Back to Journals » OncoTargets and Therapy » Volume 12
Overexpressed long noncoding RNA TUG1 affects the cell cycle, proliferation, and apoptosis of pancreatic cancer partly through suppressing RND3 and MT2A
Authors Hui B, Xu Y, Zhao B, Ji H, Ma Z, Xu S, He Z, Wang K, Lu J ![]()
Received 21 September 2018
Accepted for publication 9 January 2019
Published 5 February 2019 Volume 2019:12 Pages 1043—1057
DOI https://doi.org/10.2147/OTT.S188396
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Arseniy Yuzhalin
This paper has been retracted.
Bingqing Hui, 1,2,* Yetao Xu, 3,* Benpeng Zhao, 4,* Hao Ji, 1,2 Zhonghua Ma, 1,2 Shufen Xu, 1,2 ZhenYu He, 1,5 Keming Wang, 1,2 Jianwei Lu 6
1Department of Oncology, Second Affiliated Hospital, Nanjing Medical University, Nanjing 210000, Jiangsu, China; 2Department of Oncology, Second Clinical Medical College of Nanjing Medical University, Nanjing 210000, Jiangsu, China; 3Department of Obstetrics and Gynecology, The First Affiliated Hospital of Nanjing Medical University, Nanjing 210029, Jiangsu, China; 4Basic Medicine Faculty of Shanghai Jiaotong University, Core Facility of Basic Medical Sciences, Shanghai 200000, China; 5Department of General Surgery, Second Affiliated Hospital, Nanjing Medical University, Nanjing 210000, Jiangsu, China; 6Department of Medical Oncology, Jiangsu Cancer Hospital, Jiangsu Institute of Cancer Research, Nanjing Medical University Affiliated Cancer Hospital, Nanjing 210000, Jiangsu, China
*These authors contributed equally to this work
Background: Long noncoding RNAs (lncRNAs) are involved in various human diseases, including cancers. However, their mechanisms remain undocumented. We investigated alterations in lncRNA that may be related to pancreatic cancer (PC) through analysis of microarray data.
Methods: In the present study, quantitative real-time PCR analysis was used to examine the expression of taurine upregulated 1 (TUG1) in PC tissue samples and PC cell lines. In PC cell lines, MTT assays, colony formation assays, and flow cytometry were used to investigate the effects of TUG1 on proliferation, cell cycle regulation, and apoptosis. Moreover, we established a xenograft model to assess the effect of TUG1 on tumor growth in vivo. The molecular mechanism of potential target genes was detected through nuclear separation experiments, RNA immunoprecipitation (RIP), chromatin immunoprecipitation assays (ChIP), and other experimental methods.
Results: The findings suggest that the abnormally high expression of TUG1 in PC tissues was associated with tumor size and pathological stage. Knockdown of TUG1 blocked the cell cycle and accelerated apoptosis, thereby inhibiting the proliferation of PC cells. In addition, RIP experiments showed that TUG1 can recruit enhancer of zeste homolog 2 (EZH2) to the promoter regions of Rho family GTPase 3 (RND3) and metallothionein 2A (MT2A) and inhibit their expression at the transcriptional level. Furthermore, ChIP experiments demonstrated that EZH2 could bind to the promoter regions of RND3 and MT2A. The knockdown of TUG1 reduced this binding capacity.
Conclusion: In conclusion, our data suggest that TUG1 may regulate the expression of PC-associated tumor suppressor genes at the transcriptional level and these may become potential targets for the diagnosis and treatment of PC.
Keywords: LncRNA, ncRNA, regulate, mechanism, cancer, EZH2, transcriptional level, tumor suppressor genes
Introduction
Pancreatic cancer (PC) is one of the most aggressive malignant tumors in the world and the fourth most common cause of death. The most common sites for metastasis in PC are the common bile duct, duodenum, stomach, and celiac artery. PC is a disease characterized by rapid progression, high mortality, and a high degree of malignancy. In recent years, its morbidity and mortality have increased steadily.1–3 Although the diagnostic and therapeutic strategies have rapidly progressed over the past two decades, the achievement in diagnosing and curing PC remains inadequate. The 5-year survival rate of patients with PC is ~6%.4 Although the combination of tumor markers and imaging modalities has facilitated prompt and accurate diagnosis of this disease, the absence of early clinical symptoms continues to delay diagnosis.2 Therefore, the pathogenesis of early PC has become an important research topic.
In recent years, noncoding RNAs (ncRNAs) have been shown to act as key regulators of gene expression.5 There is a class of ncRNAs with a length between 200 and 100,000 nucleotides exhibiting limited or no protein-coding capacity. These are called long noncoding RNAs (lncRNAs) and they play essential roles in human diseases like metabolic diseases and cancers.6 lncRNAs are involved in many biological processes, such as Th-cell differentiation,7 embryonic stem cell differentiation,8 cell senescence,9 cancer cell apoptosis and metastasis,10 autophagy and myocardial infarction,11 and resistance to chemotherapy.12 Recent studies have shown that dysregulation in lncRNAs is characterized by specificity for certain tissues. In addition, its abnormally high expression in the serum or tumor tissues of some cancer patients is closely related to tumor metastasis and poor prognosis.13 These lncRNAs participate in tumor occurrence and development via the activation of tumor promoters or silencing of tumor suppressors. Different mechanisms, such as epigenetic modification, RNA decay, alternative splicing, and regulation of posttranslational modifications have been identified to explain the regulatory effect.14 Collectively, it is increasingly obvious that different lncRNAs may function as tumor suppressors or oncogenes in tumorigenesis.
To date, many lncRNAs have been demonstrated to be involved in PC, such as HOTAIR, HOTTIP, MALAT-1, AFAP1-AS1, H19, PVT1, and AF339813.15 Upregulated HOTAIR could promote resistance to tumor necrosis factor-related apoptosis inducing ligands in PC cell lines.16 HOTTIP changes the biological characteristics of cancer stem cells in PC by regulating HOXA9.17 Enhancer of zeste homolog 2 (EZH2) binds to MALAT-1, a combination that inhibits E-cadherin and promotes cell migration and invasion without altering cell proliferation.18 H19 promotes metastasis of PC cells by inhibiting let-7 against its target HMGA2-mediated epithelial–mesenchymal transition (EMT) inhibition.19 lncRNA-PVT1 competitively binds miR-448 to regulate translation of downstream target genes to promote proliferation and migration of PC cells.20 As we look into the future, we recognize the imperative need for further study on the PC-related lncRNAs.
We conjectured that there are still numerous undiscovered lncRNAs involved in PC and their molecular processes remain undocumented. We downloaded the microarray data set (GSE16515; 52 pairs of tumor and normal tissue samples) from the Gene Expression Omnibus (GEO; https://www.ncbi.nlm.nih.gov/sites/GDSbrowser?acc=GDS4102) and analyzed the data to obtain a set of lncRNAs that were abnormally expressed in PC. We found that one of the upregulated lncRNAs, namely taurine upregulated 1 (TUG1), also showed significantly increased expression levels in PC tissues. In addition, we demonstrated its biological functions, potential molecular mechanisms, and target genes in our study.
The TUG1 gene is 8,330 bp in length, located at GRCh38.p7, and consists of three exons. It has been shown that TUG1 promotes the proliferation of cells of cholangiocarcinoma and cervical cancer.21,22 Qin and Zhao and Zhao et al demonstrated that TUG1 is capable of facilitating proliferation and migration of PC cell lines through EMT or through sponging miR-382.23,24 However, there have been no reports regarding the regulatory function of TUG1 at the transcriptional level in PC cells. In this study, we aimed to examine the relationship between the expression of TUG1 in PC and the clinicopathological features of patients with PC. We focused on exploring its effect on the biological behavior of PC cell lines in vitro and in vivo. We investigated the molecular mechanisms that may explain this effect, providing a theoretical basis for the clinical genetic diagnosis and treatment of PC.
Materials and methods
Tissue collection and ethics statement
PC tissues and adjacent normal tissues (42 pairs) were collected from patients with PC. None of the patients received any local or systemic therapy prior to surgery and they provided written informed consent prior to their participation in this study. According to the WHO classification guidelines, clinical features such as pathological staging, grading, and lymph node status were determined by experts with extensive clinical experience. All the experiments described in this article have been approved by the ethics committee of Nanjing Medical University. The national guidelines for care and use of laboratory animals were strictly enforced during the animal experiments. All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 declaration of Helsinki and its later amendments or comparable ethical standards.
Cell lines and culture conditions
We purchased human PC cells (AsPC-1 and BxPC-3) and human normal pancreatic cells HPDE6-C7 from the American Type Culture Collection (Manassas, VA, USA). The cells were cultured in DMEM (Thermo Fisher Scientific, Waltham, MA, USA) at 37°C, with 5% CO2 in humid air. All media were supplemented with 10% FBS, 100 U/mL penicillin, and 100 mg/mL streptomycin (Thermo Fisher Scientific).
RNA extraction and qRT-PCR analyses
We extracted total RNA using TRIzol reagent (Thermo Fisher Scientific) according to the manufacturer’s instructions, and subsequently, reverse transcribed the RNA into cDNA using the Reverse Transcription System Kit (Takara Biotechnology, Dalian, China). Real-time PCR was performed to determine the expression level of mRNA in PC cells or tissues with GAPDH as a control according to the manufacturer’s standard procedure (Takara Biotechnology). The relative level of gene expression is in the form of ΔCt, and the fold change in gene expression was calculated using the 2-ΔΔCt method. All experiments were performed in triplicate.
Transfection of PC cells
To prevent off target effects, three separate siRNAs and scrambled negative control siRNA were designed for different sites and purchased from Thermo Fisher Scientific. According to the manufacturer’s instructions, we used Lipofectamine 3000 (Thermo Fisher Scientific) to transfect siRNA and plasmids into PC cell lines. Following transfection (48 hours), all the transfected cells were collected for analysis.
Cell proliferation assays
Cell viability was tested using the MTT kit (Sigma-Aldrich Co, St Louis, MO, USA) according to the manufacturer’s instructions and the transfected cells were grown in 96-well plates. We recorded the proliferation of cells every 24 hours after transfection of cells according to the manufacturer’s instructions. The cells were treated with 20 μL MTT and then cultured at 37°C for 4 hours. After removing the medium, 150 μL of dimethyl sulfoxide were added to each well to lyse the cells. Finally, the absorbance was measured at 490 nm. All experiments were performed in triplicate.
Colony formation and clonogenic assays
The PC cells were trypsinized into single-cell suspensions 48 hours following transfection. For the colony formation assay, 500 cells were plated into each well of a six-well plate and maintained in media containing 10% FBS to allow colony formation. The medium was replaced every 4 days. The plates were incubated for 1–2 weeks at 37°C in a 5% CO2 atmosphere until colonies were formed. The colonies were immobilized with methanol and stained with 0.1% crystal violet (Sigma-Aldrich Co.) in PBS for 15 minutes. The visible colonies were manually counted. All measurements were performed in triplicate.
Flow cytometry
Cell cycle and apoptosis were analyzed by flow cytometry and the transfected cells were harvested by trypsin digestion. The FITC-Annexin V Apoptosis Detection Kit was purchased from BD Biosciences (San Jose, CA, USA). FITC-Annexin V and propidium iodide were used for double staining in accordance with the manufacturer’s instructions, followed by flow cytometry (FACScan; BD Biosciences). We first distinguished AsPC-1 and BxPC-3 cells by living cells, dead cells, early apoptotic cells, and apoptotic cells. The relative proportion of early apoptotic cells in the transfection group and the control group was the target of our comparison. When analyzing the cell cycle, we calculated and compared the percentage of cells in the G0/G1, S, and G2/M phase in the transfected and control groups through FACScan analysis using the CycleTEST PLUS DNA kit (BD Biosciences) according to the instructions. All samples were assayed in triplicate.
Xenotransplantation mouse model
We purchased 4-week-old male nude mice from the Animal Center of Nanjing University (Nanjing, China) and maintained all mice pathogen-free in the laminar flow cabinet. For the in vivo cell proliferation assay, we stably transfected the BxPC-3 cell line with shRNA and an empty vector. After collecting the cells, both groups were resuspended at a density of 2×107 cells/mL. Subsequently, 100 μL of the shRNA-transfected cells and 100 μL of the empty vector cells were subcutaneously transplanted to both sides of the BALB/c male nude mice, respectively. We examined the growth of xenograft tumors every 2 days and the tumor volume was measured as length × width2 ×0.5. Sixteen days after the injection, the mice were sacrificed through asphyxiation using CO2 and the tumors were peeled off from the nude mice for further analysis. This study was conducted in strict accordance with the guidelines of the National Institutes of Health on the use of experimental animals. Our program was approved by the Animal Experimental Ethics Committee of Nanjing Medical University.
Subcellular fractionation location
A PARIS Kit (Thermo Fisher Scientific) was used to isolate the nuclear and cytosolic portions of PC cells according to the manufacturer’s instructions. The levels of TUG1, GAPDH, and U1 RNA in the cytoplasm and nuclear components were detected using qRT-PCR. GAPDH was used as a cytoplasmic control, while U1 was used as nuclear control. The relative ratios of TUG1, GAPDH, and U1 in the cytoplasm or nucleus are presented as percentages of the total RNA.
RIP
In accordance with the manufacturer’s instructions, we performed RIP experiments using the Magna RIP RNA Binding Protein Immunoprecipitation Kit (EMD Millipore, Billerica, MA, USA). AsPC-1 and BxPC-3 cells were lysed in complete RIP lysis buffer; the cell extracts were mixed with magnetic beads conjugated with specific antibodies or control IgG (EMD Millipore), and incubated for 6 hours at 4°C. To remove the protein, we incubated the extracts with proteinase K after washing the beads. Finally, the purified RNA was subjected to qRT-PCR analysis. The EZH2 RIP assay antibody was purchased from Abcam (Cambridge, UK).
ChIP
ChIP assays were performed using the EZ-ChIP kit according to the manufacturer’s instructions (EMD Millipore). Immunoprecipitation was performed using anti-EZH2 and anti-H3K27me3 antibodies (EMD Millipore) with normal mouse IgG as a negative control. The primers were designed according to the promoter sequences of RND3 and MT2A, referring to the upstream of the RND3 and MT2A gene transcription start sites. The corresponding primers were subsequently then used for qRT-PCR according to the manufacturer’s instructions. Using the formula 2(InputCt-TargetCt) ×0.1×100, the ChIP data were calculated as a percentage with respect to the input DNA.
Western blotting analysis and antibodies
Transfected AsPC-1 and BxPC-3 cells were treated with RIPA protein extraction reagent (Beyotime, Beijing, China) containing the protease inhibitor and phenylmethylsulfonyl fluoride. After determining the protein concentration, ~50 μg of the protein extract were separated using 10% SDS-PAGE and then transferred to a nitrocellulose membrane (Sigma-Aldrich Co). Subsequently, the nitrocellulose membranes were incubated with specific antibodies (Cell Signaling Technology, Danvers, MA, USA). The intensity of the bands was observed and determined through densitometry (Quantity One software; Bio-Rad Laboratories Inc, Hercules, CA, USA), while GAPDH was used as a control.
Statistical analysis
We performed statistical analysis using the SPSS software package (SPSS Inc., Chicago, IL, USA) and GraphPad Prism 5 software (GraphPad Software Inc., La Jolla, CA, USA). The significance of the differences observed between the experimental and control groups was estimated using the Student’s t-test or chi-squared test. The OS of PC patients was calculated using the Kaplan–Meier method and compared using the log-rank test. Pearson correlation coefficients were calculated using the Prism 5 software (GraphPad Software Inc). P<0.05 was considered statistically significant.
Results
TUG1 expression is increased in human PC tissues and cell lines
To identify lncRNAs that may be involved in the development of PC, we first downloaded the GEO data set (GSE16515) and analyzed the microarray data. The results showed that the lncRNA TUG1 was abnormally expressed in PC tissues compared with normal tissues (Figure 1A). In addition, we determined the expression levels of 42 TUG1 in PC tissues and adjacent normal tissues using quantitative reverse transcription PCR (qRT-PCR). The results showed that 39 of the 42 pairs of tissues showed high levels of TUG1 expression (fold change: >2, P<0.001) (Figure 1B). The expression levels of TUG1 were subsequently measured in human PC cell lines (AsPC-1, BxPC-3) and a human normal pancreatic cell line (HPDE6-C7). As shown in Figure 2A, the expression of TUG1 was significantly higher in PC cell lines compared with that observed in human normal pancreatic cells (both P<0.05). We then focused on detecting the biological function of this overexpressed lncRNA in PC cells to assess its diagnostic or therapeutic potential for PC.
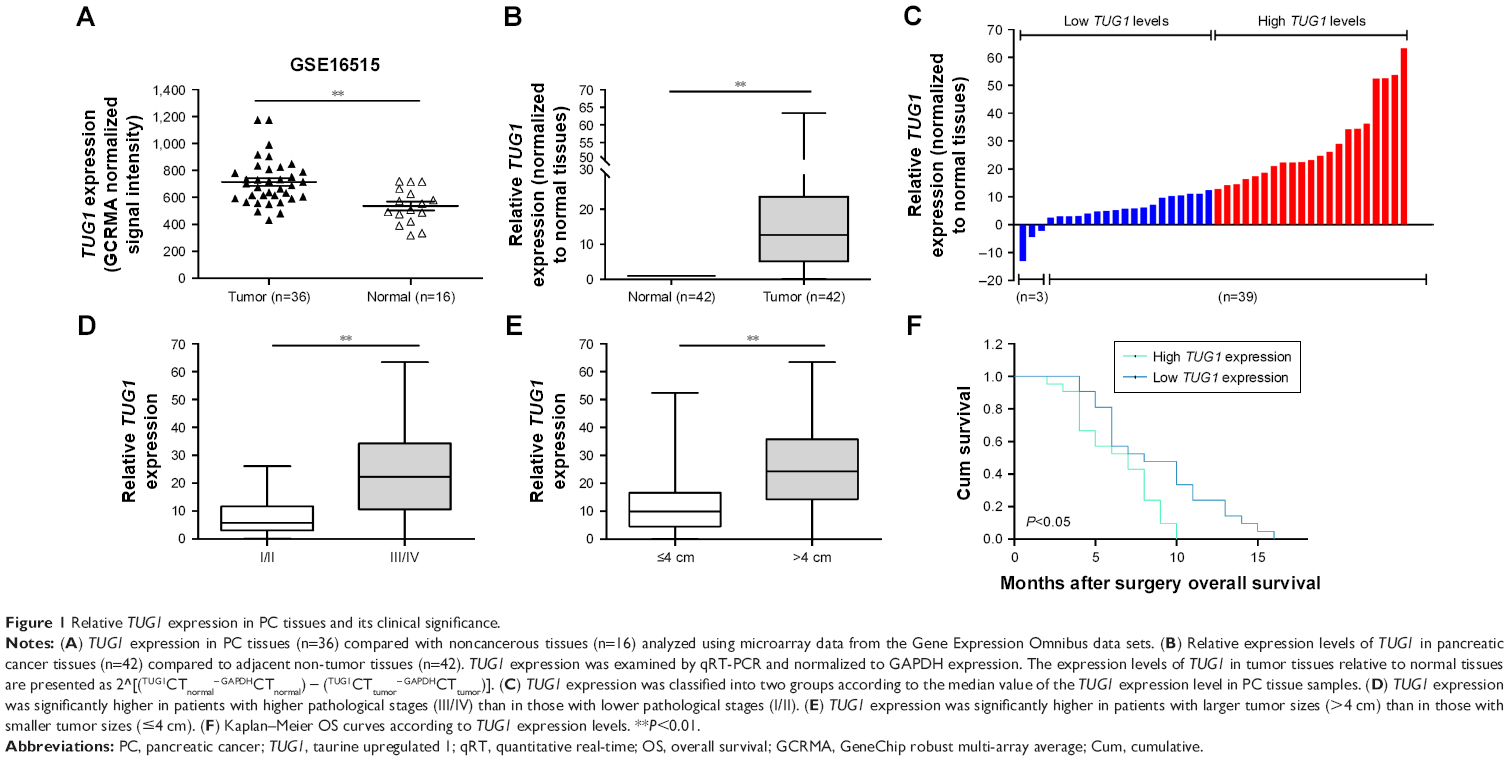 | Figure 1 Relative TUG1 expression in PC tissues and its clinical significance. |
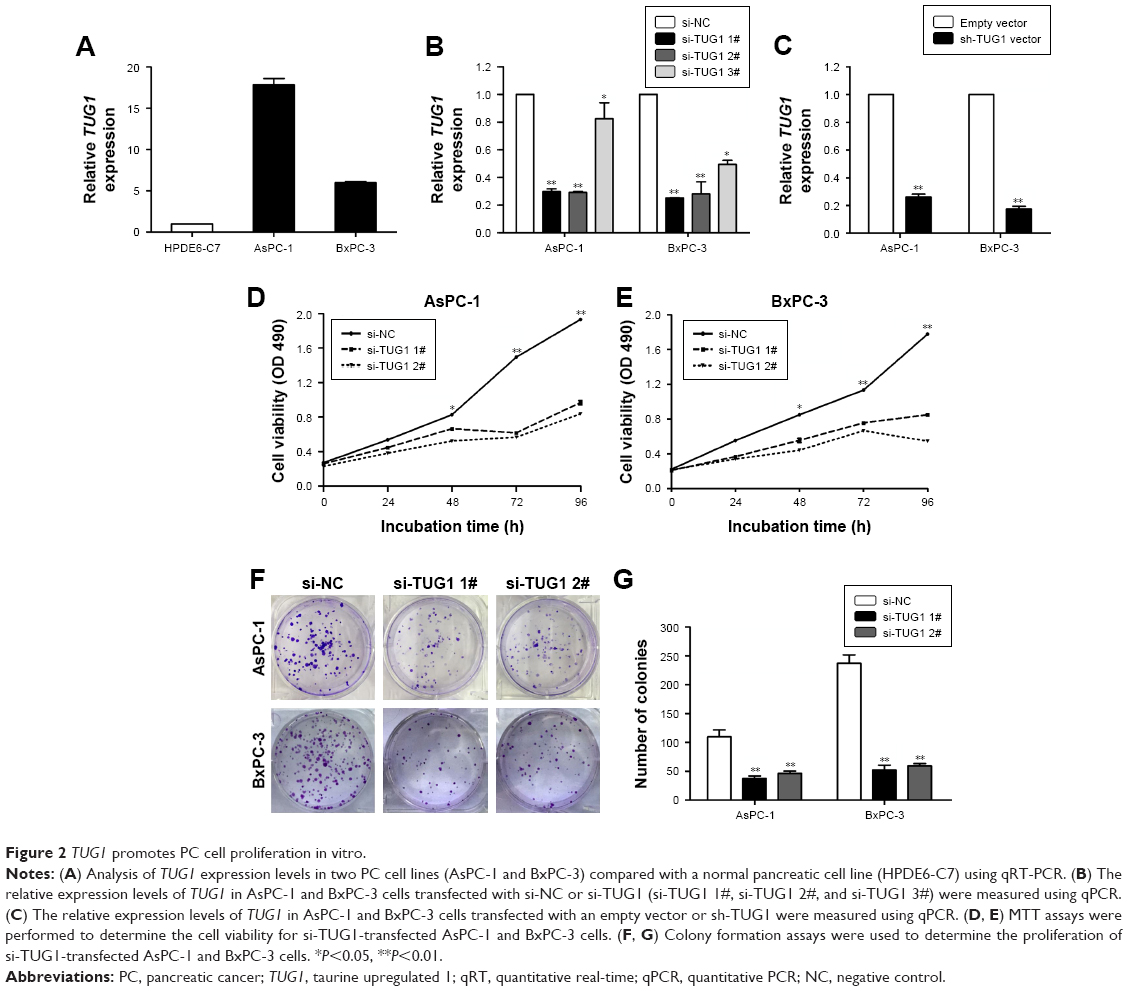 | Figure 2 TUG1 promotes PC cell proliferation in vitro. |
High expression of TUG1 is associated with tug-lymph node metastasis (TNM) stage, tumor size, lymphatic metastasis, poor prognosis
In order to further understand the importance of the abnormally high expression of TUG1 in PC, we assessed potential correlations between the level of TUG1 expression and the clinicopathologic features of patients with PC. The results showed that an increased level of TUG1 expression was positively correlated with an advanced TNM stage (P<0.001) and tumor size (P<0.01). The expression of TUG1 was higher in patients with stage III/IV or tumor size >4 cm, whereas it was lower in patients with stage I/II or tumor size <4 cm (Figure 1D and E). However, in our study, there was no significant relationship between the expression of TUG1 and other clinical factors such as gender (P=0.352) and age (P=0.537) (Table 1). To further evaluate the effect of TUG1 expression on the prognosis of PC patients, the samples were divided according to the median level of TUG1 expression into a high TUG1 expression group (above median value, N=21) and a low TUG1 expression group (below median value, N=21) (Figure 1C). The Kaplan–Meier survival analysis and logarithmic rank test were used to determine the overall survival (OS). As shown in Figure 1F, the OS rate in the high TUG1 expression group >8 months was 23.8%, while that of the low TUG1 expression group was 47.6%. Notably, the overexpression of TUG1 was associated with shorter OS (P=0.017). These results suggest that TUG1 may be a useful marker of PC prognosis or progression.
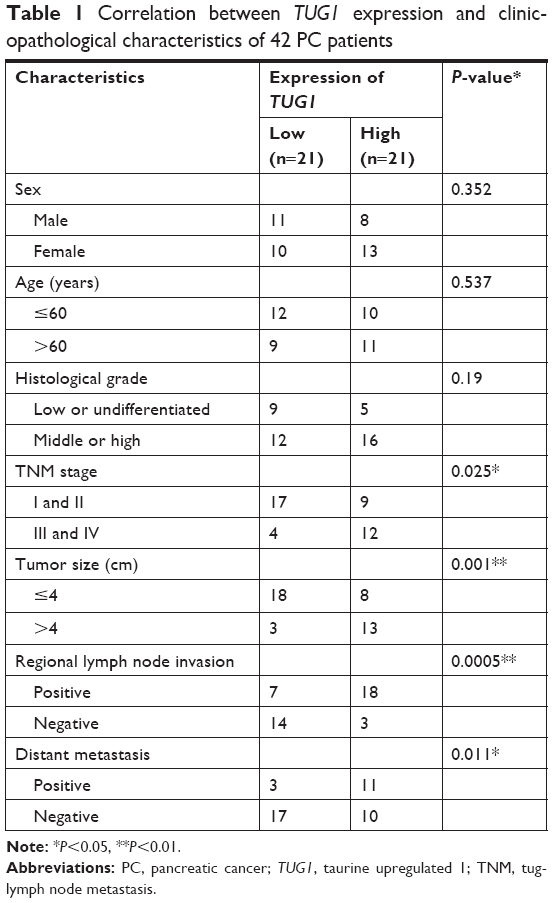 | Table 1 Correlation between TUG1 expression and clinicopathological characteristics of 42 PC patients |
TUG1 promotes proliferation of PC cells in vitro
To study the function of TUG1 in PC cells, we first performed qRT-PCR analysis to detect its expression in multiple human PC cell lines. As shown in Figure 2A, the expression of TUG1 was significantly upregulated in two PC cell lines (AsPC-1 and BxPC-3) compared with that observed in human normal pancreatic cells HPDE6-C7. Subsequently, we designed three different TUG1 siRNAs for transfection into cell lines. qRT-PCR analysis was performed 48 hours after transfection, and the data showed that all TUG1 siRNAs were effectively introduced into the cells. Of note, si-TUG1 1# and 2# showed more effective interference than si-TUG1 3# (Figure 2B). Therefore, we chose si-TUG1 1# and 2# for subsequent experiments. The sh-TUG1 we designed was successfully introduced into cells (Figure 2C). MTT assays showed that the knockdown of TUG1 expression significantly inhibited the growth of AsPC-1 and BxPC-3 cells compared with the corresponding randomized control (Figure 2D and E). Similarly, colony formation assays showed a significant reduction in the survival rate of clonal formation after downregulation of TUG1 in AsPC-1 and BxPC-3 cells (Figure 2F and G). Apoptosis and cell cycle regulation were identified as two factors leading to the growth of PC cells. Thus, we performed flow cytometry analysis to characterize these factors. In order to examine whether the effect of TUG1 on the proliferation of PC cells reflects the change in cell cycle, flow cytometry analysis was performed to study the cell cycle progression. The results showed that AsPC-1 and BxPC-3 cells transfected with si-TUG1 stagnated at the G1/ G0 phase (Figure 3A). In addition, flow cytometry was performed to determine whether apoptosis involved in TUG1 knockdown induces cell growth arrest. As shown in Figure 3B, the rate of early apoptosis (upper right) and late apoptosis (lower right) with low TUG1 in AsPC-1 and BxPC-3 cells was higher than that reported in control cells. In conclusion, it was found that the knockdown of TUG1 expression significantly reduced the proliferation rate of cells, arrested the cell cycle, and induced apoptosis. These findings suggest that TUG1 may be an oncogene involved in promoting the proliferation of PC.
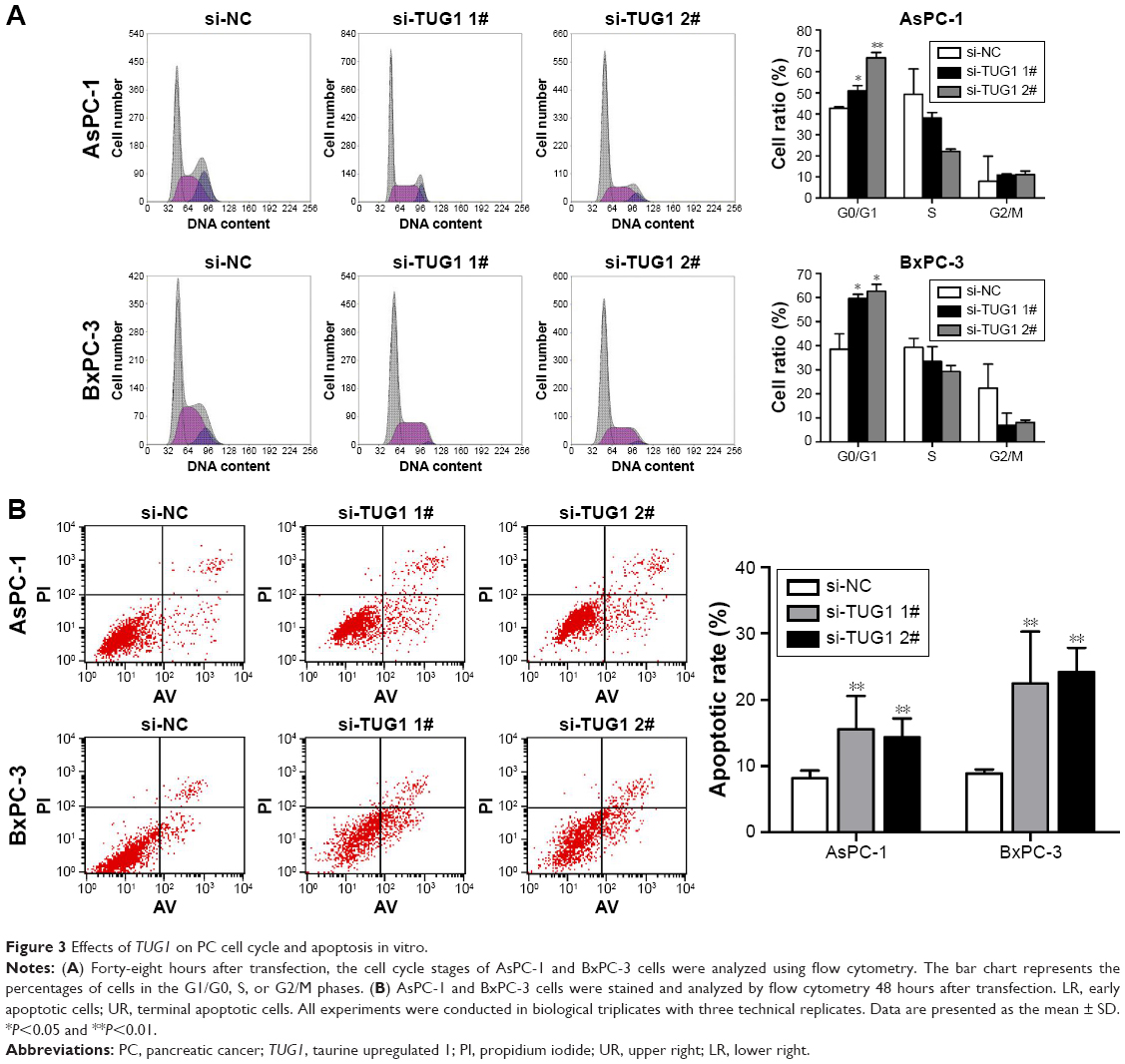 | Figure 3 Effects of TUG1 on PC cell cycle and apoptosis in vitro. |
Knockdown of TUG1 inhibits PC cells tumorigenesis in vivo
To investigate whether TUG1 can also affect tumor development in vivo, BxPC-3 cells were stably transfected with sh-TUG1 or an empty vector (Figure 2C). MTT assays showed that sh-TUG1 vector transfection impaired BxPC-3 cell growth in vitro. In addition, colony formation assays showed that BxPC-3 decreased colony formation following transfection with the sh-TUG1 vector. Subsequently, sh-TUG1 or BxPC-3 cells stably transfected with an empty vector were injected into mice. As shown in Figure 4A, silencing of TUG1 inhibited tumor growth compared with the control group. Twenty days after injection, the tumors formed in the sh-TUG1 group were significantly smaller than those formed in the control group (Figure 4B). Meanwhile, the weight of the tumor in the sh-TUG1 group was significantly reduced compared with that observed in the empty vector group (Figure 4C). In addition, the qRT-PCR assay showed that levels of TUG1 expression in tumor tissues formed by sh-TUG1 cells were lower than those observed in the control group (Figure 4D). In addition, tumors formed from BxPC-3 cells transfected with sh-TUG1 showed a decreased positivity for Ki-67 compared with the control cells (Figure 4E). These data suggest that the knockdown of TUG1 inhibits tumor growth in vivo.
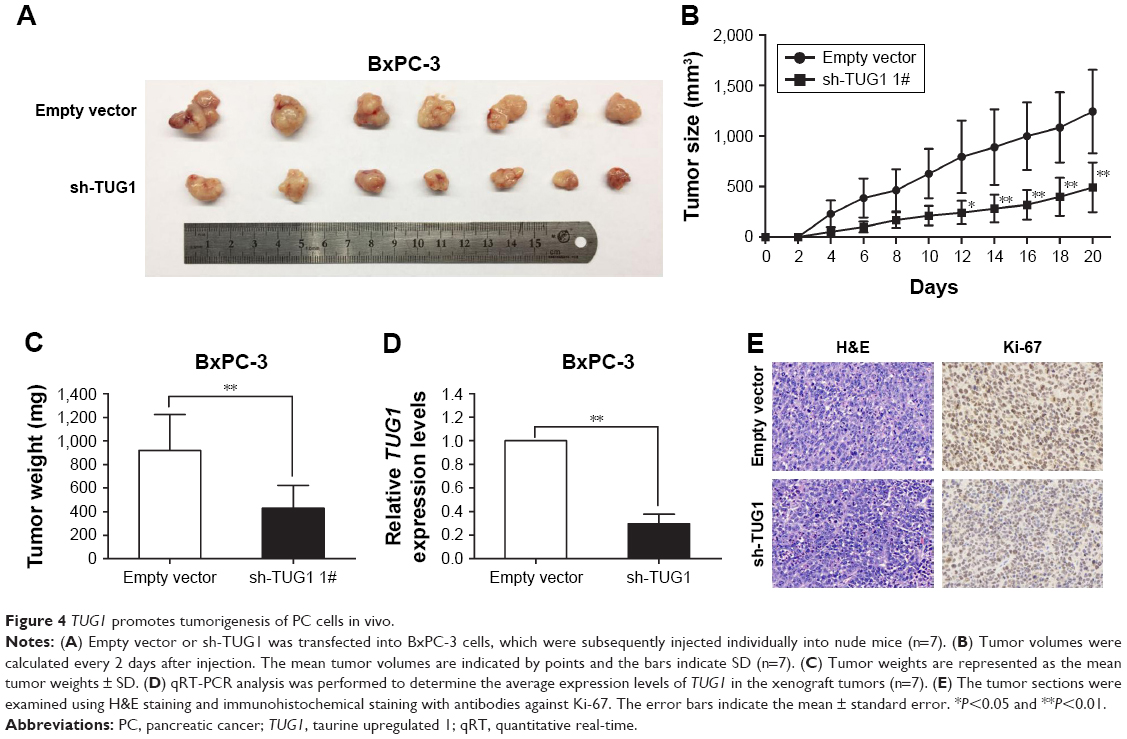 | Figure 4 TUG1 promotes tumorigenesis of PC cells in vivo. |
TUG1 suppresses the transcription of Rho family GTPase 3 (RND3)/metallothionein 2A (MT2A) in PC
In order to explore the molecular mechanism of TUG1 in the phenotype of PC cells, we investigated potential targets for the regulation of tumor cell proliferation and apoptosis. Therefore, we performed qRT-PCR to determine the gene expression that may negatively regulate tumor initiation and progression. Interestingly, the expression levels of RND3 and MT2A increased in AsPC-1 and BxPC-3 cells transfected with si-TUG1 (Figure 5A). The expression of the RND3/MT2A protein was determined through Western blotting analysis. After transfection with si-TUG1, the levels of RND3 were 3.4-fold higher in AsPC-1 cells, 2.7-fold higher in BxPC-3 cells, 2.3-fold higher in AsPC-1 cells, and 2.9-fold higher in BxPC-3 cells (Figure 5B). Meanwhile, the expression of RND3/MT2A in 42 PC tissues and PC cell lines was determined using qRT-PCR. The results showed that the mRNA levels of RND3/MT2A in PC tissues and cell lines (AsPC-1 and BxPC-3) were generally lower than those observed in matched normal tissues and cell lines (Figure 5C and D). These data showed that RND3 and MT2A were negatively regulated by the mRNA and protein levels of TUG1 in PC cells. Moreover, the inhibition of TUG1 contributed to the activation of RND3/MT2A, confirming our earlier findings that TUG1 may be involved in promoting the proliferation of PC cells.
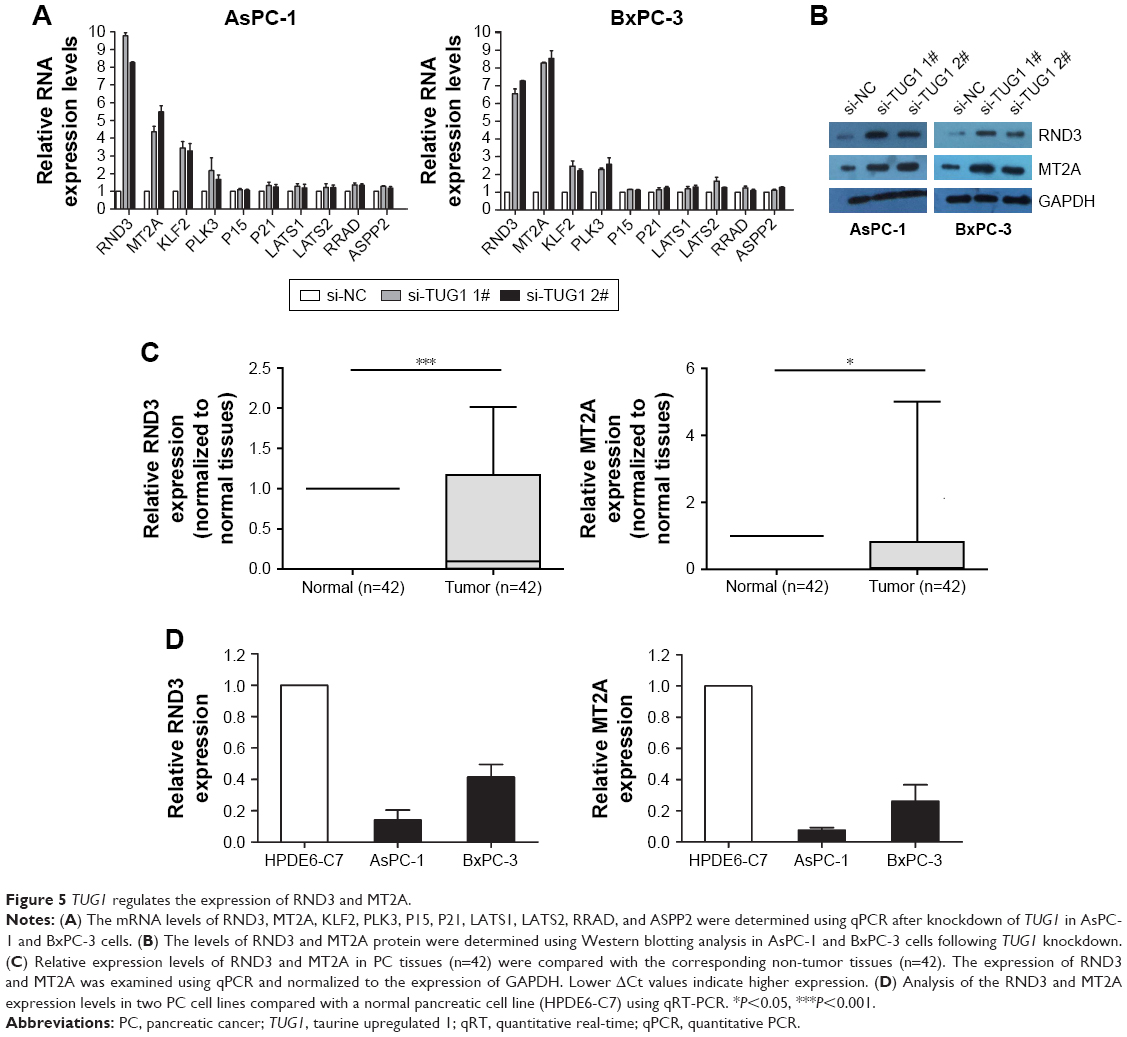 | Figure 5 TUG1 regulates the expression of RND3 and MT2A. |
Tumor-suppressive function of RND3 and MT2A in PC
In the present study, we specifically observed the effect of RND3 and MT2A overexpression on the proliferation of PC cells. The expression of RND3 and MT2A in AsPC-1 and BxPC-3 cells was induced by using pcDNA-RND3, pcDNA-MT2A, or an empty vector. Compared with each control group, the expression of RND3 and MT2A was significantly upregulated in AsPC-1 and BxPC-3 cells transfected with pcDNA-RND3 or pcDNA-MT2A at the mRNA and protein levels (Figure 6A–C). After transfection with pcDNA-RND3, pcDNA-MT2A, or an empty vector, MTT and colony formation assays were used to investigate the cellular activity in AsPC-1 and BxPC-3 cells. The MTT and colony formation assays showed that overexpression of RND3 or MT2A could inhibit cell viability in PC (Figure 6D and E). Therefore, it was concluded that RND3 and MT2A play a role in tumor inhibition in PC.
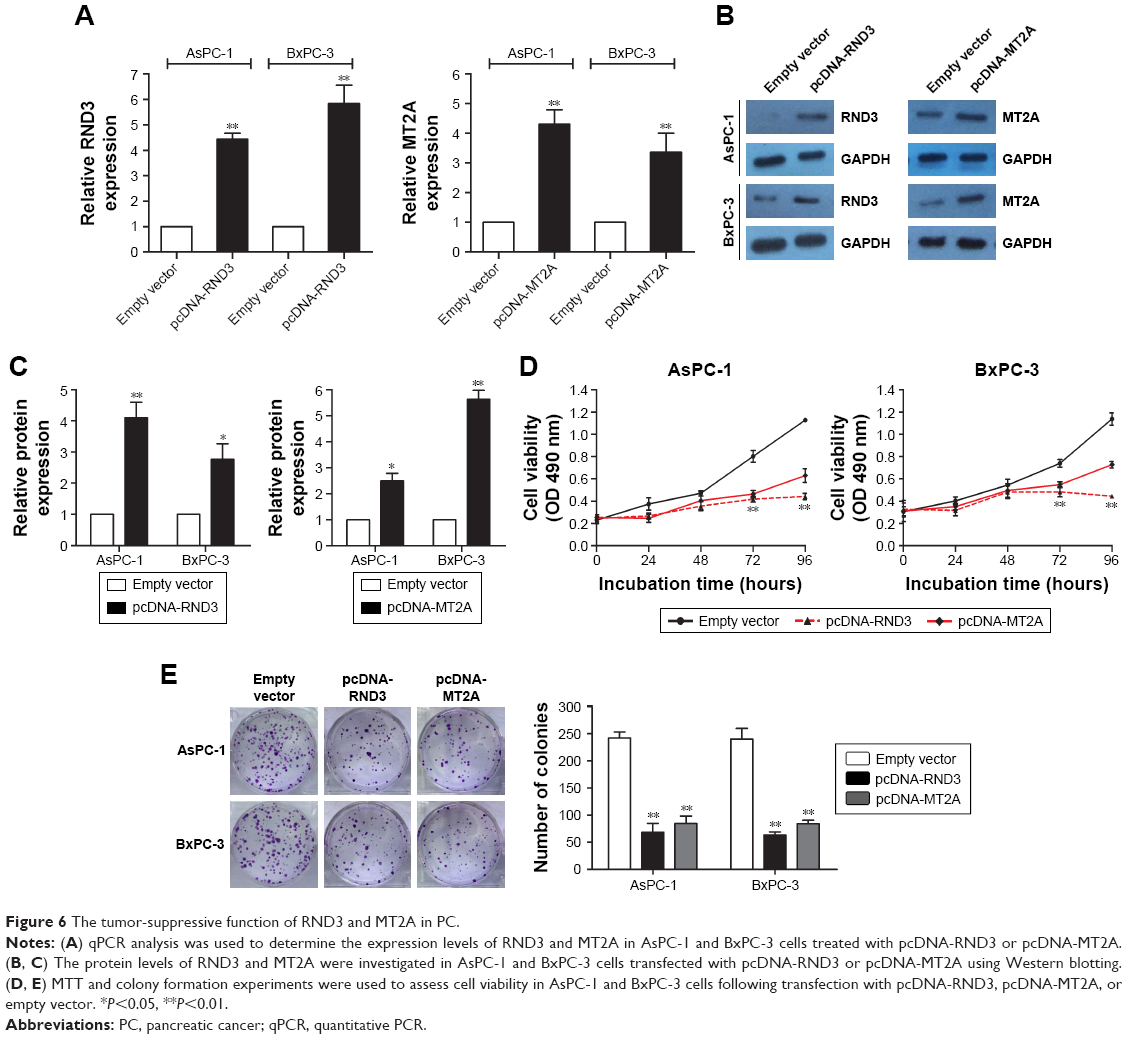 | Figure 6 The tumor-suppressive function of RND3 and MT2A in PC. |
TUG1 suppresses the transcription of RND3/MT2A by binding with EZH2 at the transcriptional level
In order to determine the distribution of TUG1 in PC cells, we performed hierarchical separation of PC cell lines and obtained nuclear and cytoplasmic grades. We found that the TUG1 RNA was mainly located in the nucleus rather than the cytoplasm (Figure 7A), indicating that it plays a regulatory role at the transcriptional level. Excessive levels of GAPDH or U1 RNA were used as an indicator of successful grading. Recent studies have concluded that ~20% of lncRNAs regulate downstream target genes by binding to the polycomb repressive complex 2 (PRC2).25 PRC2 is a methyltransferase that trimethylates H3K27 to suppress the transcription of specific genes; one of its major components is EZH2.26 A previous study demonstrated that HOXA-AS2 can epigenetically silence the expression of P21/PLK3/DDIT3 via binding to EZH2.27 In addition, ANRIL was shown to be able to cross talk with microRNAs by binding to PRC2, thus regulating the growth of PC cells.28 In view of this background, RNA immunoprecipitation (RIP) analysis was performed to confirm the binding of TUG1 to PRC2. As shown in Figure 7B, endogenous TUG1 was enriched in anti-EZH2 RIP level in PC AsPC-1 and BxPC-3 cells. Our results suggest that TUG1 may be genetically suppressed by binding to EZH2.
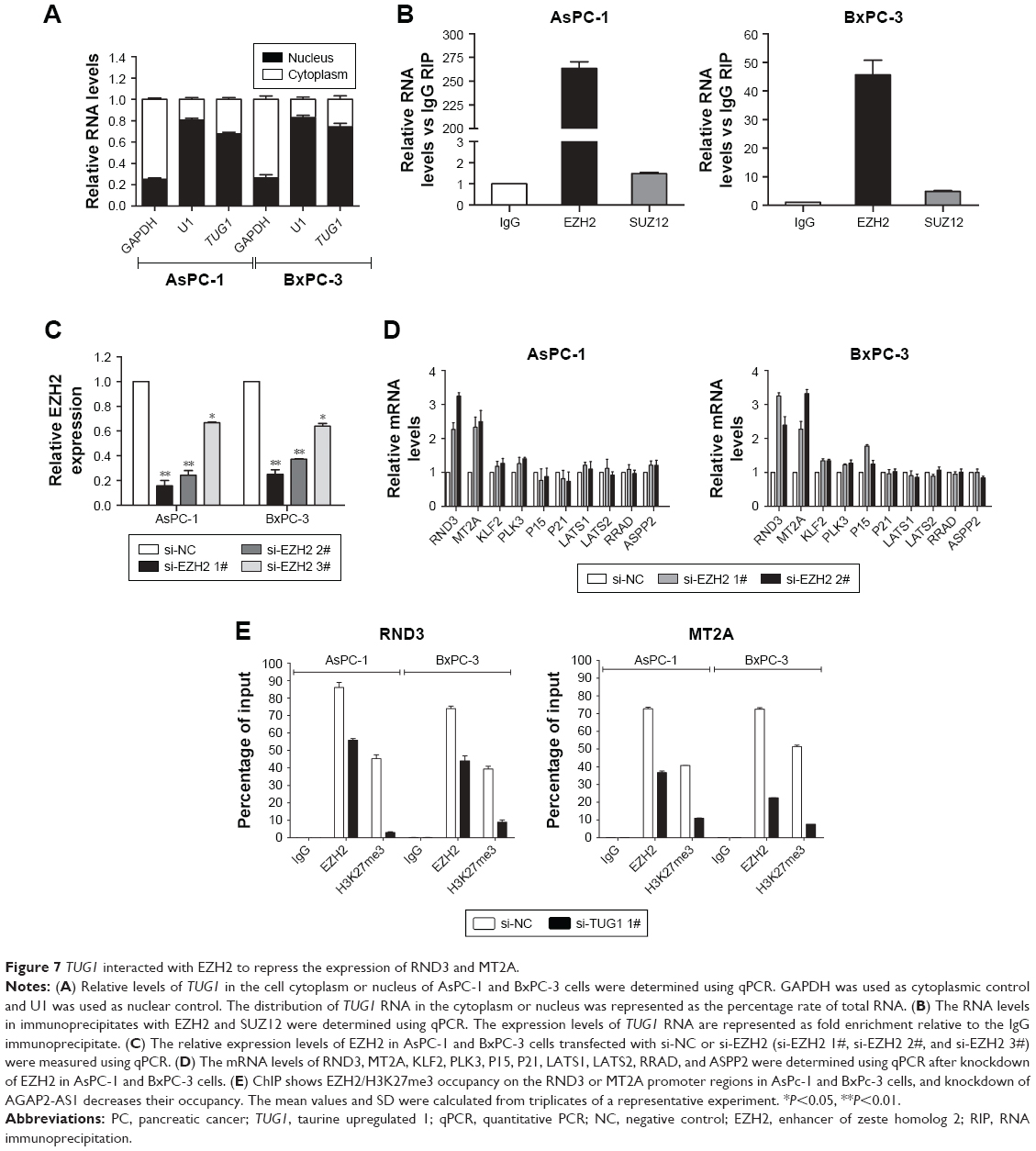 | Figure 7 TUG1 interacted with EZH2 to repress the expression of RND3 and MT2A. |
Transfection of EZH2 siRNA into PC AsPC-1 and BxPC-3 cells with si-EZH2 1# and 2# showed more effective interference than si-EZH2 3# (Figure 7C). Furthermore, we detected increased expression of RND3 and MT2A in EZH2-depleted PC cells (Figure 7D). Based on our qRT-PCR data (Figures 5A and 7D), RND3 and MT2A are the most upregulated mRNAs in TUG1-depleted PC cells and EZH2-depleted cells. Collectively, these findings suggest that RND3 and MT2A may be key downstream genes of TUG1 and that TUG1 can inhibit its expression by binding to EZH2.
In addition, the results of a chromatin immunoprecipitation (ChIP) analysis showed that EZH2 could bind to the RND3 and MT2A promoter regions to induce histone lysine 27 trimethylation (H3K27me3) modification in PC AsPC-1 and BxPC-3 cells. Knockdown of TUG1 results in binding of the RND3 and MT2A initiators by EZH2 and reduction in H3K27me3 occupancy (Figure 7E). These results showed that TUG1 can promote the growth of PC cells and regulate transcription of RND3 and MT2A by binding to EZH2.
Discussion
Recent findings have suggested that many lncRNAs (such as HOTTIP,29 NORAD,30 PVT1,31 and MEG332) play important biological roles in PC. Our previous investigation also identified that lncRNA SNHG15 inhibits the expression of P15 and KLF2 to promote the proliferation of PC cells through EZH2-mediated H3K27me3.33 Generally, lncRNAs are involved in the regulation of cancer cells, phenotypes by regulating the expression of target genes through different molecular mechanisms, including chromatin modification, genomic imprinting, RNA decay, sponging miRNAs, and binding with RNA binding protein.34,35
As databases were established and populated, more lncRNAs were identified and their abnormal expressions were revealed in a variety of human cancers, including PC. In this study, we determined the overexpression of the lncRNA TUG1 in human PC tissues by analyzing data from the GEO database and confirming the findings in paired cancer tissues and adjacent non-tumor tissues obtained from patients who had not undergone drug therapy prior to surgery. In addition, the knockdown of TUG1 expression led to significant inhibition of cell proliferation and promotion of apoptosis in vitro and in vivo. These findings suggest that TUG1 plays a direct role in regulating cell proliferation and progression of PC, and may be a useful novel marker of prognosis or progression for PC.36,37 As additional lncRNAs are studied, many have been shown to function by binding to PRC2 and silencing downstream target genes involved in multiple cancers, including PC. TUG1 has been reported to be involved in the proliferation of cancer cells by silencing the expression of KLF2,38 P57,39 and BAX.40 In this study, we found that TUG1 is mostly located in the cell nucleus and could bind to EZH2, a core subunit of PRC2, resulting in suppressing the transcription of RND3 and MT2A.
RND3 (also known as RhoE) encodes proteins belonging to the superfamily of small GTPase proteins, including Rnd1, Rnd2, and RND3, which are involved in cell migration, invasion, and cell responses to nerve processes extension and branching.41 RND3, also known as RhoE, has been shown to play a separate role in the oncogenesis of human cancer. Previous studies have shown it to be an antiproliferative protein. Tang et al reported that RND3 is downregulated in lung cancer cell lines, and its reintroduction can block the proliferation of cancer cells. Mechanistically, Notch intracellular domain (NICD) protein abundance in H358 cells was regulated by Rnd3-mediated NICD proteasome degradation. Rnd3 regulated H358 and H520 cell proliferation through a Notch1/NICD/Hes1 signaling axis independent of Rho Kinase.42 Zhu et al showed that wild-type TP53 significantly increased the expression of RND3, while the enhanced expression of RND3 significantly inhibited proliferation. These findings indicated that RND3 is a tumor suppressor regulated by TP53.43 In addition, downregulation of RND3 in esophageal squamous cell carcinoma cells promotes cell proliferation and cell cycle progression, whereas upregulation of RND3 inhibits cell proliferation and leads to cell cycle arrest at the G0/G1 phase. Also, overexpression of RND3 increased PTEN and CDKN1B/p27, and decreased pAKT and CCND1 (cell cycle protein D1).44 It has been reported that RND3 prevents the release of EIF4E from EIF4EBP1/4e-bp1 and inhibits cap-dependent translation. Therefore, RND3 also inhibits the expression and transcription activity of the EIF4E target MYC/c-myc.45 Poch et al confirmed that RND3 inhibits the activation of ERK, thereby reducing CCND1 expression and leading to decreased inactivation of RB1/retinoblastoma 1. This mechanism is involved in the inhibition of glioblastoma cell growth induced by RND3.46 RND3 induces inhibition of the proliferation of fibroblasts and serum-induced s-entry. In addition, human papillomavirus E7, adenovirus E1A, and CCNE (cell cycle protein E) can rescue cell cycle progression in RND3 expressing cells, indicating that RND3 can inhibit cell cycle progression upstream of the phosphorylated RB1 checkpoint.47 Therefore, the underlying mechanism for the anti-proliferation capability of RND3 is context-dependent. Moreover, we found that upregulation of RND3 also inhibits the proliferation of PC cells and its upregulation could be caused by TUG1 knockdown in PC cells.
Metallothionein (MT) is a low molecular weight, heavy metal-binding protein. Human MT consists of four isoforms, namely MT1, MT2A (or MT2), MT3, and MT4.48 In contrast to the histologically specific expression of MT3 and MT4, MT1 and MT2A are the major MT isoforms, which are highly conserved and present in almost all types of soft tissue. The expression of MT can be induced by many mediators and regulated in a cell/tissue-specific manner in response to external signals. The human MT genes are highly homologous and clustered in the q13 region of chromosome 16, containing one set of MT1 genes (MT1A, B, E, F, G, H, and X genes) and another of MT isomers (MT2A, MT3, and MT4). MT is involved in a variety of cellular functions, such as metal ion homeostasis, cell differentiation, apoptosis, inflammation, carcinogenesis, and chemical sensitization. The abnormal expression of MT may change its functional characteristics related to tumor and neurodegeneration.49 The effects of MTs on pathophysiological processes, particularly on the development of cancer, are the subject of numerous studies. However, the complexity of MT expression has been shown to be associated with tumorigenesis, tumor progression, and patient prognosis in different types of cancer. For example, the expression of MT is increased in breast, kidney, bladder, and ovarian cancers.49–51 In contrast, the expression of MT is low due to epigenetic silencing and plays a role in tumor inhibition in a range of other human tumors, such as thyroid, esophagus, liver, colon, and prostate cancer.52–57 The present study found that upregulation of MT2A can inhibit the proliferation of PC cells and TUG1 knockdown could induce MT2A upregulation in PC cells. However, the regulatory mechanism of MT2A in PC remains elusive.
Although to date only a few lncRNAs have been well characterized, they have been shown to regulate various levels of gene expression, including chromatin modification and posttranscriptional processing.58,59 Despite the observation of TUG1-induced proliferation of PC cells, other possible targets and mechanisms that highlight such regulatory behavior need to be fully elucidated.
Conclusion
In summary, the expression of TUG1 was significantly increased in PC tissue. This finding suggests that its upregulation may be a prognostic factor in patients with PC, indicating a lower survival rate and a higher risk of metastasis. We found that TUG1 may regulate the proliferation capacity of PC cells, probably through the regulation of RND3 and MT2A. These results suggest that lncRNAs may regulate the expression of different target genes at the transcriptional level and contribute to the biological function of different cancer cells. Our findings shed light on the pathogenesis of PC and facilitate the development of targeted lncRNAs for the diagnosis and treatment of cancer.
Acknowledgments
We thank Xuezhen Hu (Jiangsu Province Hospital of Traditional Chinese Medicine [TCM], Affiliated Hospital of Nanjing University of TCM, Nanjing 210000, Jiangsu, China) for collecting PC tissues and adjacent normal tissues from patients with PC. This study was funded by the National Natural Science Foundation of China (81772603) and Natural Science Foundation of Jiangsu Province (BK20151578).
Disclosure
The authors report no conflicts of interest in this work.
References
Petrushnko W, Gundara JS, de Reuver PR, O’Grady G, Samra JS, Mittal A. Systematic review of peri-operative prognostic biomarkers in pancreatic ductal adenocarcinoma. HPB (Oxford). 2016;18(8):652–663. | ||
Yadav D, Lowenfels AB. The epidemiology of pancreatitis and pancreatic cancer. Gastroenterology. 2013;144(6):1252–1261. | ||
Ji BL, Xia LP, Zhou FX, Mao GZ, Xu LX. Aconitine induces cell apoptosis in human pancreatic cancer via NF-κB signaling pathway. Eur Rev Med Pharmacol Sci. 2016;20(23):4955–4964. | ||
Hidalgo M. Pancreatic cancer. N Engl J Med. 2010;362(17):1605–1617. | ||
Djebali S, Davis CA, Merkel A, et al. Landscape of transcription in human cells. Nature. 2012;489(7414):101–108. | ||
Wapinski O, Chang HY. Long noncoding RNAs and human disease. Trends Cell Biol. 2011;21(6):354–361. | ||
Spurlock CF, Tossberg JT, Guo Y, Collier SP, Crooke PS, Aune TM. Expression and functions of long noncoding RNAs during human T helper cell differentiation. Nat Commun. 2015;6(1):6932. | ||
Yin Y, Yan P, Lu J, et al. Opposing roles for the lncRNA Haunt and its genomic locus in regulating HOXA gene activation during embryonic stem cell differentiation. Cell Stem Cell. 2015;16(5):504–516. | ||
Montes M, Nielsen MM, Maglieri G, et al. The lncRNA MIR31HG regulates p16(INK4A) expression to modulate senescence. Nat Commun. 2015;6(1):6967. | ||
Yarmishyn AA, Kurochkin IV. Long noncoding RNAs: a potential novel class of cancer biomarkers. Front Genet. 2015;6:145. | ||
Wang K, Liu CY, Zhou LY, et al. APF lncRNA regulates autophagy and myocardial infarction by targeting miR-188-3p. Nat Commun. 2015;6(1):6779. | ||
Shi SJ, Wang LJ, Yu B, Li YH, Jin Y, Bai XZ. LncRNA-ATB promotes trastuzumab resistance and invasion-metastasis cascade in breast cancer. Oncotarget. 2015;6(13):11652–11663. | ||
Kim HS, Minna JD, White MA. GWAS meets TCGA to illuminate mechanisms of cancer predisposition. Cell. 2013;152(3):387–389. | ||
Rinn JL. lncRNAs: linking RNA to chromatin. Cold Spring Harb Perspect Biol. 2014;6(8):a018614. | ||
Huang X, Zhi X, Gao Y, Ta N, Jiang H, Zheng J. LncRNAs in pancreatic cancer. Oncotarget. 2016;7(35):57379–57390. | ||
Yang SZ, Xu F, Zhou T, Zhao X, Mcdonald JM, Chen Y. The long non-coding RNA HOTAIR enhances pancreatic cancer resistance to TNF-related apoptosis-inducing ligand. J Biol Chem. 2017;292(25):10390–10397. | ||
Fu Z, Chen C, Zhou Q, et al. LncRNA HOTTIP modulates cancer stem cell properties in human pancreatic cancer by regulating HOXA9. Cancer Lett. 2017;410:68–81. | ||
Han T, Jiao F, Hu H, et al. EZH2 promotes cell migration and invasion but not alters cell proliferation by suppressing E-cadherin, partly through association with MALAT-1 in pancreatic cancer. Oncotarget. 2016;7(10):11194–11207. | ||
Ma C, Nong K, Zhu H, et al. H19 promotes pancreatic cancer metastasis by derepressing let-7’s suppression on its target HMGA2-mediated EMT. Tumour Biol. 2014;35(9):9163–9169. | ||
Zhao L, Kong H, Sun H, Chen Z, Chen B, Zhou M. LncRNA-PVT1 promotes pancreatic cancer cells proliferation and migration through acting as a molecular sponge to regulate miR-448. J Cell Physiol. 2018;233(5):4044–4055. | ||
Xu Y, Leng K, Li Z, et al. The prognostic potential and carcinogenesis of long non-coding RNA TUG1 in human cholangiocarcinoma. Oncotarget. 2017;8(39):65823–65835. | ||
Zhu J, Shi H, Liu H, Wang X, Li F. Long non-coding RNA TUG1 promotes cervical cancer progression by regulating the miR-138-5p-SIRT1 axis. Oncotarget. 2017;8(39):65253–65264. | ||
Qin CF, Zhao FL. Long non-coding RNA TUG1 can promote proliferation and migration of pancreatic cancer via EMT pathway. Eur Rev Med Pharmacol Sci. 2017;21(10):2377–2384. | ||
Zhao L, Sun H, Kong H, Chen Z, Chen B, Zhou M. The Lncrna-TUG1/EZH2 axis promotes pancreatic cancer cell proliferation, migration and EMT phenotype formation through sponging Mir-382. Cell Physiol Biochem. 2017;42(6):2145–2158. | ||
Khalil AM, Guttman M, Huarte M, et al. Many human large intergenic noncoding RNAs associate with chromatin-modifying complexes and affect gene expression. Proc Natl Acad Sci U S A. 2009;106(28):11667–11672. | ||
Marchese FP, Huarte M. Long non-coding RNAs and chromatin modifiers: their place in the epigenetic code. Epigenetics. 2014;9(1):21–26. | ||
Xie M, Sun M, Zhu YN, et al. Long noncoding RNA HOXA-AS2 promotes gastric cancer proliferation by epigenetically silencing P21/PLK3/DDIT3 expression. Oncotarget. 2015;6(32):33587–33601. | ||
Zhang EB, Kong R, Yin DD, et al. Long noncoding RNA ANRIL indicates a poor prognosis of gastric cancer and promotes tumor growth by epigenetically silencing of miR-99a/miR-449a. Oncotarget. 2014;5(8):2276–2292. | ||
Fu Z, Chen C, Zhou Q, et al. LncRNA HOTTIP modulates cancer stem cell properties in human pancreatic cancer by regulating HOXA9. Cancer Lett. 2017;410:68–81. | ||
Li H, Wang X, Wen C, et al. Long noncoding RNA NORAD, a novel competing endogenous RNA, enhances the hypoxia-induced epithelial-mesenchymal transition to promote metastasis in pancreatic cancer. Mol Cancer. 2017;16(1):169. | ||
Yoshida K, Toden S, Ravindranathan P, Han H, Goel A. Curcumin sensitizes pancreatic cancer cells to gemcitabine by attenuating PRC2 subunit EZH2, and the lncRNA PVT1 expression. Carcinogenesis. 2017;38(10):1036–1046. | ||
Iyer S, Modali SD, Agarwal SK. Long noncoding RNA MEG3 is an epigenetic determinant of oncogenic signaling in functional pancreatic neuroendocrine tumor cells. Mol Cell Biol. 2017;37(22):e00278-17. | ||
Ma Z, Huang H, Wang J, et al. Long non-coding RNA SNHG15 inhibits p15 and KLF2 expression to promote pancreatic cancer proliferation through EZH2-mediated H3K27me3. Oncotarget. 2017;8(48):84153–84167. | ||
Schmitt AM, Chang HY. Long noncoding RNAs in cancer pathways. Cancer Cell. 2016;29(4):452–463. | ||
Xie X, Tang B, Xiao YF, et al. Long non-coding RNAs in colorectal cancer. Oncotarget. 2016;7(5):5226–5239. | ||
Sun M, Nie F, Wang Y, et al. LncRNA HOXA11-AS promotes proliferation and invasion of gastric cancer by scaffolding the chromatin modification factors PRC2, LSD1, and DNMT1. Cancer Res. 2016;76(21):6299–6310. | ||
Yoshida K, Toden S, Ravindranathan P, Han H, Goel A. Curcumin sensitizes pancreatic cancer cells to gemcitabine by attenuating PRC2 subunit EZH2, and the lncRNA PVT1 expression. Carcinogenesis. 2017;38(10):1036–1046. | ||
Huang M-D, Chen W-M, Qi F-Z, et al. Long non-coding RNA TUG1 is up-regulated in hepatocellular carcinoma and promotes cell growth and apoptosis by epigenetically silencing of KLF2. Mol Cancer. 2015;14(1):165. | ||
Zhang E, He X, Yin D, et al. Increased expression of long noncoding RNA TUG1 predicts a poor prognosis of gastric cancer and regulates cell proliferation by epigenetically silencing of p57. Cell Death Dis. 2016;7(2):e2109. | ||
Liu H, Zhou G, Fu X, et al. Long noncoding RNA TUG1 is a diagnostic factor in lung adenocarcinoma and suppresses apoptosis via epigenetic silencing of Bax. Oncotarget. 2017;8(60):101899–101910. | ||
Riou P, Villalonga P, Ridley AJ. Rnd proteins: multifunctional regulators of the cytoskeleton and cell cycle progression. Bioessays. 2010;32(11):986–992. | ||
Tang Y, Hu C, Yang H, et al. Rnd3 regulates lung cancer cell proliferation through Notch signaling. PLoS One. 2014;9(11):e111897. | ||
Zhu Y, Zhou J, Xia H, et al. The Rho GTPase RhoE is a p53-regulated candidate tumor suppressor in cancer cells. Int J Oncol. 2014;44(3):896–904. | ||
Zhao H, Yang J, Fan T, Li S, Ren X. RhoE functions as a tumor suppressor in esophageal squamous cell carcinoma and modulates the PTEN/PI3K/AKT signaling pathway. Tumour Biol. 2012;33(5):1363–1374. | ||
Villalonga P, Fernández de Mattos S, Ridley AJ. RhoE inhibits 4E-BP1 phosphorylation and eIF4E function impairing cap-dependent translation. J Biol Chem. 2009;284(51):35287–35296. | ||
Poch E, Miñambres R, Mocholí E, et al. RhoE interferes with Rb inactivation and regulates the proliferation and survival of the U87 human glioblastoma cell line. Exp Cell Res. 2007;313(4):719–731. | ||
Villalonga P, Guasch RM, Riento K, Ridley AJ. RhoE inhibits cell cycle progression and Ras-induced transformation. Mol Cell Biol. 2004;24(18):7829–7840. | ||
Babula P, Masarik M, Adam V, et al. Mammalian metallothioneins: properties and functions. Metallomics. 2012;4(8):739–750. | ||
Pedersen MØ, Larsen A, Stoltenberg M, Penkowa M. The role of metallothionein in oncogenesis and cancer prognosis. Prog Histochem Cytochem. 2009;44(1):29–64. | ||
Werynska B, Pula B, Muszczynska-Bernhard B, et al. Metallothionein 1F and 2A overexpression predicts poor outcome of non-small cell lung cancer patients. Exp Mol Pathol. 2013;94(1):301–308. | ||
Yap X, Tan HY, Huang J, et al. Over-expression of metallothionein predicts chemoresistance in breast cancer. J Pathol. 2009;217(4):563–570. | ||
Arriaga JM, Greco A, Mordoh J, Bianchini M. Metallothionein 1G and zinc sensitize human colorectal cancer cells to chemotherapy. Mol Cancer Ther. 2014;13(5):1369–1381. | ||
Datta J, Majumder S, Kutay H, et al. Metallothionein expression is suppressed in primary human hepatocellular carcinomas and is mediated through inactivation of CCAAT/enhancer binding protein alpha by phosphatidylinositol 3-kinase signaling cascade. Cancer Res. 2007;67(6):2736–2746. | ||
Huang Y, de La Chapelle A, Pellegata NS. Hypermethylation, but not LOH, is associated with the low expression of MT1G and CRABP1 in papillary thyroid carcinoma. Int J Cancer. 2003;104(6):735–744. | ||
Mao J, Yu H, Wang C, et al. Metallothionein MT1M is a tumor suppressor of human hepatocellular carcinomas. Carcinogenesis. 2012;33(12):2568–2577. | ||
Peng D, Hu TL, Jiang A, et al. Location-specific epigenetic regulation of the metallothionein 3 gene in esophageal adenocarcinomas. PLoS One. 2011;6(7):e22009. | ||
Wei H, Desouki MM, Lin S, Xiao D, Franklin RB, Feng P. Differential expression of metallothioneins (mts) 1, 2, and 3 in response to zinc treatment in human prostate normal and malignant cells and tissues. Mol Cancer. 2008;7:7. | ||
Ponting CP, Oliver PL, Reik W. Evolution and functions of long noncoding RNAs. Cell. 2009;136(4):629–641. | ||
Nagano T, Fraser P. No-nonsense functions for long noncoding RNAs. Cell. 2011;145(2):178–181. |
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.