very low in rats, and most unchanged forms are rapidly eliminated through the bile.9 Meanwhile, the chemical structure of periplocin (Figure 1) is similar to that of digoxin, which is actively transported across the sinusoidal membrane into hepatocytes through OATPs and secreted into biliary canaliculi through P-gp.10 As a result, efflux transporters present in the intestinal membrane may play a major role in periplocin absorption. Furthermore, the efflux transporter P-gp and the uptake transporter OATP may also be involved in the biliary excretion of periplocin. These transporters may account for the low bioavailability and biliary excretion of periplocin. Therefore, the purpose of the present study was to determine the relationship between periplocin and drug transporters and to investigate the metabolic mechanism of periplocin in liver S9. These investigations will facilitate in understanding the potential DDIs/DHIs between xiangjiapi and other herbs or compounds. The MDCK-MDR1 cell line and in situ liver/intestine-perfusion models were used to evaluate the action of the efflux transporters P-gp, MRP2, BCRP, and OATPs, which are involved in the transport of periplocin in rats. Meanwhile, the hepatic metabolism mechanism of periplocin was also investigated by incubation with liver S9 homogenate fractions (S9).
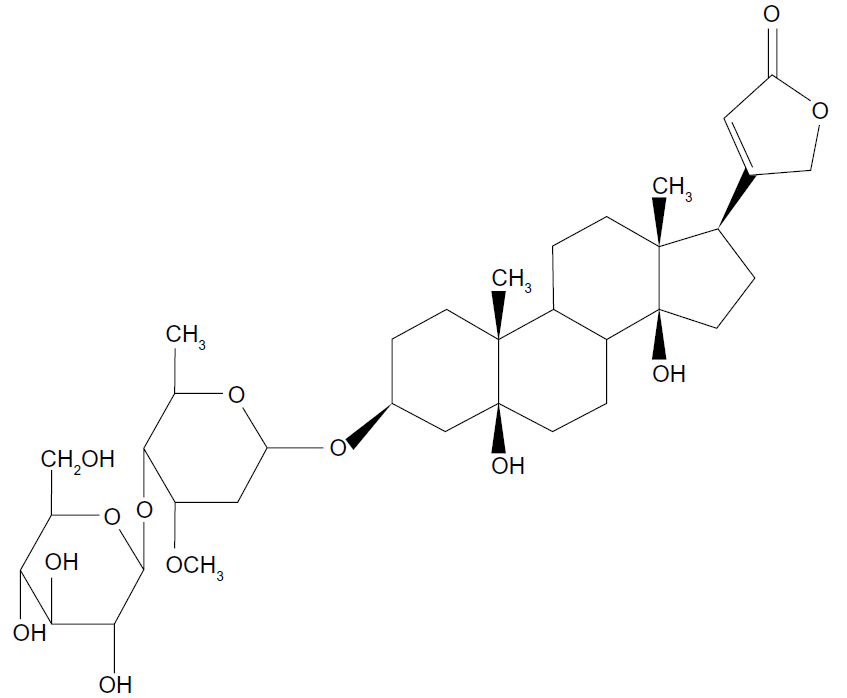 | Figure 1 Chemical structure of periplocin. |
Materials and methods
Materials
Peristaltic pumps and thermal insulation blankets were purchased from Ji’ande’er (Shanghai, People's Republic of China); the MDCK and MDCK-MDR1 cell lines were donated by Zhejiang University (Hangzhou, Zhejiang, People's Republic of China). Dulbecco’s modified Eagle’s medium (DMEM; high glucose), penicillin–streptomycin, Geneticin® (G-418), 0.25% trypsin–ethylenediaminetetraacetic acid (EDTA), fetal bovine serum (FBS), and nonessential amino acids (NEAAs) were purchased from Life Technologies (Carlsbad, CA, USA). A cell-proliferation kit (XTT) was purchased from Roche (Basel, Switzerland). A bicinchoninic acid assay kit was purchased from the Beyotime Institute of Biotechnology (Shanghai, People's Republic of China). Human/rat liver S9 homogenate fractions were purchased from the Research Institute for Liver Diseases (Shanghai, People's Republic of China). Millicell® inserts with polycarbonate, 0.4 μM pore size, and 0.6 cm2 surface areas were purchased from EMD Millipore (Billerica, MA, USA). Cell-culture plates (24 and 96 wells) were acquired from Shanghai Bioleaf Biotech Co, Ltd, (Shanghai, People's Republic of China). Rhodamine 123 (R123), cyclosporine A (CsA), verapamil HCl, Fluorescent thiocyanate, and Hank’s balanced salt solution (HBSS) were purchased from Sigma-Aldrich (St Louis, MO, USA). All other chemicals were purchased from Sigma-Aldrich.
Cell culture
MDR1-MDCK cells were maintained in DMEM supplemented with 10% FBS, 300 μg/mL G-418, and 1% NEAA. MDCK cells were also maintained in DMEM containing 10% FBS, 100 μg/mL–100 U/mL penicillin–streptomycin, and 1% NEAA at 37°C in an atmosphere of 5% CO2 and 95% relative humidity. Cells were passaged at 80%–90% confluence every 2–3 days using trypsin–EDTA solution.11 Cells were passaged three times before seeding for further experiments. For the transport studies, 24-well Millicell inserts were preincubated in the culture medium (30 minutes at 37°C), seeded at a density of approximately 1.66–2×105 cells/cm2, and cultured for 5 days as a monolayer. The culture media of the MDCK and MDCK-MDR1 cells were replaced 24 hours after seeding and again every other day. Transepithelial electrical resistance values across the monolayers were measured using the Millicell ERS Volt-Ohm Meter (EMD Millipore) before and after each experiment to ensure monolayer integrity. MDCK cells and MDCK-MDR1-cell monolayers with transepithelial electrical resistance values ≥80 Ωcm2 and 90 Ωcm2, respectively, were used for the transport assay. All transport studies were conducted at 37°C. Periplocin cytotoxicity on MDCK cells was evaluated using an XTT-based colorimetric assay, according to a published protocol.12
Bidirectional transport of periplocin
Transport assays were conducted as previously described.13 Prior to experimentation, cell monolayers were washed twice with HBSS (with 25 mM 4-[2-hydroxyethyl]-1-piperazineethanesulfonic acid and 0.35 g/L NaHCO3, pH 7.4). Monolayers were equilibrated for 30 minutes at 37°C with HBSS. Triplicate sets of wells were used to assess the transport of test compounds. Periplocin solutions (75 and 100 μM) were added in either the apical (AP, 0.4 mL) or basolateral (BL, 0.6 mL) side of the inserts. Plates were incubated at 37°C, with shaking at 60 rpm. At 20-, 40-, 60-, 80-, 100-, and 120-minute time points, a 50 μL aliquot of the periplocin solution was withdrawn from the receiver chamber, and the chamber was immediately replenished with prewarmed HBSS. The concentrations of periplocin were immediately analyzed by high performance liquid chromatography (HPLC; 600E; Waters, Milford, MA, USA). The inhibition studies using cell monolayers were performed by using HBSS containing 10 μM CsA, instead of a blank HBSS.
In situ intestinal perfusion
The in situ single-pass intestinal perfusion procedure has been optimized.14,15 Rats were anesthetized by administration of urethane (140 mg/kg, intraperitoneally), and then each rat was placed on a thermal insulation blanket to maintain their body temperature at 37°C, which was measured by inserting a thermometer into the rectum. The rats were kept under a warming light during all experiments. A 3–4 cm midline abdominal incision was made, and approximately 10 cm of the proximal jejunal segment (15 cm average distance of the outlet from the pylorus) was carefully exposed; one end was connected to flexible polyvinyl chloride tubing, and one end was connected to a smooth glass tube. The isolated segment was rinsed using a syringe containing a 37°C Krebs–Ringer buffer (pH 6.8) that consisted of 7.8 g/L NaCl, 1.37 g/L NaHCO3, 1.4 g/L d-glycose, 0.35 g/L KCl, 0.32 g/L NaH2PO4, 0.37 g/L CaCl2, and 0.042 g/L MgCl2.
After initiation of perfusion, periplocin solution with or without CsA was perfused at a flow rate of 1 mL/minute using a peristaltic pump. After 10 minutes, the flow rate of the perfusion solution was changed to 0.2 mL/minute. A steady-state condition was achieved after 30 minutes of perfusion at a flow rate of 0.2 mL/minute. The perfused samples were collected at 10-minute intervals for a total of 90 minutes by using an autosampler. The surgical area was covered with a wet pledget, and drops of saline (37°C) were added to the pledget to avoid disturbing the circulatory system during the whole perfusion process. The test tubes used in the sample collection were weighed before and after collection. The samples were transferred into 1.5 mL Eppendorf tubes and stored at 4°C until analysis. At the end of the experiment, the perfused intestinal segment was sliced along the axis without stretching the organ, and carefully opened along the mesenteric root. The width and the length of the intestinal segment was measured and recorded as accurately as possible. All the decisions on the selection of the inhibitors of P-gp, MRP2, and BCRP were based on previous reports.16–18
In situ liver perfusion
A previously developed in situ liver perfusion method was used with minor modifications.19,20 Briefly, male Sprague Dawley rats were anesthetized using urethane (140 mg/kg) prior to surgery. After a “V” cut was made on the abdomen to expose the liver, the hepatic portal vein, superior vena cava, and bile duct were exposed and cannulated. The livers were perfused in situ with oxygenated Krebs–Henseleit buffer (pH 7.4; NaCl [6.92 g/L], KCl [0.35 g/L], CaCl2 [0.28 g/L], KH2PO4 [0.16 g/L], MgSO4 [0.223 g/L], NaHCO3 [2.1 g/L], and glucose [3.0g/L]) at a flow rate of 20 mL/minute to provide sufficient oxygen-carrying capacity. After 20 minutes, which was the optimal time for the liver to stabilize, the inhibitors (verapamil/rifampicin) were added to the reservoir 10 minutes before the addition of periplocin. Liver viability was assessed by observing the organ’s gross morphology and by monitoring bile flow during the perfusion. Taurocholate (0.5 μmol/minute, in saline) was infused into the perfusate reservoir to maintain bile flow, and the temperature was maintained at 37.5°C through the use of a warming light and a heating carpet. Bile samples were collected at 30-minute intervals, and the volume was determined gravimetrically (specific gravity 1.0). The perfusate (0.5 mL) was sampled every 10 minutes for 2 hours, and immediately centrifuged to obtain the supernatant for analysis. After perfusion, the livers were weighed and processed for analysis. All samples collected from this experiment were stored at −80°C until further analysis.
In vitro incubation of periplocin with human/rat liver S9
This procedure was conducted according to the manufacturer’s instructions, with minor modifications.21 The incubation mixture with liver S9 had a total volume of 200 μL: 25 μL of phosphate-buffered saline, 25 μL of the periplocin solution, 50 μL of 8 mg/mL S9 solution, and 100 μL of 2 mM nicotinamide adenine dinucleotide phosphate and reduced nicotinamide adenine dinucleotide solution. Prior to being mixed together, all the solutions were preincubated at 37°C for 5 min. Aliquots (200 μL) of the incubates were collected at 0, 30, 60, and 120-minute time points. Samples were mixed on a vortex mixer and centrifuged at 14,000 rpm for 10 minutes. The supernatant was transferred into a clean centrifuge tube and stored at −80°C until analysis.
HPLC analysis
Quantification of periplocin in the supernatant was conducted by reversed-phase HPLC. A Waters 1100 HPLC system consisting of a quaternary pump with a degasser, a thermostated column compartment, a variable wavelength detector operated at 220 nm, an autoinjector, and Waters Empower software. The HPLC separation was performed on a Waters Xterra C18 column (5 μm, 150×4.6 mm) with a guard C18 column maintained at 30°C. The mobile phase consisted of acetonitrile–water at a ratio of 25:75 (v/v) and a flow rate of 1.0 mL/minute. The injection volume was 60 μL. A calibration curve was constructed by performing a regression linear analysis of the area under the concentration curve versus concentration. Method validation of the selectivity, precision, accuracy, linearity, and stability were conducted to ensure the feasibility of the analytical method.
Calculation of the efficient permeability coefficient
The efficient permeability (Peff) through the rat intestinal wall in the single-pass intestine-perfusion studies was determined using the “plug flow” model and expressed using the following equation with a derivation.22 The net water flux, resulting from both water absorption and efflux in the intestinal segment, was determined by the gravimetric method. The equation was expressed as follows:

Apparent permeability, efflux ratio, and net efflux ratio
The apparent permeability (Papp), efflux ratio (ER), and net efflux ratio (NER) were calculated as follows:

where dC/dt (μM/second) is the amount of drug transported per time, VR (cm3) is the volume in the receiver chamber, Co (μM) is the concentration in the donor chamber at zero time, and A is the Millicell insert surface area,

where Papp(B–A) is the Papp value measured in the BL to the AP direction, and Papp(A–B) is the Papp value measured in the AP to the BL direction, and

where ER(MDR1–MDCK) is the efflux ratio obtained from the P-gp-transfected cells, and MDCK-MDR1 is the efflux ER(MDCK) obtained from the wild-type control cells – MDCK.
The passive transport of drugs in MDCK and MDCK-MDR1 cells was estimated using Equation 7, including its derivation. Unidirectional flux values (absorptive Papp(AP→BL) and secretory Papp(BL→AP)) were calculated according to following equations. Assuming that an active efflux affected the transport of a test compound, the active efflux was calculated for each concentration according to the following equations. No evidence showed whether uptake transporters took part in the absorption of periplocin, and thus uptake transport Papp(uptake) was taken into account:

and

From equations 5 and 6, Papp(passive) was deduced using the following equation:

Results
In situ intestinal perfusion
As shown in Figure 2, the results of the permeability evaluation showed that the Peff of periplocin in the proximal jejunum was concentration-dependent. Compared with the permeability coefficient of the reference drugs, the Peff (1.90±8.63)×10−5 cm/second was similar to the Peff of atenolol (a low-permeability drug), but significantly lower than that of metoprolol (a high-permeability drug). In Figure 3, both the P-gp inhibitors verapamil and CsA significantly increased the permeability of periplocin, whereas the MRP2 inhibitors indometacin and probenecid and the BCRP inhibitor novobiocin showed no obvious effects on the intestinal absorption of periplocin. The efflux transporter P-gp showed involvement in the transport of periplocin.
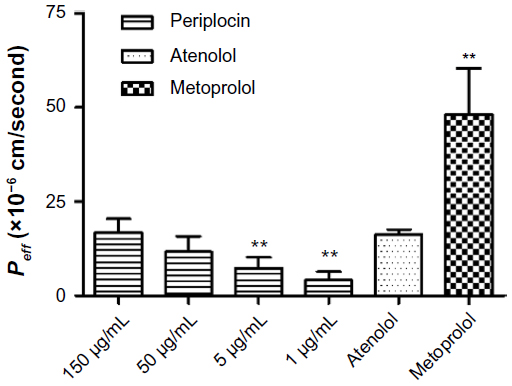 | Figure 2 Effective permeability coefficient (Peff) of periplocin obtained from in situ intestinal perfusion experiments with different concentrations. The reference drugs used were low-permeability compound atenolol and high-permeability compound metoprolol. C=150 μg/mL as a control group. Values are expressed as means ± standard deviation, n=5 per group. |
 | Figure 3 Effective permeability coefficient (Peff) of periplocin obtained from in situ intestinal perfusion experiments with or without P-glycoprotein inhibitors (verapamil and cyclosporine A), breast cancer-resistance protein (novobiocin), and multidrug-resistance protein 2 (indomethacin and probenecid). Values are expressed as means ± standard deviation, n=5 per group. |
Bidirectional transport
The permeability (Papp) of periplocin in MDCK cell monolayers was Papp=(2.980±0.003)×10−6 cm/second using a concentration of c=75 μM and Papp=(3.430±1.200)×10−6 cm/second at a concentration of c=100 μM. These experiments showed that periplocin has weak permeability. The transport-rate values of periplocin in MDCK-MDR1 cell monolayers in the BL–AP direction were much higher than those in the AP–BL direction at both 75 μM and 100 μM concentrations. The inhibition effects on the efflux of periplocin across MDCK and MDCK/MDR1 cell monolayers were investigated by incubating reaction mixtures together with the P-gp inhibitor CsA. By co-incubating with 10 μM CsA, the efflux ratio of periplocin at 75 μM and 100 μM significantly decreased (Table 1). After CsA administration, the net efflux ratios decreased, which suggests that the efflux transport of periplocin was inhibited by CsA through the action of P-gp.
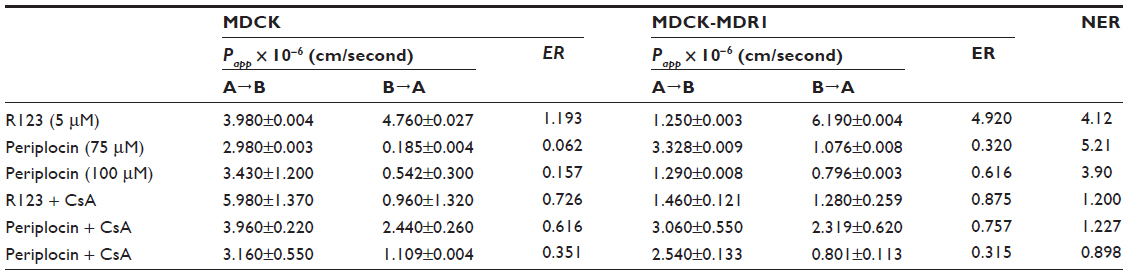 | Table 1 Efflux ratio of periplocin in the absence and presence of cyclosporine A across MDCK and MDCK-MDR1 cells |
In situ liver perfusion
Both Noé et al23 and Funakoshi et al24 reported that digoxin is a substrate of OATP2. Considering the structures of digoxin and periplocin are similar, we assume that the periplocin may also be transported by OATP2. The results demonstrated our hypothesis. Our results (Figures 4 and 5) showed that verapamil (a P-gp inhibitor) and rifampicin (an OATP inhibitor; it is not only an inhibitor of OATP2 but also some other OATP isoforms, at least for OATP1A2/1B1/1B3/2B1) significantly reduced the elimination rate of periplocin in the perfusate and biliary excretion of periplocin within 120 minutes.
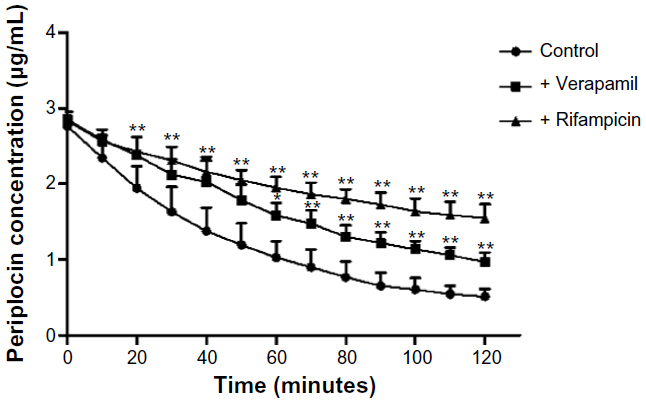 | Figure 4 Influence of the P-glycoprotein inhibitor verapamil and the organic anion-transporting polypeptide inhibitor rifampicin on the concentration of periplocin in the perfusate. Values are expressed as means ± standard deviation, n=5. |
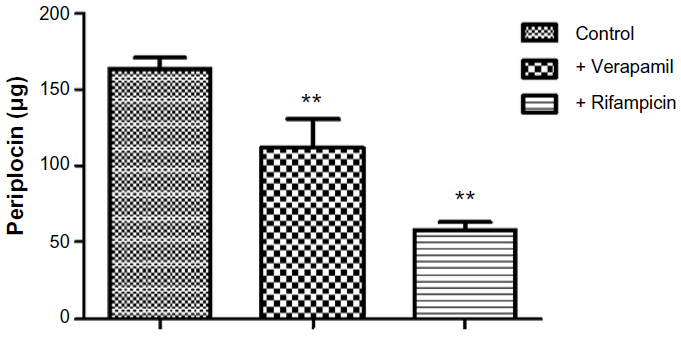 | Figure 5 Biliary excretion of periplocin with or without the P-glycoprotein inhibitor verapamil and the organic anion-transporting polypeptide inhibitor rifampicin. Values are expressed as means ± standard deviation, n=5 per group. |
In vitro metabolism of periplocin in liver S9
In vitro incubation results showed that periplocin (1 μg/mL and 3 μg/mL) in the human/rat liver S9 incubation system was stable for 120 minutes (relative standard deviation <3%). The concentration of periplocin in the incubation mixture remained the same (data not given). Although periplocin might have been metabolized to some extent, it could not be detected by HPLC, which suggests that periplocin is not a compound that is mainly metabolized by liver enzymes, and as a glycoside, hydrolysis by intestinal flora might be the main elimination route for periplocin.25
Discussion
Herbal medicine and their active ingredients have been widely used around the world. In clinical medicine, a growing number of DDIs/DHIs involving drug metabolic enzymes and transporters have been identified. In fact, herbal medicine is not as safe as previously thought, and frequently result in serious clinical accidents.26,27 Potential inhibition or activation effects of a herb or its ingredients on the drug transporters or metabolic enzymes may lead to herb–drug/herb–herb interactions. It is thus essential to examine the main active ingredients of specific herbal medicines in relation to clinical safety.
In situ intestine perfusion has been proven to be an accurate method for evaluating the permeability of chemicals and a common model for studying P-gp-mediated transport.16,28 In the present work, all the processes were in accordance with US Food and Drug Administration guidelines.29 Briefly, atenolol was chosen as the low-permeability reference compound, and metoprolol was selected as a critical compound with high permeability.30 The results verified that periplocin is a low-permeability chemical and is transported by P-gp but not by MRP2 or BCRP. Two concentrations of the P-gp inhibitor CsA (10 μM and 20 μM) were used to elucidate the permeability of periplocin, and the Peff of periplocin significantly increased after full inhibition of P-gp. These results suggest that P-gp plays an important role in the absorption of periplocin. The uptake transporters were not investigated in this study.
In situ liver perfusion showed that OATPs were potential drug transporters of periplocin, which thus accounts for the biliary excretion of periplocin. Meanwhile, the Papp(uptake) of periplocin at concentrations of 75 μM and 100 μM were lower than that of the reference drugs, showing low permeability as previous reported. In our study, periplocin was also transported by P-gp; therefore, periplocin is likely to be mediated by P-gp in both humans and rats. The present work supports the view that P-gp and OATPs inhibitors could increase the absorption and decrease the biliary excretion of periplocin. That means the bioavailability of periplocin would be increased. It is known that the high serum concentration of cardiac glycoside would increase the toxicity risk. Tuncok et al31 reported that the toxicity-reaction rate was 5%–15% after administration of digoxin at a conventional dose. Mao et al32 found the increase of the toxicity-reaction rate depends on the increase of digoxin blood concentration after investigating 288 patients. Clinical data have also shown that 90% of patients receiving quinidine (P-gp inhibitor) treatment showed an obvious increase in the blood concentration of digoxin;33 as a result, coadministered P-gp and or OATP inhibitors with periplocin should be avoided.
Drug metabolic enzymes play an important role in the transformation of drugs. The liver S9 homogenate fractions are very important in vitro tools for investigating drug metabolism because these are rich in metabolic enzymes such as flavin monooxygenases, monoamine oxidases, CO-related enzymes, and cytochrome P450 enzymes.34,35 In this study, periplocin showed no indications of metabolism in incubation systems using human and rat liver S9 fractions, which indicates that drug metabolic enzymes in the liver S9 were minimally involved in the transformation of periplocin. Previous studies on the metabolism of periplocin showed that hydrolysis under the function of intestinal bacterium was a main metabolic pathway of periplocin.25 Wang et al9 stated that the total excretion of periplocin and its hydrolysate in urine and feces is lower than 14%,9 which means other metabolic pathways like phase II metabolism may play an important role in the metabolism of periplocin. Phase II metabolism of periplocin should be checked in further research.
In summary, periplocin is a compound with low permeability and strong metabolic stability, as observed in in vitro metabolic systems using liver S9. P-gp was involved in the transport of periplocin in vivo. Furthermore, OATP1A2, OATP1B1, OATP1B3, and OATP2B1 may also play roles in the transport of periplocin. As a result, DDIs/DHIs based on the drug transporters OATP and P-gp should be considered when using P. sepium Bge in the clinic. Additional clinical or experimental studies should be conducted to elucidate the exact influence of OATPs and P-gp on the pharmacological effects of periplocin.
Acknowledgments
This work was supported by the Research Fund for the Doctoral Program of Higher Education (20121210110011), the National Natural Science Foundation of China (81373890), and the Natural Science Foundation of Tianjin (12JCZDJC26100).
Disclosure
The authors report no conflicts of interest in this work.
References
Zhang J, Shan BE, Liu GS, Zhao XT, Chen SH. Anti-tumor activity of extract from Periploca sepium Bge. Teratog Carcinog Mutagen. 2006;18(2):108–111. | |
Li YH, Gao XM, Zhang BL, Xu Q, Liu H, Pan GX. Effect of the Chinese medicine xiangjiapi on cardiac function of isolated hearts. J Liaoning Coll Tradit Chin Med. 2005;7(4):396–397. | |
Xu LQ, Lu HZ, Zhang YL. Clinical observation on treatment of 147 cases of chronic congestive heart-failure by bei wujiapi mixture. Yunnan J Tradit Chin Med Mater Med. 1998;19(4):29–31. | |
Egawa K, Kurihara Y, Ito T, Matsumoto M, Nose K. Induction of p16 INK4a transcription and of cellular senescence by aclacinomycin-derivatives and cardiac glycosides. Biol Pharm Bull. 2002;25(4):461–465. | |
Shugarts S, Benet LZ. The role of transporters in the pharmacokinetics of orally administered drugs. Pharm Res. 2009;26(9):2039–2054. | |
Giacomini, KM, Huang. SM. Transporters in drug development and clinical pharmacology. Clin Pharmacol Ther. 2013;94(1):3–9. | |
Ye L, Yang X, Yang Z, et al. The role of efflux transporters on the transport of highly toxic aconitine, mesaconitine, hypaconitine, and their hydrolysates, as determined in cultured Caco-2 and transfected MDCKII cells. Toxicol Lett. 2012;216(2–3):86–99. | |
Liu CX, Yi XL, Si DY, Xiao XF, He X, Li YZ. Herb-drug interactions involving drug metabolizing enzymes and transporters. Curr Drug Metab. 2011;12(9):835–849. | |
Wang Q, Ren XL, Wang Y. Study on excretion of periplocin in rats. J Tianjin Univ Tradit Chin Med. 2008;27(1):29–32. | |
Shitara Y, Sugiyama D, Kusuhara H, et al. Comparative inhibitory effects of different compounds on rat Oatp1 (Slc21a1)-and Oatp2 (Slc21a5)-mediated transport. Pharm Res. 2002;19(2):147–153. | |
Irvine JD, Takahashi L, Lockhart K, et al. MDCK (Madin-Darby canine kidney) cells: a tool for membrane permeability screening. J Pharm Sci. 1999;88(1):28–33. | |
Mahar Dohan KM, Humphreys JE, Webster LO, et al. Passive permeability and P-glycoprotein-mediated efflux differentiate central nervous system (CNS) and non-CNS marketed drugs. J Pharmacol Exp Ther. 2002;303(3):1029–1037. | |
Keogh JP, Kunta JR. Development, validation and utility of an in vitro technique for assessment of potential clinical drug-drug interactions involving P-glycoprotein. Eur J Pharm Sci. 2006;27(5):543–554. | |
Dahan A, Altman H. Food-drug interaction: grapefruit juice augments drug bioavailability – mechanism, extent and relevance. Eur J Clin Nutr. 2004;58(1):1–9. | |
Kim JS, Mitchell S, Kijek P, Tsume Y, Hilfinger J, Amidon GL. The suitability of an in situ perfusion model for permeability determinations: utility for BCS class I biowaiver requests. Mol Pharm. 2006;3(6):686–694. | |
Dahan A, Amidon GL. MRP2 mediated drug drug interaction: indomethacin increases sulfasalazine absorption in the small intestine, potentially decreasing its colonic targeting. Int J Pharm. 2010;386(1):216–220. | |
Rabba AK, Si L, Xue K, Li M, Li G. In situ intestinal perfusion of irinotecan: application to P-gp mediated drug interaction and introduction of an improved HPLC assay. J Pharm Pharm Sci. 2011;14(2):138–147. | |
Lindahl A, Frid S, Ungell AL, Lennernäs H. No evidence for the involvement of the multidrug resistance-associated protein and/or the monocarboxylic acid transporter in the intestinal transport of fluvastatin in rats. AAPS PharmSci. 2000;2(3):62–68. | |
Inoue H, Yokota H, Makino T, Yuasa A, Kato S. Bisphenol A glucuronide, a major metabolite in rat bile after liver perfusion. Drug Metab Dispos. 2001;29(8):1084–1087. | |
Lau YY, Wu CY, Okochi H, Benet LZ. Ex situ inhibition of hepatic uptake and efflux significantly changes metabolism: hepatic enzyme-transporter interplay. J Pharmacol Exp Ther. 2004;308(3):1040–1045. | |
Wen X, Walle T. Methylated flavonoids have greatly improved intestinal absorption and metabolic stability. Drug Metab Dispos. 2006;34(10):1786–1792. | |
Fagerholm U, Johansson M, Lennernäs H. Comparison between permeability coefficients in rat and human jejunum. Pharm Res. 1996;13(9):1336–1342. | |
Noé B, Hagenbuch B, Stieger B, Meier PJ. Isolation of a multispecific organic anion and cardiac glycoside transporter from rat brain. Proc Natl Acad Sci U S A. 1997;94(19):10346–10350. | |
Funakoshi S, Murakami T, Yumoto R, Kiribayashi Y, Takano M. Role of organic anion transporting polypeptide 2 in pharmacokinetics of digoxin and β-methyldigoxin in rats. J Pharm Sci. 2005;94(6):1196–1203. | |
Ren XL, Xie YS, Pan GX, Gao XM, Wang Z, Zuo QA. Research of periplocin on the intestinal bacterium metabolism in vitro. Tianjin J Tradit Chin Med. 2007;(6):515–518. | |
Mir O, Dessard-Diana B, Louet AL, et al. Severe toxicity related to a pharmacokinetic interaction between docetaxel and ritonavir in HIV-infected patients. Br J Clin Pharmacol. 2010;69(1):99–101. | |
Daniel WA, Wójcikowski J. Lysosomal trapping as an important mechanism involved in the cellular distribution of perazine and in pharmacokinetic interaction with antidepressants. Eur Neuropsychopharmacol. 1999;9(6):483–491. | |
Varma MV, Panchagnula R. Prediction of in vivo intestinal absorption enhancement on P-glycoprotein inhibition, from rat in situ permeability. J Pharm Sci. 2005;94(8):1694–1704. | |
US Food and Drug Administration. Guidance for industry: Drug interaction studies – study design, data analysis, implications for dosing, and labeling recommendations. 2012. Available from: http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm292362.pdf. Accessed April 4, 2014. | |
Meier J. Pharmacokinetic comparison of pindolol with other beta-adrenoceptor-blocking agents. Am Heart J. 1982;104(2 Pt 2):364–373. | |
Tuncok Y, Hazan E, Oto O, Guven H, Catalyurek H, Kalkan S. Relationship between high serum digoxin levels and toxicity. Int J Clin Pharmacol Ther. 1997;35(9):366–368. | |
Mao XQ, Jia XF, Zhang Y. [Monitoring the blood concentration of digoxin and its efficacy and toxicity]. 2013;40(20):3813–3814. Chinese. | |
Pei G, Yishi L, Donghua C. Study on relationship between serum digoxin concentration and digoxin toxicity. Chin J Hosp Pharm. 2000;20(3):137–138. | |
Dias C, Testa B, Soveral G, Dias M, Constantino L. Screening of the inhibitory effect of xenobiotics on alcohol metabolism using S9 rat liver fractions. Toxicol In Vitro. 2012;26(7):1177–1180. | |
Tang L, Zhou J, Yang CH, Xia BJ, Hu M, Liu ZQ. Systematic studies of sulfation and glucuronidation of 12 flavonoids in the mouse liver S9 fraction reveal both unique and shared positional preferences. J Agric Food Chem. 2012;60(12):3223–3233. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.