")
Back to Journals » International Journal of Nanomedicine » Volume 9 » Issue 1
Overendocytosis of gold nanoparticles increases autophagy and apoptosis in hypoxic human renal proximal tubular cells
Authors Ding F, Li Y, Liu J, Liu L, Yu W, Wang Z, Ni H, Liu B, Chen P
Received 31 May 2014
Accepted for publication 19 July 2014
Published 12 September 2014 Volume 2014:9(1) Pages 4317—4330
DOI https://doi.org/10.2147/IJN.S68685
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Fengan Ding,1 Yiping Li,1 Jing Liu,1 Lei Liu,1 Wenmin Yu,1 Zhi Wang,1 Haifeng Ni,2 Bicheng Liu,2 Pingsheng Chen1,2
1School of Medicine, Southeast University, Nanjing, People’s Republic of China; 2Institute of Nephrology, The Affiliated Zhongda Hospital, Southeast University, Nanjing, People’s Republic of China
Background: Gold nanoparticles (GNPs) can potentially be used in biomedical fields ranging from therapeutics to diagnostics, and their use will result in increased human exposure. Many studies have demonstrated that GNPs can be deposited in the kidneys, particularly in renal tubular epithelial cells. Chronic hypoxic is inevitable in chronic kidney diseases, and it results in renal tubular epithelial cells that are susceptible to different types of injuries. However, the understanding of the interactions between GNPs and hypoxic renal tubular epithelial cells is still rudimentary. In the present study, we characterized the cytotoxic effects of GNPs in hypoxic renal tubular epithelial cells.
Results: Both 5 nm and 13 nm GNPs were synthesized and characterized using various biophysical methods, including transmission electron microscopy, dynamic light scattering, and ultraviolet–visible spectrophotometry. We detected the cytotoxicity of 5 and 13 nm GNPs (0, 1, 25, and 50 nM) to human renal proximal tubular cells (HK-2) by Cell Counting Kit-8 assay and lactate dehydrogenase release assay, but we just found the toxic effect in the 5 nm GNP-treated cells at 50 nM dose under hypoxic condition. Furthermore, the transmission electron microscopy images revealed that GNPs were either localized in vesicles or free in the lysosomes in 5 nm GNPs-treated HK-2 cells, and the cellular uptake of the GNPs in the hypoxic cells was significantly higher than that in normoxic cells. In normoxic HK-2 cells, 5 nm GNPs (50 nM) treatment could cause autophagy and cell survival. However, in hypoxic conditions, the GNP exposure at the same condition led to the production of reactive oxygen species, the loss of mitochondrial membrane potential (ΔΨM), and an increase in apoptosis and autophagic cell death.
Conclusion/significance: Our results demonstrate that renal tubular epithelial cells presented different responses under normoxic and hypoxic environments, which provide an important basis for understanding the risks associated with GNP use–especially for the potential GNP-related therapies in chronic kidney disease patients.
Keywords: gold nanoparticles (GNPs), toxicity, autophagy, apoptosis
Introduction
Gold nanoparticles (GNPs), one of the most well-known nanomaterials, have several advantages, including simple synthetic protocols and stability, especially unique physical, chemical and optical properties. These extraordinary features have made them to be widely applied in biomedicine for diagnostic and therapeutic purposes in the recent past, particularly for biosensors, biomarkers, molecular imaging, photothermal therapy, and targeted delivery of drugs and antigens.1,2 Due to the rapid development of GNP applications, human safety concerns are gaining increased attention, which makes it necessary to better understand the biodistribution and circulation of GNPs in the blood, their elimination from the body, and their potential toxicity to humans.
GNPs are thought to be relatively nontoxic3–7 in most previous studies. Among them, some groups reported that the extent of the toxic nature of these particles depended on the different diameter sizes,8–13 the modifications,14,15 and the surface functional attachments16,17 of the GNPs. In the articles, GNPs were reported to induce various cellular responses, such as necrosis, autophagy, apoptosis, altered gene expression, cell cycle disruption, and oxidative stress.15,18–22 Autophagy (generally thought to be a survival mechanism) could be induced by many cytotoxic stimuli, including various types of nanoparticles,23–26 but it is still unclear whether autophagy plays a prosurvival or a prodeath role under some specific circumstances, such as hypoxic.
Until now, studies on the biological effects of GNPs are often focused on liver, spleen, and pulmonary toxicity.27–32 However, little attention has been paid to kidneys, which are particularly susceptible to xenobiotics due to their high blood supply and ability to concentrate toxins. Sereemaspun et al33 demonstrated that GNPs were heavily taken up by kidney cells, causing nephrotoxicity. To the best of our knowledge, renal tubules, particularly proximal tubules, are susceptible to different types of injuries, such as environmental toxins, nephrotoxic drugs, ischemia/reperfusion, and hypoxic. Abdelhalim and Jarrar clearly observed damage to proximal tubular epithelial cells in rats exposed to GNPs.34,35 Furthermore, numerous metallic nanoparticles are selective nephrotoxicants that preferentially accumulate in the kidneys, further to induce renal injury.36–39 It has been known for some time that chronic kidney diseases (CKD) are often accompanied by hypoxic, which may play an important pathogenic role in the development of renal damage;40 whereas, no study has been performed concerning GNP interactions with hypoxic kidney cells, especially proximal tubules.
Overall, many questions about GNP interactions with hypoxic cells need to be answered. For instance, do hypoxic cells take up GNPs at the same rate as normoxic cells? Do GNPs affect cellular proliferation of hypoxic cells? Do GNPs cause autophagy and/or apoptosis in hypoxic proximal tubular epithelial cells, and is autophagy protective against or detrimental to stress in hypoxic cells? Based on these critical questions, we employed the human renal proximal tubular cells (HK-2) to evaluate the uptake and cytotoxicity of GNPs in normoxic and hypoxic conditions and explore the relative mechanisms of toxicity to determine the biosafety of using gold or gold-derived nanoparticles in patients with CKD.
Materials and methods
Synthesis and characterization of GNPs
GNPs of 5 nm were produced via NaBH4 reduction of a starting solution of sodium citrate and HAuCl4 in water. Typically, 1.0 mL of 1% HAuCl4 (Sigma–Aldrich, St Louis, MO, USA) was added to 97 mL of Milli-Q water (EMD Millipore, Billerica, MA, USA) with vigorous stirring for 1 minute, then 1 mL of 1% sodium citrate (Shanghai Chemical Reagent Co., Ltd., Shanghai, People’s Republic of China) was added under rapid stirring for another 1 minute before the reduction was initiated through the addition of 1 mL of a freshly prepared 1% NaBH4 solution (Sinopharm Chemical Reagent Co., Ltd. People’s Republic of China). GNP formation occurred immediately, as shown by the rapid color change from pale yellow to wine red while stirring vigorously. The 13 nm GNP synthesis was performed using the classical citrate reduction route.41
Briefly, 1 mL of 1% HAuCl4 was boiled in 97 mL of Milli-Q water (EMD Millipore), followed by the addition of 2 mL of 1% sodium citrate under vigorous stirring. The mixed solution was boiled for 15 minutes and then left to cool to room temperature naturally. The GNP precipitates were collected using centrifugation and rinsed with Milli-Q water three times, and then the products were filtered through a 0.22 μm filter and kept at 4°C before use. Transmission electron microscope (TEM) (JEM-1011; JEOL, Tokyo, Japan) and ultraviolet–visible spectrophotometry (UV-2450; Shimadzu, Kyoto, Japan) were used to determine the size, shape, and aggregation state of the nanoparticles, as previously described.13 To investigate the stability of dispersed GNPs under the conditions of biological exposure, the zeta potential and the hydrodynamic diameter were measured after dispersion of GNPs (10 mg/mL) in citrate-stabilized solution, water, phosphate buffered saline (PBS), Dulbecco’s Modified Eagle’s Medium (DMEM), and DMEM medium with 10% fetal bovine serum (FBS) using dynamic light scattering (DLS) (Zetasizer 3000; Malvern Instruments, Malvern, UK).
Cell culture and hypoxic treatment
Human renal proximal tubular epithelial cell line HK-2 was purchased from the Cell Bank of Type Culture Collection of Chinese Academy of Sciences (Shanghai, People’s Republic of China). The HK-2 cells were cultured in high-glucose DMEM (Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 10% FBS, 100 units/mL penicillin, and 100 μg/mL streptomycin (Thermo Fisher Scientific) in humidified air containing 5% CO2 at 37°C. After the cell doubled and redoubled, the vitality of cells was the best; this phase can be identified as the logarithmic growth phase. Cells in the logarithmic growth phase were used in all of the experiments.
To mimic hypoxic conditions, the cells and the GNP-containing medium were then incubated under low-oxygen conditions. The oxygen concentrations were maintained at 1%–3% using a compact gas oxygen controller, which was held under positive pressure in an atmosphere of 94%–92% N2/5% CO2/1%–3% O2 for 24 hours. Hypoxic was confirmed using an oxygen meter. In the hypoxic studies, the cells were incubated with GNPs within the hypoxic workstation for 24 hours until the assays were completed. The control cells were cultured in the normoxic conditions.
Cytotoxicity assays
The cytotoxicity induced by the GNPs was determined using the Cell Counting Kit-8 (CCK-8) assay and the lactate dehydrogenase (LDH) release assay. The cells were seeded on a 96-wellplate at a density of 1×104 cells/well and incubated for 24 hours before the nanoparticles were applied. The determined concentrations of 5 nm and 13 nm GNPs were 0, 1, 25, and 50 nM. To remove the GNPs’ influence with the assay due to their inherent absorbance, cell-free controls containing different concentrations of GNPs were included in the assay in parallel to cells exposed to the nanoparticles of corresponding doses. At the end of the treatment, CCK-8 dye (Dojindo Laboratories, Kamimashiki-gun, Kumamoto, Japan) was added to each well, and the plates were incubated for another 2 hours at 37°C. Subsequently, the absorbency of the solution was measured at a wavelength of 450 nm using an enzyme-linked immunosorbent assay reader (Thermo Fisher Scientific). The cell viability was calculated as the percentage of viable cells compared with the untreated controls.
The activity of the LDH released from the damaged cells was measured using a cytotoxicity detection kit (Jiancheng Bioengineering Research Institute, Nanjing, People’s Republic of China), according to the manufacturer’s protocol. The cells were plated at a density of 1×104 cells/well on a 96-well plate and cultured for 24 hours. The medium was replaced with fresh medium containing the GNPs of various concentrations, while media alone without GNPs was a control. After treatment for 24 hours, the supernatant from each well was transferred into the corresponding wells of an optically clear 96-well plate; the wells were then washed with PBS three times, and 0.1% triton X-100 in PBS was added to each well to lyse the cell membranes for 10 minutes. Next, the lysed solution was transferred to the 96-well plate with supernatant, followed by the addition of the substrate mix (reagent A and reagent B; 5:1). The plate was incubated for 15 minutes at 37°C, followed by the addition of reagent C. After incubation at 37°C for another 15 minutes, a termination solution was added to each well. The absorbance was recorded at 450 nm using an enzyme-linked immunosorbent assay reader. The cell viability was calculated as the percentage of treated wells relative to the untreated controls. Three independent experiments and three replicates for all viability experiments were performed.
Subcellular ultrastructure and localization of GNPs using TEM
The TEM analysis was used to investigate the uptake and intracellular localization of the GNPs inside the cells. The cells were seeded on 6-well plates at a density of 5×105 cells/well and cultured for 24 hours. Then, the cells were exposed to the GNPs (50 nM) for 24 hours. At the predetermined time, the cells were collected, washed three times with PBS, pelleted using centrifugation, and fixed in 2.5% glutaraldehyde for 2 hours. After pellets were washed by PBS, the postfixation with 1% osmium tetroxide was performed.
Finally, the pellets were dehydrated with an ascending series of alcohols before embedding the samples in Araldite® (Huntsman Advanced Materials, The Woodlands, TX, USA). The specimens were cut into ultrathin sections (50~70 nm), which were placed onto copper grids and stained with uranyl acetate and lead citrate. The ultrastructural analysis was performed using a JEM-1011EX instrument (JEOL, Tokyo, Japan).
Annexin V–fluorescein isothiocyanate (FITC)/propidium iodide (PI) apoptosis assay
Cell apoptosis was assessed by flow cytometry using the Annexin V-FITC/PI assay kit (Nanjing KeyGen Biotech, Nanjing, China) to differentiate the survival and apoptotic cells. The HK-2 cells were plated on 6-well plates at initial densities of 5×105 cells/well and incubated for 24 hours prior to the addition of the GNPs. Fresh medium containing GNPs (50 nM) was added to the cells and incubated for another 24 hours. Thereafter, the cells were trypsinized, washed with PBS, resuspended in binding buffer, and incubated with staining solution (annexin V/PI =1:2) in the dark for 20 minutes at room temperature. Immediately after the annexin V/PI staining, fluorescence-activated cell sorting (FACS) analysis was performed using a FACSCalibur ™ flow cytometer (BD Biosciences, San Jose, CA, USA).
Immunofluorescence and confocal microscopy analysis
Cells were plated on cover slips in a 24-well plate and cultured with standard medium containing 10% FBS. After treatment with various concentrations of GNPs (0, 1, 25, and 50 nM ) for 24 hours, the cells were washed with PBS at 4°C, fixed with 4% paraformaldehyde in PBS for 15 minutes at room temperature and permeabilized using 0.1% Triton X-100 in PBS for 15 minutes at room temperature. After blocking unspecific binding sites in 2% bovine serum albumin, the cells were incubated with 1 μg/mL rabbit antihuman LC3B antibody (Sigma–Aldrich Co.; dilution 1:1,000) at 4°C overnight. On the following day, the slides were washed three times with PBS and treated with the appropriate FITC-labeled secondary antibody (Santa Cruz Biotechnology Inc., Dallas, TX, USA; dilution 1:1,000) for 1 hour at 37°C in the dark. Then, the cells were stained with nuclear stain Hoechst 33342 (10 μg/mL) for 5 minutes at room temperature. Finally, the images were taken using a Confocal Microscope (FV1000; Olympus Corporation, Tokyo, Japan).
Determination of intracellular reactive oxygen species (ROS) generation
The level of intracellular ROS was determined using 2′, 7′-dichlorofluorescin diacetate (H2DCFDA; Sigma–Aldrich), which is one of the most widely used techniques for directly measuring the redox state of a cell. After exposure to various concentrations of GNPs for 24 hours, the cells were rinsed with PBS and then stained with H2DCFDA diluted in serum-free medium and incubated for 30 minutes at 37°C in the dark. After incubation, the dichlorofluorescin diacetate (DCF-DA) reagent was removed, and the cells were washed three times with serum-free medium and then resuspended in PBS. The fluorescence was measured using a FACSCalibur ™ flow cytometer at excitation and emission wavelengths of 488 nm and 525 nm, respectively, for DCF fluorescence within 30 minutes.
Western blotting
The cells were plated on 24-well plates and cultured in DMEM supplemented with 10% FBS for 24 hours. After the cells were treated with GNPs of determined concentrations for another 24 hours, they were washed three times with ice-cold PBS and lysed with radio immunoprecipitation assay buffer containing protease inhibitors. After 15 minutes of incubation on ice to ensure complete lysis, the cell lysates were centrifuged at 13,000× g for 10 minutes at 4°C. The supernatant containing the cytoplasmic protein fraction was transferred to a new tube. The protein concentrations of the lysates were examined using the Bradford protein assay kit. The cell lysates were boiled and separated using sodium dodecyl sulfate polyacrylamide gel electrophoresis and transferred onto a polyvinylidene difluoride membrane via semidry transfer (Bio-Rad Laboratories Inc., Hercules, CA, USA). The membranes were washed in Tris-buffered saline containing 0.1% Tween 20 (TBS-T), blocked with 5% nonfat milk in TBS-T for 1 hour at room temperature, and incubated with a primary rabbit monoclonal antibody against LC3B (Sigma–Aldrich; dilution 1:1,000), anti-Beclin-1 (Cell Signaling Technology, USA; dilution 1:1,000) or β-actin (Bioworld Technology Inc, USA; dilution 1:5,000) overnight at 4°C. The membranes were washed three times in TBS-T, followed by incubation with the appropriate horseradish peroxidase-linked secondary antirabbit antibodies (Santa Cruz Biotechnology Inc.; dilution 1:5,000) for 1 hour at room temperature. The specific proteins on the blots were developed with enhanced chemiluminescence (ECL; Amersham Biosciences, Piscataway, NJ, USA) and visualized as the bands on an CL-XPosure Film (Thermo Fisher Scientific). The optical densities of the bands were measured on the GS710 Densitometer and analyzed with Quantity One image analysis software (Bio-Rad Laboratories Inc.).
Detection of the changes in the mitochondrial membrane potential (ΔΨM)
The mitochondrial membrane potential was determined using a JC-1 Apoptosis Detection Kit (Nanjing KeyGen Biotech, Nanjing, China). The HK-2 cells were plated on 6-well plates and treated with 0 nM and 50 nM of GNPs for 24 hours. Then, the ΔΨM was processed as per the manufacturer’s instructions and analyzed using flow cytometry at an excitation wavelength of 488 nm and an emission wavelength of 530 nm.
Statistical analysis
All experiments were performed at least three times, and the results were shown as the mean ± standard deviation. The statistical analysis was performed using the Student’s t-test with the SPSS 16.0 statistical program (SPSS Inc., Chicago, IL, USA). *P<0.05 was considered statistically significant.
Results
Synthesis and characterization of GNPs
Two particle sizes (5 nm and 13 nm) of citrate GNPs were synthesized in situ and characterized as previously described. The TEM and DLS results for the GNP samples are summarized in Table 1. Our TEM study showed that particles had either a spherical or slightly elliptical shape with a narrow size distribution (Figure 1A), and the average particle’s size was 5.07±0.45 nm and 13.41±0.94 nm, and the corresponding size distribution histograms were shown in Figure 1B. Comparable dimensions – 11.12 nm and 20.60 nm, 19.98 nm and 24.19 nm, 28.14 nm and 34.80 nm for the 5 nm and 13 nm GNPs – were examined after a 24-hour incubation in water solution, citrate stabilized solution, and cell culture media (DMEM supplemented with 10% FBS) using DLS, separately. Significant agglomeration was observed after GNPs dispersed in PBS and DMEM medium, but not in water solution. The surface charges based on the zeta potential measurements were −47.7±10.6 mV and −44.6±14.7 mV for the 5 nm and 13 nm GNPs in water solution, −40.2±9.7 mV and −36.8±15.9 mV in citrate stabilized solution and −25.2±4.6 mV and −28.4±1.96 mV in cell culture medium (DMEM supplemented with 10% FBS) after a 24-hour incubation. The presence of a single peak with a low polydispersity index (Table 1) in the size distribution confirmed that the GNPs were monodispersed. Our results demonstrated that the sizes of both GNPs in the cell culture media were greater than those in the water solution, while the zeta potentials in the cell culture media were lower than in the water solution (confirmed by DLS). This result may reflect the formation of a protein corona around the GNPs, as it has already been observed for other types of nanoparticles. In addition, the peaks in the UV-vis spectra of the 5 nm and 13 nm GNPs were detected at 519 nm and 520 nm, respectively, further indicating the formation of GNPs (Figure 1C).
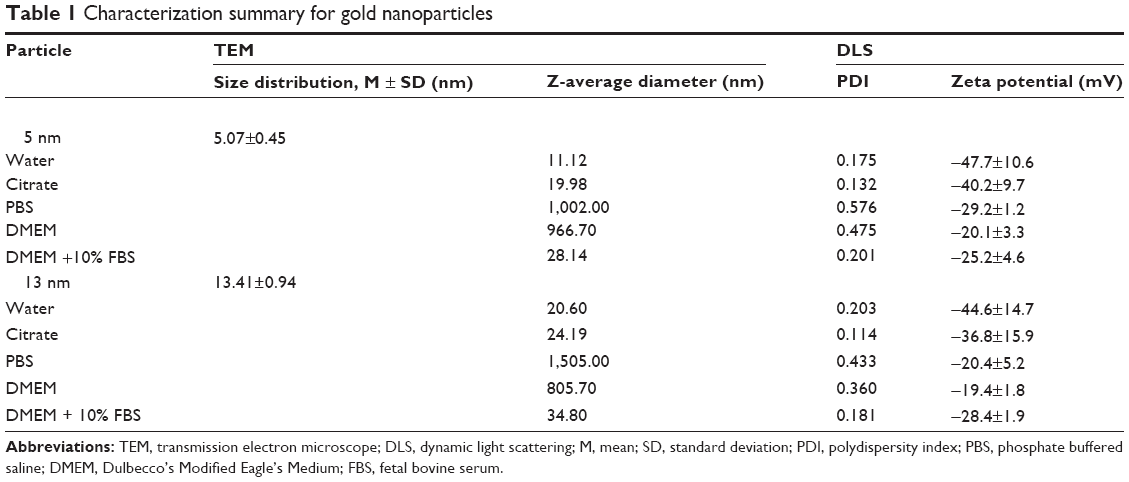 | Table 1 Characterization summary for gold nanoparticles |
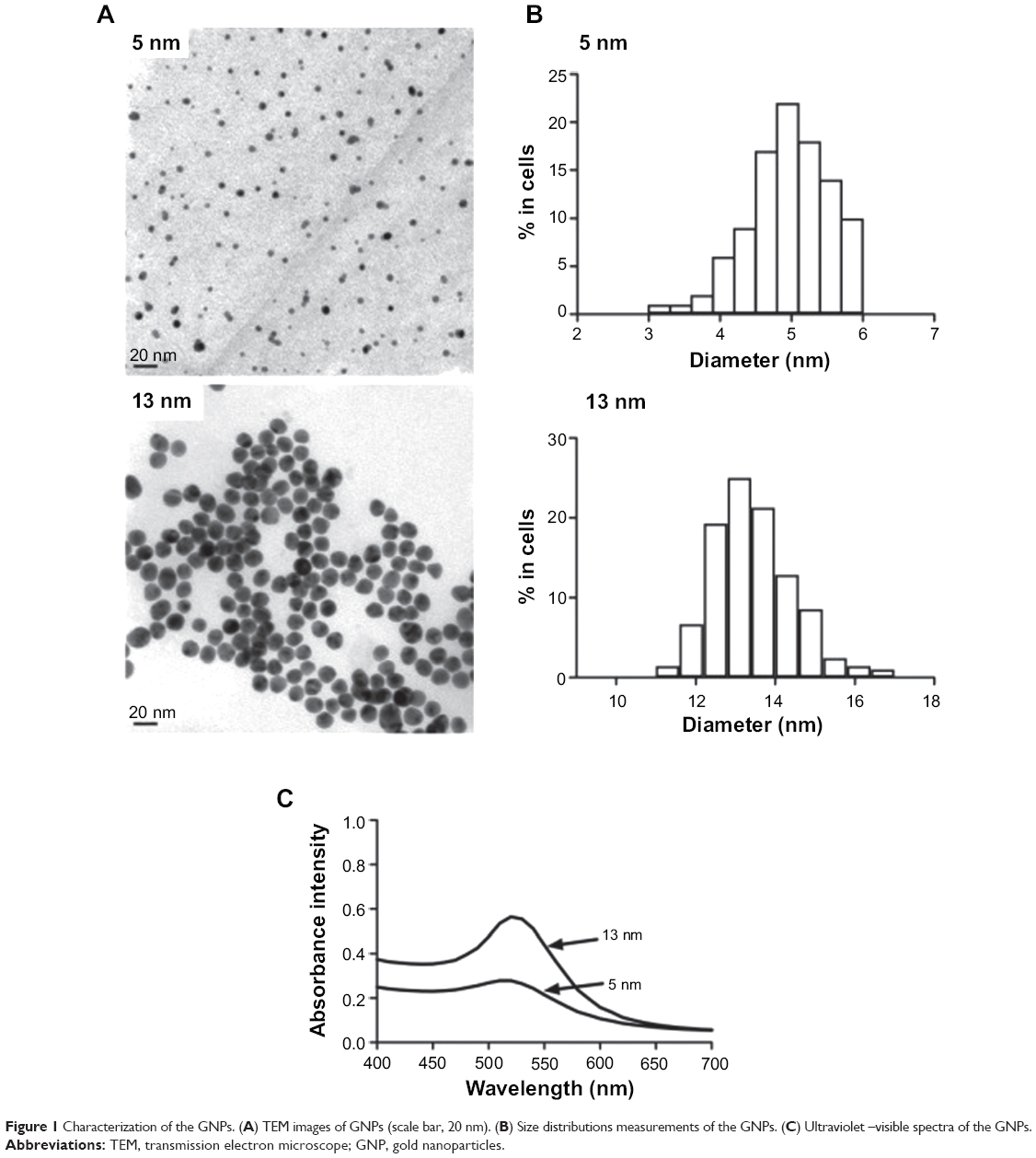 | Figure 1 Characterization of the GNPs. (A) TEM images of GNPs (scale bar, 20 nm). (B) Size distributions measurements of the GNPs. (C) Ultraviolet –visible spectra of the GNPs. |
Cytotoxicity of the gold nanoparticles
The cytotoxicity of the GNPs in both hypoxic and normoxic HK-2 cells was evaluated using both the CCK-8 assay and the LDH detection. The cell viability was examined using the CCK-8 assay and reflected the cell metabolic activity in living cells, and the data were expressed as the percentage of the untreated control. The results in Figure 2A indicated that the cell viability was decreased as the concentration of GNPs was increased, and the change of the small size GNPs was more pronounced than that of the large size. Furthermore, the cell viability had a greater GNP-induced reduction in hypoxic conditions than in air. As shown in Figure 2A, compared with cells grown in 21% oxygen, a much higher cytotoxicity was examined for hypoxic cells treated with the 5 nm GNPs at a concentration of 50 nM (P=0.038), while the cell viability of the 13 nm GNPs group did not change, even at the same concentration of 50 nM (P=0.152).
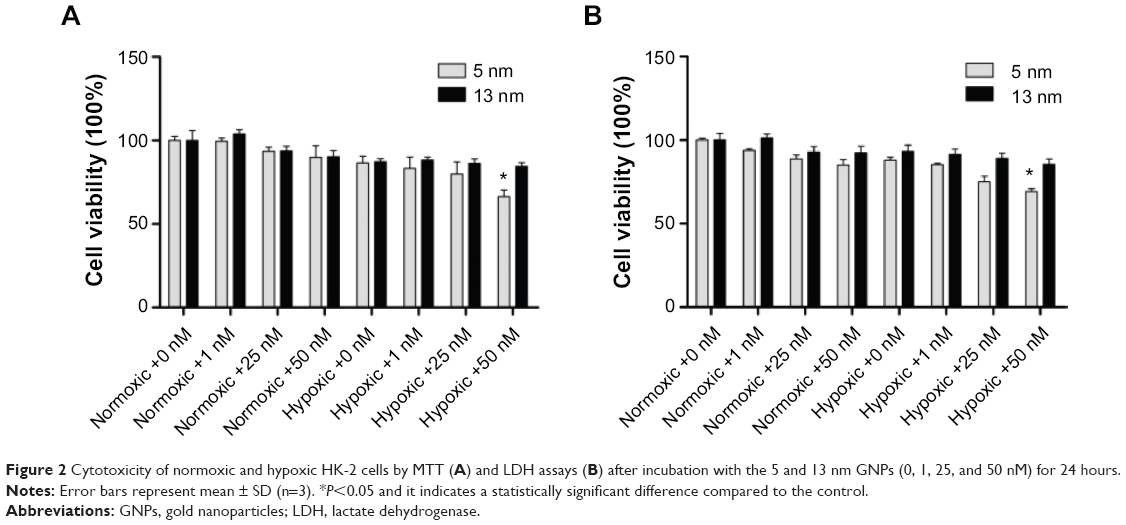 | Figure 2 Cytotoxicity of normoxic and hypoxic HK-2 cells by MTT (A) and LDH assays (B) after incubation with the 5 and 13 nm GNPs (0, 1, 25, and 50 nM) for 24 hours. |
To further investigate GNP-mediated cytotoxicity on the hypoxic HK-2 cells, the amount of LDH released, which is a sensitive marker for cell membrane integrity, was analyzed after the cells were exposed to GNPs. We found that the 5 nm GNPs presented higher damage than did the 13 nm GNPs and that the 5 nm GNPs showed slightly higher cytotoxicity in hypoxic conditions than that in natural air (Figure 2B), which further confirmed the results from the CCK-8 assay. Now that 50 nM of 5 nm GNPs could induce a significant cytotoxicity under hypoxic conditions, we chose the concentration of 50 nM of 5 nm GNPs for the following experiments to explore the related mechanisms.
GNP uptake and intracellular localization
The cellular uptake of GNPs was observed both in hypoxic and normoxic conditions treated with 5 nm GNPs (50 nM) for 24 hours. GNPs appeared as bright spots within the cells, and most of them located around the nuclei under light microscopy. More particles were observed inside the hypoxic cells compared to the normoxic cells. However, no obvious morphological alterations for GNP-exposed cells were found (Figure 3A), whether in normoxic (Figure 3B) or hypoxic conditions (Figure 3C).
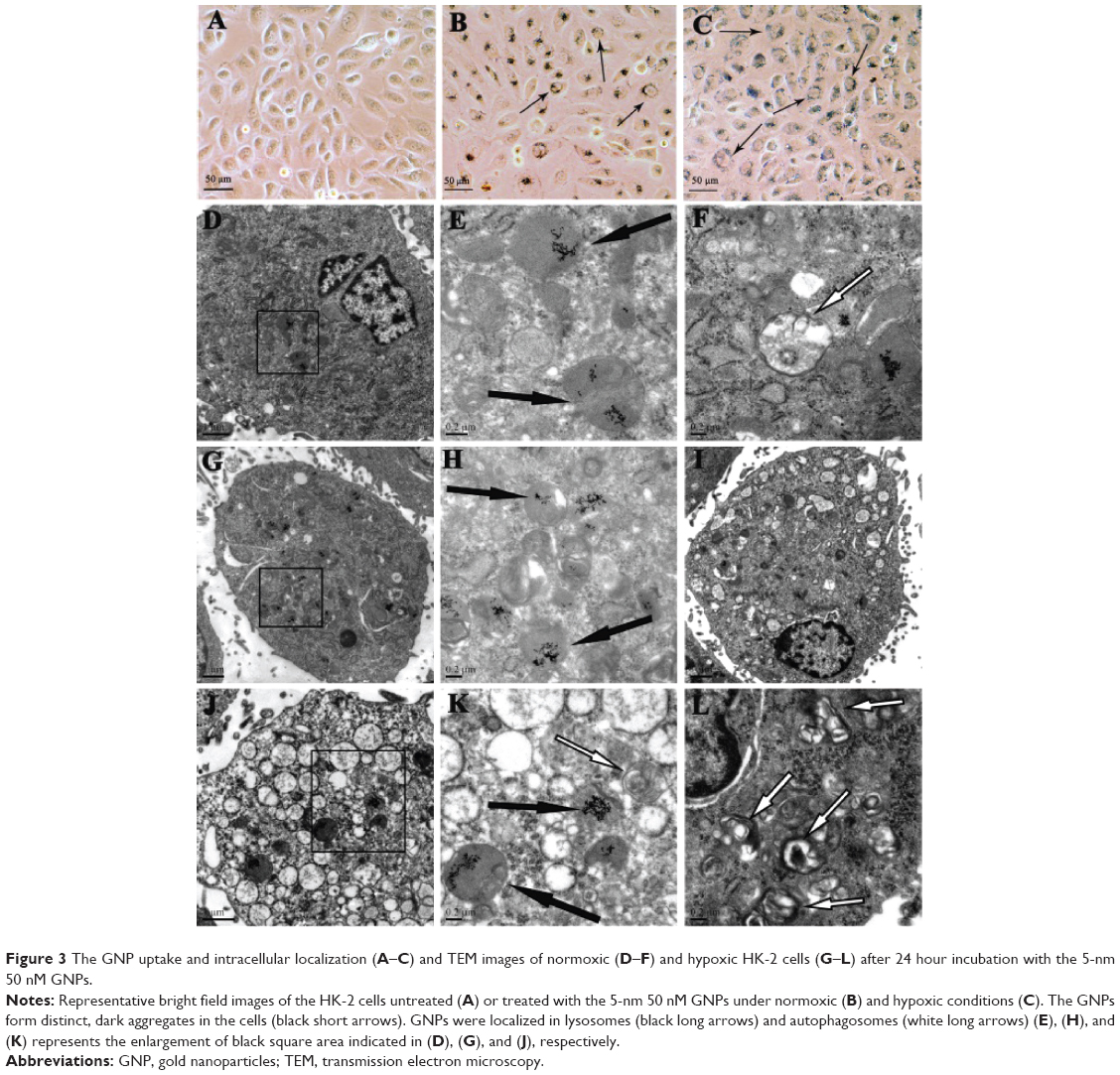 | Figure 3 The GNP uptake and intracellular localization (A–C) and TEM images of normoxic (D–F) and hypoxic HK-2 cells (G–L) after 24 hour incubation with the 5-nm 50 nM GNPs. |
To further clarify the intracellular fate of the GNPs in the hypoxic cells, TEM analysis of ultrathin sections was used to visualize the GNP uptake and the intracellular distribution both in normoxic and hypoxic cells after incubation with GNPs. As shown in Figure 3D–L, more GNPs were internalized into the hypoxic cells (Figure 3G and I) than those into the normoxic cells (Figure 3D), similar to the results from the light microscopy. It also indicated that most of the internalized GNPs was localized inside the lysosomes as small or large aggregates (Figure 3E,F,H,J,K), while few GNP aggregates localized in the intercellular compartments, such as phagosomes for both the normoxic and hypoxic cells. Moreover, compared with normoxic cells, the GNPs were more prone to be internalized in hypoxic cells, which caused the formation of an increase in autophagosome (Figure 3L), together with a greater number of vacuoles and increasing diameters of the existing vacuoles (Figure 3I,J,K).
GNPs induce apoptosis in hypoxic cells
To understand the modes of cell death, the annexin V-FITC/PI double staining was analyzed using flow cytometry. The percentage of apoptotic cells death was presented as the sum of the percentage of early apoptotic cells and late apoptotic cells.
As shown in Figure 4, only few apoptotic cells were detected in the normoxic control group (6.56%±1.01%). However, a significant increase in the percentage of apoptotic cells was detected in the GNPs and the hypoxic cotreatment group (21.11%±1.20%) compared to that in the hypoxic control group (10.45%±2.33%) and the GNP-treated normoxic group (8.48%±1.26%), which indicated that the 5-nm GNPs (50 nM) were more efficient to induce cellular apoptosis in 1% oxygen environments than in 21% oxygen conditions. The data were consistent with the CCK-8 and LDH results.
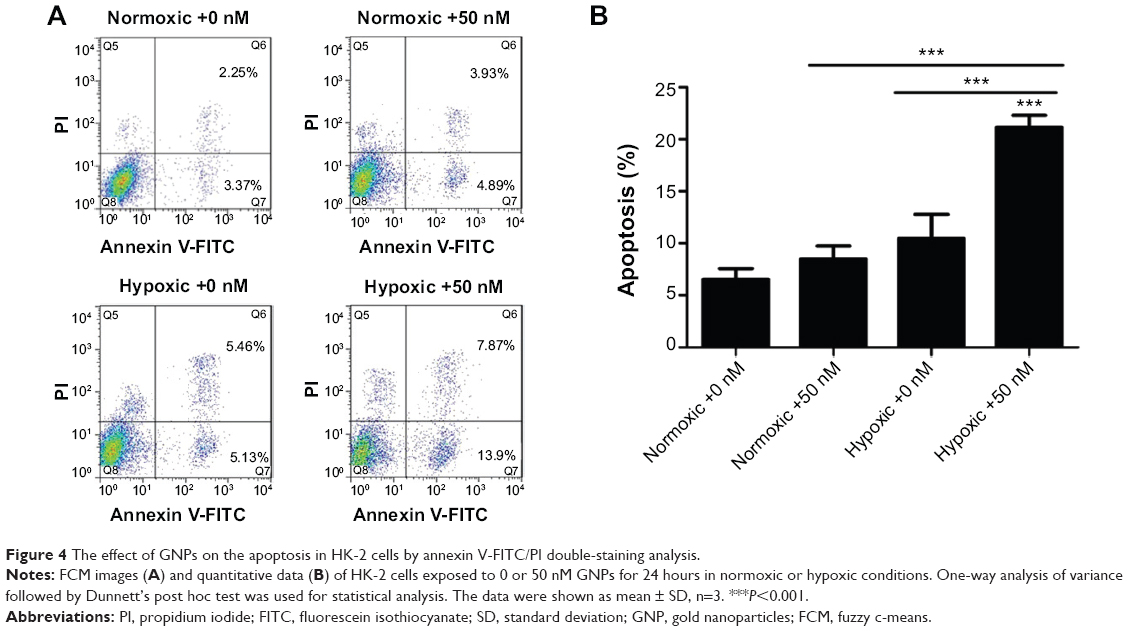 | Figure 4 The effect of GNPs on the apoptosis in HK-2 cells by annexin V-FITC/PI double-staining analysis. |
GNPs enhance autophagy in hypoxic cells
Autophagy is generally thought to be a cell survival mechanism, but it is also suggested to be an alternative cell death pathway. Few works have demonstrated that GNPs can induce autophagy.18,21 Based on this finding, we proceeded to measure whether autophagy played a role in the examined GNP-induced cell toxicity under hypoxic conditions. Therefore, we studied the ultrastructural changes in cells from the different groups using TEM (Figure 3F and L). As shown in Figure 3L, hypoxic cells treated with GNPs clearly exhibited an accumulation of autophagic vacuoles. Next, the induction of autophagy was assessed using LC3 (one autophagy biomarker) immunofluorescence.
In Figure 5A and B, the GNP-treated hypoxic cells induced a significant accumulation of LC3-positive vesicles compared with the control. Simultaneously, we performed Western blotting analysis of the protein extracts from the different cell groups for autophagy marker proteins, microtubule-associated protein 1 light chain 3 (MAP–LC3B) and Beclin-1 (Figure 5C). A quantitative analysis of the proteins was shown in Figure 5D, and β-actin was used as a loading control. We used the ratio of LC3-II/LC3-I and Beclin-1 to reflect the autophagosome formation, and the results indicated that GNP-treated hypoxic cell presented significantly autophagosome formation compared to other groups. Besides, the ratio of LC3-II/LC3-I and Beclin-1 expression was also enhanced compared with the control group in normoxic conditions. To sum up, these results strongly suggested that the 5 nm GNPs (50 nM) could cause autophagy in normoxic conditions, while hypoxic treatment could exaggerate the effect.
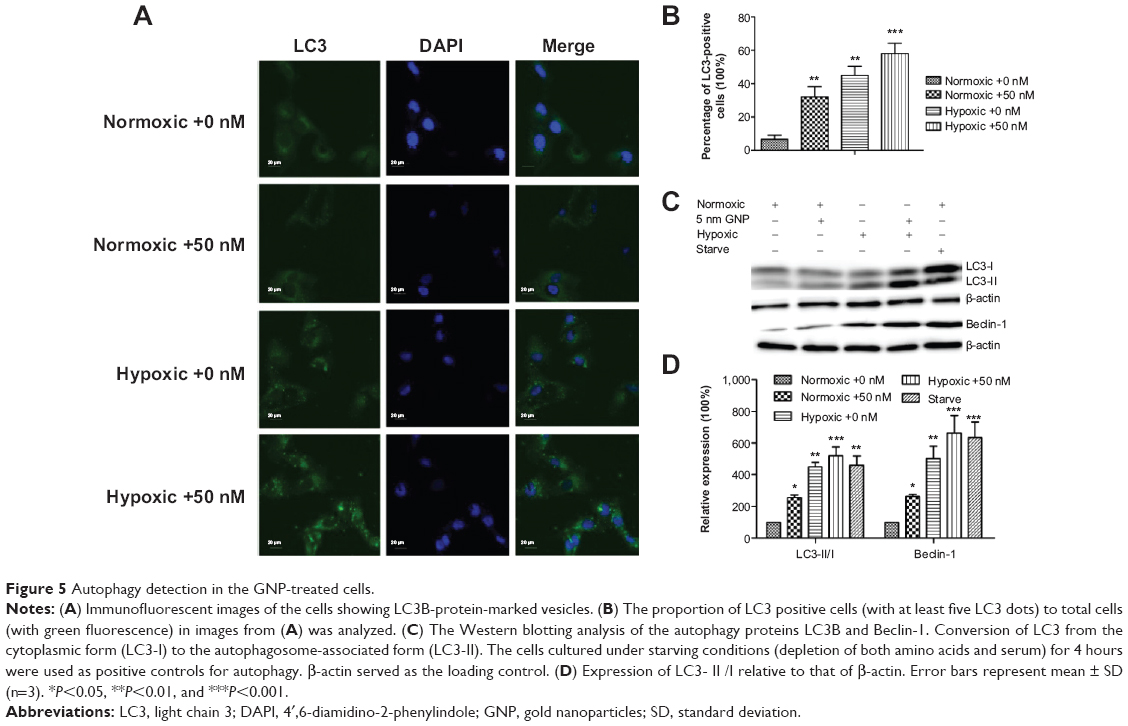 | Figure 5 Autophagy detection in the GNP-treated cells. |
GNPs enhance ROS generation in hypoxic cells
Oxidative stress is believed to be one of the most important mechanisms of nanotoxicity, and the levels of intracellular ROS serve as reliable indicators of oxidative stress, which is essential for the induction of autophagy. To assess the potential role of oxidative stress, intracellular ROS generation was measured after 24 hours of exposure to 5 nm GNPs at 50 nM in both normoxic and hypoxic conditions. As shown in Figure 6, we observed that there was an approximately two-fold increase in ROS in the GNP-treated normoxic group and a nearly seven-fold increase in the GNP-treated hypoxic group over the normoxic control cells. We also found that the GNP-induced ROS was 1.63-fold increase over the control level under hypoxic conditions. The ROS generation was also examined using fluorescence microscopy, and the brightest fluorescence intensity was detected in hypoxic cells exposed to GNPs. Therefore, the previously stated data suggested that the GNPs resulted in a significant increase of intracellular ROS level, and hypoxic condition enlarged the phenomenon.
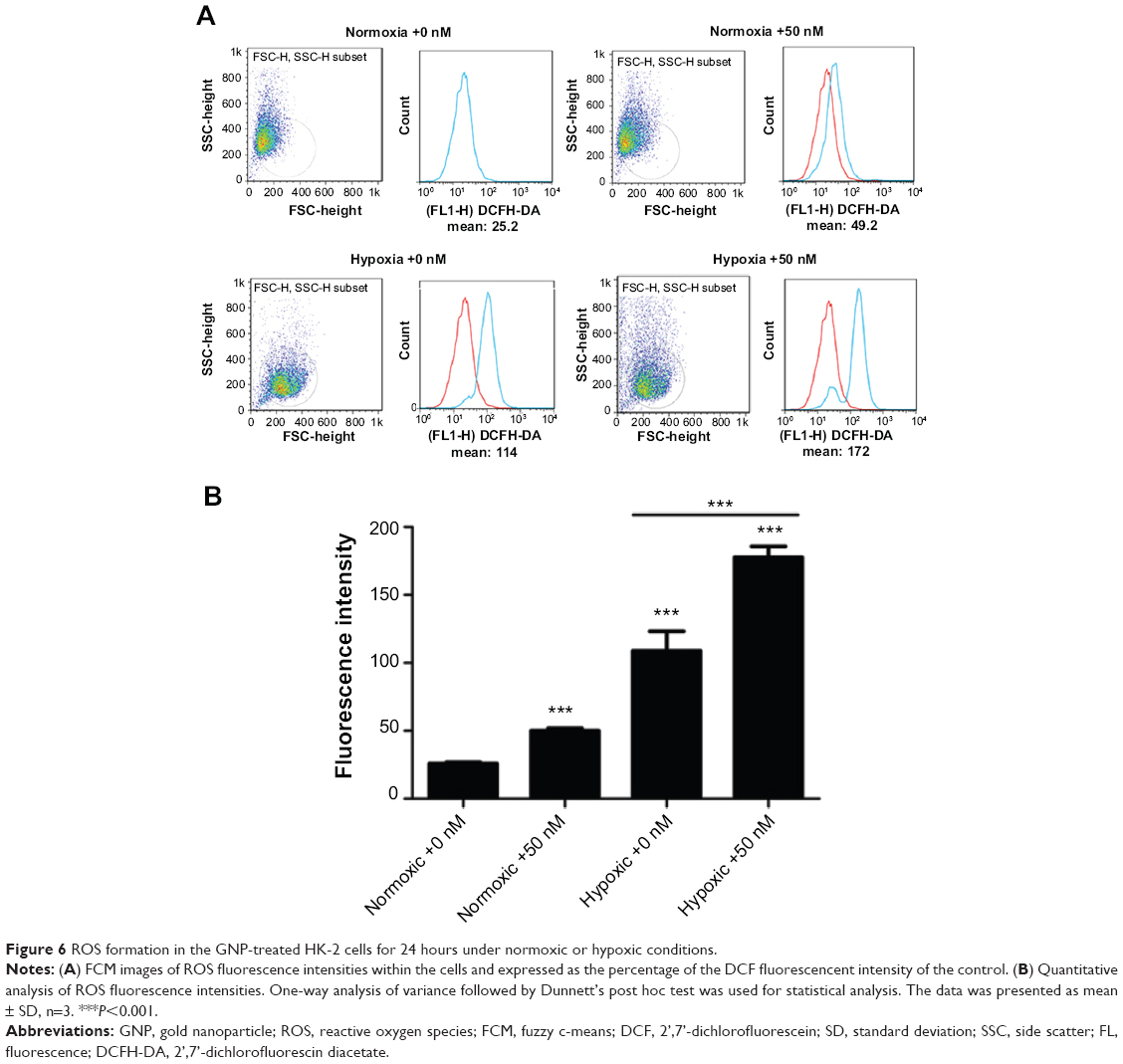 | Figure 6 ROS formation in the GNP-treated HK-2 cells for 24 hours under normoxic or hypoxic conditions. |
Crosstalk between GNP-induced autophagy and apoptosis
To clarify the interaction between GNP-induced autophagy and apoptosis, we studied the changes of apoptosis using 3-methyladenine (3-MA), a well-known autophagy inhibitor.42 As we expected, 3-MA significantly inhibited the autophagy induced by the GNPs (Figure 7A). This treatment led to a 35.85% decrease in the viability and a 29.2% increase in the apoptosis of the GNP-treated cells in normoxic conditions, and a 61.26% enhancement in the viability (Figure 7B) and a 51.48% reduction in the apoptosis (Figure 7C) in hypoxic conditions. These results indicated that autophagy could protect the GNP-treated cells against apoptosis in normoxic conditions; however, in hypoxic conditions, autophagy has a beneficial role in the apoptosis of GNP-treated cells.
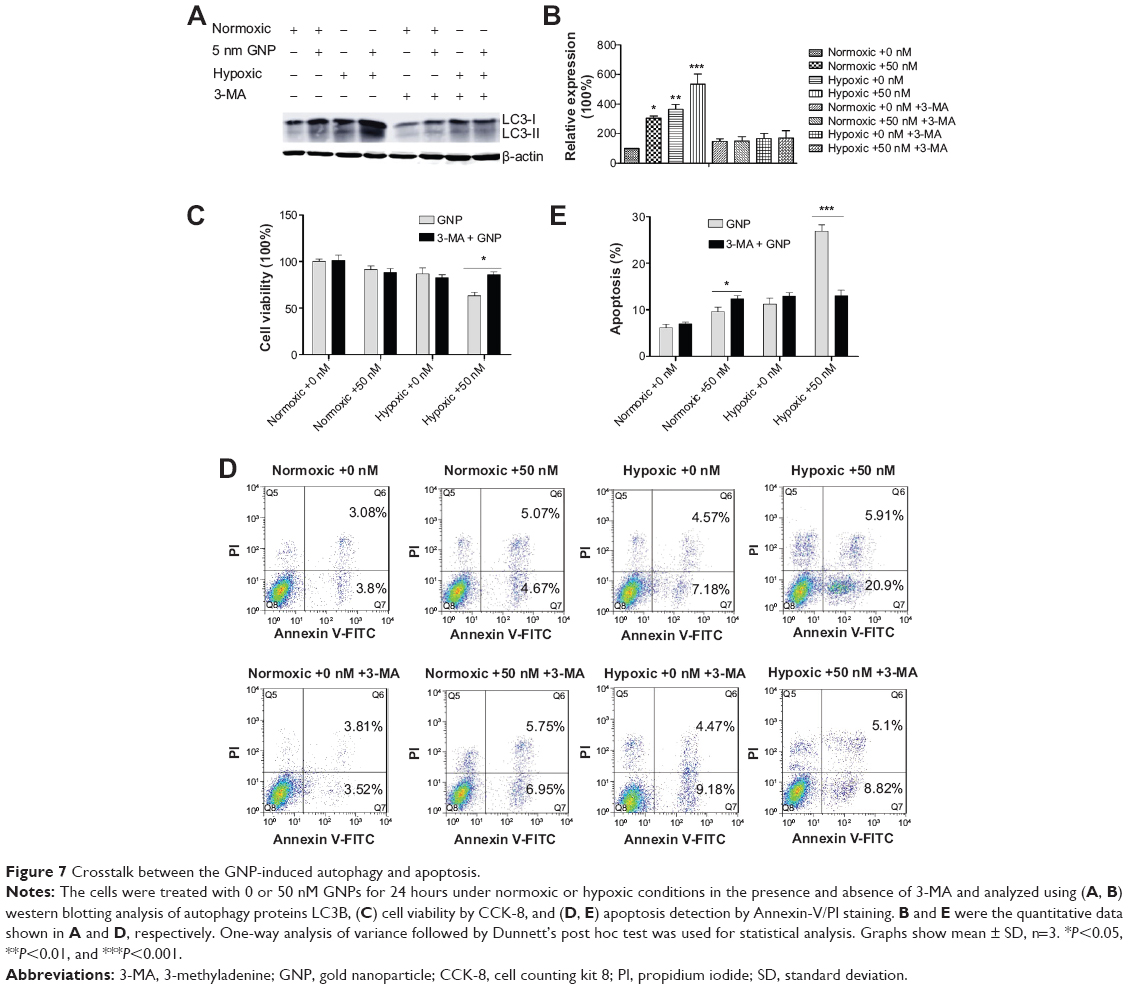 | Figure 7 Crosstalk between the GNP-induced autophagy and apoptosis. |
Mitochondrial membrane potential
Because the autophagy inhibitor 3-MA led to only a 51.48%, not complete reduction in the GNP-treated cell apoptosis, we further explored other mechanisms of apoptosis to better understand this process. It has been reported that the disruption of the mitochondrial membrane potential (ΔΨM) is an early event in apoptosis; therefore, we used JC-1 to assess the ΔΨM by examining its fluorescence intensity. As indicated in Figure 8, 0 nM and 50 nM GNPs treatment led to slight decreases (3.98% and 5.93%) of ΔΨM in normoxic conditions, while in hypoxic conditions, the decrease of ΔΨM was 16.54% and 28.29%, respectively. The GNP-treated hypoxic cells induced a much higher reduction of ΔΨM, which was statistically different from that of any other groups. This result demonstrates that GNPs were able to trigger mitochondrial damage, which led to the enhancement of apoptosis under hypoxic conditions.
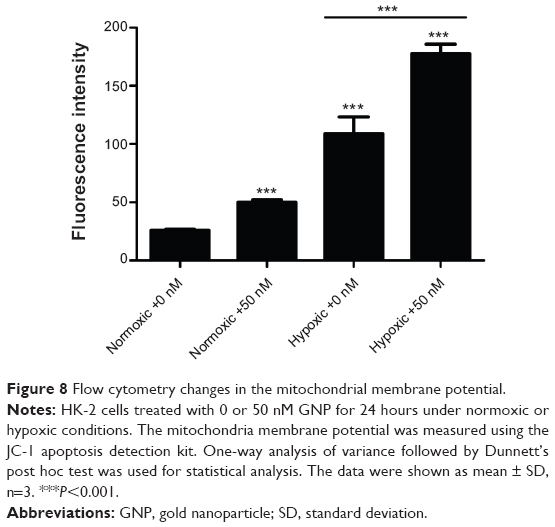 | Figure 8 Flow cytometry changes in the mitochondrial membrane potential. |
Discussion
This experiment was designed to determine the effects of GNPs on hypoxic human HK-2 cells. We noticed that the cellular uptake of GNPs was significantly increased at hypoxic conditions in HK-2 cells compared with normoxic conditions. However, Jain et al43 reported that the GNP cellular uptake was lower under hypoxic than normoxic conditions. These results seem contradictory, but it is important to understand that the experimental conditions of these two studies are different. Their cellular uptake experiment was performed under 0.1%–1%, while we did it under 1%–3% oxygen. In addition, these two studies used different cell lines. Thus, based on these findings, we hypothesize that mild hypoxic (1%–3% oxygen) is able to induce an increase in GNPs uptake, but severe hypoxic conditions or near anoxia (0.1% oxygen) often causes a reduction in GNPs uptake. Basically, hypoxic has previously been accepted to enhance receptor-mediated endocytosis, such as Na, K-ATPase, vascular endothelial growth factor receptors, and Notch.44–46 Various studies have also demonstrated that GNPs are taken into cells via the receptor-mediated endocytosis pathway.11,47 Others have reported that hypoxic can increase the cellular uptake of glucose,48 5-HT,49 Ca2+,50 and some drugs, such as fludeoxyglucose.51,52 Further studies on the mechanisms by which hypoxic may affect the cellular uptake of GNPs are warranted.
Previous studies have shown that cellular or subcellular distributions of the nanoparticles have the potential to exert an influence on cytotoxicity. Our data obtained from CCK-8 and LDH assays showed a significant cytotoxic response after a 24-hour exposure to 5 nm GNPs (50 nM) in hypoxic cells, but no obvious cytotoxicity was observed in the other groups. At the same dosage, the viability of the 5 nm GNPs-treated HK-2 cells was lower than that of the 13 nm GNPs-treated cells. The 5 nm particles showed much higher cytotoxicity than the 13 nm particles, whether in normoxic or in hypoxic conditions, which is consistent with a previously reported trend.8,10,13 In addition, only a slightly reduced proliferation was detected in the hypoxic group compared with the corresponding normoxic group (Figure 2), which may be correlated with the amount of GNPs taken up by hypoxic cells because more GNPs localizing inside cells could induce greater cytotoxicity.
Because the effects of the 5 nm GNPs (50 nM) in hypoxic conditions were assured, we primarily focused on the potential molecular mechanisms. In this study, we demonstrated that the GNP-treated normoxic cells were able to induce autophagy and cell survival, while the hypoxic cells treated with GNPs often underwent autophagic cell death and apoptosis.
The TEM analysis showed that most of particle clusters accumulated in vesicles of lysosomes or phagosomes, which is a direct evidence for the formation of autophagosomes. Moreover, multiple assays, including transfection of GFP-LC3 and expression of the autophagy biomarker of LC3 and Beclin-1, conversion of LC3-I to LC3-II, and autophagy inhibition assay using 3-MA, demonstrated that GNPs could induce autophagy in normoxic cells, while hypoxic treatment could significantly enhance GNPs-induced autophagy. To the best of our knowledge, autophagy can promote cell survival and avert apoptosis under certain cellular conditions. However, it has also been suggested that autophagy may eventually lead to cell death, either independently or in concert with apoptosis.23,24
In our study, the role of autophagy in cell death was further confirmed by the fact that pretreatment with the autophagy inhibitor 3-MA resulted in a 61.26% reduction in cell death. Furthermore, the percentage of cells that underwent apoptosis showed a 51.48% reduction after pretreatment with 3-MA, indicating that apoptosis was partly mediated by autophagy. Moreover, the autophagy inhibition induced an approximately 10% decrease in cell death. These results suggested that the GNPs upregulated autophagy could simultaneously lead to cell apoptosis and autophagic cell death in hypoxic HK-2 cells. A role for autophagy in cell death has been proposed, but the mechanisms controlling autophagy in hypoxic HK-2 cells are unclear. It is widely recognized that ROS play an important role in the induction of nanoparticle-mediated autophagy. Our results show that autophagy activation was accompanied by a corresponding increase in ROS generation. In addition, GNP-induced hypoxic cell autophagy may occur through the phosphatidylinositol 3-kinase (PI3K) pathway (an important pathway in autophagy regulation),53 because it could be efficiently inhibited by 3-MA, which is considered to be a specific inhibitor of PI3K activity.
Taken together, it is probable that the ROS induced by the GNPs can trigger autophagy both directly and indirectly via inhibition of the classical autophagy signaling pathway, phosphatidylinositol 3-kinase/protein kinase B/mammalian target of rapamycin (PI3K/Akt/mTOR), but its mechanism is still unknown. Therefore, further studies should be performed to better understand this process.
Apoptosis is an essential method for maintaining homeostasis in terms of cell division and cell death, and our results from the flow cytometry analysis identified that exposure to 5 nm GNPs (50 nM) increased apoptosis in both hypoxic and normoxic HK-2 cells, while hypoxic treatments resulted in a much higher rate of apoptosis in HK-2 cells, which is consistent with the previously mentioned reports.
Mitochondrial dysfunction plays a principal role in nanoparticle-mediated toxicity, and the loss of ΔΨM can be enhanced or inhibited by many key regulators of apoptosis. In the present study, we found that GNPs induced a significant loss in ΔΨM under hypoxic conditions, and this loss of ΔΨM suggests that the cell death observed in our experiments may be the synergetic effect of mitochondrial dysfunction and autophagy.
An overview of the GNP-induced cell death pathway was summarized in Figure 9. Briefly, the GNPs act as triggers in the early phases of the autophagic process, inducing cytoprotection by eliminating the potential sources of proapoptotic stimuli in normoxic conditions. However, in hypoxic conditions, the prosurvival attempt fails, and GNPs cause cell death, which involves both the autophagic and apoptotic pathways. Autophagy appears to be the major death pattern, because autophagy inhibition causes up to a 61.26% reduction in cell death. We also demonstrated GNP-induced hypoxic cell death was activated through the autophagy-induced and mitochondrial-mediated pathways.
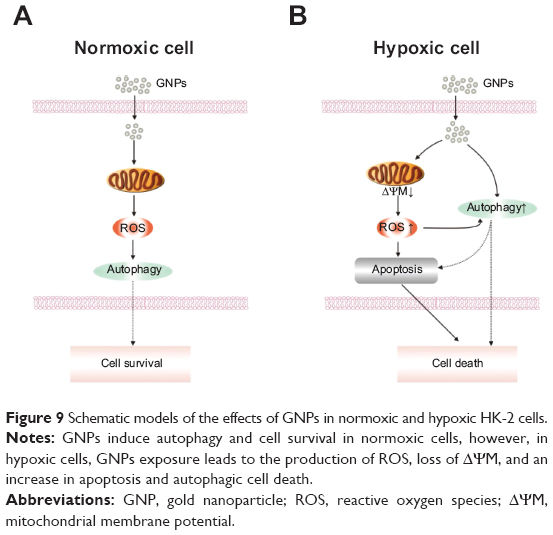 | Figure 9 Schematic models of the effects of GNPs in normoxic and hypoxic HK-2 cells. |
As we know, CKD is increasingly recognized as a worldwide public health problem,54,55 and the patients with CKD unavoidably suffer from chronic hypoxic. Based on all of the data discussed here, we suggest that GNPs could be a potential hazard to CKD patients, and care must be taken when using GNPs in CKD patients to avoid this potential toxicity. More studies are required to determine the GNP biodistribution–accumulation–retention–clearance effects and their toxicity in CKD patients.
Conclusion
In the present study, using HK-2 cells as the assay system, we performed a systematic investigation of the cellular response to the GNPs’ exposure in both normoxic and hypoxic conditions. Our data indicate that GNPs treatment could cause autophagy and cell survival in normoxic conditions, while inducing cell apoptosis and autophagic cell death in hypoxic conditions. Considering the potential toxicity of GNPs in hypoxic HK-2 cells, the knowledge obtained here will be useful for guiding the development of GNP-related therapies in CKD patients and guaranteeing the safe applications of other nanotechnologies.
Acknowledgments
This work was financially supported by National Key Basic Research Program 973 of the People’s Republic of China (number 2012CB517700), the National Nature Science Foundation of the People’s Republic China (number 6524000052) and Jiangsu Province Scientific Research Innovation Project for Graduate Students (number CXLX13_121). We thank Dr Hongtao Wang of Southeast University, Nanjing, People’s Republic of China, for technical assistance.
Disclosure
The authors report no conflicts of interest in this work.
References
Boisselier E, Astruc D. Gold nanoparticles in nanomedicine: preparations, imaging, diagnostics, therapies and toxicity. Chem Soc Rev. 2009;38(6):1759–1782. | ||
Khlebtsov N, Dykman L. Biodistribution and toxicity of engineered gold nanoparticles: a review of in vitro and in vivo studies. Chem Soc Rev. 2011;40(3):1647–1671. | ||
Connor EE, Mwamuka J, Gole A, Murphy CJ, Wyatt MD. Gold nanoparticles are taken up by human cells but do not cause acute cytotoxicity. Small. 2005;1(3):325–327. | ||
Shukla R, Bansal V, Chaudhary M, Basu A, Bhonde RR, Sastry M. Biocompatibility of gold nanoparticles and their endocytotic fate inside the cellular compartment: a microscopic overview. Langmuir. 2005; 21(23):10644–10654. | ||
Gannon CJ, Patra CR, Bhattacharya R, Mukherjee P, Curley SA. Intracellular gold nanoparticles enhance non-invasive radiofrequency thermal destruction of human gastrointestinal cancer cells. J Nanobiotechnology. 2008;6:2. | ||
Simon-Deckers A, Gouget B, Mayne-L’hermite M, Herlin-Boime N, Reynaud C, Carrière M. In vitro investigation of oxide nanoparticle and carbon nanotube toxicity and intracellular accumulation in A549 human pneumocytes. Toxicology. 2008;253(1–3):137–146. | ||
Zhang Q, Hitchins VM, Schrand AM, Hussain SM, Goering PL. Uptake of gold nanoparticles in murine macrophage cells without cytotoxicity or production of pro-inflammatory mediators. Nanotoxicology. 2011;5(3): 284–295. | ||
Boyoglu C, He Q, Willing G, et al. Microscopic studies of various sizes of gold nanoparticles and their cellular localizations. International Scholarly Research Notices Nanotechnology. 2013;2013:1–13. | ||
Cui W, Li J, Zhang Y, Rong H, Lu W, Jiang L. Effects of aggregation and the surface properties of gold nanoparticles on cytotoxicity and cell growth. Nanomedicine. 2012;8(1):46–53. | ||
Coradeghini R, Gioria S, García CP, et al. Size-dependent toxicity and cell interaction mechanisms of gold nanoparticles on mouse fibroblasts. Toxicol Lett. 2013;217(3):205–216. | ||
Mironava T, Hadjiargyrou M, Simon M, Jurukovski V, Rafailovich MH. Gold nanoparticles cellular toxicity and recovery: effect of size, concentration and exposure time. Nanotoxicology. 2010;4(1):120–137. | ||
Pan Y, Neuss S, Leifert A, et al. Size-dependent cytotoxicity of gold nanoparticles. Small. 2007;3(11):1941–1949. | ||
Gao W, Xu K, Ji L, Tang B. Effect of gold nanoparticles on glutathione depletion-induced hydrogen peroxide generation and apoptosis in HL7702 cells. Toxicol Lett. 2011;205(1):86–95. | ||
Liu X, Huang N, Li H, Jin Q, Ji J. Surface and size effects on cell interaction of gold nanoparticles with both phagocytic and nonphagocytic cells. Langmuir. 2013;29(29):9138–9148. | ||
Pan Y, Leifert A, Ruau D, et al. Gold nanoparticles of diameter 1.4 nm trigger necrosis by oxidative stress and mitochondrial damage. Small. 2009;5(18):2067–2076. | ||
Yen HJ, Hsu SH, Tsai CL. Cytotoxicity and immunological response of gold and silver nanoparticles of different sizes. Small. 2009;5(13): 1553–1561. | ||
Goodman CM, McCusker CD, Yilmaz T, Rotello VM. Toxicity of gold nanoparticles functionalized with cationic and anionic side chains. Bioconjugate Chem. 2004;15(4):897–900. | ||
Li JJ, Hartono D, Ong CN, Bay BH, Yung LY. Autophagy and oxidative stress associated with gold nanoparticles. Biomaterials. 2010;31(23): 5996–6003. | ||
Kang B, Mackey MA, El-Sayed MA. Nuclear targeting of gold nanoparticles in cancer cells induces DNA damage, causing cytokinesis arrest and apoptosis. J Am Chem Soc. 2010;132(5):1517–1519. | ||
Li JJ, Zou L, Hartono D, Ong CN, Bay BH, Lanry Young LYl. Gold nanoparticles induce oxidative damage in lung fibroblasts in vitro. Advanced Materials. 2008;20(1):138–142. | ||
Ma X, Wu Y, Jin S, et al. Gold nanoparticles induce autophagosome accumulation through size-dependent nanoparticle uptake and lysosome impairment. ACS Nano. 2011;5(11):8629–8639. | ||
Liu M, Gu X, Zhang K, et al. Gold nanoparticles trigger apoptosis and necrosis in lung cancer cells with low intracellular glutathione. Journal of Nanoparticle Research. 2013;15(8):1–14. | ||
Afeseh Ngwa H, Kanthasamy A, Gu Y, Fang N, Anantharam V, Kanthasamy AG. Manganese nanoparticle activates mitochondrial dependent apoptotic signaling and autophagy in dopaminergic neuronal cells. Toxicol Appl Pharmacol. 2011;256(3):227–240. | ||
Sun T, Yan Y, Zhao Y, Guo F, Jiang C. Copper oxide nanoparticles induce autophagic cell death in A549 cells. PLoS One. 2012;7(8):e43442. | ||
Chen L, Miao Y, Chen L, et al. The role of elevated autophagy on the synaptic plasticity impairment caused by CdSe/ZnS quantum dots. Biomaterials. 2013;34(38):10172–10181. | ||
Zhao Y, Howe JL, Yu Z, et al. Exposure to titanium dioxide nanoparticles induces autophagy in primary human keratinocytes. Small. 2013; 9(3):387–392. | ||
Cho WS, Cho M, Jeong J, et al. Size-dependent tissue kinetics of PEG-coated gold nanoparticles. Toxicol Appl Pharmacol. 2010;245(1): 116–123. | ||
Zhang XD, Wu D, Shen X, et al. Size-dependent radiosensitization of PEG-coated gold nanoparticles for cancer radiation therapy. Biomaterials. 2012;33(27):6408–6419. | ||
Sadauskas E, Wallin H, Stoltenberg M, et al. Kupffer cells are central in the removal of nanoparticles from the organism. Part Fibre Toxicol. 2007;4:10. | ||
Terentyuk GS, Maslyakova GN, Suleymanova LV, et al. Circulation and distribution of gold nanoparticles and induced alterations of tissue morphology at intravenous particle delivery. J Biophotonics. 2009;2(5):292–302. | ||
Gosens I, Post JA, de la Fonteyne LJ, et al. Impact of agglomeration state of nano- and submicron sized gold particles on pulmonary inflammation. Part Fibre Toxicol. 2010;7(1):37. | ||
Abdelhalim MA, Jarrar BM. Gold nanoparticles induced cloudy swelling to hydropic degeneration, cytoplasmic hyaline vacuolation, polymorphism, binucleation, karyopyknosis, karyolysis, karyorrhexis and necrosis in the liver. Lipids Health Dis. 2011;10:166. | ||
Sereemaspun A, Rojanathanes R, Wiwanitkit V. Effect of gold nanoparticle on renal cell: an implication for exposure risk. Ren Fail. 2008;30(3):323–325. | ||
Abdelhalim MA, Jarrar BM. Renal tissue alterations were size-dependent with smaller ones induced more effects and related with time exposure of gold nanoparticles. Lipids Health Dis. 2011;10:163. | ||
Abdelhalim MA, Jarrar BM. The appearance of renal cells cytoplasmic degeneration and nuclear destruction might be an indication of GNPs toxicity. Lipids Health Dis. 2011;10:147. | ||
Kermanizadeh A, Vranic S, Boland S, et al. An in vitro assessment of panel of engineered nanomaterials using a human renal cell line: cytotoxicity, pro-inflammatory response, oxidative stress and genotoxicity. BMC Nephrol. 2013;14:96. | ||
Pujalté I, Passagne I, Brouillaud B, et al. Cytotoxicity and oxidative stress induced by different metallic nanoparticles on human kidney cells. Part Fibre Toxicol. 2011;8:10. | ||
Pujalté I, Passagne I, Brouillaud B, et al. Study of different metallic nanoparticles on human kidney cells: toxicity and oxidative stress. Toxicol Lett. 2011;205:S172. | ||
Passagne I, Morille M, Rousset M, Pujalté I, L’azou B. Implication of oxidative stress in size-dependent toxicity of silica nanoparticles in kidney cells. Toxicology. 2012;299(2–3):112–124. | ||
Erwig LP. Macrophages and hypoxic in human chronic kidney disease. Kidney International. 2008;74(4):405–406. | ||
Lee PC, Melsel D. Adsorption and surface-enhanced Raman of dyes on silver and gold sols. J Phys Chem. 1982;86(17):3391–3395. | ||
Klionsky DJ, Abeliovich H, Agostinis P, et al. Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy. 2008;4(2):151–175. | ||
Jain S, Coulter JA, Butterworth KT, et al. Gold nanoparticle cellular uptake, toxicity and radiosensitisation in hypoxic conditions. Radiother Oncol. 2014;110(2):342–347. | ||
Dada LA, Chandel NS, Ridge KM, Pedemonte C, Bertorello AM, Sznajder JI. Hypoxic-induced endocytosis of Na, K-ATPase in alveolar epithelial cells is mediated by mitochondrial reactive oxygen species and PKC-zeta. J Clin Invest. 2003;111(7):1057–1064. | ||
Mukherjee T, Kim WS, Mandal L, Banerjee U. Interaction between Notch and Hif-alpha in development and survival of Drosophila blood cells. Science. 2011;332(6034):1210–1213. | ||
Dada LA, Welch LC, Zhou G, Ben-Saadon R, Ciechanover A, Sznajder JI. Phosphorylation and ubiquitination are necessary for Na, K-ATPase endocytosis during hypoxic. Cell Signal. 2007;19(9):1893–1898. | ||
Chithrani BD, Chan WC. Elucidating the mechanism of cellular uptake and removal of protein-coated gold nanoparticles of different sizes and shapes. Nano Lett. 2007;7(6):1542–1550. | ||
Tchirikov M, Tchirikov M, Buchert R, Wilke F, Brenner W. Glucose uptake in the placenta, fetal brain, heart and liver related to blood flow redistribution during acute hypoxic. J Obstet Gynaecol Res. 2011; 37(8):979–985. | ||
Lee SL, Fanburg BL. Glycolytic activity and enhancement of serotonin uptake by endothelial cells exposed to hypoxic/anoxia. Circ Res. 1987; 60(5):653–658. | ||
Zanelli SA, Spandou E, Mishra OP, Delivoria-Papadopoulos M. Hypoxic modifies nuclear calcium uptake pathways in the cerebral cortex of the guinea-pig fetus. Neuroscience. 2005;130(4):949–955. | ||
Burgman P, Odonoghue JA, Humm JL, Ling CC. Hypoxic-Induced increase in FDG uptake in MCF7 cells. J Nucl Med. 2001;42(1): 170–175. | ||
Matsui T, Nakata N, Nagai S, et al. Inflammatory cytokines and hypoxic contribute to 18F-FDG uptake by cells involved in pannus formation in rheumatoid arthritis. J Nucl Med. 2009;50(6):920–926. | ||
Park HJ, Lee SJ, Kim SH, et al. IL-10 inhibits the starvation induced autophagy in macrophages via class I phosphatidylinositol 3-kinase (PI3K) pathway. Mol Immunol. 2011;48(4):720–727. | ||
Levey AS, Atkins R, Coresh J, et al. Chronic kidney disease as a global public health problem: approaches and initiatives – a position statement from Kidney Disease Improving Global Outcomes. Kidney Int. 2007;72(3):247–259. | ||
Schieppati A, Remuzzi G. Chronic renal diseases as a public health problem: epidemiology, social, and economic implications. Kidney Int Suppl. 2005;98:S7–S10. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.