Back to Journals » International Journal of Nanomedicine » Volume 13
Overcoming tumor cell chemoresistance using nanoparticles: lysosomes are beneficial for (stearoyl) gemcitabine-incorporated solid lipid nanoparticles
Authors Chen Z, Zheng Y ![]() , Shi Y
, Shi Y ![]() , Cui Z
, Cui Z
Received 15 August 2017
Accepted for publication 6 November 2017
Published 9 January 2018 Volume 2018:13 Pages 319—336
DOI https://doi.org/10.2147/IJN.S149196
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Thomas Webster
Zhe Chen,1,* Yuanqiang Zheng,1,* Yanchun Shi,1 Zhengrong Cui1,2
1Inner Mongolia Key Lab of Molecular Biology, School of Basic Medical Sciences, Inner Mongolia Medical University, Hohhot, Inner Mongolia, China; 2Division of Molecular Pharmaceutics and Drug Delivery, College of Pharmacy, The University of Texas at Austin, Austin, TX, USA
*These authors contributed equally to this work
Abstract: Despite recent advances in targeted therapies and immunotherapies, chemotherapy using cytotoxic agents remains an indispensable modality in cancer treatment. Recently, there has been a growing emphasis in using nanomedicine in cancer chemotherapy, and several nanomedicines have already been used clinically to treat cancers. There is evidence that formulating small molecular cancer chemotherapeutic agents into nanomedicines significantly modifies their pharmacokinetics and often improves their efficacy. Importantly, cancer cells often develop resistance to chemotherapy, and formulating anticancer drugs into nanomedicines also helps overcome chemoresistance. In this review, we briefly describe the different classes of cancer chemotherapeutic agents, their mechanisms of action and resistance, and evidence of overcoming the resistance using nanomedicines. We then emphasize on gemcitabine and our experience in discovering the unique (stearoyl) gemcitabine solid lipid nanoparticles that are effective against tumor cells resistant to gemcitabine and elucidate the underlying mechanisms. It seems that lysosomes, which are an obstacle in the delivery of many drugs, are actually beneficial for our (stearoyl) gemcitabine solid lipid nanoparticles to overcome tumor cell resistance to gemcitabine.
Keywords: gemcitabine, chemoresistance, chemotherapeutic agents, nanomedicine
Nanomedicine and cancer chemotherapy
Cancer is a major public health problem worldwide and the second most common cause of death.1,2 Cancer chemotherapy, the treatment of cancer with one or a combination of chemotherapeutic agents, is one of the mainstream anticancer therapies.3–5 Nanomedicines are nanometer-sized medicinal entities. They are actively explored to diagnose, prevent, or treat cancer.6 Indeed, a few nanomedicines have already been approved by the United States Food and Drug Administration for cancer treatment and more are currently in various stages of preclinical and clinical development.7 Compared to conventional formulations/medicines, nanomedicines have numerous advantages; for example, they can exhibit prolonged systemic circulation time, sustained drug release kinetics, and increased tumor accumulation.8,9 Nanomedicines can be prepared using various materials, including liposomes, micelles, polymeric nanoparticles, solid lipid nanoparticles, inorganic nanoparticles, drug–polymer conjugates, drug–antibody conjugates, and supramolecular vesicular aggregates, etc.
Cancer chemotherapeutic agents and mechanisms of chemoresistance
The first modern cancer chemotherapeutic agent was discovered serendipitously. During World War I (1914–1918), accidental releases of mustard gas led to the discovery of the effect of nitrogen mustard on lymphoma.10 Historically, anticancer drugs were derived from available chemical sources. Synthetic molecules from the chemical industry, in particular dyestuffs and chemical warfare agents, and natural products from plants, bacteria, and fungi are all sources of anticancer agents.11 The breadth of cancer chemotherapeutic agents is vast, which is actually beneficial as most cancer patients receive multi-drug regimens. This is due to the inherent complexity of cancer12 – a non-responder to one chemotherapeutic agent may respond to another. In this review, we focus on traditional cytotoxic chemotherapeutic drugs. Despite the increasing desire by cancer patients for targeted therapies and immunotherapies with reduced adverse effects, cytotoxic drugs still play an indispensable role in systemic cancer therapy, and for many cancers, targeted therapy is not available.
Tumor chemoresistance is a major clinical obstacle to successful tumor therapy.13 Tumor chemoresistance can be divided into intrinsic resistance and acquired resistance.14 Intrinsic resistance indicates that before receiving chemotherapy, resistance factors already pre-exist in tumor cells. Acquired resistance develops during treatment.14,15 Cancer cell resistance to chemotherapy is the main cause of recurrence or relapse and has gained clinical attention.4 Cancer cells evade chemotherapy efficiently through a number of different mechanisms and strategies, such as decrease in drug uptake, increase in drug efflux, alteration of drug metabolism, activation of DNA repair pathways, and induction of the anti-apoptotic machinery.14,16,17 In addition, it is increasingly recognized that the tumor microenvironment plays a critical role in tumor cell response, or lack of response, to chemotherapy.18
Cytotoxic chemotherapeutic drugs can be roughly divided into alkylating agents, antimetabolites, natural products, hormones and hormone antagonists, and other miscellaneous agents.10,12,19
Alkylating agents
Alkylating agents are commonly used as cancer chemotherapeutic agents and have a long history of clinical applications. Alkylating agents, including carmustine, lomustine, and temozolomide, can easily cross the blood–brain barrier and have thus shown the most activity against malignant glioma.20 The general mode of action of alkylating agents is the in vivo formation of electron-deficient active intermediates, which are highly unstable and form covalent bonds with DNA bases. The most vulnerable to attack is the 7-N-atom of guanine.21,22 Moreover, alkylating agents can react with other molecules to produce extensive cellular damages.
The cytotoxicity of alkylating agents depends on DNA repair pathways, and thus enhancing DNA-repair capacity can lead to tumor resistance to alkylating agents.23 Mechanisms of resistance to alkylating agents mainly involve O6-methylguanine methyltransferase (MGMT), DNA mismatch repair (MMR) pathway, and base excision repair (BER) pathway. One important mechanism of resistance to alkylating agents is mediated by the DNA repair enzyme MGMT, which repairs O6-methylguanine adducts.20 MGMT covalently transfers the methyl group from O6-methylguanine to an internal cysteine residue, yielding an inactive S-alkylcysteine-modified protein and guanine.24 The effects of alkylating agents on DNA can be repaired by MGMT, leading to alkylating agent resistance. DNA MMR pathway is critical for mediating the cytotoxic effect of O6-methylguanine, which is programed to correct errors in DNA base pairing, and defects in this system cause resistance to temozolomide.20 Another mechanism of resistance to alkylating agents is the BER pathway that can repair N7-methylguanine and N3-methyladenine DNA adducts. Cells that are defective in MMR are generally resistant to temozolomide.23
Antimetabolites
Antimetabolites are widely used for the treatment of many types of cancer. Antimetabolites have molecular structures similar to the substrates of enzymes that are involved in DNA and RNA synthesis. Inhibition of DNA or RNA synthesis ultimately destroys the structure and function of DNA or RNA and leads to tumor cell death. Antimetabolites such as 5-fluorouracil, cytarabine, methotrexate, hydroxyurea, and gemcitabine are generally analogs of the natural building blocks of DNA.25,26 For example, gemcitabine is a deoxycytidine analog and is widely used in the treatment of solid tumors.27–29 However, tumor resistance of gemcitabine often seriously limits its effect.30 These drugs may interact with DNA in two ways: by acting as structural analogs of the precursors and intermediates for the synthetic pathway, and therefore interfering with the synthesis of purines and pyrimidines, or by acting as false bases in the assembly of the DNA double helix during replication and transcription.
Antimetabolites can be divided into pyrimidine analogs, purine analogs, and folic acid analogs. Research on chemoresistance to nucleoside analogs such as pyrimidine analogs and purine analogs shows that deficiency of nucleoside transporters or nucleoside kinases such as deoxycytidine kinase (dCK), increased activity of ribonucleotide reductase (RR) or cytidine deaminase (CDA), and expression of 5′-nucleotidases are related to decrease in the cytotoxicity of nucleoside analogs.31–33 In addition, folic acid analog resistance may result from decreased cellular influx or increased efflux of the analogs, impaired polyglutamation, increased expression and various alterations in target enzymes, and intracellular accumulation of tetrahydrofolate cofactors.34,35
Natural products
Natural products, molecules discovered and isolated from living organisms and possessing biological or pharmacological activity, are commonly utilized for cancer chemotherapy.36,37 In addition, natural products can also be synthesized, with chemical property equivalent to their natural counterparts. Many anticancer drugs such as paclitaxel, vinblastine, etoposide, and hydroxycamptothecine are all natural products. Anticancer antibiotics, produced by microorganisms, are also valuable natural products.10,38 These drugs tend to be cell-cycle non-specific and therefore are used in the treatment of slow-growing tumors that have a low growth fraction, including daunorubicin, doxorubicin (DOX), epirubicin, idarubicin, valrubicin, mitoxantrone, bleomycin, and mitomycin c.10
Taxanes are important natural product antitumor drugs. Paclitaxel and docetaxel interfere with spindle microtubules, causing cell apoptosis. Paclitaxel enters cells and binds to β-tubulin on the inner surface of microtubules.39 Paclitaxel resistance is mainly associated with the following factors: multidrug resistance caused by the overexpression of P-glycoprotein (P-gp), tubulin mutations or alterations in microtubule stability, and reduced function of significant apoptotic proteins, such as Bcl-2 and p53.39–41
DOX is a widely used chemotherapeutic agent. The most common mechanism of resistance to DOX is the overexpression of ATP-binding cassette (ABC) transporters such as P-gp.42 In addition, alterations of the drug target, topoisomerase, and modulation of programed cell death pathways are also important contributors to DOX resistance.43,44
Hormones and hormone antagonists
Tumors sometimes arise from hormone-sensitive cells. Tumor grows vigorously in the presence of hormones, and even depending on these hormones. Anticancer hormone therapy exploits these features to limit the availability of the hormones to cells in different ways.10 Drugs in this category include selective estrogen receptor modulators (SERMS), progestins (megestrol acetate), luteinizing hormone-releasing hormone agonists, and androgenic agonists.
Glucocorticoids (GCs) such as dexamethasone (DEX) are a class of steroid hormones frequently used as a supportive care co-medication to suppress the side effects of other chemotherapeutic agents.45,46 Hormones are carried into cells, where they interact with hormone receptors, regulating transcription and protein synthesis of target genes in tumor cells. Therefore, interference of hormone–hormone receptor interaction can lead to cancer cell death.10 Main mechanisms of GC resistance include ligand-induced downregulation of the receptors, the dominant-negative inhibition by the β-isoform of the receptors and repression by the transcription factor NF-κB.47
Tamoxifen, a SERMS, is widely used to treat estrogen receptor (ER)-positive breast tumors. However, tamoxifen therapy often fails due to de novo and acquired tamoxifen resistance.48 Tamoxifen resistance is associated with altered ER expression, especially on the plasma membrane, or altered expression of microRNAs and signaling pathways that regulate epithelial–mesenchymal transition in the tumor microenvironment.48,49
Miscellaneous agents
This group of agents includes several cancer chemotherapeutic agents that are difficult to categorize, mainly platinum analogs and enzymes. Platinum analogs are widely used in human neoplasia therapy, alone or in combination with other agents.50 Platinum adducts induce distortion of DNA double helix and cellular DNA damage. Cisplatin, carboplatin, and oxaliplatin are the main platinum analogs used for chemotherapy.19 The mechanisms of cellular resistance to platinum analogs can be classified in two groups: those that limit the formation of cytotoxic platinum-DNA adducts and those that prevent cell death occurring after platinum-DNA adduct formation.51,52
Asparaginase, an enzyme, is an important chemotherapeutic agent for the management of acute leukemia and other blood-related cancers.53 The mechanisms of asparaginase resistance include increased asparagine synthetase activity, genomic modulations and alterations, epigenetic changes, and so on.54,55 Other miscellaneous drugs include hydroxyurea, procarbazine, and dacarbazine.10
Nanomedicine in overcoming cancer cell chemoresistance
There is evidence that nanomedicines can help overcome cancer cell resistance to all the classes of chemotherapeutic agents mentioned above. Examples of using nanomedicines to overcome tumor cell resistance to representative drugs in different classes of cancer chemotherapeutic agents are showed in Table 1.
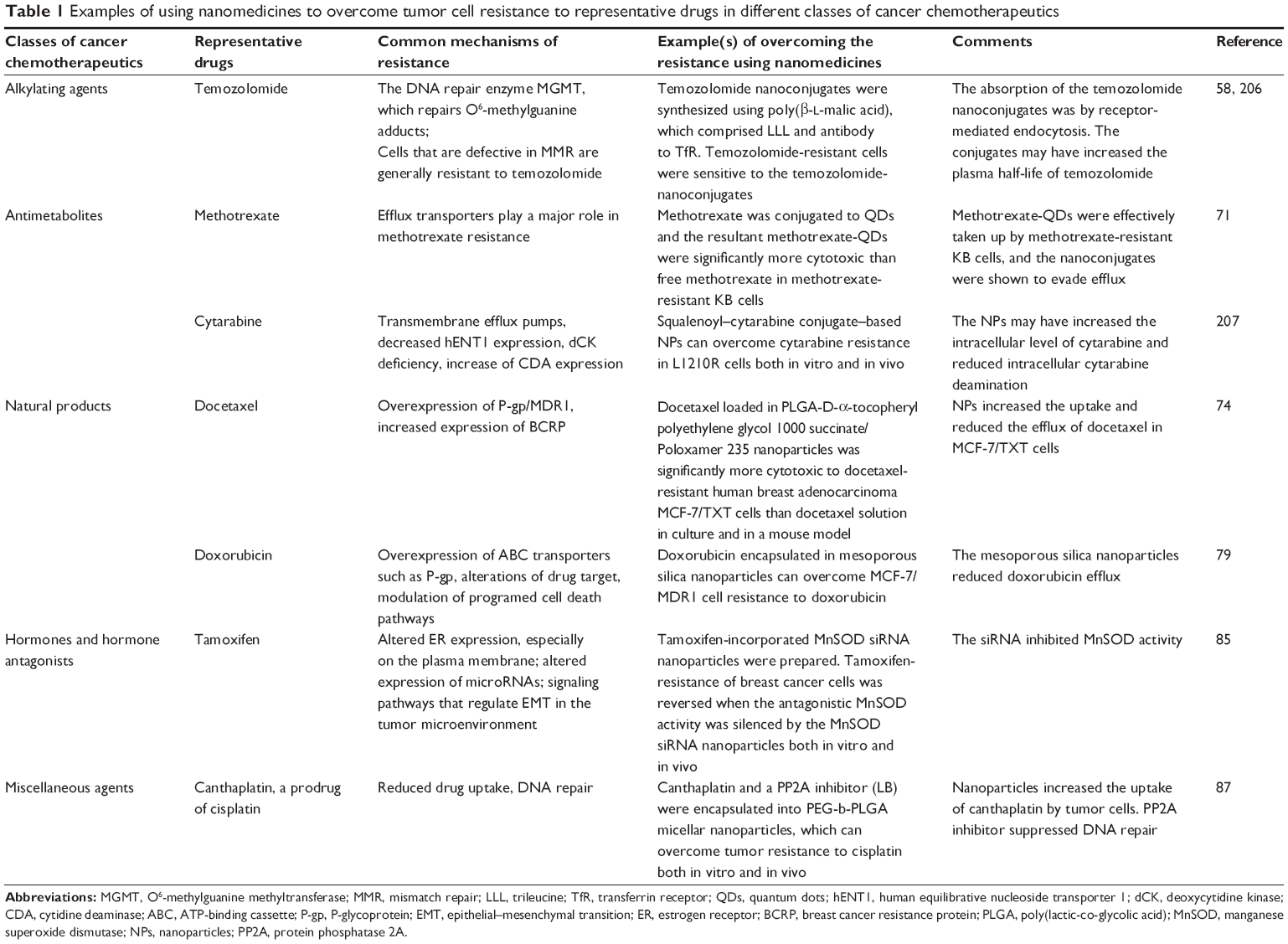 | Table 1 Examples of using nanomedicines to overcome tumor cell resistance to representative drugs in different classes of cancer chemotherapeutics |
Alkylating agents
Alkylating agents are a major class of cancer chemotherapeutic drugs. Clinical chemoresistance is a common complication in alkylating agent treatment of malignant tumors.56,57 Studies have shown that nanomedicines can overcome tumor chemoresistance to alkylating agents.58–60 For example, temozolomide, a commonly used alkylating agent, is considered the gold standard for the treatment of glioblastoma.61,62 However, growing resistance to temozolomide remains a major clinical challenge. The DNA repair enzyme MGMT plays a critical role in primary resistance to alkylating agents such as tomozolomide.20 In addition, overexpression of epidermal growth factor receptor (EGFR) and Galectin-1 by tumor cells also significantly contributes to temozolomide resistance.63,64 Messaoudi et al developed chitosan-grafted lipid nanocapsules to deliver both anti-EGFR and anti-Galectin-1 siRNA to tumor cells, which represents a promising strategy to overcome temozolomide resistance.60,65,66 Patil et al synthesized multifunctional temozolomide nanoconjugates (6.5–14.8 nm) using poly(β-L-malic acid), which contained trileucine (LLL) and antibody to transferrin receptor.58 It was found that temozolomide-resistant cells were sensitive to the temozolomide nanoconjugates,58 clearly demonstrating the feasibility of overcoming tumor cell resistance to the alkylating agent temozolomide by formulating it into nanomedicine.
Antimetabolites
Antimetabolites are widely used cancer chemotherapeutic agents, mainly including purine analogs, pyrimidine analogs, and antifolate agents. There is evidence that chemoresistance to nucleoside analogs such as gemcitabine, cytarabine, and fluorouracil can be overcome by using nanomedicines.67–70 The application of nanomedicine in overcoming gemcitabine resistance will be discussed in detail later.
Methotrexate, an antifolate agent, is indicated for the treatment of rheumatic disorders and malignant tumors. However, cancer cell resistance to methotrexate limits its applications. Johar-Ahar et al conjugated methotrexate to quantum dots (QDs) and showed that methotrexate–QDs were significantly more cytotoxic than free methotrexate in methotrexate-resistant KB cells (ie, IC50 values, 12.0 vs 105.0 μg/mL).71
Natural products
Nanomedicines have shown promise in combating chemoresistance to natural products such as paclitaxel, docetaxel, and vincristine.72–75 For example, Yuan et al reported that paclitaxel-incorporated poly(D,L-lactic-co-glycolic) acid (PLGA)-Tween 80 nanoparticles can reverse multidrug resistance to paclitaxel.72 Tang et al showed that docetaxel loaded in PLGA-D-α-tocopheryl polyethylene glycol (PEG) 1000 succinate/Poloxamer 235 nanoparticles was significantly more cytotoxic to docetaxel-resistant human breast adenocarcinoma MCF-7/TXT cells than Taxotere®, a commercial docetaxel solution, in culture and in a mouse model.74
DOX, an anthracycline antibiotic, is widely used in solid tumor therapy. However, tumor cell resistance to DOX reduces its therapeutic efficacy.76 Many studies exploited the feasibility of using nanomedicines to reverse DOX resistance.76–81 For example, Wang et al showed that DOX encapsulated in mesoporous silica nanoparticles (MSNPs) can overcome MCF-7/MDR1 cell resistance to DOX.79 Unsoy et al synthesized chitosan-coated magnetic nanoparticles for targeted delivery of DOX. DOX-loaded nanoparticles were efficiently taken up by DOX-resistant MCF-7 breast cancer cells (MCF-7/1 μM, MCF-7/S) and were more cytotoxic than free DOX in DOX-resistant MCF-7 cells.81 Yu et al also designed DOX nanoparticles that exhibited higher cytotoxic than free DOX in DOX-resistant MCF-7/ADR cells (ie, 54.4% viability vs 66.8% for free DOX).80
Hormones and hormone antagonists
In this class of chemotherapeutic agents, many researchers have reported the use of nanomedicines to overcome tamoxifen chemoresistance. Aromatase inhibitors are used to treat hormone receptor-positive, locally advanced, or metastatic breast cancer. Letrozole is a potent non-steroidal aromatase inhibitor that is indicated to treat hormone-responsive breast cancer after surgery, but some patients develop resistance to letrozole during treatment.82,83 Nair et al developed hyaluronic acid-bound letrozole nanoparticles (HA-Letr-NPs) and showed that the HA-Letr-NPs can restore the sensitivity of tumors to letrozole in the LTLT-Ca letrozole-resistant breast tumor model.84 Cho et al developed tamoxifen-incorporated manganese superoxide dismutase (MnSOD) siRNA nanoparticles that have an siRNA/poly(amidoamine) dendriplex core and an acid-sensitive polyketal shell and showed that the tamoxifen resistance of breast cancer cells was reversed when the antagonistic MnSOD activity was silenced by the MnSOD siRNA nanoparticles both in vitro and in vivo.85
Miscellaneous agents
Tumor resistance to platinum analogs is very common, and the clinical efficacy of platinum analogs is limited by intrinsic and acquired resistance.86 There is an increasing interest in developing new platinum anticancer agents, but new platinum agents have been very slow to enter clinics. There is evidence that nanomedicine may offer an effective alternative to overcome the resistance. For example, Zhou et al designed canthaplatin and PP2A inhibitor (LB) encapsulated PEG-b-PLGA micelles (ie, polymeric micelles) and showed that the micelles can overcome tumor resistance to cisplatin.87 In addition, micelles prepared with PCL-b-PABPA-b-POEGMEA (ie, polycaprolactone, PCL; 3-((tert-butoxycarbonyl)amino)propyl acrylate, ABPA; oligo(ethylene glycol) methyl ether acrylate, OEGMEA) and incorporated with curcumin and platinum were shown to be able to overcome tumor cell resistance to platinum.88
Limitations in using nanomedicine to overcome cancer cell chemoresistance
It is exciting that formulating cancer chemotherapeutic agents into nanomedicines can help overcome cancer cell chemoresistance. However, it is worth noting that to successfully overcome cancer chemoresistance, having nanomedicine formulations that can kill resistant cancer cells is often not sufficient. In vivo, the reticuloendothelial system (RES) and other physiological barriers can significantly impede the efficient delivery of nanomedicines to tumor cells, or even tumor tissues.89,90 Opsonization of nanomedicines by nonspecific adsorption of plasma proteins, such as opsonins, facilitates their phagocytosis and clearance from the circulation by RES.90,91 Many approaches have been utilized to limit the clearance of nanomedicines, either by delaying of RES clearance or by altering the surface properties of nanomedicines.92,93 The physiological barriers are another obstacle that needs to be overcome for nanomedicines to effectively overcome cancer chemoresistance in vivo.89 The accessibility of nanomedicines to solid tumors is determined by various mechanisms, such as the efficiency of the blood and lymphatic networks in tumor tissues, the permeability of the vascular barriers in tumors, and the constitution of the tumor stroma.89,94 Several strategies have been explored to enhance the delivery of nanomedicines into tumor tissues such as normalizing the tumor vasculature or reducing tumor desmoplasia.89
Nucleoside analogs and gemcitabine
Nucleoside analogs are structurally similar antimetabolites that have a broad range of actions and are clinically effective in both solid tumors and hematological malignancies.95 These agents may interact with DNA or RNA by inhibiting the ability of cancer cells to synthesize precursors of nucleic acids required to ensure sustained growth or by directly interfering with DNA or RNA synthesis. Nucleoside analogs have a generalized structure consisting of a purine or pyrimidine base linked to a deoxyribose sugar. Examples of purine nucleosides and related inhibitors include cladribine, fludarabine, and clofarabine; and examples of pyrimidine nucleoside analogs include the deoxycytidine analogs gemcitabine, fluorouracil, cytarabine, capecitabine.10
Gemcitabine (2′,2′-difluorodeoxycytidine, dFdC) is a deoxycytidine analog with antitumor activity against a wide variety of solid tumors such as pancreatic, non-small-cell lung cancer (NSCLC), breast cancer, and ovarian cancer, alone or in combination with other chemotherapeutic agents.27–29,96 Moreover, gemcitabine is indicated in several hematological disorders such as acute leukemia.97 Furthermore, gemcitabine enhances the cytotoxicity of cisplatin by increasing the formation of cytotoxic platinum-DNA adducts and is also a potent radiosensitizer used in radiation therapy.98 However, drug resistance to gemcitabine often limits its efficacy in clinics,30 and overcoming gemcitabine chemoresistance remains a challenge.99
Gemcitabine and its mechanisms of action
As a hydrophilic nucleoside analog, cellular uptake of gemcitabine is mediated by nucleoside transporters such as the human equilibrative nucleoside transporter 1 (hENT1).100 Once taken up into cells, gemcitabine is phosphorylated by dCK to gemcitabine monophosphate (dFdCMP), gemcitabine diphosphate (dFdCDP), and gemcitabine triphosphate (dFdCTP).101 The active metabolite, dFdCTP, can terminate DNA elongation by incorporating into DNA, finally leading to cell death.99,102,103 In addition, dFdCDP can inhibit RR by binding to the large subunit (RRM1).104,105 RRs catalyze the conversion of nucleoside 5′-diphosphates (ie, NDPs) to their corresponding deoxynucleotides (ie, dNDPs), which are phosphorylated to dNTPs for DNA synthesis.105,106 Inhibition of RRs will reduce the dNTP pool, allowing dFdCTP to more effectively compete with dNTPs and inhibit DNA replication and repair.104–107
Mechanisms of gemcitabine resistance
The mechanisms of resistance to gemcitabine are in many aspects different from those of the other classes of cancer chemotherapeutic agents. Multiple factors, including decreased expression of nucleoside transporters,108,109 changes in the expression of gemcitabine-activating or degradation enzymes and target molecules,33,110–112 some signaling molecules (eg, NF-κB, P53) affecting cells that are resistant to apoptosis,113,114 and the expression of efflux transporters commonly resulted in MDR,115,116 have been reported to cause gemcitabine resistance.
Nucleoside transporters
Gemcitabine is a hydrophilic compound and cannot readily diffuse across cell membrane. Therefore, it requires nucleoside transporters to enter cells.117,118 The concentrative nucleoside transporters (hCNTs) and hENTs are implicated in tumor cell uptake of gemcitabine.118,119 Among these transporters, hENT1 is a major transporter involved in gemcitabine cellular uptake.120 In fact, hENT1 has been reported as a vital predictive marker of tumor response to gemcitabine-based therapy.32,121,122 Clinical data showed that cancer patients with a decreased tumor expression of hENT1 have a significantly lower survival rate after gemcitabine treatment than those with tumors that express a higher level of hENT1.119,121–124 In culture, tumor cells lacking hENT1 expression become resistant to gemcitabine-mediated cytotoxicity.108 For example, in the hENT1-deficient CCRF CEM-AraC-8C cells, the IC50 value of gemcitabine was reported to be 471-fold greater than that in the parent CCRF-CEM cells.68
dCK
Once gemcitabine enters cells, dCK is the rate-limiting enzyme responsible for the conversion of gemcitabine to its active metabolites.125,126 Studies have indicated that in vitro and in vivo, dCK deficiency is related to gemcitabine resistance to pancreatic cancer, sarcoma, lymphoma, and leukemia.127–131 For example, Ohmine et al showed that the attenuation of gemcitabine phosphorylation is likely a key process for the acquisition of resistance by the RPK9 human pancreatic adenocarcinoma (PDAC) cells.101 Lower expression of dCK was shown to be associated with shorter overall survival in pancreatic cancer patients who received gemcitabine as an adjuvant therapy.132 It is suggested that dCK expression in both protein and mRNA levels may serve as a biomarker to predict tumor cell sensitivity to nucleoside analogs such as gemcitabine.133
CDA
CDA, a key enzyme involved in gemcitabine metabolism, was identified in the early 1990s.134 Deamination is the main mechanism by which gemcitabine is inactivated, and it is estimated that 90% of gemcitabine is inactivated to difluorodeoxyuridine by CDA intracellularly and extracellularly.96 In vitro, macrophage-induced CDA upregulation in human Panc-1 pancreatic tumor cell line has been shown to confer gemcitabine resistance.111 Data from ample studies have indicated that CDA polymorphisms alter CDA enzyme activity and the pharmacokinetics of gemcitabine,135–138 and three functional polymorphisms of CDA (rs2072671, CDA 79A > C; rs60369023, CDA 208G > A; and rs1048977, CDA 435C > T) could predict the clinical outcomes of gemcitabine-based tumor chemotherapy.33,135,136
RR or RNR
As mentioned above, RRs catalyze the conversion of NDPs to their corresponding dNDPs for DNA synthesis.139,140 In mammals, RR is a heterodimeric tetramer consisting of two large subunits (RRM1) and two small subunits (RRM2 and RRM2B).141,142 RRM1 catalyzes the rate-limiting step in the production of dNTPs and is an essential enzyme for DNA replication and repair.139,143 The catalysis activity of RRs requires the binuclear iron center and a tyrosyl-free radical located in RRM2.142 RRM2B was identified as a critical p53-inducible RR subunit that can be regulated by p53 and p73 genes/proteins.144,145
Gemcitabine self-potentiates its own effect by directly inhibiting RRM1. Therefore, upregulation of RRM1 can lead to gemcitabine resistance.110,146 Conversely, RRM1 knockdown in the resistant MIA PaCa-2 pancreatic cancer cell line completely restored gemcitabine sensitivity.147 The relationship between RRM2 mRNA expression and response to gemcitabine in clinical setting has been investigated in various cancers. For example, the response rate to gemcitabine is significantly higher in pancreatic cancer patients with low RRM2 mRNA expression in biopsy specimens.148 Similarly, high RRM2 expression was found to be correlated with poor clinical outcome in patients with lower-risk prostate cancer.112
Other mechanisms of resistance to gemcitabine
The aforementioned are the main mechanisms of gemcitabine resistance. There are other factors associated with gemcitabine resistance as well. For example, excision repair cross-complementing protein 1 can repair gemcitabine-induced strand breaks, and its overexpression is well documented in poor gemcitabine responders.149,150 Kozinn et al reported that microRNAs 1290, 138, let-7i, and let-7b are involved in gemcitabine resistance in bladder carcinoma cell lines.151 In addition, a tumor microenvironment that favors cancer progression and metastasis can elicit drug resistance.152 For example, Xu et al showed that sonic hedgehog (SHH) signaling in tumor microenvironment protects PDAC cells against gemcitabine-induced apoptosis and that overexpression of SHH in PDAC cells enhances drug resistance.153 Other factors that contribute to gemcitabine resistance include NF-κB,114 heat shock proteins, and the presence of highly resistant tumor stem cells.154,155
Overcoming gemcitabine resistance using nanomedicines
Nanomedicines have unique advantages in overcoming tumor cell resistance to gemcitabine, and various types of gemcitabine nanomedicine formulations, such as micelles,156 liposomes,157 supramolecular vesicular aggregates, and nanovesicles,158–160 have been shown to circumvent gemcitabine resistance. The general mechanisms by which gemcitabine nanomedicines overcome gemcitabine chemoresistance are discussed below.
Reducing RR expression
Accumulating evidence indicates that increased expression of RRM1 is associated with a poor response of cancer patients to gemcitabine.161,162 Higher levels of RRM1 were detected in tumors of various patients who respond poorly to gemcitabine.139,143,163–165 For example, in gemcitabine-treated advanced NSCLC patients, those with RRM1-positive tumors were shown to have worse overall survival and disease control than those with RRM1-negative tumors.163 Similarly, in patients with advanced nasopharyngeal carcinoma (NPC) and treated with gemcitabine-based regimens, high RRM1 expression is correlated with shorter progression-free survival, compared to patients with RRM1-negative expression.161 In a recent study, RRM1 siRNA was used to downregulate RRM1 expression in tumor cells, and it was shown that pre-exposure of A549 lung cancer cells to RRM1 siRNA nanoconstructs significantly decreased the IC50 value of gemcitabine in the tumor cells compared to gemcitabine alone.166 Previously, we have also shown in a mouse model with TC-1 mouse lung cancer cells that overexpress RRM1 (ie, TC-1-GR cells) that treatment with RRM1 siRNA-polyethylenimine (PEI) nanocomplexes (122±5 nm) significantly increased the effect of gemcitabine against the tumors, compared to treatment with control siRNA-PEI nanocomplexes.167
Increasing cellular uptake of gemcitabine
As mentioned above, gemcitabine depends on nucleoside transporters to enter cells.117,168 Therefore, reduced expression of nucleoside transporters on tumor cells causes tumor cell resistance to gemcitabine.123,169 For example, it was reported that high hENT1 expression in resected specimen of patients with PDAC who received postoperative gemcitabine therapy is correlated with increased overall survival,170–173 whereas low hENT1 expression was linked to gemcitabine resistance and shorter overall survival.109,123,169 In addition, low levels of hENT1 expression were also detected in tumors in gallbladder adenocarcinoma patients who respond poorly to gemcitabine.122 Nanomedicine formulations of gemcitabine can overcome gemcitabine resistance caused by reduced expression of nucleoside transporters by delivering gemcitabine into tumor cells independent of the transporters. For example, we have data showing that our previously developed stearoyl gemcitabine solid lipid nanoparticles (ie, 4-(N)-GemC18-SLNs) enter tumor cells by clathrin-mediated endocytosis.174 Hung et al showed that a nanoparticle formulation of gemcitabine has significantly smaller IC50 values, compared to free gemcitabine, in ovarian cancer cells that express low levels of hCNT1,175 indicating that the nanoparticle formulation can bypass nucleoside transporter defects.
Reducing deamination of gemcitabine
Stromal and cellular CDAs convert gemcitabine to an inactive metabolite.96,176 Preclinical and clinical studies have suggested that upregulation of CDAs increases gemcitabine resistance, while CDA deficient is associated with increased gemcitabine activity.33,96 To protect gemcitabine from rapid deamination, many attempts have been made by chemically modifying gemcitabine.177–180 For example, it was shown that conjugation of a fatty acid, such as stearic acid, to gemcitabine at the 4-N position decreases the sensitivity of the later to deaminase.181,182 In addition, gemcitabine–fatty acid conjugates formulated into nanoparticles also become less sensitive to deamination, as they are no longer good substrates of CDAs.182 Meng et al developed a MSNP for co-delivery of gemcitabine and paclitaxel to take advantage of paclitaxel’s ability to inhibit CDA expression to increase tumor response to gemcitabine.183
Enhancing distribution and/or accumulation of gemcitabine in tumor tissues
The enhanced permeability and retention (EPR) effect discovered by Matsumura and Maeda has been exploited for passive targeting of anticancer drugs into tumors.184,185 The discovery of EPR effect is of great significance to the design of antitumor nanomedicines.186–188 Nanomedicine formulations of gemcitabine can take advantage of the EPR effect to increase gemcitabine content within tumor tissues upon intravenous injection.189–192 Having more gemcitabine distributed into tumor tissues will provide them the chance to kill tumor cells or inhibit tumor cell growth.
Examples of using nanomedicine formulations of gemcitabine to overcome tumor cell resistance to gemcitabine
In their effort in finding a solution to overcome gemcitabine resistance, scientists showed that nanomedicine formulations of gemcitabine have promising potentials.68,174,180,193 Examples of overcoming tumor cell resistance to gemcitabine using nanomedicines are shown in Table 2.
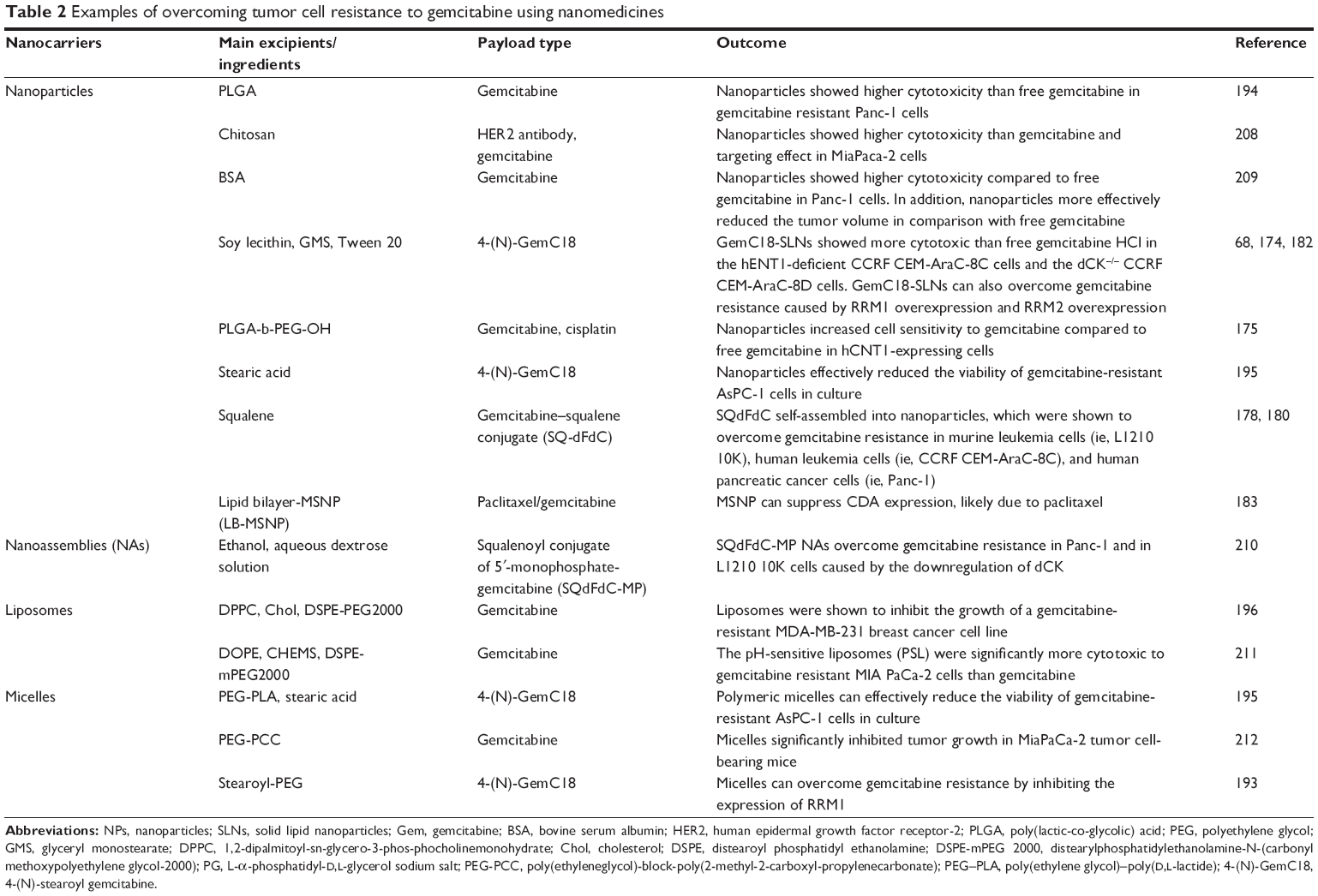 | Table 2 Examples of overcoming tumor cell resistance to gemcitabine using nanomedicines |
Polymeric nanoparticles
Recognizing that defective hCNT1 contributes to gemcitabine chemoresistance in ovarian cancer, Hung et al created PLGA-b-PEG-OH nanoparticles incorporated with gemcitabine. The gemcitabine–PLGA-b-PEG-OH nanoparticles effectively delivered gemcitabine into hCNT1-decreased ES-2 and TOV-21G tumor cells and were significantly more cytotoxic to those cells than free gemcitabine.175 Papa et al reported gemcitabine-loaded PLGA nanoparticles (PLGem) and evaluated their cytotoxicity to aggressive human Panc-1 cells, which are well-known to exhibit gemcitabine resistance.194 The PLGem was significantly more cytotoxic than free gemcitabine to Panc-1 cells.194
In another approach, gemcitabine nanoparticles were prepared by loading GemC18, a stearic acid amide derivative of gemcitabine, in PEG–poly(D,L-lactic) acid (PEG–PLA) polymeric micelles or by GemC18 self-assembling.195 Both of the nanomedicines effectively reduced the viability of gemcitabine-resistant AsPC-1 cells in culture (IC50 values, 58.88 and 46.34 μM, respectively), whereas the molar equivalent free gemcitabine did not show any significant cytotoxicity to AsPC-1 cells. The GemC18 self-assembled nanoparticles showed greater in vitro cellular uptake and cytotoxicity than the GemC18-PEG-PLA polymeric micellar nanoparticles (ie, drug uptake in Panc-1 cells, 37.55%±2.21% for GemC18 self-assembled nanoparticles, 28.60%±1.85% for GemC18-PEG-PLA polymeric micelles, and 30.11%±1.98% for GemC18 in solution).195
Gemcitabine nanoparticles were also prepared based on a gemcitabine–squalene conjugate (SQ-dFdC or SQ-Gem), which displayed a stronger antiproliferative and cytotoxic activity than gemcitabine. After orthotopic Panc-1 tumor-bearing mice were treated two times at a 4-day interval with either gemcitabine (20 mg/kg) or SQ-Gem (20 mg/kg), SQ-Gem was more effective than gemcitabine in inhibiting the tumor growth.178 The SQ-dFdC nanoparticles were also shown to overcome gemcitabine resistance in murine and human leukemia cells (ie, L1210-10K and CEM/AraC-8C, respectively).180
Liposomes
Xu et al developed pH-sensitive liposomes (PSLs) with a high content of gemcitabine. The cytotoxicity values of the various gemcitabine-PSLs developed were evaluated in the gemcitabine-resistant MIA PaCa-2 cell line.67 Gemcitabine-PSLs with drug loading of 0.5% and 4.5% had similar IC50 values (ie, 1.1±0.1 versus 0.7±0.1 μM), but both were significantly smaller than that of free gemcitabine in solution and gemcitabine in non-PSLs. In an animal model, the gemcitabine-PSLs were significantly more effective than free gemcitabine in controlling the growth of gemcitabine-resistant pancreatic cancer cells.67 Papa et al also reported gemcitabine-encapsulated nanoliposomes (GemPo), which were shown to be more cytotoxic than free gemcitabine in gemcitabine-resistant MDA-MB-231 breast cancer cells and sensitive 4T1 cells.196
Overcoming tumor cell resistance to gemcitabine using stearoyl gemcitabine-incorporated solid lipid nanoparticles
In our effort to improve the antitumor activity of gemcitabine, we previously developed a stearoyl gemcitabine nanoparticle formulation by incorporating 4-(N)-stearoyl gemcitabine (ie, 4-(N)-GemC18) into solid lipid nanoparticles engineered from lecithin/glycerol monostearate-in-water emulsions.181 In animal models (ie, C57BL/6 mice with TC-1 mouse lung cancer cells, nude mice with BxPC-3 human pancreatic cancer cells), the 4-(N)-GemC18-SLNs were significantly more effective than the molar equivalent of free gemcitabine or 4-(N)-GemC18 in a Tween 20 solution in inhibiting tumor growth.181 More importantly, we discovered that the 4-(N)-GemC18-SLNs can overcome tumor cell resistance to gemcitabine.68 For example, 4-(N)-GemC18-SLNs were 15-fold more cytotoxic than gemcitabine HCl in the hENT1-deficient CCRF CEM-AraC-8C cells, and ~8-fold more cytotoxic in the dCK−/− CCRF CEM-AraC-8D cells.68 In the gemcitabine resistant human Panc-1 tumor cells that overexpress RRM2, 4-(N)-GemC18-SLNs were >17-fold more cytotoxic than gemcitabine HCl.68 In the RRM1-overexpresssing, gemcitabine-resistant TC-1-GR cells, the IC50 value of 4-(N)-GemC18-SLNs was only about 5% of that of gemcitabine HCl.68 Importantly, although both 4-(N)-GemC18-SLNs and free gemcitabine HCl can significantly inhibit the growth of the highly gemcitabine-sensitive TC-1 tumor cells in a mouse model, only 4-(N)-GemC18-SLNs, but not free gemcitbaine HCl, can significantly inhibit the growth of the gemcitabine-resistant TC-1-GR tumors.68 When elucidating the mechanisms underlying the 4-(N)-GemC18-SLNs’ ability to overcome gemcitabine resistance, we discovered that the unique composition of the 4-(N)-GemC18-SLNs and the way by which the gemcitabine in the 4-(N)-GemC18-SLNs enters tumor cells are likely responsibe for their ability to more effectively kill tumor cells, especially tumor cells that are otherwise resistant to gemcitabine.174 We concluded that for gemcitabine to effectively kill tumor cells, especially those resistant to gemcitaine, entering tumor cells is important, but not enough.174
The unique composition of the 4-(N)-GemC18-SLNs is critical for their ability to kill tumor cells resistant to gemcitabine
The conclusion that the unique composition of the 4-(N)-GemC18-SLNs is critical for their ability to overcome gemcitabine resistance is supported by the following findings: 1) a 3′-(O)-GemC18 ester synthesized by conjugating gemcitabine in the 3′-O position with stearic acid, when incorporated into the same solid lipid nanoparticles engineered from lecithin/glyceroyl monostearate-in-water emulsions, was not significantly more effective than free gemcitabine in controlling the growth of the gemcitabine-resistant TC-1-GR tumor cells in culture and in a mouse model; 2) the same 4-(N)-GemC18 amide when incorporated into polymeric PLGA nanoparticles was not more effective than gemcitabine in inhibiting the growth of the gemcitabine-resistant TC-1-GR cells; and 3) 4-(N)-GemC8, another amide gemcitabine derivative synthesized by conjugating gemcitaine in its 4-N position with a medium chain fatty acid, caprylic acid (C8), incorporated into the same solid lipid nanoparticles engineered from lecithin/glyceroyl monostearate-in-water emulsions was not more cytotoxic than free gemcitabine against the gemcitabine-resistant TC-1-GR cells.174 Therefore, it seems that the amide nature of the 4-(N)-GemC18, the long chain fatty acid (ie, stearic acid) derivative nature of the 4-(N)-GemC18, and the solid lipid nanoparticles in which the 4-(N)-GemC18 is incorporated in are all critical for the resultant 4-(N)-GemC18-SLNs to overcome tumor cell resistance to gemcitabine.
It is noted that the 4-(N)-GemC18 needs to be incorporated into the solid lipid nanoparticles, as our results showed that 4-(N)-GemC18 alone and the physical mixture of it and blank solid lipid nanoparticles were not as cytotoxic as 4-(N)-GemC18-SLNs against the gemcitabine-resistant TC-1-GR cells.174 Furthermore, we showed that our 4-(N)-GemC18-SLNs, but not free gemcitabine, nor 4-(N)-GemC18, significantly downregulate RRM1 expression in the gemcitabine-resistant TC-1-GR cells in culture and in a mouse model and increased the level of dFdCTP in TC-1-GR cells in culture.
The pathway by which the 4-(N)-GemC18 in the 4-(N)-GemC18-SLNs enters tumor cells is critical for its ability to kill tumor cells resistant to gemcitabine
The exact mechanisms why our 4-(N)-GemC18-SLNs can more effectively kill the TC-1-GR tumors that overexpress RRM1 than other gemcitabine formulations, including free gemcitabine, free 4-(N)-GemC18, 3′-(O)-GemC18-SLNs, 4-(N)-GemC8-SLNs, and 4-(N)-GemC18-PLGA-nanoparticles remains unknown, but we hypothesize that it is very likely related to the pathway by which 4-(N)-GemC18-SLNs deliver the gemcitabine into tumor cells. Free gemcitabine depends on nucleoside transporters such as hENT1 to enter tumor cells, and free 4-(N)-GemC18 enters cells by passive diffusion due to its high lipophilicity. Our 4-(N)-GemC18-SLNs, however, enter cells by clathrin-mediated endocytosis.174 Alkalization of lysosomes (ie, increasing pH) did not affect the uptake and intracellular degradation of 4-(N)-GemC18 when it was taken up as free 4-(N)-GemC18 in solution. However, when cells were incubated with the 4-(N)-GemC18-SLNs, alkalization of lysosomal pH significantly inhibited the intracellular degradation of 4-(N)-GemC18.174 Lysosomal acidification is required for the activation of many enzymes in lysosomes, indicating that the acidic lysosomal environment and thus many pre-enzymes in lysosomes activated in acidic environment are important for the degradation of the solid lipid nanoparticles, the release of the 4-(N)-GemC18 from the nanoparticles, and the hydrolysis of 4-(N)-GemC18 to free gemcitabine, when 4-(N)-GemC18 was brought into cells by the endocytosis of the 4-(N)-GemC18-SLNs.174 The 3′-(O)-GemC18-SLNs were not as effective as 4-(N)-GemC18-SLNs because the ester bond in the 3′-(O)-GemC18 was readily hydrolyzed even before the 3′-(O)-GemC18-SLNs were endocytized.174 Similarly, the 4-(N)-GemC8-SLNs were not as effective as 4-(N)-GemC18-SLNs because the 4-(N)-GemC8 was readily released or leaked from the 4-(N)-GemC8-SLNs before the nanoparticles were endocytized.174
Proposed mechanism by which 4-(N)-GemC18-SLNs overcome tumor cells resistant to gemcitabine
Based on the findings mentioned above, we hypothesized that the 4-(N)-GemC18-SLNs take advantage of the salvage nucleotide synthesis pathway and “channel” the 4-(N)-GemC18 into a “natural” pathway that has evolved for cells to efficiently reuse bases and nucleosides from within or outside cells. When cells take up the apoptotic bodies or foreign pathogens by endocytosis, the nucleic acids are enzymatically degraded into nucleosides and bases for reuse.197 As shown in Figure 1, it is likely that after our 4-(N)-GemC18-SLNs enter tumor cells by clathrin-mediated endocytosis, enzymes in lysosomes catalyze the degradation of the solid lipid nanoparticles, and the degradation facilitates the release of 4-(N)-GemC18 from the nanoparticles. Lysosomal enzymes such as cathepsin B catalyze the hydrolysis of 4-(N)-GemC18 to free gemcitabine, a nucleoside analog. Gemcitaine released into the lysosomes can then be exported out of the lysosomes to the cytoplasm by nucleoside transporters, such as the lysosome-specific hENT3,198 into the proper intracellular compartment for efficient phosphorylation to its active metabolites, dFdCDP and dFdCTP. In contrast, when free 4-(N)-GemC18 diffuses into tumor cells by passive diffusion, it may be hydrolyzed to release gemcitabine intracellularly, but not in the proper intracellular compartment for efficient phosphorylation, due to its high lipophilicity. Exposing gemcitabine in the “wrong” compartment in cells will likely subject it to deamination by CDA, considering that nucleotides normally do not enter cells in the form of long chain fatty acid conjugates. Free gemcitabine enters tumor cells with the help of nucleoside transporters, but it is subjected to extensive deamination intracellularly and extracellularly before being phosphorylated (Figure 1).174 The small amount of dFdCTP generated may be sufficient to inhibit tumor cells that are sensitive to gemcitabine, but not against tumor cells that developed various resistant mechanisms (eg, overexpression of RRM1). Of course, more experiments will have to be carried out to generate data to fully support the hypothesized mechanism, but designing nanomedicine formulations of anticancer drugs that mimic or take advantage of a natural pathway, such as the nucleotide salvage pathway in the case of nucleoside analogs such as gemcitabine, likely represents a desirable strategy to improve the activity of the drugs and to overcome chemoresistance.
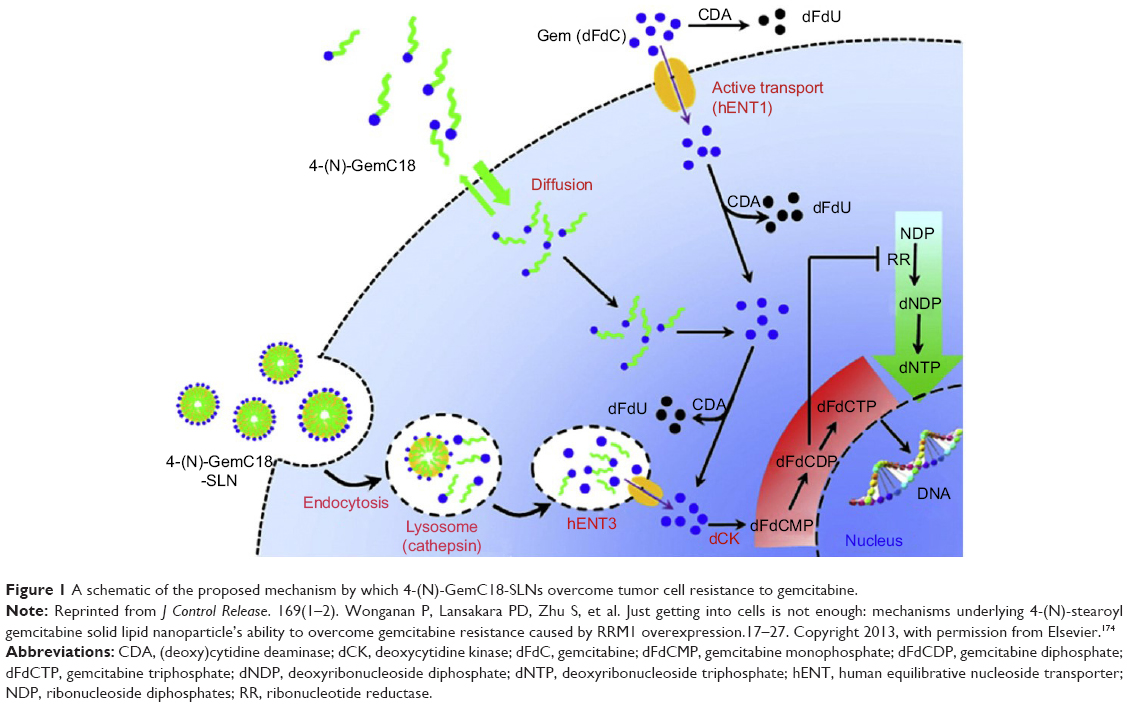 | Figure 1 A schematic of the proposed mechanism by which 4-(N)-GemC18-SLNs overcome tumor cell resistance to gemcitabine. |
Finally, it is worth noting that the mechanism mentioned above was largely based on cell culture data.174 In a mouse model with tumor cells that are resistant to gemcitabine due to the overexpression of RRM1, our 4-(N)-GemC18-SLNs significantly inhibited the tumor growth, although the molar equivalent dose of free gemcitabine did not show any significant antitumor effect.68 Moreover, we have also engineered 3′-(O)-GemC18-SLNs by incorporating 3′-(O)-GemC18 into the same solid lipid nanoparticles; 3′-(O)-GemC18 was synthesized by conjugating stearic acid to gemcitabine in the 3′-O position to form an ester, which is more sensitive to hydrolysis than 4-(N)-GemC18. Therefore, 3′-(O)-GemC18-SLNs cannot as effectively as 4-(N)-GemC18-SLNs kill the RRM1-overexpressing tumor cells in culture and in vivo.174 The data from our in vivo studies in a mouse model indicated that the mechanism we proposed above is also applicable in vivo. In other words, in the tumor-bearing mouse model we tested, some of our 4-(N)-GemC18-SLNs should have reached the tumor cells after intravenous injection and entered the endolysosomes of the tumor cells by endocytosis.
It is not easy for nanoparticles to evade uptake by the RES and overcome other physiological barriers to successfully reach tumor cells. For nanoparticles that reach tumor tissues intact, besides the aforementioned endocytosis by tumor cells, there are other potential mechanisms for the chemotherapeutic agents carried by the nanoparticles to enter tumor cells. One obvious mechanism is that the chemotherapeutic agents are released from the nanoparticles in the tumor tissues, diffuse to tumor cell surface, and then enter tumor cells by passive diffusion or transporter-mediated uptake. There are strategies to facilitate the release of chemotherapeutic agents from nanoparticles within tumor tissues. For example, secretory phospholipase A2 (sPLA2) are enzymes overexpressed in various tumors.199–201 Liposomes that are responsible for sPLA2 (SPRL) were engineered to facilitate liposomal degradation and drug release in tumor tissues.202–204 In a study comparing the uptakes and cytotoxicities of the SPRL encapsulated with DOX and sterically stabilized liposomes encapsulated with DOX, Moch et al suggested that the efficacy of the sPLA2 liposomes are mediated by cell-dependent mechanisms.203 Recently, Hofmann et al provided evidence supporting the existence of drug delivery into cells without cellular uptake of the nanoparticles through a new “kiss-and-run” mechanism between (polymeric) nanoparticles and the cell membrane.205
Conclusion
Despite recent advances in targeted therapies and immunotherapies, chemotherapy using cytotoxic agents remains an indispensable modality in cancer treatment. Formulating cancer chemotherapeutic agents into nanomedicines represents an attractive approach to modify their pharmacokinetics, efficacy, and toxicity profiles. Moreover, cancer cells often develop resistance to chemotherapeutic agents prior to or during treatment, and there is encouraging evidence that formulating cancer chemotherapeutic agents into nanomedicines may also represent a viable approach to overcome cancer cell chemoresistance. However, better nanomedicine formulations of chemotherapeutical agents are often the results of rational mechanism-based design, and occasionally by accident.
Acknowledgments
This work was supported in part by the National Natural Science Foundation of China (81660272, 81460454, 81460248, 81260457) (to YZ, ZC, and/or YS), the Inner Mongolia Natural Science Fund (2017ZD08, 2014ZD05, 2016MS0318, 2016MS0368, 2015MS0820, 2013MS1138, 2012MS1121, 2011MS1110) (to YS, ZC, and/or YZ), the National “Twelfth Five-Year” Plan for Science & Technology Support of China (2014BAI13B03) (to YS), the Inner Mongolia Science and Technology Plan (20120101, 20120402, 20110501) (to YS), and the US National Institutes of Health (CA135274, CA179362) (to ZC).
Disclosure
The authors report no conflicts of interest in this work.
References
Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65(2):87–108. | ||
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin. 2016;66(1):7–30. | ||
Jonckheere N, Skrypek N, Van Seuningen I. Mucins and tumor resistance to chemotherapeutic drugs. Biochim Biophys Acta. 2014;1846(1):142–151. | ||
Zhao J. Cancer stem cells and chemoresistance: the smartest survives the raid. Pharmacol Ther. 2016;160:145–158. | ||
Du B, Shim JS. Targeting epithelial-mesenchymal transition (EMT) to overcome drug resistance in cancer. Molecules. 2016;21(7):pii:E965. | ||
Naguib YW, Cui Z. Nanomedicine: the promise and challenges in cancer chemotherapy. Adv Exp Med Biol. 2014;811:207–233. | ||
Bobo D, Robinson KJ, Islam J, Thurecht KJ, Corrie SR. Nanoparticle-based medicines: a review of FDA-approved materials and clinical trials to date. Pharm Res. 2016;33(10):2373–2387. | ||
Wang AZ, Gu F, Zhang L, et al. Biofunctionalized targeted nanoparticles for therapeutic applications. Expert Opin Biol Ther. 2008;8(8):1063–1070. | ||
Zhang L, Gu FX, Chan JM, Wang AZ, Langer RS, Farokhzad OC. Nanoparticles in medicine: therapeutic applications and developments. Clin Pharmacol Ther. 2008;83(5):761–769. | ||
Matray-devoti J. Cancer Drugs. New York: Chelsea House Publications; 2006. | ||
Teicher BA. Cancer Therapeutics: Experimental and Clinical Agents. Totowa, NJ: Humana Press; 1997. | ||
Livshits Z, Rao RB, Smith SW. An approach to chemotherapy-associated toxicity. Emerg Med Clin North Am. 2014;32(1):167–203. | ||
Kibria G, Hatakeyama H, Harashima H. Cancer multidrug resistance: mechanisms involved and strategies for circumvention using a drug delivery system. Arch Pharm Res. 2014;37(1):4–15. | ||
Holohan C, Van Schaeybroeck S, Longley DB, Johnston PG. Cancer drug resistance: an evolving paradigm. Nat Rev Cancer. 2013;13(10):714–726. | ||
Drinberg V, Bitcover R, Rajchenbach W, Peer D. Modulating cancer multidrug resistance by sertraline in combination with a nanomedicine. Cancer Lett. 2014;354(2):290–298. | ||
Geng M, Wang L, Chen X, Cao R, Li P. The association between chemosensitivity and Pgp, GST-pi and Topo II expression in gastric cancer. Diagn Pathol. 2013;8:198. | ||
Turrini E, Ferruzzi L, Fimognari C. Natural compounds to overcome cancer chemoresistance: toxicological and clinical issues. Expert Opin Drug Metab Toxicol. 2014;10(12):1677–1690. | ||
Provenzano PP, Hingorani SR. Hyaluronan, fluid pressure, and stromal resistance in pancreas cancer. Br J Cancer. 2013;108(1):1–8. | ||
Priestman T. Cancer Chemotherapy in Clinical Practice. London: Springer; 2008. | ||
Sarkaria JN, Kitange GJ, James CD, et al. Mechanisms of chemoresistance to alkylating agents in malignant glioma. Clin Cancer Res. 2008;14(10):2900–2908. | ||
Fu D, Calvo JA, Samson LD. Balancing repair and tolerance of DNA damage caused by alkylating agents. Nat Rev Cancer. 2012;12(2):104–120. | ||
Bordin DL, Lima M, Lenz G, et al. DNA alkylation damage and autophagy induction. Mutat Res. 2013;753(2):91–99. | ||
Srinivasan A, Gold B. Small-molecule inhibitors of DNA damage-repair pathways: an approach to overcome tumor resistance to alkylating anticancer drugs. Future Med Chem. 2012;4(9):1093–1111. | ||
Kaina B, Christmann M, Naumann S, Roos WP. MGMT: key node in the battle against genotoxicity, carcinogenicity and apoptosis induced by alkylating agents. DNA Repair (Amst). 2007;6(8):1079–1099. | ||
Tiwari M. Antimetabolites: established cancer therapy. J Cancer Res Ther. 2012;8(4):510–519. | ||
DeVita VT Jr, Chu E. A history of cancer chemotherapy. Cancer Res. 2008;68(21):8643–8653. | ||
Tsai S, Evans DB. Therapeutic advances in localized pancreatic cancer. JAMA Surg. 2016;151(9):862–868. | ||
Nagourney RA, Flam M, Link J, et al. Carboplatin plus gemcitabine repeating doublet therapy in recurrent breast cancer. Clin Breast Cancer. 2008;8(5):432–435. | ||
Pfisterer J, Plante M, Vergote I, et al. Gemcitabine plus carboplatin compared with carboplatin in patients with platinum-sensitive recurrent ovarian cancer: an intergroup trial of the AGO-OVAR, the NCIC CTG, and the EORTC GCG. J Clin Oncol. 2006;24(29):4699–4707. | ||
Long J, Zhang Y, Yu X, et al. Overcoming drug resistance in pancreatic cancer. Expert Opin Ther Targets. 2011;15(7):817–828. | ||
Jordheim LP, Dumontet C. Review of recent studies on resistance to cytotoxic deoxynucleoside analogues. Biochim Biophys Acta. 2007;1776(2):138–159. | ||
Marechal R, Bachet JB, Mackey JR, et al. Levels of gemcitabine transport and metabolism proteins predict survival times of patients treated with gemcitabine for pancreatic adenocarcinoma. Gastroenterology. 2012;143(3):664–674.e1–e6. | ||
Ciccolini J, Dahan L, Andre N, et al. Cytidine deaminase residual activity in serum is a predictive marker of early severe toxicities in adults after gemcitabine-based chemotherapies. J Clin Oncol. 2010;28(1):160–165. | ||
Hagner N, Joerger M. Cancer chemotherapy: targeting folic acid synthesis. Cancer Manag Res. 2010;2:293–301. | ||
Purcell WT, Ettinger DS. Novel antifolate drugs. Curr Oncol Rep. 2003;5(2):114–125. | ||
Siddiqui IA, Sanna V. Impact of nanotechnology on the delivery of natural products for cancer prevention and therapy. Mol Nutr Food Res. 2016;60(6):1330–1341. | ||
Orlikova B, Legrand N, Panning J, Dicato M, Diederich M. Anti-inflammatory and anticancer drugs from nature. Cancer Treat Res. 2014;159:123–143. | ||
Airley R. Cancer Chemotherapy: Basic Science to the Clinic. West Sussex, UK: Wiley-Blackwell; 2009. | ||
Smoter M, Bodnar L, Duchnowska R, Stec R, Grala B, Szczylik C. The role of Tau protein in resistance to paclitaxel. Cancer Chemother Pharmacol. 2011;68(3):553–557. | ||
Vergara D, Tinelli A, Iannone A, Maffia M. The impact of proteomics in the understanding of the molecular basis of paclitaxel-resistance in ovarian tumors. Curr Cancer Drug Targets. 2012;12(8):987–997. | ||
Barbuti AM, Chen ZS. Paclitaxel through the ages of anticancer therapy: exploring its role in chemoresistance and radiation therapy. Cancers (Basel). 2015;7(4):2360–2371. | ||
Sheng Y, You Y, Chen Y. Dual-targeting hybrid peptide-conjugated doxorubicin for drug resistance reversal in breast cancer. Int J Pharm. 2016;512(1):1–13. | ||
Cox J, Weinman S. Mechanisms of doxorubicin resistance in hepatocellular carcinoma. Hepat Oncol. 2016;3(1):57–59. | ||
Vatsyayan R, Chaudhary P, Lelsani PC, et al. Role of RLIP76 in doxorubicin resistance in lung cancer. Int J Oncol. 2009;34(6):1505–1511. | ||
Simko V, Takacova M, Debreova M, et al. Dexamethasone downregulates expression of carbonic anhydrase IX via HIF-1alpha and NF-kappaB-dependent mechanisms. Int J Oncol. 2016;49(4):1277–1288. | ||
Cook AM, McDonnell AM, Lake RA, Nowak AK. Dexamethasone co-medication in cancer patients undergoing chemotherapy causes substantial immunomodulatory effects with implications for chemo-immunotherapy strategies. Oncoimmunology. 2016;5(3):e1066062. | ||
Schaaf MJM, Cidlowski JA. Molecular mechanisms of glucocorticoid action and resistance. J Steroid Biochem Mol Biol. 2002;83(1–5):37–48. | ||
Rondon-Lagos M, Villegas VE, Rangel N, Sanchez MC, Zaphiropoulos PG. Tamoxifen resistance: emerging molecular targets. Int J Mol Sci. 2016;17(8):pii:E1357. | ||
Viedma-Rodriguez R, Baiza-Gutman L, Salamanca-Gomez F, et al. Mechanisms associated with resistance to tamoxifen in estrogen receptor-positive breast cancer (review). Oncol Rep. 2014;32(1):3–15. | ||
Macerelli M, Ganzinelli M, Gouedard C, et al. Can the response to a platinum-based therapy be predicted by the DNA repair status in non-small cell lung cancer? Cancer Treat Rev. 2016;48:8–19. | ||
Tapia G, Diaz-Padilla I. Molecular mechanisms of platinum resistance in ovarian cancer. In: Diaz-Padilla I, editor. Ovarian Cancer – A Clinical and Translational Update. InTechOpen; 2013:205–223. | ||
Rabik CA, Dolan ME. Molecular mechanisms of resistance and toxicity associated with platinating agents. Cancer Treat Rev. 2007;33(1):9–23. | ||
Shrivastava A, Khan AA, Khurshid M, Kalam MA, Jain SK, Singhal PK. Recent developments in L-asparaginase discovery and its potential as anticancer agent. Crit Rev Oncol Hematol. 2016;100:1–10. | ||
Andrade AF, Borges KS, Silveira VS. Update on the use of l-asparaginase in infants and adolescent patients with acute lymphoblastic leukemia. Clin Med Insights Oncol. 2014;8:95–100. | ||
Chen SH. Asparaginase therapy in pediatric acute lymphoblastic leukemia: a focus on the mode of drug resistance. Pediatr Neonatol. 2015;56(5):287–293. | ||
Kwa FA, Cole-Sinclair M, Kapuscinski M. Chlorambucil-sensitive and -resistant lymphoid cells display different responses to the histone deacetylase inhibitor, sodium butyrate. Biochem Biophys Res Commun. 2010;403(3–4):288–292. | ||
Xiao S, Yang Z, Qiu X, et al. miR-29c contribute to glioma cells temozolomide sensitivity by targeting O6-methylguanine-DNA methyltransferases indirectly. Oncotarget. 2016;7(31):50229–50238. | ||
Patil R, Portilla-Arias J, Ding H, et al. Temozolomide delivery to tumor cells by a multifunctional nano vehicle based on poly(beta-L-malic acid). Pharm Res. 2010;27(11):2317–2329. | ||
Orza A, Soritau O, Tomuleasa C, et al. Reversing chemoresistance of malignant glioma stem cells using gold nanoparticles. Int J Nanomedicine. 2013;8:689–702. | ||
Danhier F, Messaoudi K, Lemaire L, Benoit JP, Lagarce F. Combined anti-galectin-1 and anti-EGFR siRNA-loaded chitosan-lipid nanocapsules decrease temozolomide resistance in glioblastoma: in vivo evaluation. Int J Pharm. 2015;481(1–2):154–161. | ||
Balca-Silva J, Matias D, do Carmo A, et al. Tamoxifen in combination with temozolomide induce a synergistic inhibition of PKC-pan in GBM cell lines. Biochim Biophys Acta. 2015;1850(4):722–732. | ||
Yoshimoto K, Mizoguchi M, Hata N, et al. Complex DNA repair pathways as possible therapeutic targets to overcome temozolomide resistance in glioblastoma. Front Oncol. 2012;2:186. | ||
Yang RY, Yang KS, Pike LJ, Marshall GR. Targeting the dimerization of epidermal growth factor receptors with small-molecule inhibitors. Chem Biol Drug Des. 2010;76(1):1–9. | ||
Lefranc F, Sadeghi N, Camby I, Metens T, Dewitte O, Kiss R. Present and potential future issues in glioblastoma treatment. Expert Rev Anticancer Ther. 2006;6(5):719–732. | ||
Messaoudi K, Saulnier P, Boesen K, Benoit JP, Lagarce F. Anti-epidermal growth factor receptor siRNA carried by chitosan-transacylated lipid nanocapsules increases sensitivity of glioblastoma cells to temozolomide. Int J Nanomedicine. 2014;9:1479–1490. | ||
Messaoudi K, Clavreul A, Danhier F, Saulnier P, Benoit JP, Lagarce F. Combined silencing expression of MGMT with EGFR or galectin-1 enhances the sensitivity of glioblastoma to temozolomide. Eur J Nanomed. 2015;7(2):97–107. | ||
Xu H, Paxton JW, Wu Z. Development of long-circulating pH-sensitive liposomes to circumvent gemcitabine resistance in pancreatic cancer cells. Pharm Res. 2016;33(7):1628–1637. | ||
Chung WG, Sandoval MA, Sloat BR, Lansakara PD, Cui Z. Stearoyl gemcitabine nanoparticles overcome resistance related to the over-expression of ribonucleotide reductase subunit M1. J Control Release. 2012;157(1):132–140. | ||
Wang J, Yin C, Tang G, Lin X, Wu Q. Glucose-functionalized multidrug-conjugating nanoparticles based on amphiphilic terpolymer with enhanced anti-tumorous cell cytotoxicity. Int J Pharm. 2013;441(1–2):291–298. | ||
Cheng M, Gao X, Wang Y, et al. Synthesis of glycyrrhetinic acid-modified chitosan 5-fluorouracil nanoparticles and its inhibition of liver cancer characteristics in vitro and in vivo. Mar Drugs. 2013;11(9):3517–3536. | ||
Johari-Ahar M, Barar J, Alizadeh AM, Davaran S, Omidi Y, Rashidi MR. Methotrexate-conjugated quantum dots: synthesis, characterisation and cytotoxicity in drug resistant cancer cells. J Drug Target. 2016;24(2):120–133. | ||
Yuan X, Ji W, Chen S, et al. A novel paclitaxel-loaded poly(d,l-lactide-co-glycolide)-Tween 80 copolymer nanoparticle overcoming multidrug resistance for lung cancer treatment. Int J Nanomedicine. 2016;11:2119–2131. | ||
Wang L, Jia E. Ovarian cancer targeted hyaluronic acid-based nanoparticle system for paclitaxel delivery to overcome drug resistance. Drug Deliv. 2016;23(5):1810–1817. | ||
Tang X, Liang Y, Feng X, Zhang R, Jin X, Sun L. Co-delivery of docetaxel and Poloxamer 235 by PLGA-TPGS nanoparticles for breast cancer treatment. Mater Sci Eng C Mater Biol Appl. 2015;49:348–355. | ||
Song XR, Zheng Y, He G, et al. Development of PLGA nanoparticles simultaneously loaded with vincristine and verapamil for treatment of hepatocellular carcinoma. J Pharm Sci. 2010;99(12):4874–4879. | ||
Tacar O, Sriamornsak P, Dass CR. Doxorubicin: an update on anticancer molecular action, toxicity and novel drug delivery systems. J Pharm Pharmacol. 2013;65(2):157–170. | ||
Kopecka J, Porto S, Lusa S, et al. Zoledronic acid-encapsulating self-assembling nanoparticles and doxorubicin: a combinatorial approach to overcome simultaneously chemoresistance and immunoresistance in breast tumors. Oncotarget. 2016;7(15):20753–20772. | ||
Yang H, Deng L, Li T, et al. Multifunctional PLGA nanobubbles as theranostic agents: combining doxorubicin and P-gp siRNA co-delivery into human breast cancer cells and ultrasound cellular imaging. J Biomed Nanotechnol. 2015;11(12):2124–2136. | ||
Wang X, Teng Z, Wang H, et al. Increasing the cytotoxicity of doxorubicin in breast cancer MCF-7 cells with multidrug resistance using a mesoporous silica nanoparticle drug delivery system. Int J Clin Exp Pathol. 2014;7(4):1337–1347. | ||
Yu C, Zhou M, Zhang X, Wei W, Chen X, Zhang X. Smart doxorubicin nanoparticles with high drug payload for enhanced chemotherapy against drug resistance and cancer diagnosis. Nanoscale. 2015;7(13):5683–5690. | ||
Unsoy G, Khodadust R, Yalcin S, Mutlu P, Gunduz U. Synthesis of doxorubicin loaded magnetic chitosan nanoparticles for pH responsive targeted drug delivery. Eur J Pharm Sci. 2014;62:243–250. | ||
Generali D, Berruti A, Cappelletti MR, et al. Effect of primary letrozole treatment on tumor expression of mTOR and HIF-1alpha and relation to clinical response. J Natl Cancer Inst Monogr. 2015;2015(51):64–66. | ||
Hoeflich KP, Guan J, Edgar KA, et al. The PI3K inhibitor taselisib overcomes letrozole resistance in a breast cancer model expressing aromatase. Genes Cancer. 2016;7(3–4):73–85. | ||
Nair HB, Huffman S, Veerapaneni P, et al. Hyaluronic acid-bound letrozole nanoparticles restore sensitivity to letrozole-resistant xenograft tumors in mice. J Nanosci Nanotechnol. 2011;11(5):3789–3799. | ||
Cho SK, Pedram A, Levin ER, Kwon YJ. Acid-degradable core-shell nanoparticles for reversed tamoxifen-resistance in breast cancer by silencing manganese superoxide dismutase (MnSOD). Biomaterials. 2013;34(38):10228–10237. | ||
Dilruba S, Kalayda GV. Platinum-based drugs: past, present and future. Cancer Chemother Pharmacol. 2016;77(6):1103–1124. | ||
Zhou D, Cong Y, Qi Y, et al. Overcoming tumor resistance to cisplatin through micelle-mediated combination chemotherapy. Biomater Sci. 2015;3(1):182–191. | ||
Scarano W, de Souza P, Stenzel MH. Dual-drug delivery of curcumin and platinum drugs in polymeric micelles enhances the synergistic effects: a double act for the treatment of multidrug-resistant cancer. Biomater Sci. 2015;3(1):163–174. | ||
Jain RK, Stylianopoulos T. Delivering nanomedicine to solid tumors. Nat Rev Clin Oncol. 2010;7(11):653–664. | ||
Owens DE 3rd, Peppas NA. Opsonization, biodistribution, and pharmacokinetics of polymeric nanoparticles. Int J Pharm. 2006;307(1):93–102. | ||
Gao H, He Q. The interaction of nanoparticles with plasma proteins and the consequent influence on nanoparticles behavior. Expert Opin Drug Deliv. 2014;11(3):409–420. | ||
Rattan R, Bhattacharjee S, Zong H, et al. Nanoparticle-macrophage interactions: a balance between clearance and cell-specific targeting. Bioorg Med Chem. 2017;25(16):4487–4496. | ||
Chrastina A, Massey KA, Schnitzer JE. Overcoming in vivo barriers to targeted nanodelivery. Wiley Interdiscip Rev Nanomed Nanobiotechnol. 2011;3(4):421–437. | ||
Stylianopoulos T. Intelligent drug delivery systems for the treatment of solid tumors. Eur J Nanomed. 2016;8(1):9–16. | ||
Ewald B, Sampath D, Plunkett W. Nucleoside analogs: molecular mechanisms signaling cell death. Oncogene. 2008;27(50):6522–6537. | ||
Wong A, Soo RA, Yong WP, Innocenti F. Clinical pharmacology and pharmacogenetics of gemcitabine. Drug Metab Rev. 2009;41(2):77–88. | ||
Kim TM, Kim S, Ahn YO, Lee SH, Kim DW, Heo DS. Anti-cancer activity of gemcitabine against natural killer cell leukemia/lymphoma. Leuk Lymphoma. 2014;55(4):940–943. | ||
Edward C. Physicians’ Cancer Chemotherapy Drug Manual 2015. Burlington, MA: Jones and Bartlett Publishers; 2015. | ||
Fan P, Liu L, Yin Y, et al. MicroRNA-101-3p reverses gemcitabine resistance by inhibition of ribonucleotide reductase M1 in pancreatic cancer. Cancer Lett. 2016;373(1):130–137. | ||
Orlandi A, Calegari MA, Martini M, et al. Gemcitabine versus FOLFIRINOX in patients with advanced pancreatic adenocarcinoma hENT1-positive: everything was not too bad back when everything seemed worse. Clin Transl Oncol. 2016;18(10):988–995. | ||
Ohmine K, Kawaguchi K, Ohtsuki S, et al. Attenuation of phosphorylation by deoxycytidine kinase is key to acquired gemcitabine resistance in a pancreatic cancer cell line: targeted proteomic and metabolomic analyses in PK9 cells. Pharm Res. 2012;29(7):2006–2016. | ||
Song Y, Baba T, Mukaida N. Gemcitabine induces cell senescence in human pancreatic cancer cell lines. Biochem Biophys Res Commun. 2016;477(3):515–519. | ||
Huang P, Chubb S, Hertel LW, Grindey GB, Plunkett W. Action of 2′,2′-difluorodeoxycytidine on DNA synthesis. Cancer Res. 1991;51(22):6110–6117. | ||
Elnaggar M, Giovannetti E, Peters GJ. Molecular targets of gemcitabine action: rationale for development of novel drugs and drug combinations. Curr Pharm Des. 2012;18(19):2811–2829. | ||
Shao J, Liu X, Zhu L, Yen Y. Targeting ribonucleotide reductase for cancer therapy. Expert Opin Ther Targets. 2013;17(12):1423–1437. | ||
Gesto DS, Cerqueira NM, Fernandes PA, Ramos MJ. Gemcitabine: a critical nucleoside for cancer therapy. Curr Med Chem. 2012;19(7):1076–1087. | ||
Ciccolini J, Serdjebi C, Peters GJ, Giovannetti E. Pharmacokinetics and pharmacogenetics of Gemcitabine as a mainstay in adult and pediatric oncology: an EORTC-PAMM perspective. Cancer Chemother Pharmacol. 2016;78(1):1–12. | ||
Mori R, Ishikawa T, Ichikawa Y, et al. Human equilibrative nucleoside transporter 1 is associated with the chemosensitivity of gemcitabine in human pancreatic adenocarcinoma and biliary tract carcinoma cells. Oncol Rep. 2007;17(5):1201–1205. | ||
Farrell JJ, Elsaleh H, Garcia M, et al. Human equilibrative nucleoside transporter 1 levels predict response to gemcitabine in patients with pancreatic cancer. Gastroenterology. 2009;136(1):187–195. | ||
Wang C, Zhang W, Fu M, Yang A, Huang H, Xie J. Establishment of human pancreatic cancer gemcitabine-resistant cell line with ribonucleotide reductase overexpression. Oncol Rep. 2014;33(1):383–390. | ||
Weizman N, Krelin Y, Shabtay-Orbach A, et al. Macrophages mediate gemcitabine resistance of pancreatic adenocarcinoma by upregulating cytidine deaminase. Oncogene. 2014;33(29):3812–3819. | ||
Huang Y, Liu X, Wang YH, et al. The prognostic value of ribonucleotide reductase small subunit M2 in predicting recurrence for prostate cancers. Urol Oncol. 2014;32(1):51.e9-19. | ||
Galmarini CM, Clarke ML, Falette N, Puisieux A, Mackey JR, Dumontet C. Expression of a non-functional p53 affects the sensitivity of cancer cells to gemcitabine. Int J Cancer. 2002;97(4):439–445. | ||
Skrypek N, Duchene B, Hebbar M, Leteurtre E, van Seuningen I, Jonckheere N. The MUC4 mucin mediates gemcitabine resistance of human pancreatic cancer cells via the concentrative nucleoside transporter family. Oncogene. 2013;32(13):1714–1723. | ||
Rudin D, Li L, Niu N, et al. Gemcitabine cytotoxicity: interaction of efflux and deamination. J Drug Metab Toxicol. 2011;2(107):1–10. | ||
Wattanawongdon W, Hahnvajanawong C, Namwat N, et al. Establishment and characterization of gemcitabine-resistant human cholangiocarcinoma cell lines with multidrug resistance and enhanced invasiveness. Int J Oncol. 2015;47(1):398–410. | ||
Mackey JR, Yao SY, Smith KM, et al. Gemcitabine transport in xenopus oocytes expressing recombinant plasma membrane mammalian nucleoside transporters. J Natl Cancer Inst. 1999;91(21):1876–1881. | ||
Mackey JR, Mani RS, Selner M, et al. Functional nucleoside transporters are required for gemcitabine influx and manifestation of toxicity in cancer cell lines. Cancer Res. 1998;58(19):4349–4357. | ||
Spratlin J, Sangha R, Glubrecht D, et al. The absence of human equilibrative nucleoside transporter 1 is associated with reduced survival in patients with gemcitabine-treated pancreas adenocarcinoma. Clin Cancer Res. 2004;10(20):6956–6961. | ||
Brandi G, Deserti M, Vasuri F, et al. Membrane localization of human equilibrative nucleoside transporter 1 in tumor cells may predict response to adjuvant gemcitabine in resected cholangiocarcinoma patients. Oncologist. 2016;21(5):600–607. | ||
Liu ZQ, Han YC, Zhang X, et al. Prognostic value of human equilibrative nucleoside transporter1 in pancreatic cancer receiving gemcitabin-based chemotherapy: a meta-analysis. PLoS One. 2014;9(1):e87103. | ||
Espinoza JA, Garcia P, Bizama C, et al. Low expression of equilibrative nucleoside transporter 1 is associated with poor prognosis in chemotherapy-naive pT2 gallbladder adenocarcinoma patients. Histopathology. 2016;68(5):722–728. | ||
Giovannetti E, Del Tacca M, Mey V, et al. Transcription analysis of human equilibrative nucleoside transporter-1 predicts survival in pancreas cancer patients treated with gemcitabine. Cancer Res. 2006;66(7):3928–3935. | ||
Bird NT, Elmasry M, Jones R, et al. Immunohistochemical hENT1 expression as a prognostic biomarker in patients with resected pancreatic ductal adenocarcinoma undergoing adjuvant gemcitabine-based chemotherapy. Br J Surg. 2017;104(4):328–336. | ||
Nakano T, Saiki Y, Kudo C, et al. Acquisition of chemoresistance to gemcitabine is induced by a loss-of-function missense mutation of DCK. Biochem Biophys Res Commun. 2015;464(4):1084–1089. | ||
Shimada Y, Okumura T, Sekine S, et al. Clinicopathological significance of deoxycytidine kinase expression in esophageal squamous cell carcinoma. Mol Clin Oncol. 2013;1(4):716–720. | ||
Jordheim LP, Cros E, Gouy MH, et al. Characterization of a gemcitabine-resistant murine leukemic cell line: reversion of in vitro resistance by a mononucleotide prodrug. Clin Cancer Res. 2004;10(16):5614–5621. | ||
Galmarini CM, Clarke ML, Jordheim L, et al. Resistance to gemcitabine in a human follicular lymphoma cell line is due to partial deletion of the deoxycytidine kinase gene. BMC Pharmacol. 2004;4:8. | ||
Jordheim LP, Galmarini CM, Dumontet C. Gemcitabine resistance due to deoxycytidine kinase deficiency can be reverted by fruitfly deoxynucleoside kinase, DmdNK, in human uterine sarcoma cells. Cancer Chemother Pharmacol. 2006;58(4):547–554. | ||
Kazuno H, Sakamoto K, Fujioka A, Fukushima M, Matsuda A, Sasaki T. Possible antitumor activity of 1-(3-C-ethynyl-beta-D-ribo-pentofuranosyl)cytosine (ECyd, TAS-106) against an established gemcitabine (dFdCyd)-resistant human pancreatic cancer cell line. Cancer Sci. 2005;96(5):295–302. | ||
Kroep JR, Loves WJ, van der Wilt CL, et al. Pretreatment deoxycytidine kinase levels predict in vivo gemcitabine sensitivity. Mol Cancer Ther. 2002;1(6):371–376. | ||
Sebastiani V, Ricci F, Rubio-Viqueira B, et al. Immunohistochemical and genetic evaluation of deoxycytidine kinase in pancreatic cancer: relationship to molecular mechanisms of gemcitabine resistance and survival. Clin Cancer Res. 2006;12(8):2492–2497. | ||
Fujita H, Ohuchida K, Mizumoto K, et al. Gene expression levels as predictive markers of outcome in pancreatic cancer after gemcitabine-based adjuvant chemotherapy. Neoplasia. 2010;12(10):807–808. | ||
Bouffard DY, Laliberte J, Momparler RL. Kinetic studies on 2′,2′-difluorodeoxycytidine (gemcitabine) with purified human deoxycytidine kinase and cytidine deaminase. Biochem Pharmacol. 1993;45(9):1857–1861. | ||
Yoon KA, Woo SM, Hong EK, et al. Cytidine deaminase as a molecular predictor of gemcitabine response in patients with biliary tract cancer. Oncology. 2015;89(6):345–350. | ||
Okazaki T, Javle M, Tanaka M, Abbruzzese JL, Li D. Single nucleotide polymorphisms of gemcitabine metabolic genes and pancreatic cancer survival and drug toxicity. Clin Cancer Res. 2010;16(1):320–329. | ||
Tibaldi C, Giovannetti E, Vasile E, et al. Correlation of CDA, ERCC1, and XPD polymorphisms with response and survival in gemcitabine/cisplatin-treated advanced non-small cell lung cancer patients. Clin Cancer Res. 2008;14(6):1797–1803. | ||
Gilbert JA, Salavaggione OE, Ji Y, et al. Gemcitabine pharmacogenomics: cytidine deaminase and deoxycytidylate deaminase gene resequencing and functional genomics. Clin Cancer Res. 2006;12(6):1794–1803. | ||
Jordheim LP, Sève P, Trédan O, Dumontet C. The ribonucleotide reductase large subunit (RRM1) as a predictive factor in patients with cancer. Lancet Oncol. 2011;12(7):693–702. | ||
Iwamoto K, Nakashiro K, Tanaka H, Tokuzen N, Hamakawa H. Ribonucleotide reductase M2 is a promising molecular target for the treatment of oral squamous cell carcinoma. Int J Oncol. 2015;46(5):1971–1977. | ||
Wang X, Zhenchuk A, Wiman KG, Albertioni F. Regulation of p53R2 and its role as potential target for cancer therapy. Cancer Lett. 2009;276(1):1–7. | ||
Nordlund N, Reichard P. Ribonucleotide reductases. Annu Rev Biochem. 2006;75:681–706. | ||
Zeng C, Fan W, Zhang X. RRM1 expression is associated with the outcome of gemcitabine-based treatment of non-small cell lung cancer patients – a short report. Cell Oncol (Dordr). 2015;38(4):319–325. | ||
Nakano K, Bálint E, Ashcroft M, Vousden KH. A ribonucleotide reductase gene is a transcriptional target of p53 and p73. Oncogene. 2000;19(37):4283–4289. | ||
Cho EC, Kuo ML, Liu XY, et al. Tumor suppressor FOXO3 regulates ribonucleotide reductase subunit RRM2B and impacts on survival of cancer patients. Oncotarget. 2014;5(13):4834–4844. | ||
Lecca P. Methods of biological network inference for reverse engineering cancer chemoresistance mechanisms. Drug Discov Today. 2014;19(2):151–163. | ||
Nakahira S, Nakamori S, Tsujie M, et al. Involvement of ribonucleotide reductase M1 subunit overexpression in gemcitabine resistance of human pancreatic cancer. Int J Cancer. 2007;120(6):1355–1363. | ||
Itoi T, Sofuni A, Fukushima N, et al. Ribonucleotide reductase subunit M2 mRNA expression in pretreatment biopsies obtained from unresectable pancreatic carcinomas. J Gastroenterol. 2007;42(5):389–394. | ||
Akita H, Zheng Z, Takeda Y, et al. Significance of RRM1 and ERCC1 expression in resectable pancreatic adenocarcinoma. Oncogene. 2009;28(32):2903–2909. | ||
Ceppi P, Volante M, Novello S, et al. ERCC1 and RRM1 gene expressions but not EGFR are predictive of shorter survival in advanced non-small-cell lung cancer treated with cisplatin and gemcitabine. Ann Oncol. 2006;17(12):1818–1825. | ||
Kozinn SI, Harty NJ, Delong JM, et al. MicroRNA Profile to Predict Gemcitabine Resistance in Bladder Carcinoma Cell Lines. Genes Cancer. 2013;4(1–2):61–69. | ||
Neesse A, Michl P, Frese KK, et al. Stromal biology and therapy in pancreatic cancer. Gut. 2011;60(6):861–868. | ||
Xu M, Li L, Liu Z, et al. ABCB2 (TAP1) as the downstream target of SHH signaling enhances pancreatic ductal adenocarcinoma drug resistance. Cancer Lett. 2013;333(2):152–158. | ||
Kuramitsu Y, Taba K, Ryozawa S, et al. Identification of up- and down-regulated proteins in gemcitabine-resistant pancreatic cancer cells using two-dimensional gel electrophoresis and mass spectrometry. Anticancer Res. 2010;30(9):3367–3372. | ||
Feig C, Gopinathan A, Neesse A, Chan DS, Cook N, Tuveson DA. The pancreas cancer microenvironment. Clin Cancer Res. 2012;18(16):4266–4276. | ||
Mittal A, Chitkara D, Behrman SW, Mahato RI. Efficacy of gemcitabine conjugated and miRNA-205 complexed micelles for treatment of advanced pancreatic cancer. Biomaterials. 2014;35(25):7077–7087. | ||
Meng H, Zhao Y, Dong J, et al. Two-wave nanotherapy to target the stroma and optimize gemcitabine delivery to a human pancreatic cancer model in mice. ACS Nano. 2013;7(11):10048–10065. | ||
Licciardi M, Paolino D, Celia C, Giammona G, Cavallaro G, Fresta M. Folate-targeted supramolecular vesicular aggregates based on polyaspartyl-hydrazide copolymers for the selective delivery of antitumoral drugs. Biomaterials. 2010;31(28):7340–7354. | ||
Ventura CA, Cannava C, Stancanelli R, et al. Gemcitabine-loaded chitosan microspheres. Characterization and biological in vitro evaluation. Biomed Microdevices. 2011;13(5):799–807. | ||
Jia L. Preparation, physicochemical characterization and cytotoxicityin vitroof gemcitabine-loaded PEG-PDLLA nanovesicles. World J Gastroenterol. 2010;16(8):1008. | ||
Zhao LP, Xue C, Zhang JW, et al. Expression of RRM1 and its association with resistancy to gemcitabine-based chemotherapy in advanced nasopharyngeal carcinoma. Chin J Cancer. 2012;31(10):476–483. | ||
Xie H, Jiang W, Jiang J, et al. Predictive and prognostic roles of ribonucleotide reductase M1 in resectable pancreatic adenocarcinoma. Cancer. 2013;119(1):173–181. | ||
Lee JJ, Maeng CH, Baek SK, et al. The immunohistochemical overexpression of ribonucleotide reductase regulatory subunit M1 (RRM1) protein is a predictor of shorter survival to gemcitabine-based chemotherapy in advanced non-small cell lung cancer (NSCLC). Lung Cancer. 2010;70(2):205–210. | ||
Ulker M, Duman BB, Sahin B, Gumurdulu D. ERCC1 and RRM1 as a predictive parameter for non-small cell lung, ovarian or pancreas cancer treated with cisplatin and/or gemcitabine. Contemp Oncol (Pozn). 2015;19(3):207–213. | ||
Gong W, Zhang X, Wu J, et al. RRM1 expression and clinical outcome of gemcitabine-containing chemotherapy for advanced non-small-cell lung cancer: a meta-analysis. Lung Cancer. 2012;75(3):374–380. | ||
Khatri N, Rathi M, Baradia D, Misra A. cRGD grafted siRNA nano-constructs for chemosensitization of gemcitabine hydrochloride in lung cancer treatment. Pharm Res. 2015;32(3):806–818. | ||
Wonganan P, Chung WG, Zhu S, Kiguchi K, Digiovanni J, Cui Z. Silencing of ribonucleotide reductase subunit M1 potentiates the antitumor activity of gemcitabine in resistant cancer cells. Cancer Biol Ther. 2012;13(10):908–914. | ||
Damaraju VL, Damaraju S, Young JD, et al. Nucleoside anticancer drugs: the role of nucleoside transporters in resistance to cancer chemotherapy. Oncogene. 2003;22(47):7524–7536. | ||
Marechal R, Mackey JR, Lai R, et al. Human equilibrative nucleoside transporter 1 and human concentrative nucleoside transporter 3 predict survival after adjuvant gemcitabine therapy in resected pancreatic adenocarcinoma. Clin Cancer Res. 2009;15(8):2913–2919. | ||
Greenhalf W, Ghaneh P, Neoptolemos JP, et al. Pancreatic cancer hENT1 expression and survival from gemcitabine in patients from the ESPAC-3 trial. J Natl Cancer Inst. 2014;106(1):djt347. | ||
Nakagawa N, Murakami Y, Uemura K, et al. Combined analysis of intratumoral human equilibrative nucleoside transporter 1 (hENT1) and ribonucleotide reductase regulatory subunit M1 (RRM1) expression is a powerful predictor of survival in patients with pancreatic carcinoma treated with adjuvant gemcitabine-based chemotherapy after operative resection. Surgery. 2013;153(4):565–575. | ||
Morinaga S, Nakamura Y, Watanabe T, et al. Immunohistochemical analysis of human equilibrative nucleoside transporter-1 (hENT1) predicts survival in resected pancreatic cancer patients treated with adjuvant gemcitabine monotherapy. Ann Surg Oncol. 2012;19(Suppl 3):S558–S564. | ||
Kondo N, Murakami Y, Uemura K, et al. Combined analysis of dihydropyrimidine dehydrogenase and human equilibrative nucleoside transporter 1 expression predicts survival of pancreatic carcinoma patients treated with adjuvant gemcitabine plus S-1 chemotherapy after surgical resection. Ann Surg Oncol. 2012;19(Suppl 3):S646–S655. | ||
Wonganan P, Lansakara PD, Zhu S, et al. Just getting into cells is not enough: mechanisms underlying 4-(N)-stearoyl gemcitabine solid lipid nanoparticle’s ability to overcome gemcitabine resistance caused by RRM1 overexpression. J Control Release. 2013;169(1–2):17–27. | ||
Hung SW, Marrache S, Cummins S, et al. Defective hCNT1 transport contributes to gemcitabine chemoresistance in ovarian cancer subtypes: overcoming transport defects using a nanoparticle approach. Cancer Lett. 2015;359(2):233–240. | ||
Frese KK, Neesse A, Cook N, et al. nab-Paclitaxel potentiates gemcitabine activity by reducing cytidine deaminase levels in a mouse model of pancreatic cancer. Cancer Discov. 2012;2(3):260–269. | ||
Couvreur P, Stella B, Reddy LH, et al. Squalenoyl nanomedicines as potential therapeutics. Nano Lett. 2006;6(11):2544–2548. | ||
Rejiba S, Reddy LH, Bigand C, Parmentier C, Couvreur P, Hajri A. Squalenoyl gemcitabine nanomedicine overcomes the low efficacy of gemcitabine therapy in pancreatic cancer. Nanomedicine. 2011;7(6):841–849. | ||
Reddy LH, Ferreira H, Dubernet C, et al. Squalenoyl nanomedicine of gemcitabine is more potent after oral administration in leukemia-bearing rats: study of mechanisms. Anticancer Drugs. 2008;19(10):999–1006. | ||
Reddy LH, Dubernet C, Mouelhi SL, Marque PE, Desmaele D, Couvreur P. A new nanomedicine of gemcitabine displays enhanced anticancer activity in sensitive and resistant leukemia types. J Control Release. 2007;124(1–2):20–27. | ||
Sloat BR, Sandoval MA, Li D, et al. In vitro and in vivo anti-tumor activities of a gemcitabine derivative carried by nanoparticles. Int J Pharm. 2011;409(1–2):278–288. | ||
Lansakara PD, Rodriguez BL, Cui Z. Synthesis and in vitro evaluation of novel lipophilic monophosphorylated gemcitabine derivatives and their nanoparticles. Int J Pharm. 2012;429(1–2):123–134. | ||
Meng H, Wang M, Liu H, et al. Use of a lipid-coated mesoporous silica nanoparticle platform for synergistic gemcitabine and paclitaxel delivery to human pancreatic cancer in mice. ACS Nano. 2015;9(4):3540–3557. | ||
Iyer AK, Khaled G, Fang J, Maeda H. Exploiting the enhanced permeability and retention effect for tumor targeting. Drug Discov Today. 2006;11(17–18):812–818. | ||
Matsumura Y, Maeda H. A new concept for macromolecular therapeutics in cancer chemotherapy: mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs. Cancer Res. 1986;46(12 Pt 1):6387–6392. | ||
Acharya S, Sahoo SK. PLGA nanoparticles containing various anticancer agents and tumour delivery by EPR effect. Adv Drug Deliv Rev. 2011;63(3):170–183. | ||
Maeda H, Nakamura H, Fang J. The EPR effect for macromolecular drug delivery to solid tumors: Improvement of tumor uptake, lowering of systemic toxicity, and distinct tumor imaging in vivo. Adv Drug Deliv Rev. 2013;65(1):71–79. | ||
Fang J, Nakamura H, Maeda H. The EPR effect: unique features of tumor blood vessels for drug delivery, factors involved, and limitations and augmentation of the effect. Adv Drug Deliv Rev. 2011;63(3):136–151. | ||
Xu Y, Meng H, Du F, et al. Preparation of intravenous injection nanoformulation of VESylated gemcitabine by co-assembly with TPGS and its anti-tumor activity in pancreatic tumor-bearing mice. Int J Pharm. 2015;495(2):792–797. | ||
Sobot D, Mura S, Rouquette M, et al. Circulating Lipoproteins: A Trojan Horse Guiding Squalenoylated Drugs to LDL-Accumulating Cancer Cells. Mol Ther. 2017;25(7):1596–1605. | ||
Moysan E, Bastiat G, Benoit JP. Gemcitabine versus modified gemcitabine: a review of several promising chemical modifications. Mol Pharm. 2013;10(2):430–444. | ||
Federico C, Morittu VM, Britti D, Trapasso E, Cosco D. Gemcitabine-loaded liposomes: rationale, potentialities and future perspectives. Int J Nanomedicine. 2012;7:5423–5436. | ||
Zhu S, Wonganan P, Lansakara PD, O’Mary HL, Li Y, Cui Z. The effect of the acid-sensitivity of 4-(N)-stearoyl gemcitabine-loaded micelles on drug resistance caused by RRM1 overexpression. Biomaterials. 2013;34(9):2327–2339. | ||
Papa AL, Basu S, Sengupta P, Banerjee D, Sengupta S, Harfouche R. Mechanistic studies of Gemcitabine-loaded nanoplatforms in resistant pancreatic cancer cells. BMC cancer. 2012;12:419. | ||
Daman Z, Ostad S, Amini M, Gilani K. Preparation, optimization and in vitro characterization of stearoyl-gemcitabine polymeric micelles: a comparison with its self-assembled nanoparticles. Int J Pharm. 2014;468(1–2):142–151. | ||
Papa AL, Sidiqui A, Balasubramanian SU, et al. PEGylated liposomal gemcitabine: insights into a potential breast cancer therapeutic. Cell Oncol (Dordr). 2013;36(6):449–457. | ||
Nyhan W. Nucleotide Synthesis Via Salvage Pathway. In: eLS. Chichester: John Wiley & Sons Ltd; 2005. | ||
Kang N, Jun AH, Bhutia YD, Kannan N, Unadkat JD, Govindarajan R. Human equilibrative nucleoside transporter-3 (hENT3) spectrum disorder mutations impair nucleoside transport, protein localization, and stability. J Biol Chem. 2010;285(36):28343–28352. | ||
Jiang J, Neubauer BL, Graff JR, et al. Expression of group IIA secretory phospholipase A2 is elevated in prostatic intraepithelial neoplasia and adenocarcinoma. Am J Pathol. 2002;160(2):667–671. | ||
Abe T, Sakamoto K, Kamohara H, Hirano Y, Kuwahara N, Ogawa M. Group II phospholipase A2 is increased in peritoneal and pleural effusions in patients with various types of cancer. Int J Cancer. 1997;74(3):245–250. | ||
Oka Y, Ogawa M, Matsuda Y, et al. Serum immunoreactive pancreatic phospholipase A2 in patients with various malignant tumors. Enzyme. 1990;43(2):80–88. | ||
Quach ND, Mock JN, Scholpa NE, et al. Role of the phospholipase A2 receptor in liposome drug delivery in prostate cancer cells. Mol Pharm. 2014;11(10):3443–3451. | ||
Mock JN, Costyn LJ, Wilding SL, Arnold RD, Cummings BS. Evidence for distinct mechanisms of uptake and antitumor activity of secretory phospholipase A2 responsive liposome in prostate cancer. Integr Biol (Camb). 2013;5(1):172–182. | ||
Zhu G, Mock JN, Aljuffali I, Cummings BS, Arnold RD. Secretory phospholipase A(2) responsive liposomes. J Pharm Sci. 2011;100(8):3146–3159. | ||
Hofmann D, Messerschmidt C, Bannwarth MB, Landfester K, Mailander V. Drug delivery without nanoparticle uptake: delivery by a kiss-and-run mechanism on the cell membrane. Chem Commun (Camb). 2014;50(11):1369–1371. | ||
Markman JL, Rekechenetskiy A, Holler E, Ljubimova JY. Nanomedicine therapeutic approaches to overcome cancer drug resistance. Adv Drug Deliv Rev. 2013;65(13–14):1866–1879. | ||
Cosco D, Rocco F, Ceruti M, Vono M, Fresta M, Paolino D. Self-assembled squalenoyl-cytarabine nanostructures as a potent nanomedicine for treatment of leukemic diseases. Int J Nanomedicine. 2012;7:2535–2546. | ||
Arya G, Vandana M, Acharya S, Sahoo SK. Enhanced antiproliferative activity of herceptin (HER2)-conjugated gemcitabine-loaded chitosan nanoparticle in pancreatic cancer therapy. Nanomedicine. 2011;7(6):859–870. | ||
Li J, Di Y, Jin C, et al. Gemcitabine-loaded albumin nanospheres (GEM-ANPs) inhibit PANC-1 cells in vitro and in vivo. Nanoscale Res Lett. 2013;8(1):176. | ||
Maksimenko A, Caron J, Mougin J, Desmaele D, Couvreur P. Gemcitabine-based therapy for pancreatic cancer using the squalenoyl nucleoside monophosphate nanoassemblies. Int J Pharm. 2015;482(1–2):38–46. | ||
Xu HT, Paxton JW, Wu ZM. Development of long-circulating pH-sensitive liposomes to circumvent gemcitabine resistance in pancreatic cancer cells. Pharm Res-Dordr. 2016;33(7):1628–1637. | ||
Chitkara D, Mittal A, Behrman SW, Kumar N, Mahato RI. Self-assembling, amphiphilic polymer-gemcitabine conjugate shows enhanced antitumor efficacy against human pancreatic adenocarcinoma. Bioconjug Chem. 2013;24(7):1161–1173. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.