Back to Journals » OncoTargets and Therapy » Volume 8
Overexpression of miR-100 inhibits cell proliferation, migration, and chemosensitivity in human glioblastoma through FGFR3
Authors Luan Y, Zhang S, Zuo L, Zhou L
Received 29 March 2015
Accepted for publication 30 April 2015
Published 17 November 2015 Volume 2015:8 Pages 3391—3400
DOI https://doi.org/10.2147/OTT.S85677
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Faris Farassati
Yongxin Luan,1 Shuyan Zhang,1 Ling Zuo,2 Lixiang Zhou1
1Department of Neurosurgery, First Bethune Hospital of Jilin University, 2Department of Ophthalmology, Second Bethune Hospital of Jilin University, Changchun, People’s Republic of China
Background: Glioblastoma multiforme is one of the most deadly forms of brain cancer. We investigated the regulatory effects of microRNA-100 (miR-100) on cell proliferation, migration, and chemosensitivity in human glioblastoma.
Methods: miR-100 expression was assessed by quantitative real-time polymerase chain reaction in both glioblastoma cells and human tumors. Lentiviruses of miR-100 mimics and inhibitors were transfected into U251 and T98G cells. The regulatory effects of either overexpressing or downregulating miR-100 on glioblastoma were evaluated by a viability assay, growth assay, migration assay, chemosensitivity assay, and an in vivo tumor transplantation assay. Expression of fibroblast growth factor receptor 3 (FGFR3), the bioinformatically predicted target of miR-100, was examined by Western blot in glioblastoma. FGFR3 was then ectopically overexpressed in U251 and T98G cells, and its effects on miR-100-mediated cancer regulation were evaluated by growth, migration, and chemosensitivity assays.
Results: MiR-100 was markedly downregulated in both glioblastoma cell lines and human tumors. Overexpressing miR-100 through lentiviral transfection in U251 and T98G cells significantly inhibited cancer growth (both in vitro and in vivo) and migration and increased chemosensitivity to cisplatin and 1, 3-bis (2-chloroethyl)-l-nitrosourea, whereas downregulation of miR-100 had no effects on development of cancer. FGFR3 was directly regulated by miR-100 in glioblastoma. Ectopically overexpressing FGFR3 was able to ameliorate the anticancer effects of upregulation of miR-100 on glioblastoma growth, migration, and chemosensitivity.
Conclusion: MiR-100 was generally downregulated in glioblastoma. Overexpressing miR-100 had anticancer effects on glioblastoma, likely through regulation of FGFR3. The MiR-100/FGFR3 signaling pathway might be a biochemical target for treatment in patients with glioblastoma.
Keywords: glioblastoma, miR-100, fibroblast growth factor receptor 3, cisplatin
Introduction
Glioblastoma multiforme is one of the most deadly forms of brain cancer.1 Every year, more than 15,000 new patients are diagnosed with glioblastoma, and most have a very poor prognosis, with median survival predicted to be 15 months or less.1,2 Although great strides had been made in providing patients with the best available treatments, including advanced surgery, radiotherapy or immunotherapy, and combinational or targeted chemotherapy,3–6 the underlying neoplastic characteristics of glioblastoma, including resistance to apoptosis or chemotherapy, as well as rapid metastasis to the surrounding brain tissues, are major obstacles to overcome in order to achieve a better prognosis or survival in patients with glioblastoma.6,7 Thus, it is critical to understand the underlying molecular mechanisms involved in growth, metastasis, or chemosensitivity in glioblastoma.
MicroRNAs (miRNAs) are a group of noncoding, short-sequence RNA molecules that posttranscriptionally suppress gene expression by binding to the 3'-untranslated region (UTR) of the targeted genes to induce gene or protein degradation.8,9 In recent decades, miRNAs have been shown to be involved in almost every aspect of cancer regulation, including cell proliferation, apoptosis, metastasis, and chemosensitivity.10–13 Among the many cancer-regulating miRNAs, microRNA-100 (miR-100) has been shown to be a critical factor in the development, diagnosis, and treatment of cancer.14 In glioblastoma, miR-100 has been shown to be differentially expressed in radiosensitive and radioresistant glioblastoma cells.15 However, only limited functions of miR-100 in regulating human glioblastoma have been revealed.16
In this work, we systemically evaluated miR-100 expression levels in glioblastoma cell lines and human glioblastoma tumors. We then examined the regulatory mechanisms of miR-100 in glioblastoma, including cancer growth (both in vitro and in vivo), migration, and sensitivity to chemotherapy reagents, by either upregulating or downregulating miR-100 using lentiviral infection in glioblastoma cell lines. Moreover, we investigated the possible downstream target of miR-100 in glioblastoma, by assessing the expression profile of fibroblast growth factor receptor 3 (FGFR3), as well as the effects of ectopically upregulating FGFR3 in glioblastoma. The results of our work would help to elaborate the regulatory mechanisms of miRNA in human glioblastoma.
Materials and methods
Cell culture
Seven commonly used human glioblastoma cell lines or glioblastoma stem cell lines (U251, U89, LN229, U373, A172, HS682, and T98G) and two normal control cell lines (human astrocytes and HEK293T cells) were purchased from the Chinese Academy of Sciences Shanghai Branch, Shanghai Institute of Cell Biology, People’s Republic of China, for this study. All cells were maintained in Dulbecco’s Modified Eagle’s Medium (Invitrogen) supplemented with 10% fetal bovine serum (Invitrogen), 100 U/mL penicillin, and 100 mg/mL streptomycin, in a humidified tissue culture incubator with 95% O2/5% CO2 at 37°C. Fresh culturing medium was added every 3 or 4 days.
Patients
Tissue samples of glioblastoma as well as adjacent normal brain tissues were surgically isolated from patients during their surgical procedures at the Department of Neurosurgery, First Bethune Hospital of Jilin University in Changchun, People’s Republic of China. All clinical protocols were reviewed and approved by the Human Research and Ethics Committee at the First Bethune Hospital of Jilin University. All procedures were performed in accordance with state laws of the People’s Republic of China and commonwealth regulations of Jilin Province. All patients participating in the study signed written consent forms.
Quantitative real-time PCR
Total RNA was extracted from the glioblastoma cells and clinical tissue samples using TRIzol reagent (Invitrogen). To obtain cDNA, reverse transcription was performed with a kit from TaKaRa Biotechnology, and polymerase chain reaction (PCR) amplification was conducted using an ABI Prism 7500 cycler (Applied Biosystems). For mature miR-100 gene quantification, a Hairpin-it™ miRNA PCR quantitation kit (GenePharma) was used as per the manufacturer’s protocol. The ratios of real-time PCR results were calculated using the 2−ΔΔCT method with U6 transcripts as the internal control. For FGFR3 gene quantification, a SYBR Premix Ex Taq gene detection kit (TaKaRa Biotechnology) was used as per the manufacturer’s protocol and glyceraldehyde-3-phosphate dehydrogenase transcripts were used as the internal control.
Lentivirus transfection
To overexpress or downregulate miR-100, lentivirus of human mature miR-100 mimic (lv-miR-100), miR-100 inhibitor (lv-miR100-In), and a nonspecific control lentivirus (lv-miRC) were purchased from SunBio Medical Biotechnology. The glioblastoma cell lines were then transfected with lentiviruses by a Lipofectamine™ 2000 reagent (Invitrogen) as per the manufacturer’s protocol, followed by exchanging fresh culture medium at 24 hours.
Cell viability assay
U251 and T98G glioblastoma cells were cultured in 96-well plates (5×104 cells/well). Five days after lentiviral transfection, a CellTiter-Glo™ luminescence cell viability assay (Promega) was applied as per the manufacturer’s protocol to assess the health of the glioblastoma cells.
Growth assay
Twenty-four hours after transfection, the U251 and T98G cells were washed in free medium. The viable cells were resuspended, replated in 96-well plates (5×104 cells/well), and maintained for 5 consecutive days. On day 6, 3-(4,5-dimethylthazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT, 5 mg/mL) was added to the culture for 4 hours. The cells were then lysed with dimethyl sulfoxide. Cell growth was assessed by measuring relative optical densities at 570 nm for each well, then normalizing to the control condition.
Migration assay
Migration of human glioblastoma cells was assessed using a QCMi-FN quantitative cell migration assay (ECM500, Chemicon). First, transwell filters were coated with Matrigel (Invitrogen) for 1 hour. U251 and T98G cells were then cultured in the upper chamber (5×104 cells/well) with 200 μL of serum-free Dulbecco’s Modified Eagle’s Medium for 2 hours, followed by resuspension in 200 μL of L-Dulbecco’s Modified Eagle’s Medium for another hour. In the lower chamber, the migration medium was mixed with various treatment reagents, such as lentiviruses. The transwell was then cultured with 5% CO2 for 48 hours at 37°C, followed by immunostaining of the upper chamber (30 minutes, 10% acetic acid). Optical absorbance at 570–690 nm was measured using a VersaMax tunable microplate reader (Molecular Devices) for each well and normalized to the control.
Chemosensitivity assay
Human U251 and T98G glioblastoma cells were transfected with lentiviruses for 24 hours, followed by another 24 hours of culture in fresh medium. Chemotherapy reagents, including 1,3-bis (2-chloroethyl)-l-nitrosourea (BCNU, 25 μg/mL) and various concentrations of cisplatin (0, 1, 1.5, 2.5, and 5 μM) were then added to the culture for 3 days. Cell survival, represented as relative optical density at 570 nm, was estimated by viability assay and normalized to the optical density value under the control condition of 25 μg/mL BCNU plus 0 μM cisplatin.
In vivo assay
After lentiviral transfection for 24 hours, the T98G cells were resuspended in fresh culture medium (200 μL, 1 million cells) and subcutaneously inoculated into 2-month-old adult female athymic NCr (nu/nu) nude mice. Growth of the inoculated tumors was monitored weekly for 5 consecutive weeks. Tumor size was quantified as (length × width2)/2, and normalized to tumor size with lv-miRC transfection at week 1. At the end of the 5-week in vivo assay, the tumors were extracted and assessed by Ki67 immunohistochemistry.
Dual-luciferase reporter assays
The 3′-UTR of human FGFR3, including the possible binding site for hsa-miR-100, was cloned into a dual-luciferase reporter plasmid pMIR-REPORT (Ambion) to produce a wild-type FGFR3 reporter. A mutated sequence of FGFR3 3′-UTR at the hsa-miR-100 binding site was also cloned into the luciferase plasmid. In HEK293T cells, cotransfection of lv-miRC, lv-miR-100, and either wild-type FGFR3 or mutant FGFR3 was performed by Lipofectamine 2000 reagent, followed by examination of luciferase activity using a dual-luciferase reporter assay (Promega) in 24 hours.
Western blot assay
Human U251 and T98G glioblastoma cells were lysed in RIPA buffer (50 mM Tris-HCl, Invitrogen). The extracted proteins were electroseparated using 10% sodium dodecyl sulfate polyacrylamide gel electrophoresis, and then transferred to polyvinylidene difluoride membranes (Amersham Biosciences). The primary antibody against human FGFR3 (1:200, Santa Cruz Biotechnology) was applied overnight at 4°C. Horseradish peroxidase-linked secondary antibody was applied for 2 hours at room temperature. Glyceraldehyde-3-phosphate dehydrogenase was used as the internal control and blots were visualized by an enhanced chemiluminesence system (Amersham Biosciences) as per the manufacturer’s protocol.
FGFR3 overexpression assay
The DNA sequence of FGFR3 was amplified from the human cDNA library and cloned into a recombinant eukaryotic expression plasmid pcDNA3.1 (pcDNA3.1-FGFR3). A negative control, ie, an empty plasmid, was also generated. The U251 and T98G cells were then transfected with overexpression plasmids using Lipofectamine 2000 reagent for 24 hours. The efficiency of overexpression transfection was assessed by quantitative real-time PCR.
Statistical analysis
All data are expressed as the mean ± standard error of the mean. The statistical analysis was performed with the Student’s t-test using Statistical Package for the Social Sciences version 11.0 software. Statistical difference was marked at P<0.05. All experiments were performed in triplicate.
Results
MiR-100 was downregulated in glioblastoma cells and human tumors
The gene expression levels of miR-100 in seven human glioblastoma cell lines (U251, U89, LN229, U373, A172, HS683, and T98G) were compared against the expression level of miR-100 in normal human astrocytes. The results of quantitative real-time PCR demonstrated that miR-100 was significantly downregulated in all tested glioblastoma cells (Figure 1A; P<0.05). Next, we assessed the gene expression levels of miR-100 in human patients with glioblastoma. The extracted brain tissue included tumors and adjacent non-tumor brain tissues. We examined paired tumor samples of glioblastoma and adjacent normal brain tissue samples in 13 patients, and found that miR-100 was significantly downregulated in glioblastoma tumors when compared with non-tumor brain tissues in all patients (Figure 1B; P<0.05).
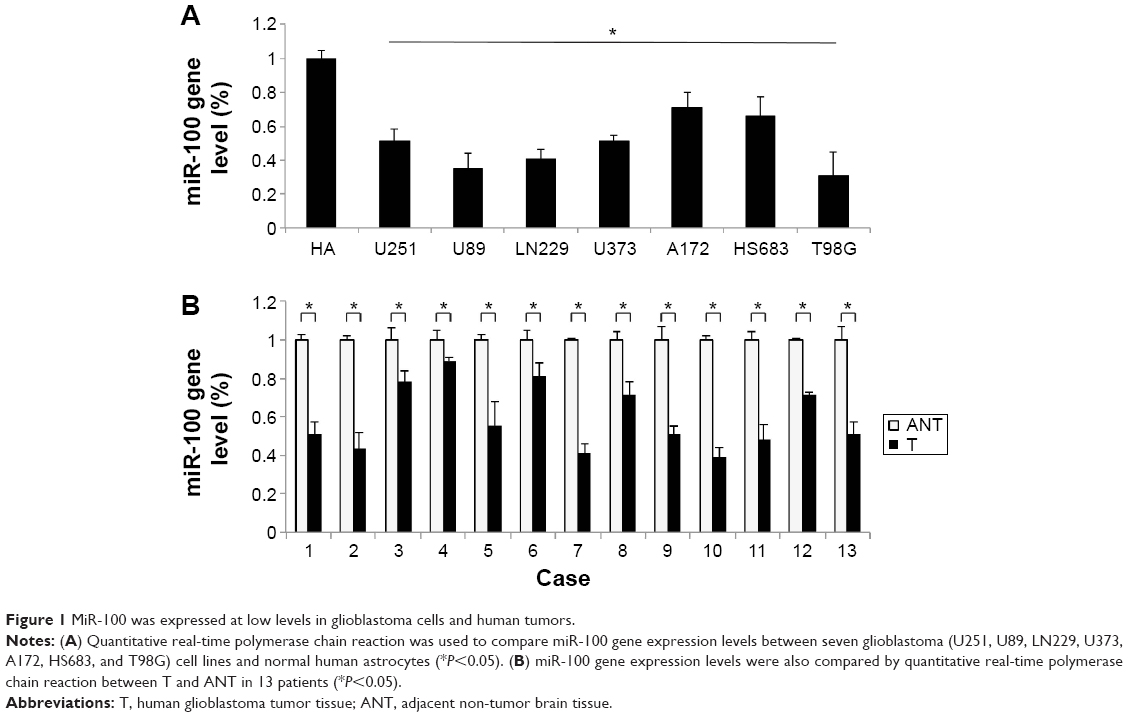 | Figure 1 MiR-100 was expressed at low levels in glioblastoma cells and human tumors. |
Ectopically overexpressing or downregulating miR-100 did not induce cytotoxicity in glioblastoma cells
Having discovered that miR-100 was downregulated in human glioblastoma cells, we then investigated its possible mechanistic role in regulation of glioblastoma. To do so, we used lentiviral vectors to ectopically manipulate the miR-100 gene expression level. We transfected human U251 and T98G glioblastoma cells with lentiviral vector containing miR-100 mimics to overexpress miR-100 (lv-miR-100) or lentiviral vector containing miR-100 inhibitor to downregulate miR-100 (lv-miR100-In). Quantitative real-time PCR demonstrated that, in both U251 and T98G cells, miR-100 gene expression levels were significantly upregulated by lv-miR-100 and significantly downregulated by lv-miR100-In when compared with miR-100 gene expression levels in cells transfected with a nonspecific control lentiviral vector (lv-miRC), as shown in Figure 2A and B (P<0.05 vs lv-miRC). We then examined whether ectopically manipulating the gene expression level of miR-100 would induce toxicity or apoptosis in glioblastoma cells. To do so, we transfected U251 and T98G cells with lv-miRC, lv-miR-100, and lv-miR100-In, and then performed a viability assay in 48 hours. This showed that overexpressing or downregulating miR-100 did not induce cytotoxicity in either cell line (Figure 2C and D; P>0.05 vs lv-miRC).
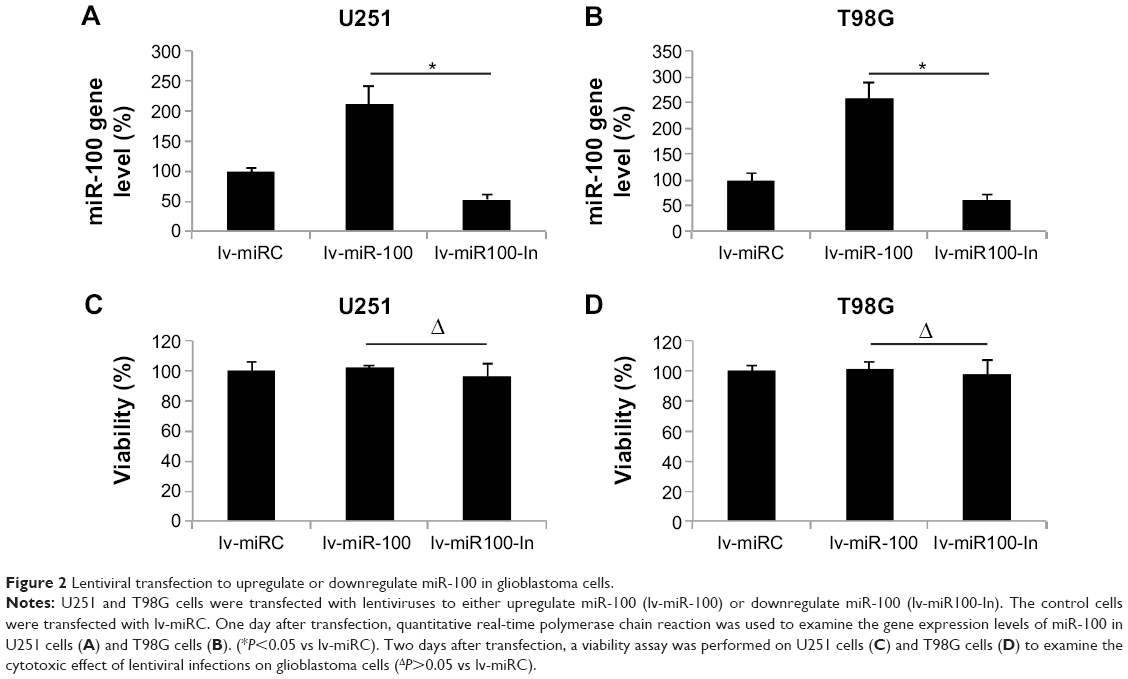 | Figure 2 Lentiviral transfection to upregulate or downregulate miR-100 in glioblastoma cells. |
MiR-100 overexpression inhibited cancer growth and migration of glioblastoma cells
We then investigated whether overexpressing or downregulating miR-100 affected growth and migration of glioblastoma cells. We transfected U251 and T98G cells with lv-miR-100 or lv-miR100-In to upregulate or downregulate miR-100. Transfection of lv-miRC as served as nonspecific control. Glioblastoma growth was then monitored by a growth assay for 5 days. The results showed that, in both U251 and T98G cells, overexpression of miR-100 (lentiviral transfection of lv-miR-100) significantly reduced cancer cell growth rates (Figure 3A and B; P<0.05 vs lv-miRC), whereas downregulation of miR-100 (lentiviral transfection of lv-miR100-In) had no effect on cell growth (Figure 3A and B; P>0.05 vs lv-miRC).
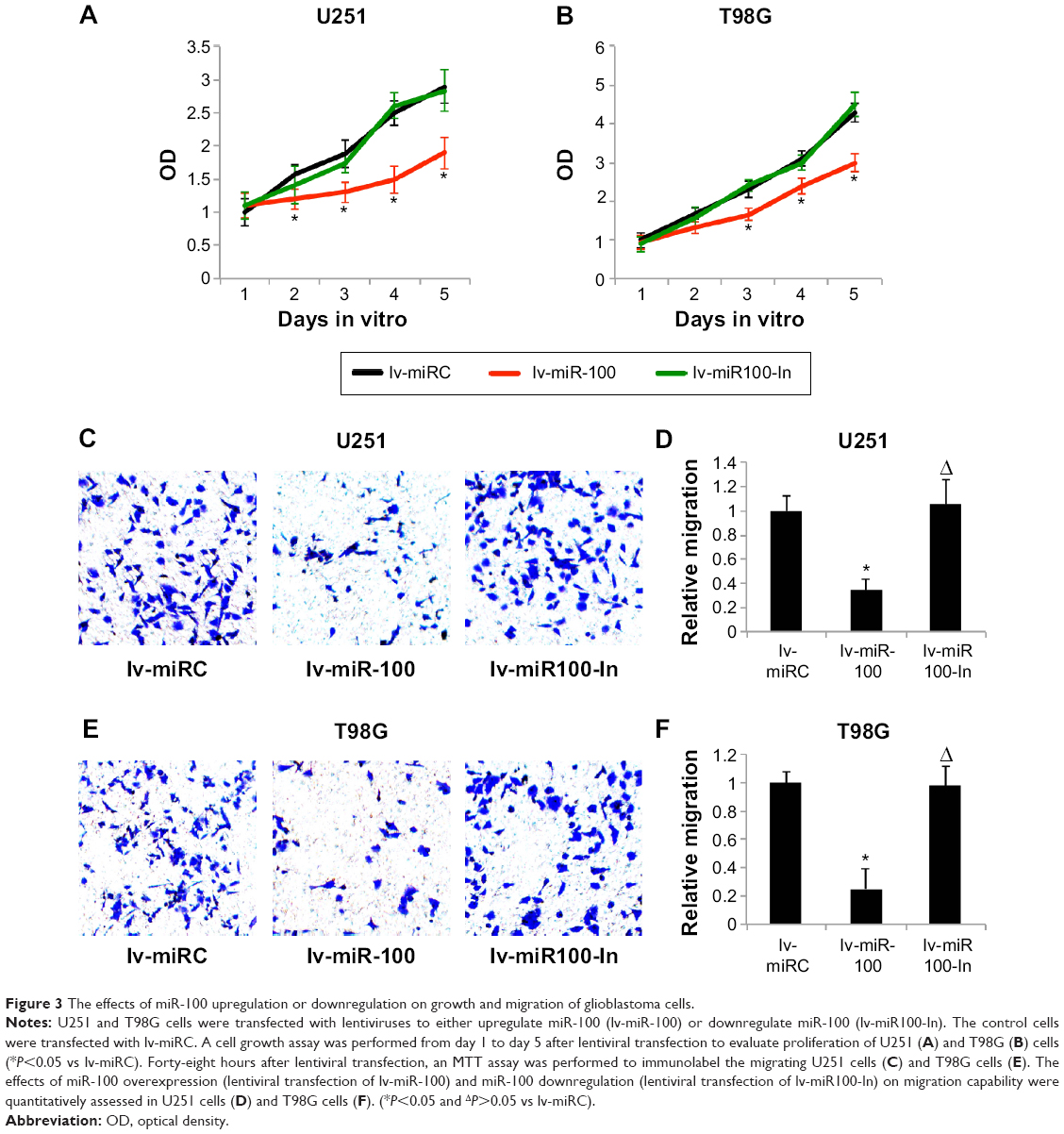 | Figure 3 The effects of miR-100 upregulation or downregulation on growth and migration of glioblastoma cells. |
We then investigated the effects of miR-100 upregulation or downregulation on migration of glioblastoma cells. The results of the MTT migration assay demonstrated that, in both U251 and T98G cells, overexpression of miR-100 significantly reduced the number of migrating (MTT-positive) cancer cells in the culture chambers, whereas downregulation of miR-100 had little effect on migration of cancer cells (Figure 3C and E). Quantitative measurement demonstrated that overexpression of miR-100 reduced migration capability by ~70% in U251 cells (Figure 3D; P<0.05) and ~80% in T98G cells (Figure 3F; P<0.05). On the other hand, downregulation of miR-100 did not alter migration capability in either cell line (Figure 3D and F; P>0.05).
Overexpression of MiR-100 increased chemosensitivity in glioblastoma cells
We then speculated if overexpressing or downregulating miR-100 might also affect the chemosensitivity of glioblastoma cells. U251 and T98G cells were transfected with lv-miR-100 to ectopically overexpress miR-100 or with lv-miR100-In to downregulate miR-100 for 24 hours. The control cells were transfected with lv-miRC. After washing out the lentiviruses with fresh culture medium, human glioblastoma cells were cultured for 24 hours, followed by another 24 hours of in vitro culture with chemoreagents, including 25 μg/mL BCNU and various concentrations of cisplatin (0, 1, 1.5, 2.5, and 5 μM). A growth assay was then used to assess the chemosensitivity of the cells over 72 hours. The results demonstrated that, in both U251 and T98G cells, overexpression of miR-100 significantly increased chemosensitivity in glioblastoma cells (Figure 4A and B; P<0.05 vs lv-miRC), whereas downregulation of miR-100 had no effect on glioblastoma chemosensitivity (Figure 4A and B, P>0.05 vs lv-miRC).
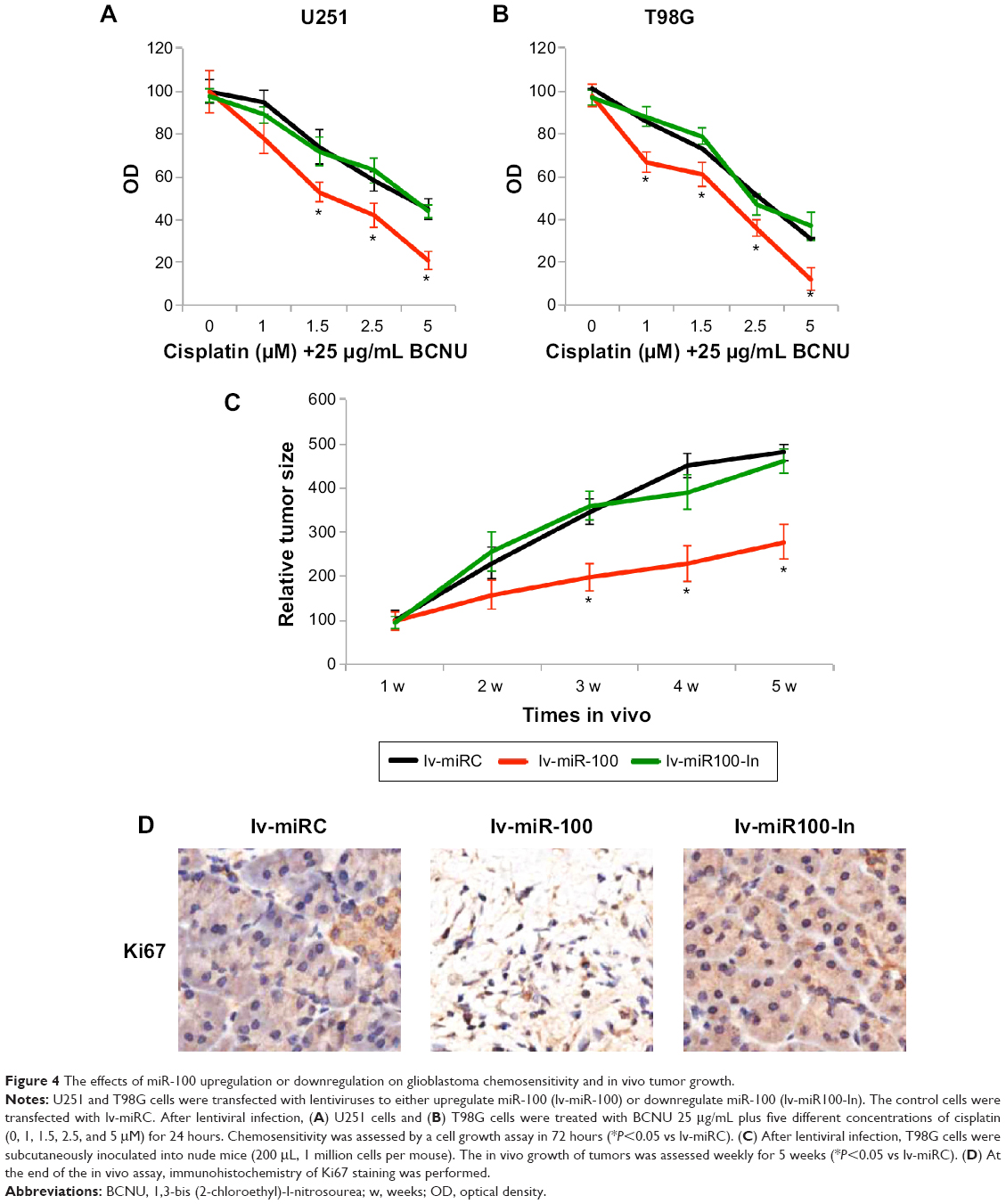 | Figure 4 The effects of miR-100 upregulation or downregulation on glioblastoma chemosensitivity and in vivo tumor growth. |
MiR-100 overexpression inhibited growth of glioblastoma tumors in vivo
We then investigated whether overexpressing or downregulating miR-100 affected growth of glioblastoma cells in vivo. To do so, we transfected T98G cells with lv-miR-100, lv-miR100-In, or lv-miRC for 24 hours. After changing with fresh medium, the viable cells were resuspended and dissociated with 0.25% trypsin to form a single cell suspension. Lentivirus-infected T98G cells (1 million per mouse) were then subcutaneously inoculated into female athymic NCr (nu/nu) nude mice. Tumor size was monitored for 5 weeks after inoculation, and quantitative measurements demonstrated that overexpression of miR-100 significantly inhibited glioblastoma tumor growth in vivo (Figure 4C; P<0.05), whereas downregulation of miR-100 had little effect on in vivo tumor growth (Figure 4C; P>0.05). Furthermore, at the end of the 5-week in vivo tumor growth assay, we extracted the tumor tissues from mice and immunolabelled them with Ki-67. The immunohistochemistry results showed less Ki-67 staining in tumor cells transfected with lv-miR-100 (Figure 4D), further confirming that overexpression of miR-100 inhibited growth of glioblastoma in vivo.
FGFR3 was the molecular target of miR-100 in glioblastoma
After discovering that miR-100 plays a critical role in regulating both in vitro and in vivo growth migration, and chemosensitivity of human glioblastoma cells, we sought to evaluate the possible molecular target of miR-100 in glioblastoma. We used online miRNA targeting software (TargetScan, www.targetscan.org) as well as a literature search to determine if FGFR3 was a possible candidate for direct regulation by miR-100 in human glioblastoma (Figure 5A). Using a dual-luciferase reporter assay in HEK293T cells, we found that wild-type FGFR3 (Figure 5B; P<0.05) but not mutant FGFR3 (Figure 5B; P>0.05) was differentially regulated by exogenously introduced miR-100, thus confirming that FGFR3 was directly regulated by miR-100. Moreover, we investigated whether direct regulation of miR-100 on FGFR3 was the case in glioblastoma cells. Western blotting assay results demonstrated that FGFR3 protein expression levels were significantly downregulated in U251 and T98G glioblastoma cells overexpressing miR-100 (Figure 5C and D).
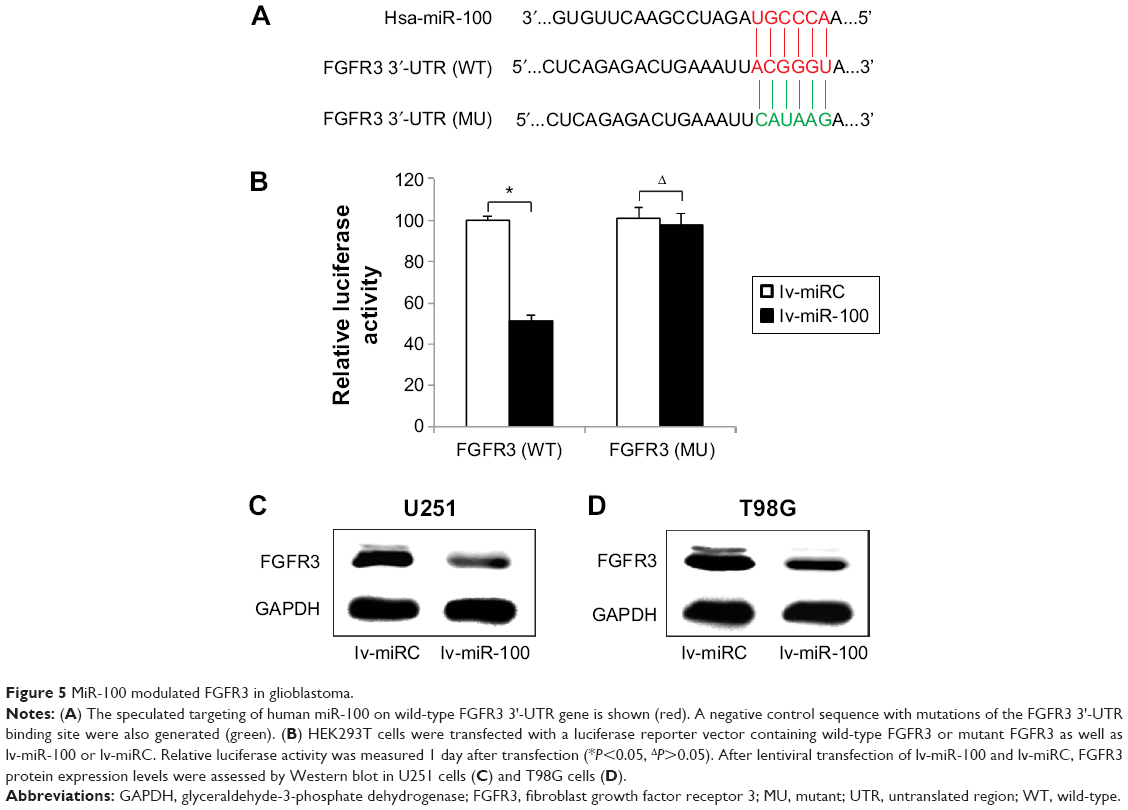 | Figure 5 MiR-100 modulated FGFR3 in glioblastoma. |
FGFR3 overexpression reversed the antitumor effects of miR-100 in glioblastoma cells
Finally, we investigated whether overexpressing FGFR3 might change the effects of miR-100 on the growth, migration, and chemosensitivity of human glioblastoma cells. We took advantage of a eukaryotic expression system to create a plasmid overexpressing FGFR3 (pcDNA3.1-FGFR3), as well as a nonspecific plasmid (pcDNA3.1-FGFR3) to be used as a control. After transfecting the plasmids into both U251 and T98G cells, quantitative real-time PCR confirmed that endogenous gene expression levels of FGFR3 were significantly upregulated by the overexpression plasmid (Figure 6A and B; P<0.05).
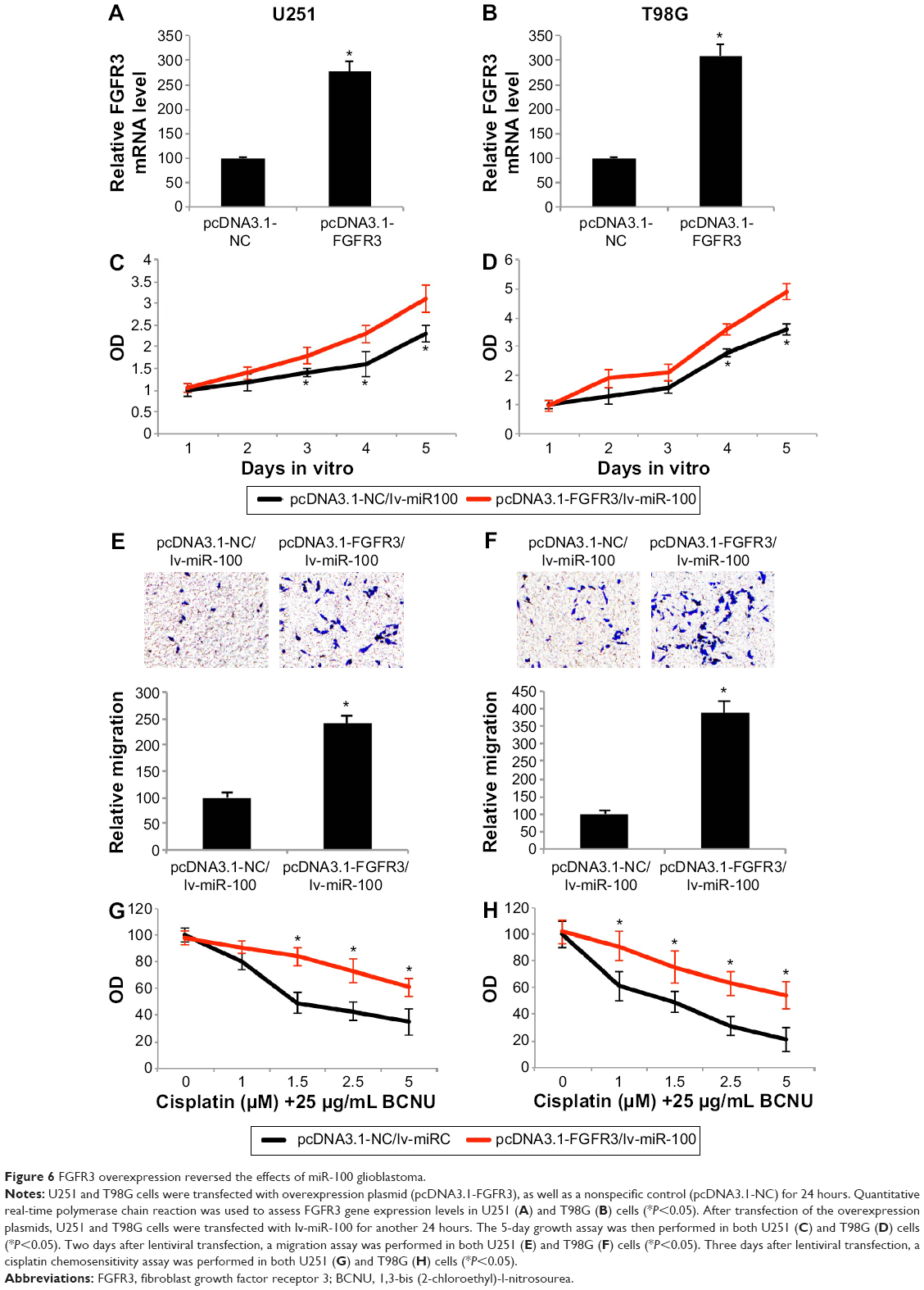 | Figure 6 FGFR3 overexpression reversed the effects of miR-100 glioblastoma. |
Twenty-four hours after the U251 and T98G cells were transfected with the overexpression plasmid, miR-100 was upregulated by lentivirus in both cell lines to assess the effects of FGFR3 and miR-100 in human glioblastoma. First, the growth assay showed that proliferation of U251 and T98G cells, initially inhibited by upregulation of miR-100, were significantly restored by overexpressing FGFR3 3 or 4 days after double-transfection (Figure 6C and D; P<0.05). Second, the migration assay demonstrated that the inhibited migrating ability of human glioblastoma cells (induced by upregulation of miR-100) was significantly reversed by overexpressing FGFR3 (Figure 6E and F; P<0.05). Finally, the chemosensitivity assay demonstrated that, while FGFR3 was overexpressed, upregulation of miR-100-induced chemosensitivity to cisplatin was significantly reduced in U251 and T98G cells (Figure 6G and H; P<0.05). Taken together, the results of our growth, migration, and chemosensitivity assays suggested that the antitumor effects of miR-100 upregulation were reversed by overexpressing FGFR3 in human glioblastoma cells.
Discussion
In this work, we evaluated miR-100 gene expression levels in seven human glioblastoma cell lines, as well as in 13 human patients with matched tumor tissue and non-tumor brain tissue. We found that miR-100 was significantly downregulated in both glioblastoma cell lines and in human tumors. In a previous study, miR-100 was shown to be differentially expressed in radiosensitive vs radioinsensitive glioblastoma cells.15 miR-100 was also found to be expressed in glioma cells in another study.17 However, miR-100 expression levels were not characterized between carcinoma and non-carcinoma tissues in either report, and nor were the functional roles of miR-100 in glioblastoma. Just recently, a further study showed that miR-100 modulated the growth of glioblastoma by regulation of SMRT/NCOR2.16 However, the full molecular profile of miR-100 in regulating glioblastoma is largely unknown.
We demonstrated that overexpression of miR-100 in human glioblastoma inhibited cancer cell growth (both in vitro and in vivo) and migration, and increased sensitivity to chemotherapy reagents. On the other hand, we demonstrated that downregulating miR-100 had little effect on growth, migration, or chemosensitivity of glioblastoma. The mechanism of miR-100 in glioblastoma revealed here is consistent with a previous report that overexpressing miR-100 suppressed tumor development and increased sensitivity to chemotherapeutics in breast cancer and ovarian cancer.18 Thus, miR-100 seems to be generally acting as an anticancer miRNA in various forms of cancers, including glioblastoma.
Another important finding in our work is that FGFR3 is the downstream target of miR-100 in regulating human glioblastoma. We showed that FGFR3 protein levels were substantially downregulated while miR-100 was upregulated in U251 and T98G glioblastoma cells. We also revealed that while FGFR3 was ectopically upregulated in U251 and T98G cells, the anticancer effects of miR-100 overexpression were ameliorated. These results are in line with previous studies demonstrating that the FGF signaling pathway is a critical oncogenic regulator in human glioblastoma.19,20 Interestingly, a recent report demonstrated that FGFR3 was also directly associated with miR-100 regulation of tumor growth and chemosensitivity in pancreatic cancer.21 Thus, it is very likely that the association between miR-100 regulation and FGF signaling pathways plays an important role in regulating various cancers, including glioblastoma.
Conclusion
Our work demonstrates that miR-100 is downregulated in glioblastoma and that overexpression of miR-100 had significant anticancer effects on the growth, migration, and chemosensitivity of glioblastoma. It is the first study to characterize miR-100 as a tumor-suppressing miRNA in glioblastoma, possible via disruption of the FGF signaling pathway. Therefore, targeting the miR-100/FGFR3 pathway may lead to an effective therapy for patients with glioblastoma.
Disclosure
The authors report no conflicts of interest in this work.
References
Adamson C, Kanu OO, Mehta AI, et al. Glioblastoma multiforme: a review of where we have been and where we are going. Expert Opin Investig Drugs. 2009;18(8):1061–1083. | ||
Omuro A, DeAngelis LM. Glioblastoma and other malignant gliomas: a clinical review. JAMA. 2013;310(17):1842–1850. | ||
Weller M. Novel diagnostic and therapeutic approaches to malignant glioma. Swiss Med Wkly. 2011;141:w13210. | ||
Thomas AA, Brennan CW, DeAngelis LM, Omuro AM. Emerging therapies for glioblastoma. JAMA Neurol. 2014;71(11):1437–1444. | ||
Noda SE, El-Jawahri A, Patel D, Lautenschlaeger T, Siedow M, Chakravarti A. Molecular advances of brain tumors in radiation oncology. Semin Radiat Oncol. 2009;19(3):171–178. | ||
Okonogi N, Shirai K, Oike T, et al. Topics in chemotherapy, molecular-targeted therapy, and immunotherapy for newly-diagnosed glioblastoma multiforme. Anticancer Res. 2015;35(3):1229–1235. | ||
Urbanska K, Sokolowska J, Szmidt M, Sysa P. Glioblastoma multiforme – an overview. Contemp Oncol. 2014;18(5):307–312. | ||
Pillai RS. MicroRNA function: multiple mechanisms for a tiny RNA? RNA. 2005;11(12):1753–1761. | ||
Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell. 2005;120(1):15–20. | ||
Lynam-Lennon N, Maher SG, Reynolds JV. The roles of microRNA in cancer and apoptosis. Biol Rev Camb Philos Soc. 2009;84(1):55–71. | ||
Hwang HW, Mendell JT. MicroRNAs in cell proliferation, cell death, and tumorigenesis. Br J Cancer. 2007;96 Suppl:R40–R44. | ||
Chan B, Manley J, Lee J, Singh SR. The emerging roles of microRNAs in cancer metabolism. Cancer Lett. 2015;356(2 Pt A):301–308. | ||
Ma J, Dong C, Ji C. MicroRNA and drug resistance. Cancer Gene Ther. 2010;17(8):523–531. | ||
Qin C, Huang RY, Wang ZX. Potential role of miR-100 in cancer diagnosis, prognosis, and therapy. Tumour Biol. 2015;36(3):1403–1409. | ||
Ng WL, Yan D, Zhang X, Mo YY, Wang Y. Over-expression of miR-100 is responsible for the low-expression of ATM in the human glioma cell line: M059J. DNA Repair. 2010;9(11):1170–1175. | ||
Alrfaei BM, Vemuganti R, Kuo JS. microRNA-100 targets SMRT/NCOR2, reduces proliferation, and improves survival in glioblastoma animal models. PLoS One. 2013;8(11):e80865. | ||
Yang CH, Yue J, Pfeffer SR, et al. MicroRNA-21 promotes glioblastoma tumorigenesis by down-regulating insulin-like growth factor-binding protein-3 (IGFBP3). J Biol Chem. 2014;289(36):25079–25087. | ||
Chen D, Sun Y, Yuan Y, et al. miR-100 induces epithelial-mesenchymal transition but suppresses tumorigenesis, migration and invasion. PLoS Genet. 2014;10(2):e1004177. | ||
Loilome W, Joshi AD, ap Rhys CM, et al. Glioblastoma cell growth is suppressed by disruption of fibroblast growth factor pathway signaling. J Neurooncol. 2009;94(3):359–366. | ||
Haley EM, Kim Y. The role of basic fibroblast growth factor in glioblastoma multiforme and glioblastoma stem cells and in their in vitro culture. Cancer Lett. 2014;346(1):1–5. | ||
Li Z, Li X, Yu C, et al. MicroRNA-100 regulates pancreatic cancer cells growth and sensitivity to chemotherapy through targeting FGFR3. Tumour Biol. 2014;35(12):11751–11759. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.