Back to Archived Journals » Hypoxia » Volume 4
Oxygen-dependent regulation of aquaporin-3 expression
Authors Hoogewijs D, Vogler M, Zwenger E, Krull S, Zieseniss A
Received 6 October 2015
Accepted for publication 26 January 2016
Published 21 April 2016 Volume 2016:4 Pages 91—97
DOI https://doi.org/10.2147/HP.S97681
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Prof. Dr. Roland Wenger
David Hoogewijs,1,2 Melanie Vogler,3 Eveline Zwenger,3 Sabine Krull,3 Anke Zieseniss3
1Institute of Physiology, University of Duisburg-Essen, Essen, Germany; 2Institute of Physiology, University of Zürich, Zürich, Switzerland; 3Institute of Cardiovascular Physiology, University Medical Center Göttingen, University of Göttingen, Göttingen, Germany
Abstract: The purpose of this study was to investigate whether aquaporin-3 (AQP3) expression is altered in hypoxia and whether hypoxia-inducible transcription factor (HIF)-1 regulates the hypoxic expression. AQP3 mRNA expression was studied in L929 fibrosarcoma cells and in several tissues derived from mice that were subjected to hypoxia. Computational analysis of the AQP3 promoter revealed conserved HIF binding sites within close proximity to the translational start site, and chromatin immunoprecipitation assays confirmed binding of HIF-1 to the endogenous hypoxia response elements. Furthermore, hypoxia resulted in increased expression of AQP3 mRNA in L929 fibrosarcoma cells. Consistently, shRNA-mediated knockdown of HIF-1 greatly reduced the hypoxic induction of AQP3. In addition, mRNA analysis of organs from mice exposed to inspiratory hypoxia demonstrated pronounced hypoxia-inducible expression of AQP3 in the kidney. Overall, our findings suggest that AQP3 expression can be regulated at the transcriptional level and that AQP3 represents a novel HIF-1 target gene.
Keywords: transcriptional regulation, oxygen, hypoxia-inducible factor, hypoxia response element
Introduction
Aquaporins (AQP) are integral membrane proteins that form pores on the membranes of cells.1 There are 13 known types of AQP in mammals. They transport water molecules in and out of the cells along an osmotic gradient, preventing the passage of ions and other solutes. AQPs are classified into three major subfamilies, ie, classical AQPs (AQPs 0, 1, 2, 4, and 5), aquaglyceroporins (AQPs 3, 7, 9, and 10), and unorthodox AQPs (AQPs 6, 8, 11, and 12).2 In contrast to the classical “water channels”, aquaglyceroporins are permeated by small uncharged molecules like glycerol and urea in addition to water; the function of the unorthodox AQP is currently being elucidated. Genetic defects involving AQP genes have been associated with several human diseases like congenital cataracts,3 nephrogenic diabetes insipidus,4 or neuromyelitis optica.5
Although some of the AQPs are expressed constitutively, there is growing knowledge of stimulus or context-dependent expression of specific AQP isoforms. The expression of AQP1 and 4 has been linked to changes in oxygen availability.6–8 If oxygen supply does not meet oxygen demand, the affected cells become hypoxic. Cells respond to hypoxia by activating a gene expression program, which is mediated by the hypoxia-inducible transcription factor (HIF). HIF contains two subunits, ie, the constitutively expressed β-subunit (HIFβ) and the oxygen-dependently regulated α-subunit (HIFα).9 Among the three described HIFα subunits (HIF-1α, HIF-2α, and HIF-3α), the HIF-1α subunit is most widely expressed. In normoxia, HIFα undergoes oxygen-dependent prolyl hydroxylation, which triggers the subsequent von Hippel Lindau-mediated ubiquitination and proteasomal degradation.10 The hydroxylation is facilitated by the three described prolyl-4-hydroxylase domain (PHD) enzymes, which are members of the oxoglutarate-dependent dioxygenase family.11,12 Compared to other members of this family, PHDs have a low affinity toward oxygen, which results in a substrate/oxygen-dependent enzymatic activity within the range of physiological pO2.13 In hypoxia, the HIFα subunits escape PHD-dependent hydroxylation and subsequent degradation. As a result, HIFα is stabilized, heterodimerizes with HIFβ, and induces HIF-dependent gene expression. For AQP1, a direct transcriptional regulation of the AQP1 promoter by HIF-1 has been demonstrated.14 The oxygen-dependent expression of the water-permeable AQP1 complex goes along with the rationale that it also contributes to the transmembrane gas permeation for CO2, NO, H2O, NH3 and most likely, also O2 in several cell types.15–17
In humans, AQP3 was found to be expressed in a variety of tissues. However, expression levels varied considerably, with weak expression in the heart, skeletal muscle, and central nervous tissue; moderate expression in the respiratory system; and high expression levels in the kidney.18 In the kidney, AQP3 constitutively localizes to the basolateral plasma membrane of collecting duct principal cells, connecting tubule cells, and inner medullary collecting duct cells. Treatment of human retinal pigment cells with CoCl2 results in increased AQP3 mRNA levels.19 CoCl2 is sometimes described as an inducer of a “chemical hypoxia”, because it can induce a HIF-response by inhibiting PHDs. The inhibitory action of cobaltous ions is thought to be based on replacement of catalytic Fe (II) and/or the effects on the availability of ascorbate.20 However, this is a PHD nonspecific effect and would also affect other Fe (II) and ascorbate-dependent processes. Therefore, we analyzed the response of AQP3 mRNA levels upon exposure to hypoxia in vitro and in vivo and tested if the expression of AQP3 is mediated by HIF-1.
Material and methods
Cell lines and cell culture
L929 cells (ATCC # CCL-1, ATCC, Manassas, VA, USA), derived from mouse, were cultivated in high glucose modified Eagle’s medium (Pan Biotech GmbH, Aidenbach, Germany) containing 10% fetal calf serum (Biochrom Ltd, Berlin, Germany), 50 units/mL penicillin G, and 50 μg/mL streptomycin (Pan Biotech). Cells were cultivated in a humidified 5% CO2, 95% air atmosphere at 37°C. For hypoxic conditions, O2 levels were decreased to 1% with N2 in an in vivo 400 workstation or SCITIVE workstation (Ruskinn Technology Ltd, Pencoed, UK). The establishment and characterization of the HIF-1α knockdown cell line was described recently.21
Hypoxic exposure of mice
Animal housing, hypoxic exposure, and organ excision for RNA isolation in male C57BL/6 mice have been described previously.22 Briefly, groups of three male C57BL/6 mice were exposed to 8% O2 inspiratory normobaric hypoxia for 0, 12, and 30 hours. All animal experiments were performed according to FELASA (Federation of Laboratory Animal Science Associations) category B and GV-SOLAS (Society for Laboratory Animal Science/Society of Laboratory Animals) standard guidelines and approved by the German animal welfare committee (Regierungspräsidium Karlsruhe, Karlsruhe, Germany) as described previously.22
RNA extraction and RT-qPCR
For RNA isolation, 2×105 L929 cells were seeded in 2 mL of culture medium and incubated for 1–48 hours at 37°C and 5% CO2 in normoxia (21% O2) or hypoxia (1% O2). RNA was isolated, and transcript levels were analyzed as described previously.23 For mouse tissue samples, 2 μg of total RNA was reverse transcribed (RT) using AffinityScript reverse transcriptase (Agilent Technologies, Santa Clara, CA, USA), and the cDNA levels were estimated by quantitative polymerase chain reaction (qPCR) using a SybrGreen qPCR reagent kit (Sigma-Aldrich, St Louis, MO, USA) in a MX3000P light cycler (Agilent Technologies). The ribosomal protein S12 (mS12) was used as a housekeeping gene. The fold change in gene expression was determined by using the delta-delta-CT data analysis method.
Primers were as follows: AQP3 (qPCR on L929 cell extracts) forward 5′-TGCCTTGCGCTAGCTACTTT-3′, AQP3 reverse 5′-AGGCCCAGATTGCATCGTAG-3′; AQP3 (qPCR on tissue extracts) forward 5′-GCTGTGACCTTCGCAATGTG-3, AQP3 reverse 5′-GTACACGAAGACACCAGCGA-3′; Glut1 forward 5′-TGGCCTTGCTGGAACGGCTG-3′, Glut1 reverse 5′-TCCTTGGGCTGCAGGGAGCA-3′; mS12 forward, 5′-GAAGCTGCCAAGGCCTTAGA-3′, mS12 reverse 5′-AACTGCAACCAACCACCTTC-3′; PHD3 forward 5′-GGCCGCTGTATCACCTGTA T-3′, PHD3 reverse 5′-TTCTGCCCTTTCTTCAGCAT-3′.
Chromatin immunoprecipitation assay
A total of 3×106 cells were incubated under normoxic (20% O2) or hypoxic (1% O2, 14 hours) conditions. Cells were treated with 1% formaldehyde for 30 minutes at room temperature, followed by the addition of glycine to a final concentration of 0.125 M. Cells were washed in 4°C phosphate-buffered saline and pelleted. Cell pellets were resuspended in 600 μL of cold cell lysis buffer (50 mM Tris-Cl, pH 8.1, 10 mM ethylenediaminetetraacetic acid [EDTA], 1% sodium dodecyl sulfate [SDS], and protease inhibitors). Cell lysates were sonicated (Bandelin Sonopuls, BANDELIN electronic GmbH and Co. KG, Berlin, Germany) for five 20-second intervals on ice to shear DNA to fragments. Cell debris was removed by centrifugation. The supernatants were diluted 10-fold in IP buffer (16.7 mM Tris [pH 8.1], 1.1% Triton X-100, 1.2 mM EDTA, 0.01% sodium dodecyl sulfate [SDS], and protease inhibitors). Chromatin solutions were precleared with protein A/G Plus Agarose slurry (Santa Cruz Biotechnology, Dallas, TX, USA) while rotating at 4°C for 2 hours. An input sample control (500 μL) was taken from the precleared supernatant, and the remaining supernatant was divided into three parts and incubated with anti-HIF-1α (NB100-479, Novus Biologicals LLC, Littleton, CO, USA), anti-Histone (ab12179, Abcam, Cambridge, UK), or anti-Tubulin (ab6046, Abcam)-antibodies overnight at 4°C. Chromatin/protein complexes were isolated with 60 μL protein A/G Agarose. Pellets were washed with low salt, high salt buffer series (low salt buffer: 20 mM Tris [pH 8.1], 150 mM NaCl, 1% Triton X-100, 2 mM EDTA, 0.1% SDS; high salt buffer: 20 mM Tris [pH 8.1], 500 mM NaCl, 1% Triton X-100, 2 mM EDTA, 0.1% SDS), LiCl buffer (10 mM Tris [pH 8.1], 0.25 mM LiCl, 1 mM EDTA, 1% NP-40, 1% Na-desoxycholate), and TE. DNA–protein complexes were eluted with 1% SDS, 0.1 M NaHCO3. DNA–protein crosslinks were reversed with 5 M NaCl at 65°C for 4 hours. Immunoprecipitated DNA segments were purified with phenol–chloroform–isoamylalcohol. PCR analysis was performed, as described in Zheng et al’s24 study, of the following gene region spanning the hypoxia response element (HRE) consensus: mGlut1 (from –2,906 to –2,689), mAQP3 (from –573 to –1,201). The sequences of the primers used in the PCRs were as follows: mGlut1prom (forward), 5′-CCGGGCTGTCTTACTCACTCTTACTCC-3′, mGlut1prom (reverse), 5′-GGGCTGTGTTCAAGCTGCGCC-3′; mAQP3prom (forward), 5′-GGAGGCAGAGACAGGTAGATCT-3′, mAQP3prom (reverse) 5′-GGAACATGTCATCACATGTTGCATG-3′.
Western blot
Western blot experiments were performed as described by Vogler et al.21 Briefly, cells were lysed in 400 mM NaCl, 1 mM EDTA, 10 mM Tris/HCl, pH 8.0, and 0.1% Triton X-100 including the protease inhibitor cocktail complete mini (Roche Applied Science, Mannheim, Germany). Protein concentrations were quantified with the Bradford method using bovine serum albumin as a standard. Protein extracts were electrophoresed through SDS–polyacrylamide gels and electrotransferred onto nitrocellulose membranes (Amersham, Freiburg, Germany) by semi-dry blotting (PeqLab, Erlangen, Germany). For detection of specific proteins, the following primary and secondary antibodies were used: anti-HIF-1α (NB100-479, Novus Biologicals LLC), anti-β-tubulin (ab6046, Abcam), and HRP-labeled anti-rabbit (Sc-2004, Santa Cruz).
Statistical analysis
Results are expressed as mean values ± standard error of the mean of at least three independent experiments. For statistical comparisons, results were analyzed by one-way analysis of variance followed by Bonferroni’s correction. Values of P<0.05 (*), P<0.01 (**), P<0.001 (***) were considered statistically significant.
Results
L929 fibrosarcoma cells were subjected to hypoxia for 1–48 hours (Figure 1). In this time-course experiment, stabilization of HIF-1α is clearly identifiable after 1 hour of hypoxia (Figure 1A), and a significant induction in AQP3 mRNA expression was observed after 4 hours (P<0.001; Figure 1B). The AQP3 mRNA expression peaked at about 24 hours of hypoxic incubation (P<0.001) and succeeded the induction of the well-established HIF-1 target gene glucose transporter-1 (Glut1). The expression of Glut1 peaked at 4 hours (P<0.001) following hypoxic cell incubation, and then declined.
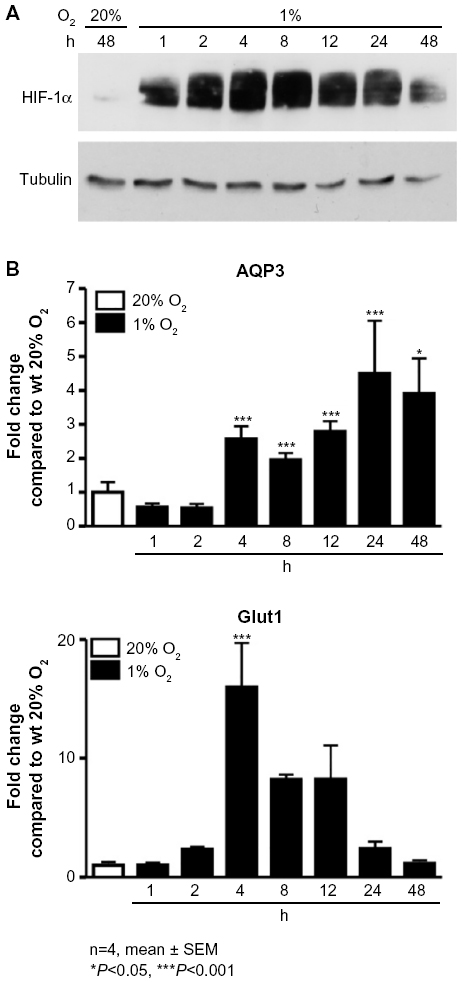 | Figure 1 Hypoxia increases the mRNA expression levels of AQP3 in L929 cells. |
To analyze whether the hypoxic induction of AQP3 mRNA expression levels is HIF-1-dependent, we employed two independent HIF-1α knockdown clones (shC1 and shC2). Generation and characterization of these stable shRNA-mediated knockdown clones have been described by Vogler et al.21 Similar to the HIF-1 target genes Glut1 and PHD3, knockdown of HIF-1α in L929 cells prevented the hypoxia-stimulated induction of AQP3 in both HIF- 1α-deficient clones. However, as seen before, in the wild-type cells, AQP3, Glut1, and PHD3 mRNA expression levels are significantly induced after hypoxic incubation (P<0.001) (Figure 2).
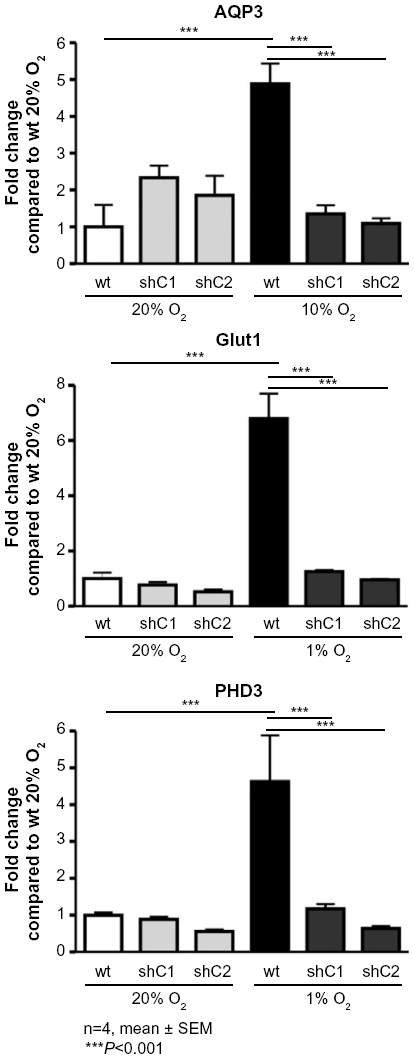 | Figure 2 The hypoxic induction of AQP3 mRNA expression is HIF-1 dependent. |
In silico analysis of the mouse AQP3 gene identified a region close to the translational start site containing two core sequences of a consensus HIF-1 response element (HRE) 5′-RCGTG-325 located at –647 and –916 (Figure 3). To test whether HIF-1α can bind to these HREs in vivo, chromatin immunoprecipitation assays were performed. L929 cells cultured at either 1% O2 or 20% O2 were fixed with formaldehyde, and sonicated cell lysates were immunoprecipitated with anti-HIF-1α antibodies. Precipitated DNA was used to amplify a fragment containing both putative HREs at –647 and –916 of mouse AQP3. The product was only amplified from hypoxia-treated cell samples immunoprecipitated with the HIF-1α antibody. Primers spanning the mouse Glut1 HRE region amplified fragments in a similar pattern and served as a positive control.
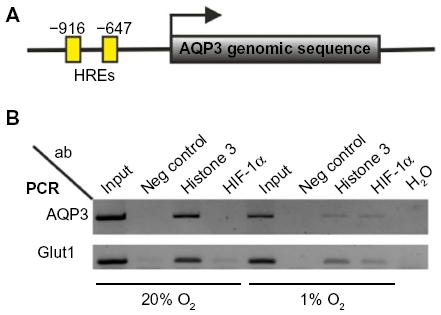 | Figure 3 HREs are located in close proximity to the AQP3 translational start site. |
To test whether a hypoxic induction of AQP3 can be observed in vivo, mice were exposed to 8% O2 for 0, 12, and 30 hours. mRNA was isolated from various organs, and AQP3 expression levels were analyzed. Compared to tissues like heart, brain, and liver, we found higher basal levels of AQP3 relative to the housekeeping gene mS12 in the kidney (Figure 4). Furthermore, in the hypoxia-treated group, a significant induction (P<0.001 after 12 hours 8% O2, P<0.01 after 30 hours 8% O2) of AQP3 was observed in the kidney samples.
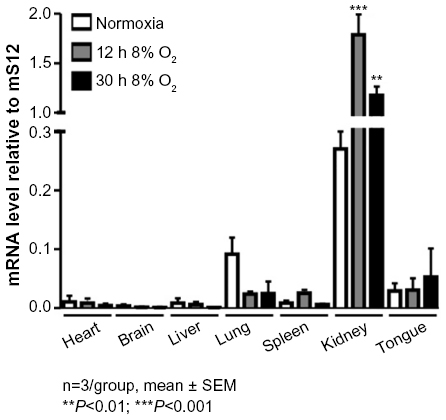 | Figure 4 AQP3 is induced in the hypoxic kidneys in vivo. |
Discussion
AQP3 belongs to the family of aquaglyceroporin and thus facilitates the transport of water and small noncharged solutes.26,27 AQP3 is expressed in the renal collecting duct,28,29 but can also be found in several extrarenal tissues, including epithelial cells of the skin and retina, and skin fibroblasts.19,30–32 Furthermore, AQP3 is elevated in various cancers such as lung, prostate, breast, and skin.33–39 AQP3 adds to tissue hydration, but it has also been discussed to have a role in cell shape, attachment, and migration.19,39 In cancer, the upregulation of AQP3 expression may promote tumorigenesis and tumor development. Hence, investigating the mechanisms underlying AQP3 expression stood at the focal point of several studies. In skin fibroblasts, AQP3 protein levels are induced by epidermal growth factor,32 and at the transcriptional level, AQP3 has been described to be regulated under hyperosmotic conditions and by stimulation with platelet-derived growth factor, prostaglandin E2, arachidonic acid, blood serum, and CoCl2 in retinal pigment epithelial cells.19 In the promoter of the AQP3 gene, a functional estrogen response element was identified, which might mediate the AQP3 expression in breast cancer cells.38
In the present study, we add to the understanding of the regulation of AQP3 expression, and we show that the AQP3 transcription is induced in hypoxia in vitro and in vivo. This induction is HIF-1α dependent, because HIF-1α deficient cells display a significant reduction in AQP3 mRNA expression levels in hypoxia compared to wild-type cells. Furthermore, our data suggest that the hypoxic AQP3 induction is directly regulated by HIF-1, and we find a cluster of two functional HREs within close proximity to the AQP3 translational start site. Analysis of a panel of murine tissues for AQP3 expression revealed high expression levels of AQP3 in the kidney and substantially lower levels in heart, brain, liver, lung, and tongue, consistent with our own in silico analyses (not shown) and previous studies.11,18 To the best of our knowledge, hypoxia-dependent regulation of renal AQP3 in mice has not been previously reported. The pronounced hypoxic induction of renal AQP3 in mice observed in the current study substantiates the physiological relevance of oxygen-dependent regulation of AQP3 expression levels in vivo.
A common characteristic of solid tumors is tissue hypoxia and the activation of the HIF-1 pathway. HIF-1 can play a major role in tumor progression, including proliferation and metastasis. Moreover, there is compelling evidence that HIF-1 is involved in resistance to cancer therapy. However, while many components of the HIF-pathway are protumorigenic, others are antitumorigenic, and the overall consequences for tumor progression and aggressiveness may be cell type- and context-specific.40 AQP3 has been reported to promote cancer cell motility and invasion.41 Accordingly, the inhibition of AQP3 reduces proliferation, migration, and invasion of non-small-cell lung cancer cells.42 Our data reveal that AQP3 is hypoxia-inducible in some cells and tissues, consistent with the hypothesis that AQP3 may represent a protumorigenic component of the HIF-pathway.
Conclusion
Collectively, we show that hypoxia enhances AQP3 expression in L929 fibrosarcoma cells in a HIF-1-dependent manner. These findings should encourage further research evaluating the hypoxic induction of AQP3 in cancer cells.
Acknowledgments
We are grateful to Hugo Marti for providing mouse tissue samples and the Swiss National Centre of Competence in Research (NCCR) Kidney Control of Homeostasis (Kidney.CH) for funding.
Disclosure
The authors report no conflicts of interest in this work.
References
Verkman AS, Anderson MO, Papadopoulos MC. Aquaporins: important but elusive drug targets. Nat Rev Drug Discov. 2014;13(4):259–277. | |
Rojek A, Praetorius J, Frokiaer J, Nielsen S, Fenton RA. A current view of the mammalian aquaglyceroporins. Annu Rev Physiol. 2008;70:301–327. | |
King LS, Kozono D, Agre P. From structure to disease: the evolving tale of aquaporin biology. Nat Rev Mol Cell Biol. 2004;5(9):687–698. | |
Loonen AJ, Knoers NV, van Os CH, Deen PM. Aquaporin 2 mutations in nephrogenic diabetes insipidus. Semin Nephrol. 2008;28(3):252–265. | |
Pereira WL, Reiche EM, Kallaur AP, Kaimen-Maciel DR. Epidemiological, clinical, and immunological characteristics of neuromyelitis optica: a review. J Neurol Sci. 2015;355(1–2):7–17. | |
Kaneko K, Yagui K, Tanaka A, et al. Aquaporin 1 is required for hypoxia-inducible angiogenesis in human retinal vascular endothelial cells. Microvasc Res. 2008;75(3):297–301. | |
Ding JY, Kreipke CW, Speirs SL, Schafer P, Schafer S, Rafols JA. Hypoxia-inducible factor-1α signaling in aquaporin upregulation after traumatic brain injury. Neurosci Lett. 2009;453(1):68–72. | |
Mou K, Chen M, Mao Q, et al. AQP-4 in peritumoral edematous tissue is correlated with the degree of glioma and with expression of VEGF and HIF-α. J Neurooncol. 2010;100(3):375–383. | |
Bishop T, Ratcliffe PJ. Signaling hypoxia by hypoxia-inducible factor protein hydroxylases: a historical overview and future perspectives. Hypoxia. 2014;2:197–213. | |
Jaakkola P, Mole DR, Tian YM, et al. Targeting of HIF-α to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science. 2001;292(5516):468–472. | |
Epstein AC, Gleadle JM, McNeill LA, et al. C. elegans EGL-9 and mammalian homologs define a family of dioxygenases that regulate HIF by prolyl hydroxylation. Cell. 2001;107(1):43–54. | |
Bruick RK, McKnight SL. A conserved family of prolyl-4-hydroxylases that modify HIF. Science. 2001;294(5545):1337–1340. | |
Hirsila M, Koivunen P, Gunzler V, Kivirikko KI, Myllyharju J. Characterization of the human prolyl 4-hydroxylases that modify the hypoxia-inducible factor. J Biol Chem. 2003;278(33):30772–30780. | |
Abreu-Rodriguez I, Sanchez Silva R, Martins AP, et al. Functional and transcriptional induction of aquaporin-1 gene by hypoxia; analysis of promoter and role of HIF-1α. PLoS One. 2011;6(12):e28385. | |
Herrera M, Hong NJ, Garvin JL. Aquaporin-1 transports NO across cell membranes. Hypertension. 2006;48(1):157–164. | |
Musa-Aziz R, Chen LM, Pelletier MF, Boron WF. Relative CO2/NH3 selectivities of AQP1, AQP4, AQP5, AmtB, and RhAG. Proc Natl Acad Sci U S A. 2009;106(13):5406–5411. | |
Echevarria M, Munoz-Cabello AM, Sanchez-Silva R, Toledo-Aral JJ, Lopez-Barneo J. Development of cytosolic hypoxia and hypoxia-inducible factor stabilization are facilitated by aquaporin-1 expression. J Biol Chem. 2007;282(41):30207–30215. | |
Mobasheri A, Wray S, Marples D. Distribution of AQP2 and AQP3 water channels in human tissue microarrays. J Mol Histol. 2005; 36(1–2):1–14. | |
Hollborn M, Ulbricht E, Reichenbach A, Wiedemann P, Bringmann A, Kohen L. Transcriptional regulation of aquaporin-3 in human retinal pigment epithelial cells. Mol Biol Rep. 2012;39(8):7949–7956. | |
Salnikow K, Donald SP, Bruick RK, Zhitkovich A, Phang JM, Kasprzak KS. Depletion of intracellular ascorbate by the carcinogenic metals nickel and cobalt results in the induction of hypoxic stress. J Biol Chem. 2004;279(39):40337–40344. | |
Vogler M, Vogel S, Krull S, et al. Hypoxia modulates fibroblastic architecture, adhesion and migration: a role for HIF-1α in cofilin regulation and cytoplasmic actin distribution. PLoS One. 2013;8(7):e69128. | |
Kaufmann MR, Barth S, Konietzko U, et al. Dysregulation of hypoxia-inducible factor by presenilin/gamma-secretase loss-of-function mutations. J Neurosci. 2013;33(5):1915–1926. | |
Swain L, Wottawa M, Hillemann A, et al. Prolyl-4-hydroxylase domain 3 (PHD3) is a critical terminator for cell survival of macrophages under stress conditions. J Leukoc Biol. 2014;96(3):365–375. | |
Zheng X, Linke S, Dias JM, et al. Interaction with factor inhibiting HIF-1 defines an additional mode of cross-coupling between the Notch and hypoxia signaling pathways. Proc Natl Acad Sci U S A. 2008;105(9):3368–3373. | |
Semenza GL, Jiang BH, Leung SW, et al. Hypoxia response elements in the aldolase A, enolase 1, and lactate dehydrogenase A gene promoters contain essential binding sites for hypoxia-inducible factor 1. J Biol Chem. 1996;271(51):32529–32537. | |
Ishibashi K, Sasaki S, Fushimi K, et al. Molecular cloning and expression of a member of the aquaporin family with permeability to glycerol and urea in addition to water expressed at the basolateral membrane of kidney collecting duct cells. Proc Natl Acad Sci U S A. 1994;91(14):6269–6273. | |
Echevarria M, Windhager EE, Frindt G. Selectivity of the renal collecting duct water channel aquaporin-3. J Biol Chem. 1996; 271(41):25079–25082. | |
Echevarria M, Windhager EE, Tate SS, Frindt G. Cloning and expression of AQP3, a water channel from the medullary collecting duct of rat kidney. Proc Natl Acad Sci U S A. 1994;91(23):10997–11001. | |
Ecelbarger CA, Terris J, Frindt G, et al. Aquaporin-3 water channel localization and regulation in rat kidney. Am J Physiol. 1995;269(5 Pt 2):F663–F672. | |
Ishibashi K, Sasaki S, Saito F, Ikeuchi T, Marumo F. Structure and chromosomal localization of a human water channel (AQP3) gene. Genomics. 1995;27(2):352–354. | |
Matsuzaki T, Suzuki T, Koyama H, Tanaka S, Takata K. Water channel protein AQP3 is present in epithelia exposed to the environment of possible water loss. J Histochem Cytochem. 1999;47(10):1275–1286. | |
Cao C, Sun Y, Healey S, et al. EGFR-mediated expression of aquaporin-3 is involved in human skin fibroblast migration. Biochem J. 2006;400(2):225–234. | |
Liu YL, Matsuzaki T, Nakazawa T, et al. Expression of aquaporin 3 (AQP3) in normal and neoplastic lung tissues. Hum Pathol. 2007;38(1):171–178. | |
Wang J, Tanji N, Kikugawa T, Shudou M, Song X, Yokoyama M. Expression of aquaporin 3 in the human prostate. Int J Urol. 2007; 14(12):1088–1092; discussion 1092. | |
Verkman AS, Hara-Chikuma M, Papadopoulos MC. Aquaporins – new players in cancer biology. J Mol Med (Berl). 2008;86(5):523–529. | |
Hara-Chikuma M, Verkman AS. Prevention of skin tumorigenesis and impairment of epidermal cell proliferation by targeted aquaporin-3 gene disruption. Mol Cell Biol. 2008;28(1):326–332. | |
Nakakoshi M, Morishita Y, Usui K, Ohtsuki M, Ishibashi K. Identification of a keratinocarcinoma cell line expressing AQP3. Biol Cell. 2006;98(2):95–100. | |
Huang YT, Zhou J, Shi S, et al. Identification of estrogen response element in aquaporin-3 gene that mediates estrogen-induced cell migration and invasion in estrogen receptor-positive breast cancer. Sci Rep. 2015;5:12484. | |
Kusayama M, Wada K, Nagata M, et al. Critical role of aquaporin 3 on growth of human esophageal and oral squamous cell carcinoma. Cancer Sci. 2011;102(6):1128–1136. | |
Ratcliffe PJ. Oxygen sensing and hypoxia signalling pathways in animals: the implications of physiology for cancer. J Physiol. 2013; 591(Pt 8):2027–2042. | |
Bui LC, Tomkiewicz C, Pierre S, Chevallier A, Barouki R, Coumoul X. Regulation of aquaporin 3 expression by the AhR pathway is critical to cell migration. Toxicol Sci. 2016;149(1):158–166. | |
Hou SY, Li YP, Wang JH, et al. Aquaporin-3 inhibition reduces the growth of NSCLC cells induced by hypoxia. Cell Physiol Biochem. 2016;38(1):129–140. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.