Back to Journals » OncoTargets and Therapy » Volume 13
Overexpression of Progerin Results in Impaired Proliferation and Invasion of Non-Small Cell Lung Cancer Cells
Authors Hu XT ![]() , Song HC
, Song HC ![]() , Yu H, Wu ZC
, Yu H, Wu ZC ![]() , Liu XG
, Liu XG ![]() , Chen WC
, Chen WC
Received 2 November 2019
Accepted for publication 26 February 2020
Published 30 March 2020 Volume 2020:13 Pages 2629—2642
DOI https://doi.org/10.2147/OTT.S237016
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Nicola Silvestris
Xiao-Ting Hu, Hao-Chang Song, Hui Yu, Zu-Chun Wu, Xin-Guang Liu, Wei-Chun Chen
Guangdong Provincial Key Laboratory of Medical Molecular Diagnostics, Institute of Aging Research, Institute of Biochemistry & Molecular Biology, Guangdong Medical University, Dongguan, Guangdong, People’s Republic of China
Correspondence: Wei-Chun Chen; Xin-Guang Liu
Guangdong Provincial Key Laboratory of Medical Molecular Diagnostics, Institute of Aging Research, Institute of Biochemistry & Molecular Biology, Guangdong Medical University, Dongguan, Guangdong, People’s Republic of China
Tel +86759 22896026
Email [email protected]; [email protected]
Purpose: The accumulation of progerin (PG) in patients is responsible for the pathogenesis of Hutchinson-Gilford Progeria Syndrome (HGPS) because it triggers accelerated aging of cells. However, there are few studies on the effects of progerin on tumor cells. Lung cancer is one of the most common malignant cancers with high global morbidity and mortality rates; non-small cell lung cancer accounts for the majority of cases. The purpose of this study was to determine the effects of progerin on A549 cell proliferation, cell cycle, invasion, migration, sensitivity to DNA damaging agents, senescence and apoptosis with a goal of exploring new ideas for lung cancer treatment.
Methods: A549 cells overexpressing progerin (A549-PG) and a corresponding blank control (A549-GFP) were constructed by lentiviral infection. A nuclear staining assay was utilized to detect abnormal nuclear morphology. The proliferation, cell cycle, colony formation, invasion and migration abilities of A549-PG were compared with those of A549-GFP via EdU assays, flow cytometry, colony formation experiments, and Matrigel invasion and migration assays, respectively. SA‐β‐gal staining was used to measure senescence in cells.
Results: The expression of progerin was significantly higher in A549-PG than A549-GFP. About 20% of A549-PG possessed abnormal nuclei. Overexpression of progerin in A549 cells inhibited cell proliferation, migration and invasion, and associated proteins (CDK4, pRB, ANLN, MMP7 and MMP9) were downregulated. DNA damage repair was also impaired. Progerin did not cause cells to senesce, and there was no difference in apoptosis.
Conclusion: A549-PG generated some cellular changes, including the nuclear skeleton, the cell cycle, DNA damage repair, and migration and invasion abilities. Our data indicate that progerin could cause an imbalance in the steady state in A549 cells and increase their sensitivity to chemotherapeutic drugs.
Keywords: A549 lung cancer cells, progerin, cell cycle, invasion, DNA damage
Introduction
Senescence refers to the cell cycle entering an irreversible state of growth arrest.1 The first description of cellular senescence was reported in 1965, when Leonard Hayflick observed that cells undergo replicative senescence in culture.2 In recent years, studies proved that cell senescence can inhibit the development of tumors. Therapeutic approaches such as p53 reactivation, inhibition of c-MYC in drug-treated tumors or treatment with cyclin-dependent kinase (CDK) inhibitors have proven to be effective by invoking a senescence response.3,4 The possibility of using therapies that promote senescence for cancer treatment has provoked considerable interest. In studies of human tissue and rodents, aging-related markers can be detected in inchoate tumor tissues, and senescence has an inhibitory effect in the early stage of tumorigenesis.5 The senescence-associated secretory phenotype (SASP) exhibited by aging cells activates the immune response and exerts immune clearance to eliminate premalignant tumor cells, thereby inhibiting tumor progression.6 It is well known that the accumulation of mutations in the nuclear protein Lamin A, which is called progerin or Lamin AΔ50, is responsible for the pathogenesis of HGPS patients. HGPS is a rare but devastating genetic disease characterized by premature aging, and it generally leading to death at approximately 13 years old due to cardiovascular disease.7,8 Currently, 90% of HGPS patients are thought to have de novo point mutations in the LMNA gene that substitute cytosine with thymine, giving rise to permanently farnesylated, mutant Lamin A that breaks the nuclear shape, disrupts chromatin organization, and ultimately leads to a widespread accumulation of DNA damage and then senescence.9,10 HGPS is an ideal model for studying human aging, and the accumulation of progerin is a key molecular mechanism that accelerates aging. However, previous studies on progerin mainly focused on normal cells and patients with premature aging. What happens if progerin were introduced into tumor cells, and are there similarities between HGPS cells and tumor cells? Lung cancer is one of the most common malignant cancers in the world, and it has high rates of recurrence and metastasis. Among lung carcinomas, non-small cell lung cancer accounts for nearly 80% of cases.11 The main treatment is chemotherapy; however, most patients develop resistance after 9 to 11 months of therapy,12 which itself is not ideal due to the many side effects. New treatment strategies are urgently needed. This study concentrated on the non-small cell lung cancer cell line A549, which overexpresses progerin, and explored phenotypes related to proliferation, sensitivity to DNA damage, apoptosis, invasion and migration. Related molecular mechanisms were identified, which provides new ideas for inhibiting the progression of A549 cells.
Materials and Methods
Culture of Human A549 Lung Cancer Cells
A549 cells (Gaining Biological, China) were cultured in an incubator (Thermo, USA) of 5% CO2 at 37°C with RPMI 1640 medium (Gibco, USA) containing 10% fetal bovine serum (FBS, Gibco, USA). Then, the cells were split at a ratio of 1:3 when they reached 80% confluency.
Gene Cloning Procedure
Incorporating the enzyme sites Xho I and EcoR I on the GV219 vector, we used the corresponding primer sequences of progerin: the upstream sequence: 5ʹ CTCTCGAGATGGAGACCCCGTCCCAGCG 3ʹ, the downstream sequence: 5ʹ TCGAATTCTTACATGATGCTGCAGTTCTGG 3ʹ. Then, the target product progerin was amplified by PCR. Next, Xho I and EcoR I were used in a double-digest of the GV219 empty vector, the progerin product was ligated into the empty vector, and the recombinant plasmid was used to transform competent E. coli. A monoclonal single colony containing the plasmid was picked carefully, and the recombinant plasmid was obtained. The company IGE performed sequencing and identification of the plasmid. Finally, the company Genechem packaged the plasmid into a lentivirus.
Lentivirus Infection to Generate Overexpressing Cell Lines
A lentiviral expression system (Genechem Company) was used according to the manufacturer’s protocol. On the day before transfection, 5×104 A549 cells were plated on 24-well plates in 1640 medium supplemented with 10% FBS. The following day, cells were infected at a MOI = 50 according to the virus titer. After 72 hrs, fluorescence was observed under a fluorescence microscope. Then, we replated cells into six-well plates, and a stable strain was selected by treatment with puromycin.
DAPI Nuclear Staining Assay
Logarithmic growth stage A549 cells were cultured to 80% confluence in 6-well plates and then were fixed for 10 min at room temperature. Two milliliters of DAPI stain solution was added and incubated with the cells for 5 min, and the morphology of the nuclei was observed by inverted fluorescence microscopy.
Flow Cytometry
Cells at a density of up to 85% were harvested from six-well plates, collected in precooled PBS, and fixed in cold 70% ethanol overnight at 4°C. After treatment with 100 μg/mL RNase (Sigma-Aldrich), the cells were stained with 50 μg/mL propidium iodide (Sigma-Aldrich) in PBS and then were incubated in the dark for 30 min at 4°C. Flow cytometry was performed on a Becton Dickinson FACScan and was analyzed by ModFit software (Verity Software House, Inc., Topsham, ME). Cells from at least 20,000 ungated cells were analyzed for DNA content.
EdU (5-Ethynyl-2´- Deoxyuridine) Assay
A total of 4×104 logarithmic growth stage cells were inoculated per well of 96-well plates and then were cultured to a normal growth stage. The medium was then replaced by RPMI 1640 containing 10% FBS and 50 μM 5-ethynyl-2´-deoxyuridine (EdU). These cells were incubated for 4 hrs according to the protocol of the EdU kit (C10310-1, RiboBio, China).
Clone Formation Experiment
A total of 1×104 logarithmic growth stage cells were inoculated per well of 6-well plates in RPMI 1640 medium containing 10% FBS. Cells were cultured for one week at 37°C and 5% CO2. After incubation, the cells were fixed and stained with 0.1% crystal violet, and the number of colonies was calculated through analysis with a microscope (Olympus, Tokyo, Japan).
Matrigel Invasion Assay
A549-PG and A549-GFP cells were grown to confluency in RPMI 1640 medium containing 10% FBS. The cells were harvested by trypsinization, washed in RPMI 1640 without the addition of serum, and suspended in RPMI 1640 medium at concentrations of 1×105 cells/mL. Before preparing the cell suspensions, a dried layer of Matrigel matrix (Corning, USA) was rehydrated with RPMI 1640 medium for 2 hrs at room temperature. RPMI 1640 medium (0.50 mL) containing 10% FBS was added to each lower section of 24-well Matrigel invasion chambers, and 0.5 mL (5×104 cells) of cell suspension was added to each insert of the upper chamber. The plates were incubated for 20 hrs at 37°C. After incubation, the cells that had invaded through the Matrigel-coated inserts were fixed and stained with 0.1% crystal violet.
Migration Experiment
A549-PG and A549-GFP were grown to confluency in RPMI 1640 medium containing 10% FBS. The cells were harvested and suspended in RPMI 1640 medium at concentrations of 1×104 cells/mL. RPMI 1640 medium (0.50 mL) containing 10% FBS was added to each lower section of 24-well Matrigel invasion chambers, and 0.50 mL (5×103 cells) of cell suspension was added to each insert of the upper chamber. The plates were incubated for 12 hrs at 37°C. After incubation, the cells migrating across the microporous film were fixed and stained with 0.1% crystal violet.
Western Blot
Initially, the cell protein lysates were separated on 6% or 12% SDS polyacrylamide gels and then were electrophoretically transferred to PVDF membranes (0.20 μm pore size; Millipore, USA). The PVDF membranes were then incubated with rabbit anti‐p53 polyclonal antibody (#9284, Cell Signaling, USA), rabbit anti‐ATM monoclonal antibody (#2873, Cell Signaling, USA), rabbit anti‐ATR monoclonal antibody (#13934, Cell Signaling, USA), mouse anti‐DNA-PKcs monoclonal antibody (#12311, Cell Signaling, USA), rabbit anti‐Phospho-p53 polyclonal antibody (#9284, Cell Signaling, USA), mouse anti-FLAG monoclonal antibody (#TA180144, OriGene, USA), rabbit anti-γ-H2AX monoclonal antibody (#2577, Cell Signaling, USA), mouse anti-Lamin A/C monoclonal antibody (sc376248, Santa Cruz Biotechnology, USA), rabbit anti‐H2AX monoclonal antibody (#7631, Cell Signaling, USA), rabbit anti-GAPDH monoclonal antibody (#5174, Cell Signaling, USA), rabbit anti‐CDK4 monoclonal antibody (ab108357, Abcam, USA), rabbit anti‐cyclin D1 monoclonal antibody (#2978, Cell Signaling, USA), mouse anti-cyclin E1 monoclonal antibody (#4129, Cell Signaling, USA), mouse anti-pRB (#9308, Cell Signaling, USA) polyclonal antibody, mouse anti-ANLN monoclonal antibody (sc271814, Santa Cruz Biotechnology, USA), rabbit anti-MMP7 monoclonal antibody (ab205525, Abcam, USA) and rabbit anti-MMP9 monoclonal antibody (ab137867, Abcam, USA). Secondary anti-mouse (A0216, Beyotime, China) and anti-rabbit (A0208, Beyotime, China) antibodies were used to visualize proteins with an ECL Western blot analysis system (Pierce, USA).
Senescence‐Associated β‐Galactosidase Staining
Senescence‐associated β‐galactosidase staining (SA‐β‐gal) of cells was performed utilizing a Senescence β-Galactosidase Staining kit (Beyotime, China). The cells were washed with PBS and fixed. The fixed cells were incubated with fresh β‐galactosidase staining solution at pH 5.8 and then were incubated at 37°C for at least 12 hrs. SA‐β‐galactosidase‐positive cells were detected by inverted bright field microscopy (Nikon, Japan) at 100× magnification.
Statistical Analysis
Data are presented as the mean ± SD from at least three separate experiments. Two‐tailed Student’s t tests were used to analyze the difference between the control group and the experimental group, and a P value of less than 0.05 was considered statistically significant.
Results
Construction and Identification of A549 Cells Overexpressing Progerin
A recombinant plasmid overexpressing progerin was fused with protein flag to label the protein of interest by gene cloning, and the plasmid was packaged into a lentiviral vector that was GFP-positive. Efficient gene delivery of progerin in A549-PG cells was assessed by fluorescence microscopy and Western blot. A549-GFP cells were transduced by a lentivirus containing only GFP as a control. The efficiency of the transfection was detected by fluorescence microscopy and was found to be more than 80% (Figure 1A), and the progerin level showed a significant increase in A549-PG cells compared with that of the A549-GFP cells (Figure 1B and C).
 |
Figure 1 Construction and characterization of A549-GFP and A549-PG cells. (A) Efficient lentivirus delivery in A549-PG and A549-GFP cells was detected by fluorescence microscopy. (B and C) The protein level of progerin in A549-PG cells was significantly increased compared with that in A549-GFP cells. **P<0.01, and ***P<0.001. |
Overexpression of Progerin Significantly Increased Abnormal Nuclear Morphology in A549 Cells
Due to the lack of a cleavage site, progerin remains permanently anchored to the membrane, causing blebbing of the nucleus in HGPS patients.13,14 To determine whether similar morphological changes occurred in the nuclei of A549-PG cells, a DAPI nuclear staining assay was applied. The nuclei of A549-GFP cells showed normal nuclear morphology, while A549-PG cells showed obvious nuclear blebbing, and the nuclear membrane was not smooth (Figure 2A). Data analysis revealed that approximately 20 ± 1.1% of A549-PG cells contained abnormal nuclei (Figure 2B). The above results indicated that the accumulation of progerin in A549 cells could cause the same abnormal nuclear morphology that was observed HGPS patients, but the proportion was lower.
 |
Figure 2 Overexpression of progerin caused abnormal nuclear morphology in A549 cells. (A) DAPI nuclear staining assay revealed that progerin significantly increased the number of A549-PG cells with abnormal nuclear morphology (arrows point to the abnormal nucleus). (B) The percentage of abnormal nuclei in A549-PG cells was approximately 20 ± 1.1% greater than that in A549-GFP cells. **p<0.01. Scale bar, 25 μm. |
Progerin Decreased Cell Proliferation and Caused G1/S Arrest Through the CDK4-pRB Pathway in A549-PG Cells
During cell culture, we found that the growth of A549-PG cells was slower than that of A549-GFP cells. Subsequently, we designed experiments to prove this phenomenon. Cloning formation experiments and EdU assays were employed to compare the proliferation ability of A549-PG and A549-GFP cells. As shown in Figure 3A and B, the cloning formation rate of A549-PG was dramatically decreased compared with that of the control. Cell proliferation was slowed by progerin, as shown by the EdU assay (Figure 3C and D). Flow cytometry indicated that A549-PG cells exhibited G1-S phase arrest; 49.6 ± 1.19% of the cells were in G1 phase, which was an increase over that of the A549-GFP cells (43.5 ± 1.24%). Further, 37.3 ± 1.37% of A549-PG cells in S phase, compared to 43.4 ± 1.31% of A549-GFP cells (Figure 4A).
 |
Figure 3 Progerin weakened the proliferation ability of A549 cells. (A and B) Cloning formation experiments demonstrated that proliferation was decreased by progerin in A549-PG cells. (C and D) EdU assays further proved that the proliferative capacity of A549-PG cells was impaired compared with that of the control group. *p<0.05. Scale bar, 100 μm. |
 |
Figure 4 Progerin impaired proliferative capacity through the CDK4-pRB pathway in A549 cells. (A) Flow cytometry showed that the G1-S phase was arrested by progerin in A549-PG cells (B and C). Western blot analysis indicated that the levels of CDK4, cyclin D1, and pRB in A549-PG cells were dramatically downregulated compared with those in A549-GFP cells (D and E). The p53 signaling pathway was not changed. *p<0.05, and **P<0.01. |
To probe into the underlying molecular mechanisms by which progerin modulates the cell cycle, Western blot was performed in A549-PG and A549-GFP The results revealed that progerin caused significant downregulation of cell cycle-associated proteins. CDK4, cyclin D1 and pRB were downregulated in A549-PG cells (Figure 4B and 4C), but cyclin E1 did not change, suggesting that progerin may cause G1/S arrest in A549 tumor cells through the CDK4-pRB pathway. The accumulation of the DNA damage product γ-H2AX is one of the hallmarks of senescence.15,16 Double-strand DNA breaks (DSBs) are powerful activators of DNA damage repair and can induce cell cycle arrest to repair damage; however, they can lead to cellular senescence when unresolved.17,18 Progerin-induced replication stress facilitates premature senescence in HGPS.19,20 Cells from HGPS patients exhibited activated p53 signaling due to the accumulation of γ-H2AX,21 so we hypothesized that γ-H2AX initiated DNA repair and caused cell cycle arrest in A549-PG cells.
As the results indicated, total p53, phosphorylated p53 and γ-H2AX levels were not any changed in A549-PG cells relative to the levels in A549-GFP cells (Figure 4D and E). These results indicated that progerin caused cell cycle arrest and impaired proliferation by downregulating the CDK4-pRB signaling pathway, but such outcomes did not occur through the p53 signaling pathway.
A549 Cells Overexpressing Progerin Exhibited Increased Sensitivity to DNA Damaging Agents
The accumulation of DNA double strand breaks is found in fibroblasts from HGPS patients, suggesting that progerin disrupts the mechanism of DNA damage repair.22,23 The main treatment for cancer is chemotherapy; this approach primarily aims to induce extensive DNA damage to kill rapidly dividing tumor cells.24 Therefore, we determined whether A549-PG could cause more DNA damage through the overexpressing progerin than A549-GFP when faced with the same dose of a DNA damage inducer. Hydrogen peroxide (H2O2) can lead to increased intracellular reactive oxygen species, mimicking in vivo oxidative stress.25,26 Therefore, we utilized H2O2 to induce cellular oxidative stress for 1 or 2 hrs. The results showed that A549-PG cells treated with 400 μM H2O2 over different time periods were more sensitive to DNA damaging agents and produced more γ-H2AX than A549-GFP cells (Figure 5A and B). Cisplatin is commonly used in the clinic as a cancer chemotherapy.24,27,28 Treatment of cells with 50 μM cisplatin produced results similar to those observed following treatment with H2O2 (Figure 5C and D). We further explored the relevant molecular mechanisms, and the upstream regulatory signaling molecules of γ-H2AX, such as DNA-PKcs and ATM, were both activated (Figure 5E and F). Further analysis of A549-PG cells treated with H2O2 for one hour conclusively showed that progerin not only caused an increase in DSBs but also activated the p53 signaling pathway; further, the cell cycle-associated proteins CDK4 and pRB were downregulated. We concluded that A549-PG cells were more sensitive to DNA damaging agents, and they accumulated a substantial amount of γ-H2AX via the activated upstream kinases ATM and DNA-PKcs; additionally, p53 was the downstream signal of γ-H2AX, which activated the CDK4-pRB pathway, giving rise to cell cycle arrest.
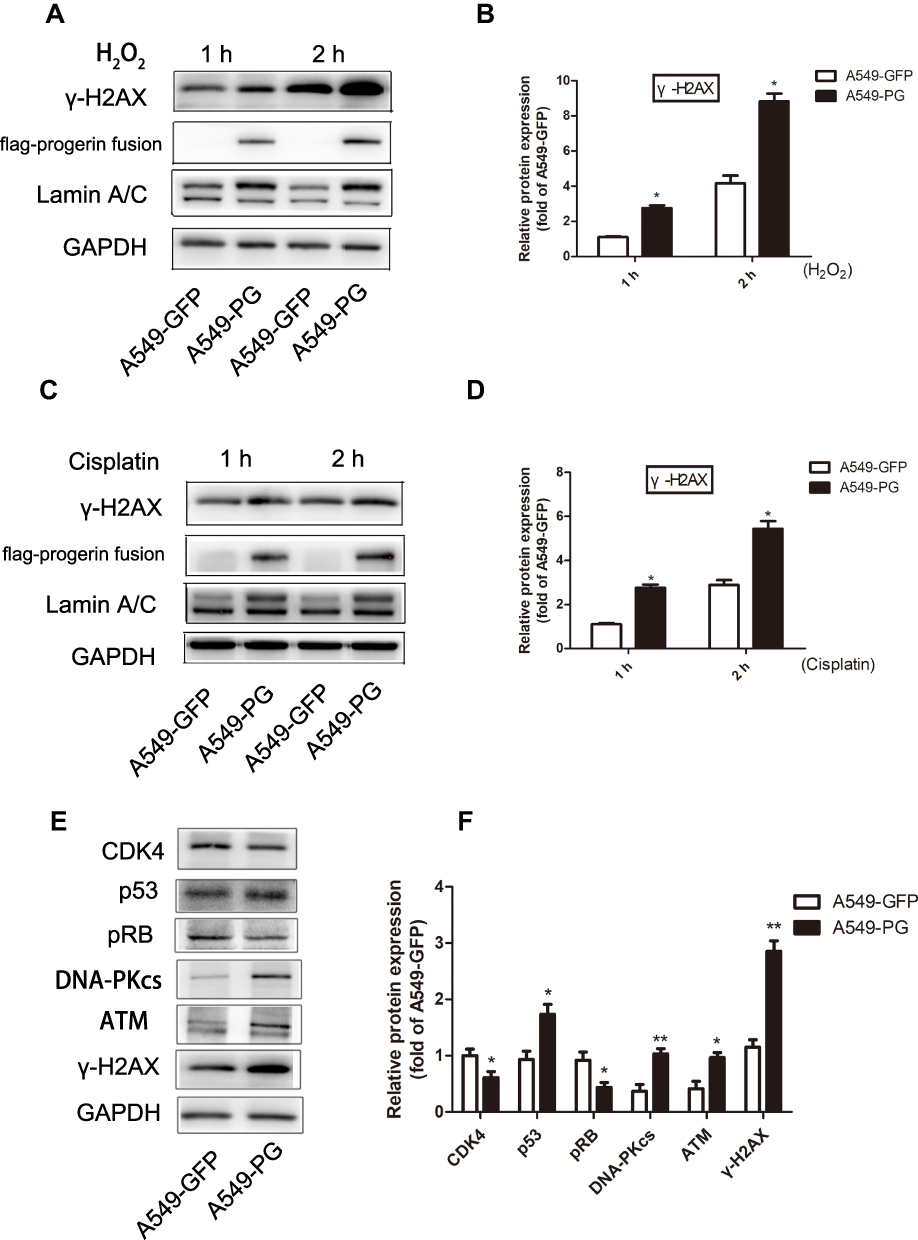 |
Figure 5 A549-PG cells exhibited increased sensitivity to DNA damage. (A and B) A549-PG cells treated with hydrogen peroxide for different times produced more γ-H2AX than A549-GFP cells did. (C and D) DNA damage was observed in A549 cells treated with cisplatin for 1 hr and 2 hrs. (E and F) 1 hr H2O2 treatment was used to observe the effect on expression of upstream kinases and cell cycle-related proteins in A549-PG and A549-GFP cells. *p<0.05, and **P<0.01. |
Progerin Significantly Impaired the Migration and Invasion Abilities of A549 Cells
Abundant evidence points to a crucial physiological role for cellular senescence in combating tumorigenesis. Patricia Fernandez performed a study with primary dermal fibroblasts from HGPS patients and normal age-matched individuals. The results showed that the presence of progerin makes it impossible for HGPS cells to trigger tumorigenesis because it prevents their reprogramming and thus resists carcinogenic factors.29 To further investigate the effects of progerin on A549 cells, wound migration experiments and transwell assays were performed, which are commonly used to explore the major features of migration and invasion in malignant tumors. As our results showed, both migration (Figure 6A–D) and invasion (Figure 6E and F) abilities were significantly downregulated in A549-PG cells relative to that of A549-GFP cells. To clarify the mechanism responsible for this phenomenon, related proteins involved in the pathway were validated. It is well known that matrix metalloproteinase families (MMPs) are essential for tumor invasion. As shown in Figure 6G, MMP7 and MMP9 were dramatically downregulated in A549-PG cells compared with their levels in A549-GFP cells. Further studies have found that the ANLN gene is significantly reduced in A549-PG cells (Figure 6G and H). The ANLN gene encodes a microfilament-binding protein that functions in cell growth, migration, and cell division.30 The results implied that progerin impaired migration and invasion in A549-PG cells by affecting ANLN, MMP7 and MMP9 proteins.
 |
Figure 6 Progerin significantly impaired the migration and invasion abilities of A549 cells. (A and B) The migration ability of A549 cells was evaluated by wound migration experiments. (C and D) Transwell assay of A549 cells. (E and F) Progerin impaired the invasive ability in A549-PG. (G and H) Representative Western blot analysis of relevant proteins showing that progerin decreased ANLN, MMP7 and MMP9 levels in A549-PG cells. *p<0.05, and **p<0.01. |
The Overexpression of Progerin Could Not Cause Senescence or Apoptosis in A549-PG Cells
Progerin is a major cause of accelerated cell aging in HGPS patients, so we examined the phenotype of senescence and the expression of related proteins in A549 tumor cells that were exposed to 10 μM cisplatin for 2 days. As our results showed, there was no difference in SA-β-gal staining (Figure 7A), and almost no cells were stained for either A549-PG or A549-GFP. Related senescent proteins, such as p16 and p21, were not significantly different from PARP1, which is an important protein in the process of apoptosis (Figure 7B and C). These results indicated that overexpression of progerin could not cause senescence and apoptosis in A549-PG cells.
 |
Figure 7 A549-PG showed no differences in cell senescence and apoptosis compared with A549-GFP cells. (A) SA‐β‐gal staining of A549-PG and A549-GFP cells, which were exposed to cisplatin for 2 days. (B and C) Proteins such as p21, p16, and PARP1 were not changed in A549 cells. |
Discussion
Lung cancer is one of the most common malignant cancers in the world, and it has high rates of recurrence and metastasis, which causes extremely high morbidity and mortality. Non-small cell lung cancer accounts for nearly 80% of lung carcinomas.31 Chemotherapy is the main method used to treat non-small cell lung cancer, but the treatment results are not ideal due to high side effects; the current situation urgently requires researchers to explore new treatments.32
In recent years, increasing evidence has demonstrated that senescence plays a key role in the physiological development of tumors.33 The accumulation of progerin is responsible for the pathogenesis of HGPS, but it is not known what function progerin would perform in tumor cells. To address this unknown, progerin was induced in A549 cells in our experiments. The results revealed that progerin accumulation in A549-PG cells exhibited a phenotype similar to that of fibroblasts from HGPS patients, and the nuclei of A549-PG cells showed abnormal nuclear morphology with obvious blebbing and an irregular nuclear membrane (Figure 2A). Data analysis revealed that approximately 20 ± 1.1% of A549-PG cells showed abnormal nuclei (Figure 2B). The above results indicated that progerin in A549 cells caused the same abnormal nuclear morphology that was observed in the cells of patients with premature aging, revealing that progerin was successfully introduced into A549-PG cells and affected their nuclear morphology.
Studies have shown that mature lamin A is closely related to various proteins in the nucleus; it regulates normal nuclear morphology, epigenetic modification, and intranuclear and extranuclear signals, and it maintains cellular homeostasis and normal cellular function.34,35 Prelamin A and progerin, however, fail to perform these functions and alter normal physiological processes, ultimately leading to a series of imbalances in cellular homeostasis. Our results suggested that overexpression of progerin in A549 cells decreased cell proliferation and caused G1/S arrest through the CDK4-pRB pathway (Figure 3). Western blot analysis indicated that the protein levels of CDK 4, cyclin D1, and pRB in A549-PG cells were dramatically downregulated compared with those in A549-GFP cells, but the p53 signaling pathway was not changed (Figure 4). Normal nuclear lamins form the major components of the nuclear lamina and participate in genome maintenance by providing a scaffold for tethering chromatin and protein complexes that regulate genome stability,36,37 progerin has no such functions, and it may disrupt the transcription of normal genes, such as CDK4. We believe that the accumulation of progerin in the nuclear fiber layer may disrupt the transcription of CDK4 and other cycle-related genes, ultimately impairing cell proliferation. In addition, lung cancer cells themselves exhibit p53 inactivation, but changes in p53 were not be obvious in A549-PG cells.
Chemotherapy is one of the essential methods used to inhibit the growth of tumors, among which cisplatin is a first-line drug for various kinds of tumors. Cisplatin is designed to cause extensive DNA damage in tumor cells to kill them. Generally, the more sensitive tumor cells are to cisplatin, the better the effect of chemotherapy. Therefore, we utilized cisplatin and H2O2 to induce cellular oxidative stress. The results showed that A549-PG cells treated with cisplatin or H2O2 for different amounts of time were more sensitive to DNA damaging agents and produced more γ-H2AX than A549-GFP cells were (Figure 5). The upstream kinases ATM and DNA-PKcs and the downstream protein p53 were significantly activated, which ultimately caused cell cycle arrest. The results revealed that H2O2 caused DNA damage by activating the ATM/DNA-PKcs-p53 pathway, giving rise to cell death and cell cycle arrest. Therefore, we concluded that progerin could increase the sensitivity of tumor cells to DNA damaging agents and improve the efficiency of chemotherapy in A549 cells.
Invasion and migration are two major characteristics of tumor cells that determine the degree of tumor malignancy and the effect of chemotherapy. Therefore, experiments were designed to analyze the effects of progerin on migration and invasion. The results implied that progerin influenced both the migration and invasion abilities of A549-PG cells by affecting the ANLN, MMP7 and MMP9 proteins (Figure 6G). MMP7 is a matrix metalloproteinases that is closely related to the growth and spread of tumors, and MMP9 is a gelatinase that is related to the metastasis of lung cancer.38 Research also found that the accumulation of prelamin A reduced invasion in cancer cell lines such as breast cancer, oral cancer and lung cancer.5 Others discovered that prelamin A may play a tumor protective role by altering the extracellular matrix; for instance, it makes changes in the proteoglycan synthesis pathway.29 Our results indicated that progerin impaired the migration and invasion abilities of A549-PG cells by downregulating the related proteins ANLN, MMP9 and MMP7.
Senescence has been shown to be markedly activated in fibroblasts from HGPS patients. However, there were no significant differences in either senescence or apoptosis in our experiments (Figure 7). We assume that normal lamin A/C also persisted in A549-PG cells since progerin was overexpressed in A549 cells, as opposed to knocking out normal lamin A/C. In addition, tumor cells themselves escape aging and can proliferate indefinitely, so it is difficult to induce aging in A549-PG cells.
We imagine future clinical applications based on our findings regarding the differences between A549 cells and normal cells, such as what was observed in the cell cycle. We propose that progerin could be introduced into tumor cells by gene targeting technology, or tumor cells could be directly induced to produce progerin to weaken the invasion and migration of tumor cells, thus achieving the purpose of treating lung cancer. Certainly, these ideas need further exploration and research.
In summary, we have developed a new cell model, A549-PG cells, by introducing progerin, and we compared these cells to control A549-GFP cells, which have normal lamin A expression. Our experiments demonstrated that, similar to HGPS cells, progerin caused disruptions of lung cancer A549 cells by causing abnormal nuclear shape, proliferation, tumor migration, invasion and increased sensitivity to chemotherapy drugs. It may be helpful to explore targeted gene therapy for lung cancer cells, but specific mechanisms and possible applications warrant further exploration. Other biological functions of A549-PG, such as migration and invasion, need to be explored because they could be regulated by the Wnt/β-catenin pathway as well. Last but not least, animal experiments are required to explore the in vivo effect of progerin on lung cancer development.
Acknowledgment
This work was supported by the grants from the National Natural Science Foundation of China (No.30900729, No.81671399, No.81971329) and the Ordinary University Innovation Team Construction Project of Guangdong Province (No.2015KCXTD022).
Author Contributions
All authors contributed toward data analysis, drafting and revising the paper, gave final approval of the version to be published and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Campisi J, D’adda DFF. Cellular senescence: when bad things happen to good cells. Nat Rev Mol Cell Biol. 2007;8(9):729–740. doi:10.1038/nrm2233
2. Hayflick L, Moorhead PS. The serial cultivation of human diploid cell strains. Exp Cell Res. 1961;25(3):585–621. doi:10.1016/0014-4827(61)90192-6
3. Acosta JC, Gil J. Senescence: a new weapon for cancer therapy. Trends Cell Biol. 2012;22(4):211–219. doi:10.1016/j.tcb.2011.11.006
4. Nardella C, Clohessy JG, Alimonti A, et al. Pro-senescence therapy for cancer treatment. Nat Rev Cancer. 2011;11(7):503–511. doi:10.1038/nrc3057
5. de la Rosa J, Freije JM, Cabanillas R, et al. Prelamin A causes progeria through cell-extrinsic mechanisms and prevents cancer invasion. Nat Commun. 2013;4(1):2268. doi:10.1038/ncomms3268
6. Xue W, Zender L, Miething C, et al. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature. 2007;445(7128):656–660. doi:10.1038/nature05529
7. Merideth MA, Gordon LB, Clauss S, et al. Phenotype and course of Hutchinson-Gilford progeria syndrome. N Engl J Med. 2008;358(6):592–604. doi:10.1056/NEJMoa0706898
8. Vidak S, Foisner R. Molecular insights into the premature aging disease progeria. Histochem Cell Biol. 2016;145(4):401–417. doi:10.1007/s00418-016-1411-1
9. Lopez-otin C, Blasco MA, Partridge L, et al. The hallmarks of aging. Cell. 2013;153(6):1194–1217. doi:10.1016/j.cell.2013.05.039
10. Ahmed MS, Ikram S, Bibi N, et al. Hutchinson-Gilford progeria syndrome: a premature aging disease. Mol Neurobiol. 2018;55:4417–4427. doi:10.1007/s12035-017-0610-7
11. Pallis AG, Serfass L, Dziadziusko R, et al. Targeted therapies in the treatment of advanced/metastatic NSCLC. Eur J Cancer. 2009;45(14):2473–2487. doi:10.1016/j.ejca.2009.06.005
12. Doroshow DB, Herbst RS. Treatment of advanced non-small cell lung cancer in 2018. JAMA Oncol. 2018;4(4):569–570. doi:10.1001/jamaoncol.2017.5190
13. Goldman RD, Shumaker DK, Erdos MR, et al. Accumulation of mutant lamin A causes progressive changes in nuclear architecture in Hutchinson-Gilford progeria syndrome. Proc Natl Acad Sci U S A. 2004;101(24):8963–8968. doi:10.1073/pnas.0402943101
14. Capell BC, Collins FS. Human laminopathies: nuclei gone genetically awry. Nat Rev Genet. 2006;7(12):940–952. doi:10.1038/nrg1906
15. Shiloh Y. The ATM-mediated DNA-damage response: taking shape. Trends Biochem Sci. 2006;31(7):402–410. doi:10.1016/j.tibs.2006.05.004
16. Zou L. Single- and double-stranded DNA: building a trigger of ATR-mediated DNA damage response. Genes Dev. 2007;21(8):879–885. doi:10.1101/gad.1550307
17. Ciccia A, Elledge SJ. The DNA damage response: making it safe to play with knives. Mol Cell. 2010;40(2):179–204. doi:10.1016/j.molcel.2010.09.019
18. Niedernhofer LJ, Gurkar AU, Wang Y, et al. Nuclear genomic instability and aging. Annu Rev Biochem. 2018;87:295–322. doi:10.1146/annurev-biochem-062917-012239
19. Wheaton K, Campuzano D, Ma W, et al. Progerin-induced replication stress facilitates premature senescence in hutchinson-gilford progeria syndrome. Mol Cell Biol. 2017;37.
20. Hilton BA, Liu J, Cartwright BM, et al. Progerin sequestration of PCNA promotes replication fork collapse and mislocalization of XPA in laminopathy-related progeroid syndromes. FASEB J. 2017;31(9):3882–3893. doi:10.1096/fsb2.v31.9
21. von Muhlinen N, Horikawa I, Alam F, et al. p53 isoforms regulate premature aging in human cells. Oncogene. 2018;37(18):2379–2393. doi:10.1038/s41388-017-0101-3
22. Gonzalo S, Kreienkamp R. DNA repair defects and genome instability in Hutchinson-Gilford progeria syndrome. Curr Opin Cell Biol. 2015;34:75–83. doi:10.1016/j.ceb.2015.05.007
23. Musich PR, Zou Y. Genomic instability and DNA damage responses in progeria arising from defective maturation of prelamin A. Aging (Albany NY). 2009;1(1):28–37. doi:10.18632/aging.v1i1
24. Chabner BA, Roberts TJ. Timeline: chemotherapy and the war on cancer. Nat Rev Cancer. 2005;5(1):65–72. doi:10.1038/nrc1529
25. Murphy E, Friedman AJ. Hydrogen peroxide and cutaneous biology: translational applications, benefits, and risks. J Am Acad Dermatol. 2019;81(6):1379–1386. doi:10.1016/j.jaad.2019.05.030
26. Lismont C, Revenco I, Fransen M. Peroxisomal hydrogen peroxide metabolism and signaling in health and disease. Int J Mol Sci. 2019;20.
27. Cepeda V, Fuertes MA, Castilla J, et al. Biochemical mechanisms of cisplatin cytotoxicity. Anticancer Agents Med Chem. 2007;7(1):3–18. doi:10.2174/187152007779314044
28. Miyagawa K. Clinical relevance of the homologous recombination machinery in cancer therapy. Cancer Sci. 2008;99(2):187–194. doi:10.1111/cas.2008.99.issue-2
29. Fernandez P, Scaffidi P, Markert E, et al. Transformation resistance in a premature aging disorder identifies a tumor-protective function of BRD4. Cell Rep. 2014;9(1):248–260. doi:10.1016/j.celrep.2014.08.069
30. Piekny AJ, Glotzer M. Anillin is a scaffold protein that links RhoA, actin, and myosin during cytokinesis. Curr Biol. 2008;18(1):30–36. doi:10.1016/j.cub.2007.11.068
31. Greenlee RT, Hill-harmon MB, Murray T, et al. Cancer statistics, 2001. CA Cancer J Clin. 2001;51(1):15–36. doi:10.3322/canjclin.51.1.15
32. Cortes-dericks L, Galetta D. The therapeutic potential of mesenchymal stem cells in lung cancer: benefits, risks and challenges. Cell Oncol (Dordr). 2019:1–2.
33. Collado M, Serrano M. Senescence in tumours: evidence from mice and humans. Nat Rev Cancer. 2010;10(1):51–57. doi:10.1038/nrc2772
34. Houben F, Ramaekers FC, Snoeckx LH, et al. Role of nuclear lamina-cytoskeleton interactions in the maintenance of cellular strength. Biochim Biophys Acta. 2007;1773(5):675–686. doi:10.1016/j.bbamcr.2006.09.018
35. Dechat T, Pfleghaar K, Sengupta K, et al. Nuclear lamins: major factors in the structural organization and function of the nucleus and chromatin. Genes Dev. 2008;22(7):832–853. doi:10.1101/gad.1652708
36. Liu B, Wang J, Chan KM, et al. Genomic instability in laminopathy-based premature aging. Nat Med. 2005;11(7):780–785. doi:10.1038/nm1266
37. Gonzalez-suarez I, Redwood AB, Perkins SM, et al. Novel roles for A-type lamins in telomere biology and the DNA damage response pathway. EMBO J. 2009;28(16):2414–2427. doi:10.1038/emboj.2009.196
38. Merchant N, Nagaraju GP, Rajitha B, et al. Matrix metalloproteinases: their functional role in lung cancer. Carcinogenesis. 2017;38(8):766–780. doi:10.1093/carcin/bgx063
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.