Back to Journals » OncoTargets and Therapy » Volume 11
Over-expression of ARHGAP18 suppressed cell proliferation, migration, invasion, and tumor growth in gastric cancer by restraining over-activation of MAPK signaling pathways
Authors Li Y, Ji S, Fu L, Jiang T, Wu D, Meng F
Received 14 December 2016
Accepted for publication 19 May 2017
Published 9 January 2018 Volume 2018:11 Pages 279—290
DOI https://doi.org/10.2147/OTT.S130255
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Geoffrey Pietersz
Yan Li,1 Shan Ji,2 Liye Fu,1 Tao Jiang,1 Di Wu,1 Fandong Meng1
1Department of Biotherapy, Cancer Research Institute, the First Affiliated Hospital of China Medical University, Shenyang City, People’s Republic of China; 2Department of Endocrinology, The Fifth People’s Hospital of Shenyang City, Shenyang City, People’s Republic of China
Abstract: Globally, gastric cancer is the second-greatest cause of cancer death. ARHGAP18 belongs to the Rho family of GTPases which is involved in cellular migration, invasion, and growth phases. The aim of the present study was to investigate whether ARHGAP18 could regulate cell proliferation, migration, invasion, and related molecular mechanisms in gastric cancer. Cell Counting Kit-8 (CCK-8) assay results showed that following transfection of a recombinant plasmid, over-expression of ARHGAP18 inhibited cell viability in MGC-803 and BGC823 cells. Using in vitro transwell analysis, migration and invasion abilities were significantly inhibited in cells with high ARHGAP18 expression. Phosphorylation levels of ERK, JNK, and p38 by Western blot analysis significantly declined after transfection of cells with the ARHGAP18 plasmid. Expression levels of ROCK, MTA1, and MMP-2/9 were detected by real-time polymerase chain reaction and Western blotting, and over-expression of ARHGAP18 decreased the expression levels of ROCK, MTA1, and MMP-9. A further in vivo tumor formation study in nude mice indicated that over-expression of ARHGAP18 delayed the progress of tumor formation. These results indicate that ARHGAP18 could act as a tumor suppressor and may serve as a promising therapeutic strategy for gastric cancer.
Keywords: ARHGAP18, gastric cancer, cell proliferation, migration, invasion, MAPK
Introduction
Gastric cancer is the most common digestive carcinoma, and its incidence and mortality rates, which are very high, show regional features. Asian countries are high prevalence regions, and major reasons are the spread and metastasis of cancer cells.1–3 On examination, we have found that more than 50% of patients do not have obvious or typical symptoms. Therefore, the patients with locally advanced gastric cancer are vulnerable to peritoneum, and more distant, metastasis and can lose the chance of surgery. Regarding well-differentiated gastric cancer, 50%–65% of patients cannot be cured by surgery.4 Although there are many therapeutic options for gastric cancer patients including surgery, chemotherapy, and radiotherapy, the therapeutic efficacy is not ideal for intermediate-advanced stage gastric cancer patients.5 Thus, detailed investigation of invasion- and migration-related molecular mechanisms to find key targets is important for gastric cancer control and prevention.
ARHGAP18 (alias SENEX) is located at chromosome 4q31.23. It encodes a protein of approximately 75 KD which is composed of 663 amino acids. ARHGAP18 has RhoGAP determinants and belongs to the GAP family.6–10 The Rho family of GTPases consists of more than 20 different members and is divided into seven subgroups including Cdc42, Rac, Rho, Rnd, RhoD, RhoH, and RhBTB, which regulate different aspects of the actin-myosin cytoskeleton including affecting cell polarity, migration, adhesion, and endocytosis. According to the previously mentioned research, we suspected that ARHGAP18 is involved in the regulation of gene transcription, cell cycle, and cell apoptosis.11–13 In the Rho GTPase family, Cdc42, Rac1, RhoA, and Rho are typical members.12 RhoA and Rac1 proteins play a key role in the whole process of cell migration. Activation of Racl regulates the contractile speed of cells and the change of cytoskeleton, PAK protein, as effectors of RhoA and Rac1, promote cell adhesion.14,15 Therefore, ARHGAP18, as a novel RhoGAP, may regulate cell proliferation, migration, and invasion in cancer.
MAPKs belong to serine/threonine protein kinase and can be activated by many stimuli including cytokines, growth factor, neurotransmitter, hormone, stress, and adhesion. Therefore, MAPKs can be involved in the process of cell division.16,17 However, more importantly, MAPKs can regulate the whole process from beginning to end in nearly all cells. In mammals, MAPK signaling pathways are involved in many reactions affecting cell proliferation, differentiation, and apoptosis. The MAPK family is divided into five subfamilies: ERK1/2, JNK, p38, ERK3/4, and ERK5.18–22 In addition, MAPKKKK or MAPKKK can interact with Ras/Rho family to activate MAPKKK and result in the phosphorylation and activation of MAPK signaling pathway.23 Many studies have confirmed that MAPK signaling pathway was activated in gastric cancer, lung cancer, ovarian cancer, and liver cancer.24–27 Therefore, we speculated that ARHGAP18 might be the downstream factor of MAPK pathways. In this study, clinical specimens and gastric cancer cell lines were used to detect ARHGAP18 expression to further verify the assumption. Our results confirmed that ARHGAP18 acted as a tumor suppressor and might serve as a promising therapeutic strategy for gastric cancer.
Materials and methods
Tissue specimen collection of gastric cancer patients
Forty-six gastric cancer patients had signed the informed consent form before surgery. The samples of gastric cancer tissues and benign gastric tumor tissues surgically removed in the First Affiliated Hospital of China Medical University were collected from 2014 to 2015. Adjacent normal tissues were collected as negative controls. Preoperative clinical and pathological follow-up data were collected, and the related analysis was done with the permission of all patients. The research was approved by the ethics committee of The Fifth People’s Hospital of Shenyang City.
Cell culture
Normal gastric cell line (GES-1) and gastric cancer cell lines (AGS, MGC-803, MKN-45, MKN-28, HGC-27, and BGC823) were obtained from the Shanghai Cell Bank, Chinese Academy of Sciences (Shanghai, People’s Republic of China) and were cultured in complete RPMI 1640 media supplemented with 10% FBS, 50 U/mL penicillin, and 50 μg/mL streptomycin (Thermo Fisher Scientific, Waltham, MA, USA). The cells were incubated in a humidified atmosphere (SHEL-LAB, Portland, OR, USA) at 37°C with 5% CO2.
Cell treatment and transfection
Cells in logarithmic growth phase were seeded in a 6-well plate and cultured for 24 h. Recombinant plasmids were transfected into cells using Lipofectamine™ LTX (Thermo Fisher Scientific) according to the manufacturer’s instructions. Briefly, 2 μg of pIRES2-ZsGreen1-vector or pIRES2-ZsGreen1-ARHGAP18 (Sangon Biotech Inc., Shanghai, People’s Republic of China), 5 μL of Lipofectamine LTX, and 250 μL Opti-MEM® were mixed well and incubated for 25 min at room temperature. Five-hundred microliters of mixture was added into a 6-well plate with RPMI 1640 medium and cultured for 48 h, after which the transfected cells were harvested and used for the next experiment.
Detection of cell viability by Cell Counting Kit (CCK-8) assay
The 4×103 cells were seeded in each well of a 96-well plate and cultured for 24 h. pIRES2-ZsGreen1-vector or pIRES2-ZsGreen1-ARHGAP18 was respectively transfected into cells and cultured for another 12, 24, and 48 h. Cell viability was determined by using CCK-8 (Dojindo Molecular Technologies, Gaithersburg, MD, USA). CCK-8 reagent was added to each well 1 h before the endpoint of incubation. Optical density at 450 nm in each well was determined using a microplate reader (Tecan Safire, Männedorf, Switzerland). Experiments were repeated at least three times, each time in triplicate.
Transwell assay in vitro
Cells in logarithmic phase were digested with 0.25% trypsin (Thermo Fisher Scientific) and then suspended in RPMI 1640 (Hyclone, Logan, UT, USA) medium containing 10% FBS (Thermo Fisher Scientific). The viable cells were then seeded in a 24-well transwell chamber with a pore size of 8 μm (Sigma-Aldrich Co., St Louis, MO, USA). The inserts were coated with 50 μL Matrigel (diluted at 1:2; BD Biosciences, San Jose, CA, USA). Cells were counted with a hemocytometer using 0.4% trypan blue (Sigma-Aldrich Co.) and seeded at a density of 1×105 cells/well in serum-free RPMI 1640. RPMI 1640 plus 10% FBS was placed in the lower chamber as a source of chemo-attractant. Cells were allowed to migrate through a porous, uncoated membrane (BD Biosciences) for 12 h at 37°C. Incubation was carried out for 48 h at 37°C in humidified air with 5% CO2. Non-migratory cells in the upper chamber were then removed with a cotton-tip applicator. Migrated cells on the lower surface were fixed with methanol and stained with hematoxylin. The number of migrating cells was determined by counting five randomly selected high power fields under a microscope (Olympus Corporation, Tokyo, Japan) on each membrane and calculated as mean number of cells/field. The procedure for the cell invasion assay was similar to the cell migration assay, except that the transwell membranes were precoated with Matrigel (BD Biosciences).
In vivo experiments
For tumor growth assay, cells transfected with pIRES2-ZsGreen1-vector or pIRES2-ZsGreen1-ARHGAP18 were trypsinized, washed, and resuspended in RPMI 1640 without FBS. Six male athymic nude mice were purchased from Medical Experimental Center of Guangdong Province (People’s Republic of China). The animals were housed at 20°C–22°C with a 12-h light/dark cycle and fed standard chow and water ad libitum. The procedures in this study were approved by the Animal Ethics Committee of the First Affiliated Hospital, China Medical University (Shenyang, People’s Republic of China) and performed in accordance with the guidelines for the use of experimental animals from the National Institutes of Health.
All mice were randomly divided into two groups (three mice/group), and 5×104 cells were subcutaneously injected into the right armpit of each mouse. The tumor size was determined every 5–7 days after injection. At 30 days after injection, the mice were euthanized and the excised tumor tissues were measured.
Reverse transcription and real-time polymerase chain reaction (RT-PCR)
The expression levels of ARHGAP18, RhoC, MTA1, and MMP-2/-9 mRNA were determined by RT-PCR. Cells were seeded at a density of 5×105 cells/well in 6-well plates, cultured overnight, and then transfected with pIRES2-ZsGreen1-vector or pIRES2-ZsGreen1-ARHGAP18 for 48 h. Total RNA was isolated from cells using Total RNA kit Maxi Kit (Omega, Dalian, People’s Republic of China) and quantified. RNA in gastric cancer tissues was extracted from 46 tumor tissues and adjacent tissues by TRIzol reagent (Thermo Fisher Scientific) on ice. cDNA was then synthesized from 5 μg RNA in cells or tissues using Avian Myeloblastosis Virus (AMV) reverse transcriptase (Fermentas, Shanghai, People’s Republic of China) according to the manufacturer’s instructions. RT-PCR was performed using SYBR® Green 10× Supermix (Takara, Dalian, People’s Republic of China) in a 25 μL total volume and on ABI 7500 Fast Thermocycler (Thermo Fisher Scientific). The PCR cycles were 95°C for 10 min, followed by 40 cycles at 95°C for 15 s and annealing/extension at 60°C for 45 s. Primers were designed by Shanghai Sangon Company (Shanghai, People’s Republic of China) and were as follows: ARHGAP18, forward: 5′-GGATCCCTGGAGCTGCCATT-3′ and reverse: 5′-GGCAACTCCCGAATGAAGAGC-3′ (product: 139 bp); RhoC, forward: 5′-ACATGGCGAACCGGATCAGT-3′ and reverse: 5′-CGCTTGTTCTTGCGGACCTG-3′ (product: 121 bp); MTA1, forward: 5′-CGAGACCGAGTCGCTCAAGT-3′ and reverse: 5′-CTGCCTGGTACCGGTTTCCT-3′ (product: 131 bp); MMP-2, forward: 5′-CGCCATGTCCACTGTTGGTG′-3′ and reverse: 5′-TGTGGTCGCACACCACATCT-3′ (product: 130 bp); MMP-9, forward: 5′-TGATTGACGACGCCTTTGCC-3′ and reverse: 5′-CCGCGACACCAAACTGGATG-3′ (product: 114 bp); GAPDH, forward: 5′-AGCTCACTGGCATGGCCTTC-3′ and reverse: 5′-CGCCTGCTTCACCACCTTCT-3′ (product: 116 bp). GAPDH was used as control of the input RNA level. The gene expression was calculated using the 2−ΔΔCt method.
Western blot assay
The expression levels of ARHGAP18, RhoC, MTA1, and MMP-2/-9 protein and the phosphorylation levels of ERK, JNK, and p38 in cells after pIRES2-ZsGreen1-vector or pIRES2-ZsGreen1-ARHGAP18 transfection were quantified by Western blot assay. Cells were seeded at a density of 5×103 cells/well in 6-well plates, cultured overnight, and then transfected with pIRES2-ZsGreen1-vector or pIRES2-ZsGreen1-ARHGAP18 for 48 h. Protein in these cells was extracted using Total Protein Extraction Kit and Phosphorylation Protein Extraction Kit (both BestBio and Bio-Technology Limited, Shanghai, People’s Republic of China). Gastric cancer tissues were harvested and washed twice with PBS and lysed in ice-cold RIPA (radioimmunoprecipitation assay; Beyotime, Shanghai, People’s Republic of China) with freshly added 0.01% protease inhibitor PMSF (phenylmethylsulfonyl fluoride; Amresco, Shanghai, People’s Republic of China) and incubated on ice for 30 min. Five-hundred microliters of protein extraction solution were added to 5×103 cells in the 6-well plates, and the lysed cells and tissues were collected after being centrifuged at 1,000× g for 10 min at 4°C and washed with 2 mL PBS (Beyotime Institute of Biotechnology) twice. The protein concentration of the extracts was measured by BCA Protein Quantitation Kit (Keygen Biotech. Co., Ltd, Nanjing, Jiangsu, People’s Republic of China). Samples containing equal amounts of proteins (25 μg) were boiled in sodium dodecyl sulfate (SDS) sample buffer and then analyzed by 10% SDS-PAGE (sodium dodecyl sulphate-polyacrylamide gel electrophoresis; Beyotime Institute of Biotechnology). The proteins were electrotransferred to nitrocellulose membranes (Merck Millipore, Billerica, MA, USA), and blocked with 5% non-fat milk in Tris-buffered saline and 0.1% Tween 20 (TBST) with shaking at room temperature for 1 h. Membranes were incubated with primary antibodies targeting ARHGAP18 (1:1,000; abcam, Cambrige, UK), RhoC (1:800; abcam), MTA1 (1:1,000; abcam), MMP-2 (1:800; abcam), MMP-9 (1:600; abcam), ERK (1:1,000; Cell Signaling Technology, Danvers, MA, USA), JNK (1:1,000; CST), p38 (1:1,500; CST), p-ERK (Thr202/Tyr204; 1:500, CST), p-JNK (Thr183/Tyr185, 1,800; CST), p-p38 (Thr180/Tyr182, 1:1,000; CST), and GAPDH (1:2,500; Beyotime Biotechnology, Wuhan, People’s Republic of China) overnight at 4°C. After washing with TBST three times, 5 min each time, the membranes were incubated with HRP-conjugated secondary antibody (1:2,000; Beyotime) in TBST at room temperature for 1 h. Equal sample loading was confirmed using GAPDH. Protein expression was detected using enhanced chemiluminescence (Thermo Fisher Scientific). The protein bands were quantified using Quantity One software v4.62 (Bio-Rad Laboratories Inc., Hercules, CA, USA).
Statistics
All data represent the mean ± SD. Statistical analyses were performed using a two-tailed Student’s t-test when comparing two groups for in vitro and in vivo studies, and one-way ANOVA test (Dunnett’s multiple comparisons test when comparing multiple groups with control group, Tukey’s multiple comparisons test when making multiple pair-wise comparisons between different groups) for multiple group comparison. P<0.05 was considered significant.
Results
Low expression of ARHGAP18 in gastric cancer specimens and cell lines
ARHGAP18 mRNA expression was detected in a panel of surgically resected gastric cancer specimens and corresponding adjacent normal gastric specimens by means of RT-PCR. As shown in Figure 1A, ARHGAP18 mRNA expression was decreased significantly in tumor tissues compared to that of normal tissues. MGC-803 and BGC823 cells, as ARHGAP18-low populations, were selected from gastric cancer cell lines including AGS, MGC-803, MKN-45, HGC-27, and BGC 325, and normal gastric cell GES-1, using RT-PCR and Western blot methods (Figure 1B–D). This showed that the low expression of ARHGAP18 might be related to gastric cancer.
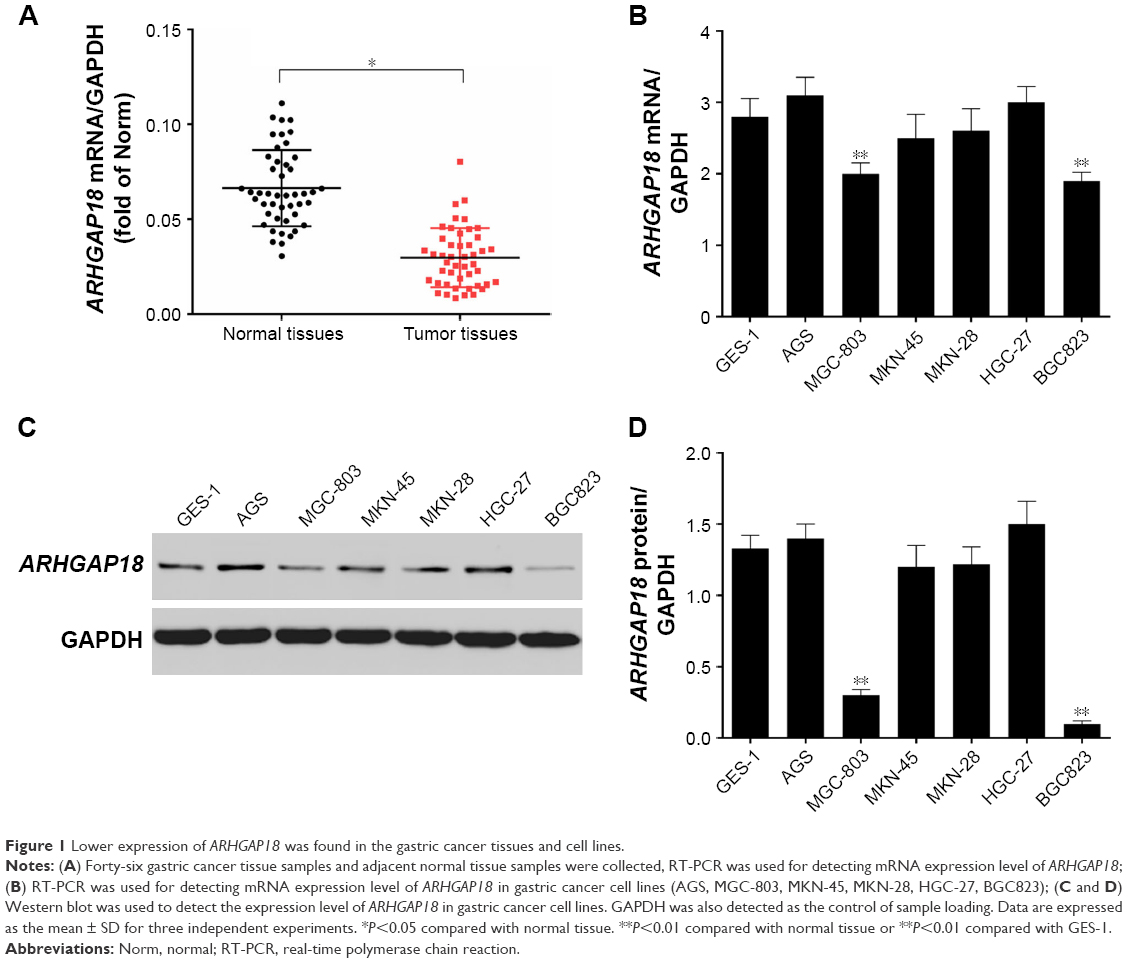 | Figure 1 Lower expression of ARHGAP18 was found in the gastric cancer tissues and cell lines. |
ARHGAP18 over-expression in MGC-803 and BGC823 cell lines inhibits cell proliferation
MGC-803 and BGC823 cell lines were used to examine changes in cell viability after ARHGAP18 over-expression; the results are shown in Figure 2. ARHGAP18 over-expression revealed abundant expression of ARHGAP18 at both mRNA and protein levels by means of RT-PCR and Western blot, respectively in MGC-803 and BGC823 cells. (Figure 2A–D). CCK-8 assay results showed that transfection with ARHGAP18 expression vector (pIRES2-ZsGreen1-ARHGAP18) resulted in a gradual and apparent decrease in cell proliferation in MGC-803 and BGC823 cells (Figure 2E and F). In MGC-803 cells, the cell viabilities of control group at 12, 24, and 48 h were respectively 100%±9.86%, 100%±7.25%, and 100%±11.3% (Figure 2E). There was no significant difference in cell viability between the control group and normal control (NC)-vector (IRES2-ZsGreen1-vector) group. After being transfected with ARHGAP18 plasmid for 12, 24, and 48 h, the cell viabilities were 79%±5.72%, 62%±3.89%, and 50%±2.77%, respectively (Figure 2E). In BGC823 cells, the cell viability had a similar trend to that of MGC-803 cells for control and NC-vector groups (Figure 2F). Following transfection with ARHGAP18 for 12 and 24 h, BGC823 cell viabilities were 71%±6.11% and 54%±3.52%, respectively, and after 48 h, cell viability was reduced to 33% (Figure 2F). These results suggested that the over-expression of ARHGAP18 could significantly reduce cell proliferation in gastric cancer cell lines.
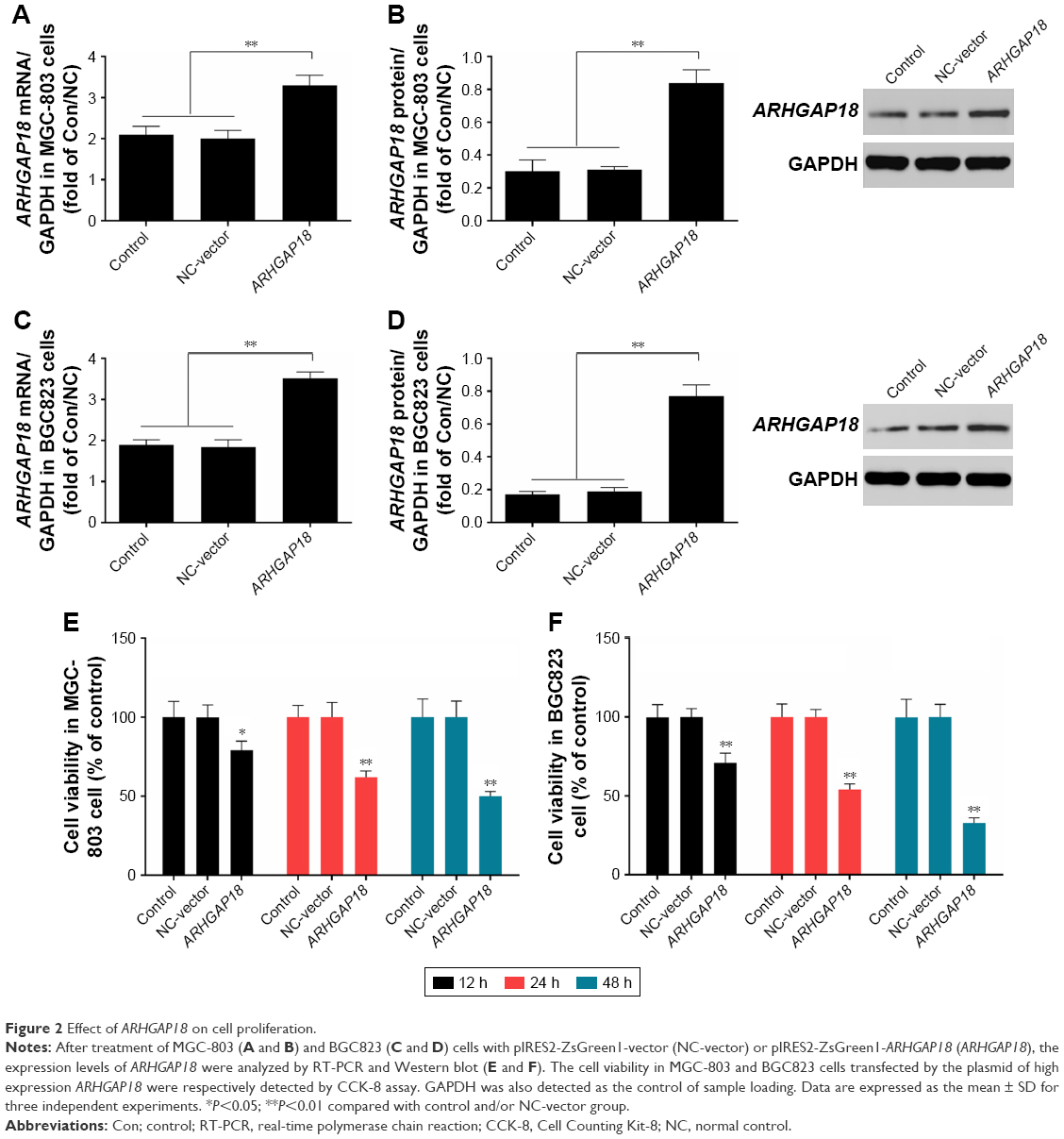 | Figure 2 Effect of ARHGAP18 on cell proliferation. |
ARHGAP18 over-expression in MGC-803 and BGC823 cell lines inhibits cell migration and invasion abilities in vitro
To investigate whether ARHGAP18 over-expression could affect protumorigenic behaviors including migration and invasion; we detected the capacity of gastric cancer cells with lowest expression of ARHGAP18 to invade through Matrigel, an artificial extracellular matrix, after transfection with a pIRES2-ZsGreen1-vector or pIRES2-ZsGreen1-ARHGAP18 plasmid. Over-expression of ARHGAP18 resulted in the inhibition of migration and invasion by 45%±3.88% and 50%±4.32%, respectively, in MGC-803 cells (Figure 3A and B). The assay revealed 55%±5.12% and 61%±5.64% inhibition in respective migration and invasion capabilities of BGC823 cells transfected with the pIRES2-ZsGreen-ARHGAP18 plasmid (Figure 3C and D). These data showed that the migration and invasion abilities of gastric cancer cell lines transfected with pIRES2-ZsGreen1-ARHGAP18 plasmid were significantly suppressed.
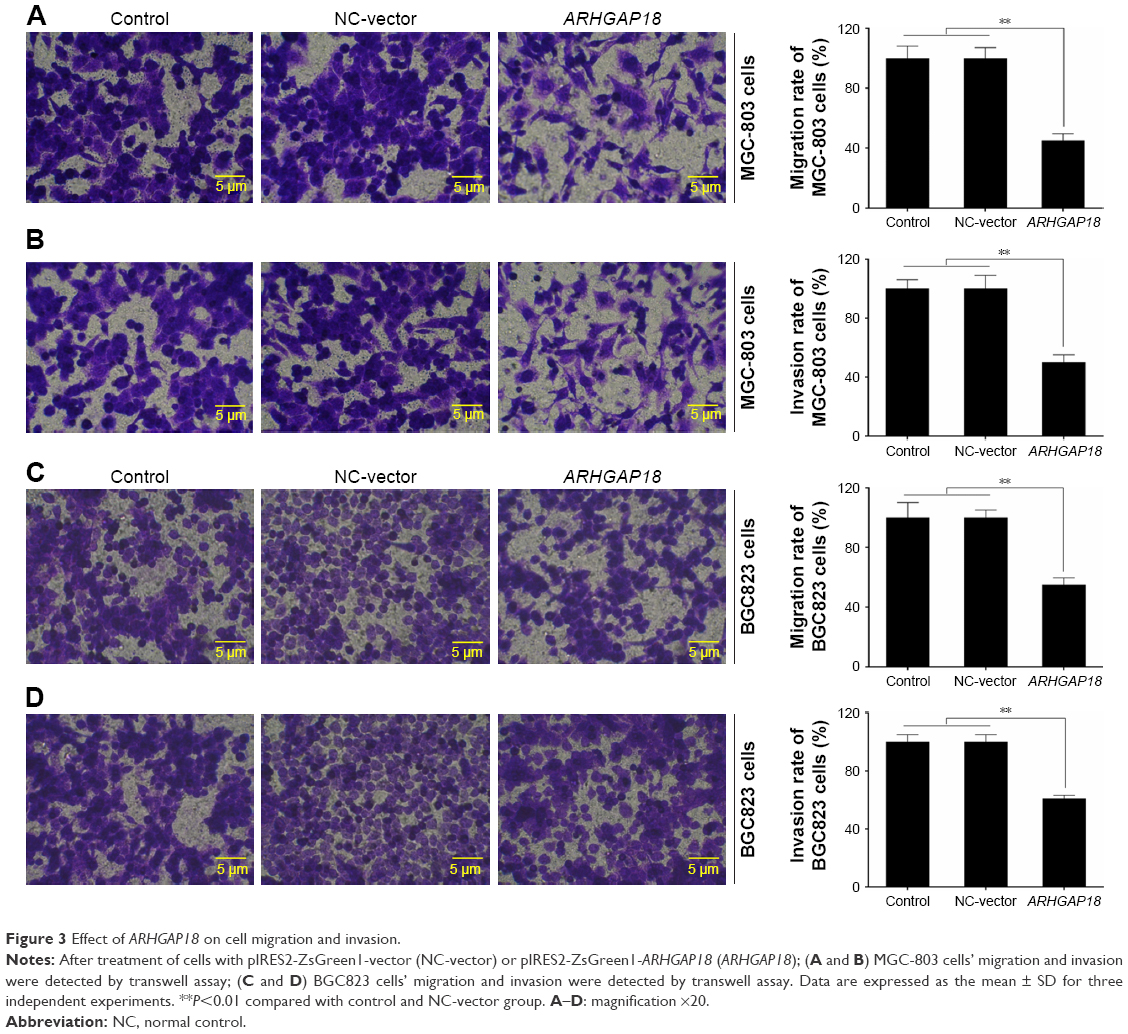 | Figure 3 Effect of ARHGAP18 on cell migration and invasion. |
Over-expressed ARHGAP18 restrained the excessive activation of MAPK signaling pathways in vitro
To further explore the mechanism of ARHGAP18 in terms of regulating cell proliferation, migration, and invasion functions of gastric cancer cell lines, signaling molecules of MAPK pathway associated with proliferation and migration, including ERK, JNK, and p38 were detected by Western blot.28,29 As shown in Figure 4, the phosphorylation levels of ERK, JNK, and p38 were dramatically decreased after transfection of MGC-803 and BGC823 cell lines with the pIRES2-ZsGreen1-ARHGAP18 plasmid, in comparison with control cells and NC-vector cells. This indicated that increased ARHGAP18 expression inhibited over-activated MAPK signaling pathways in MGC-803 and BGC823 cells.
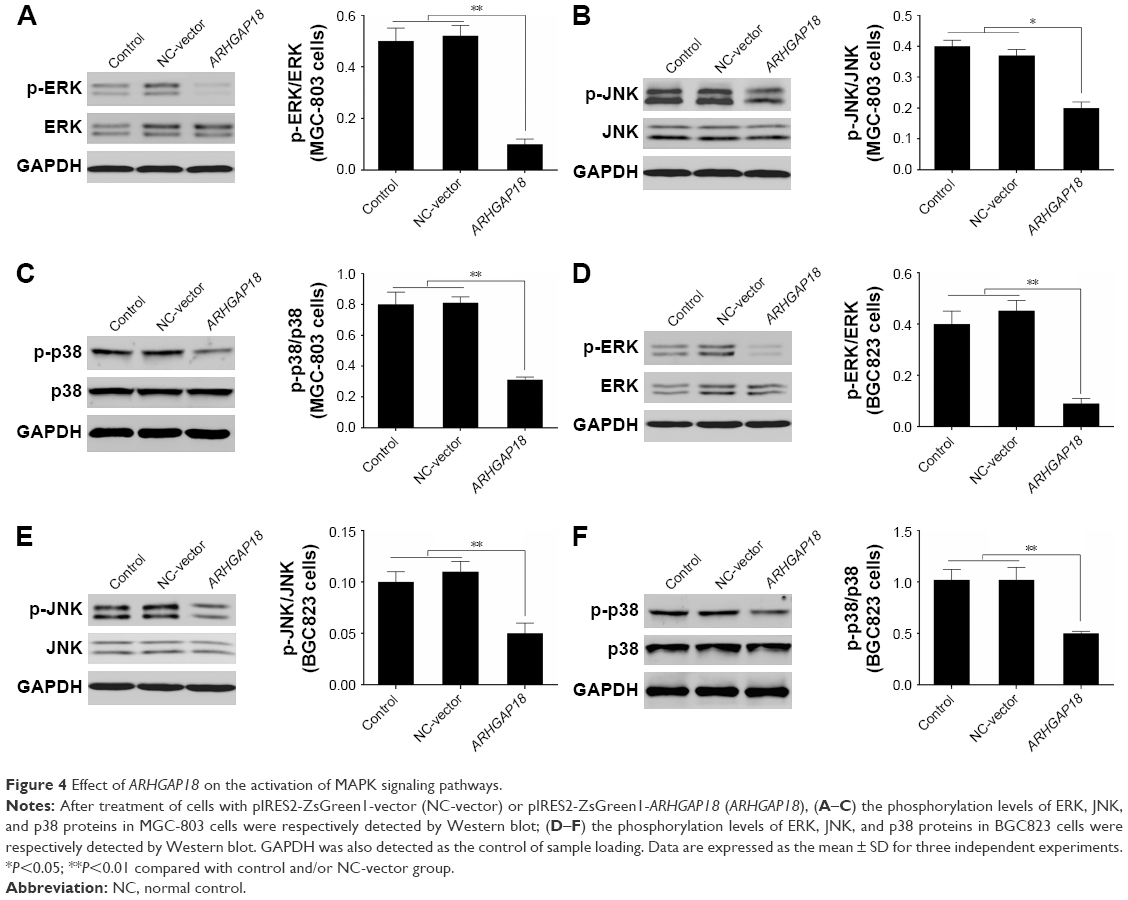 | Figure 4 Effect of ARHGAP18 on the activation of MAPK signaling pathways. |
Over-expression of ARHGAP18 regulated the expression levels of Rho, MAT1, and MMP-2/MMP-9 in vitro
In order to further validate ARHGAP18’s impact on cell proliferation, migration, and invasion by effecting the activation of MAPK signaling pathways, expression of Rho, MAT1, and MMP-2/MMP-9 associated with MAPK signaling pathways and predicated to be ARHGAP18 targets was investigated.28,30,31 As shown in Figure 5, the mRNA and protein levels of Rho, MAT1, and MMP-9 in MGC-803 and BGC823 cells were remarkably decreased after transfection with the pIRES2-ZsGreen1-ARHGAP18 plasmid compared with control cells and NC-vector cells. There were no significant changes in expression levels of MMP-2 in control cells, NC-vector cells, and ARHGAP18 over-expressing cells.
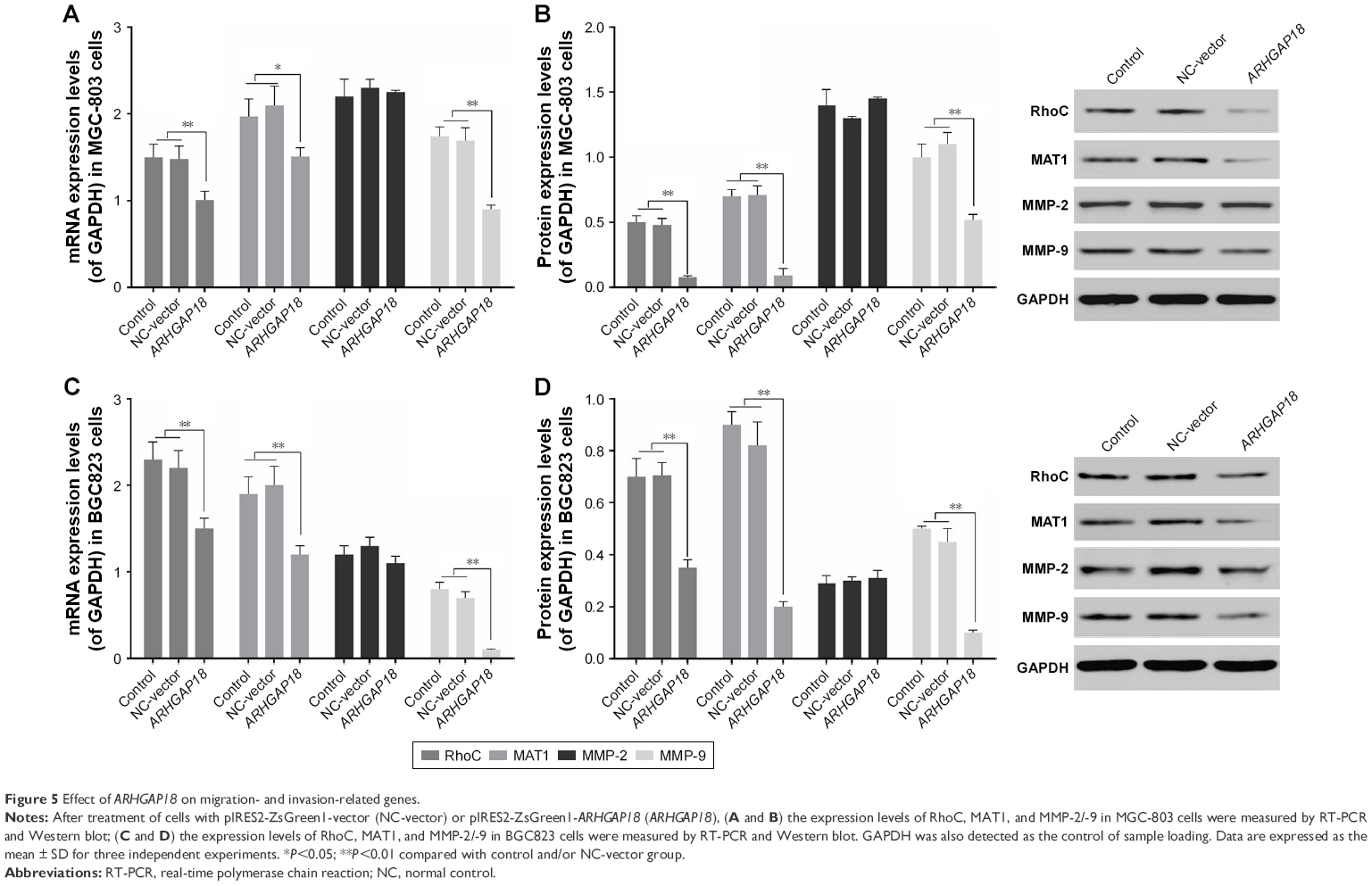 | Figure 5 Effect of ARHGAP18 on migration- and invasion-related genes. |
Over-expression of ARHGAP18 affected tumor growth of MGC-803 and BGC823 cells in nude mice
To examine the effects of ARHGAP18 on tumor growth in vivo, MGC803 and BGC823 cells transfected with ARHGAP18 plasmid were subcutaneously injected into the right armpit of nude mice. Tumors in ARHGAP18 groups grew slower in mice, whereas tumors in control group grew faster (Figure 6). Thirty-five days after injection, tumor weights in ARHGAP18 group mice were decreased compared with control groups (Figure 6). These data suggest that the over-expression of ARHGAP18 could inhibit tumor growth in nude mice.
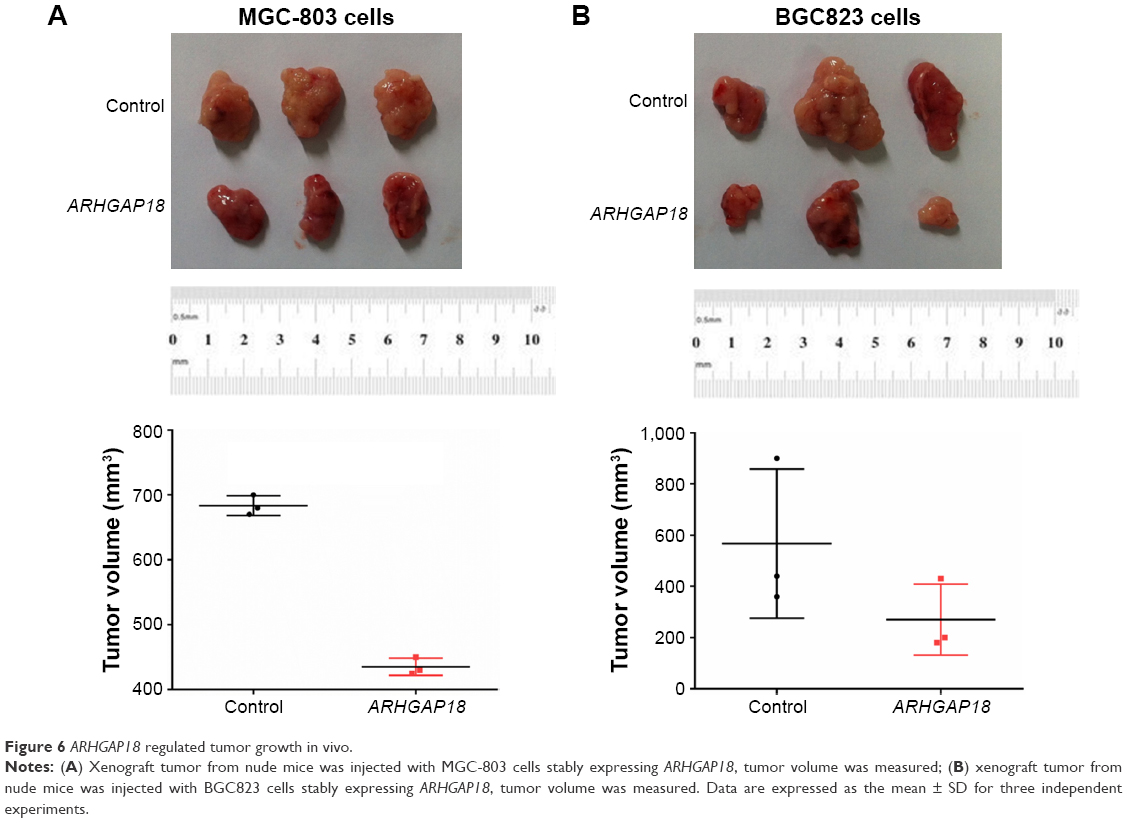 | Figure 6 ARHGAP18 regulated tumor growth in vivo. |
Discussion
In experimental and clinical investigation of gastric cancer, tumor cell over-proliferation, migration, and invasion are an important problem which urgently needs to be clarified and solved.32 This is an extremely complex process involving multiple genes and multiple pathways. ARHGAP18, a GTPase-activating protein for RhoA, was found to regulate the lifecycle of cells and to be downregulated in the early migration phase,6,8–10,33,34 but the precise molecular mechanism of ARHGAP18 involvement in gastric cancer tumorigenesis has not been specifically described to date. In this study, we found that the expression level of ARHGAP18 mRNA in tumor tissues was significantly lower than that of matched adjacent normal tissue (Figure 1A), which indicated that ARHGAP18 might affect gastric cancer tumorigenesis. Therefore, the functions of ARHGAP18 needed to be further studied in gastric cancer at the cellular level. MGC-803 cell and BGC823 cell, human gastric cancer cell lines with the lowest expression of ARHGAP18 (Figure 1B–D), were selected for further studies. Following transfection of cells with the ARHGAP18 plasmid for 48 h, RT-PCR and Western blot detections found that ARHGAP18 was highly expressed in MGC-803 and BGC823 cells (Figure 2A–D). However, cellular proliferative activity was apparently decreased in MGC-803 and BGC823 cells over-expressing ARHGA18 (Figure 2E and F). Early reports indicated that ARHGAP18 could control cell shape, spreading, and migration.34 Accordingly, several studies have provided the basis and evidence for further researching the role of ARHGAP18 in gastric cancer cells.
Metastasis and invasion are the most important biological characteristics of malignant cells and are an important stage of tumorigenesis and tumor progression. Unlimited proliferation of cancer cells can accelerate the progress of neoplasm metastasis. Moreover, invasion is an internal factor of neoplasm metastasis.35,36 In this study, the migration and invasion abilities as assessed by transwell assay were significantly reduced in MGC-803 and BGC823 cells over-expressing ARHGAP18 (Figure 3). Based on these results, over-expression of ARHGAP18 inhibited cell proliferation, migration, and invasion in MGC-803 and BGC823 cells. The related molecular mechanisms in this process need to be further researched.
MAPK is a class of serine/threonine protein kinase, which exists widely in the cytoplasm and nuclei of mammalian cells, and can regulate cell proliferation, differentiation, development, and apoptosis by communicating signals from the outside with the inside of the cell.16,17 ERK, JNK, and p38MAPK signal transduction pathways are three parallel MAPK pathways. The ERK signal pathway affects cell proliferation and differentiation; the JNK signal pathway is involved in cell apoptosis caused by oxidative stress, and the p38MAPK signal pathway is involved in the chain reaction of various protein kinases caused by extracellular stressors and proinflammatory factors which affects RNA transcription, protein synthesis, and cell surface receptor expression. Cell growth and differentiation caused by various growth factors and proto-oncogenes are mediated by MAPK pathway. Some studies have suggested that MAPK pathways were closely associated with migration and invasion of various malignant tumors.37–39 In this study, the phosphorylation levels of ERK, JNK, and p38 proteins were significantly decreased in MGC-803 and BGC823 cells over-expressing ARHGAP18 (Figure 4). This showed that MAPK pathways were activated in migration and invasion of gastric cancer cells, and the over-expression of ARHGAP18 could inhibit the activation.
The activation of MAPK signal pathway could enhance the expression levels of invasion- and migration-related genes.37–39 RhoC belongs to the Rho subgroup of small molecule G proteins and has been called the molecular switch of migration and invasion in cancer.10 RhoA and RhoC were shown to be closely related to tumor metastasis, but the difference was that RhoA activated MAPK to regulate cell proliferation and apoptosis indirectly and RhoC activated MAPK to regulate cell migration and invasion directly.40–43 In addition, the over-expression of RhoC could promote MMP-2/-9 and TIMP-2 expression.44 Therefore, in this study, we selected RhoC to test the MAPK signal pathway. MAT1, a tumor-related gene, was recently discovered to be involved in angiogenesis, invasion, and metastasis of tumors.45,46 MMP-2 and MMP-9 belong to a family of zinc-dependent proteolytic enzymes, which was shown to be over-expressed in tumors, and promoted the migration and invasion of tumor by destroying basement membranes.47 In our results, the expression levels of RhoC, MAT1, and MMP-9 were significantly decreased in the ARHGAP18 group (Figure 5). Previous studies have shown a tight correlation between MMP-2/-9 and other metastasis associated factors, with the precursor of MMP-2 most affected in degradation and destruction of the extracellular matrix.48,49 Therefore, this might be the reason that no change occurred in MMP-2 expression in this study.
To further validate our results, MGC-803 and BGC823 cells over-expressing ARHGAP18 were respectively injected subcutaneously into nude mice to observe the tumor growth. We found that over-expression of ARHGAP18 reduced the rate of tumor growth (Figure 6).
Conclusion
This study is the first to preliminarily clarify that over-expression of ARHGAP18 suppresses cell proliferation, migration, invasion, and tumor growth in gastric cancer by restraining over-activation of MAPK signaling pathways to decrease the expression of migration- and invasion-related genes. In further work, we are hopeful that an in vivo study can be carried out to illuminate the function of ARHGAP18 and related mechanisms.
Disclosure
The authors report no conflicts of interest in this work.
References
Bosolino A, Cravero A, Ratto R. [Gastric metastases: not only identified at autopsies]. Acta Gastroenterol Latinoam. 2013;43(4):316–320. Spanish. | ||
Dickson JL, Cunningham D. Systemic treatment of gastric cancer. Eur J Gastroenterol Hepatol. 2004;16(3):255–263. | ||
Yeh KH, Cheng AL. Recent advances in therapy for gastric cancer. J Formos Med Assoc. 2004;103(3):171–185. | ||
Choi Y, Park J, Choi Y, et al. C-Jun N-terminal kinase activation has a prognostic implication and is negatively associated with FOXO1 activation in gastric cancer. BMC Gastroenterology. 2016;16(1):59. | ||
Popa F, Bratucu M, Radu P. Present and future tense in operable rectal cancer. Chirurgia (Bucur). 2011;106(1):11–16. | ||
Calvisi DF, Ladu S, Conner EA, et al. Inactivation of Ras GTPase-activating proteins promotes unrestrained activity of wild-type Ras in human liver cancer. J Hepatol. 2011;54(2):311–319. | ||
Coleman PR, Hahn CN, Grimshaw M, et al. Stress-induced premature senescence mediated by a novel gene, SENEX, results in an anti-inflammatory phenotype in endothelial cells. Blood. 2010;116(19):4016–4024. | ||
Dransart E, Olofsson B, Cherfils J. RhoGDIs revisited: novel roles in Rho regulation. Traffic. 2005;6(11):957–966. | ||
Katoh M, Katoh M. Characterization of human ARHGAP10 gene in silico. Int J Oncol. 2004;25(4):1201–1206. | ||
Sahai E, Marshall CJ. RHO-GTPases and cancer. Nat Rev Cancer. 2002;2(2):133–142. | ||
Hall A. Rho GTPases and the actin cytoskeleton. Science. 1998;279(5350):509–514. | ||
Heasman SJ, Ridley AJ. Mammalian Rho GTPases: new insights into their functions from in vivo studies. Nat Rev Mol Cell Biol. 2008;9(9):690–701. | ||
Wennerberg K, Der CJ. Rho-family GTPases: it’s not only Rac and Rho (and I like it). J Cell Sci. 2004;117(Pt 8):1301–1312. | ||
Pollard TD, Blanchoin L, Mullins RD. Molecular mechanisms controlling actin filament dynamics in nonmuscle cells. Ann Rev Biophys Biomol Struct. 2000;29:545–576. | ||
Zhao ZS, Manser E, Loo TH, Lim L. Coupling of PAK-interacting exchange factor PIX to GIT1 promotes focal complex disassembly. Mol Cell Biol. 2000;20(17):6354–6363. | ||
Cuevas BD, Abell AN, Johnson GL. Role of mitogen-activated protein kinase kinase kinases in signal integration. Oncogene. 2007;26(22):3159–3171. | ||
Tibbles LA, Woodgett JR. The stress-activated protein kinase pathways. Cell Mol Life Sci. 1999;55(10):1230–1254. | ||
Jelinek T, Catling AD, Reuter CW, Moodie SA, Wolfman A, Weber MJ. RAS and RAF-1 form a signalling complex with MEK-1 but not MEK-2. Mol Cell Biol. 1994;14(12):8212–8218. | ||
Yamauchi J, Kaziro Y, Itoh H. Differential regulation of mitogen-activated protein kinase kinase 4 (MKK4) and 7 (MKK7) by signaling from G protein beta gamma subunit in human embryonal kidney 293 cells. J Biol Chem. 1999;274(4):1957–1965. | ||
Rouse J, Cohen P, Trigon S, et al. A novel kinase cascade triggered by stress and heat shock that stimulates MAPKAP kinase-2 and phosphorylation of the small heat shock proteins. Cell. 1994;78(6):1027–1037. | ||
Cheng M, Zhen E, Robinson MJ, Ebert D, Goldsmith E, Cobb MH. Characterization of a protein kinase that phosphorylates serine 189 of the mitogen-activated protein kinase homolog ERK3. J Biol Chem. 1996;271(20):12057–12062. | ||
Kato Y, Chao TH, Hayashi M, Tapping RI, Lee JD. Role of BMK1 in regulation of growth factor-induced cellular responses. Immunol Res. 2000;21(2–3):233–237. | ||
Yamashita M, Otsuka F, Mukai T, et al. Simvastatin antagonizes tumor necrosis factor-alpha inhibition of bone morphogenetic proteins-2-induced osteoblast differentiation by regulating Smad signaling and Ras/Rho-mitogen-activated protein kinase pathway. J Endocrinol. 2008;196(3):601–613. | ||
Yuan JP, Ling H, Zhang MX, Liu Y, Song Y, Su Q. [Diallyl disulfide-induced G2/M arrest of human gastric cancer MGC803 cells involves activation of p38 MAP kinase pathways]. Ai Zheng. 2004;23(2):169–172. Chinese. | ||
Zhao HJ, Liu T, Mao X, et al. Fructus phyllanthi tannin fraction induces apoptosis and inhibits migration and invasion of human lung squamous carcinoma cells in vitro via MAPK/MMP pathways. Acta Pharmacol Sin. 2015;36(6):758–768. | ||
Li L, Zheng BB, Ma LS, et al. Telekin suppresses human hepatocellular carcinoma cells in vitro by inducing G2/M phase arrest via the p38 MAPK signaling pathway. Acta Pharmacol Sin. 2014;35(10):1311–1322. | ||
Zhu J, Zheng Y, Zhang H, Sun H. Targeting cancer cell metabolism: The combination of metformin and 2-Deoxyglucose regulates apoptosis in ovarian cancer cells via p38 MAPK/JNK signaling pathway. Am J Transl Res. 2016;8(11):4812–4821. | ||
Li P, Xue WJ, Feng Y, Mao QS. Long non-coding RNA CASC2 suppresses the proliferation of gastric cancer cells by regulating the MAPK signaling pathway. Am J Transl Res. 2016;8(8):3522–3529. | ||
Yang M, Huang CZ. Mitogen-activated protein kinase signaling pathway and invasion and metastasis of gastric cancer. World J Gastroenterol. 2015;21(41):11673–11679. | ||
Lin M, DiVito MM, Merajver SD, Boyanapalli M, van Golen KL. Regulation of pancreatic cancer cell migration and invasion by RhoC GTPase and caveolin-1. Mol Cancer. 2005;4(1):21. | ||
Wang F, Bhat K, Doucette M, et al. Docosahexaenoic acid (DHA) sensitizes brain tumor cells to etoposide-induced apoptosis. Curr Mol Med. 2011;11(6):503–511. | ||
Zhao L, Han T, Li Y, et al. The lncRNA SNHG5/miR-32 axis regulates gastric cancer cell proliferation and migration by targeting KLF4. FASEB J. 2017;31(3):893–903. | ||
Chang GH, Lay AJ, Ting KK, et al. ARHGAP18: an endogenous inhibitor of angiogenesis, limiting tip formation and stabilizing junctions. Small GTPases. 2014;5(3):1–15. | ||
Maeda M, Hasegawa H, Hyodo T, et al. ARHGAP18, a GTPase-activating protein for RhoA, controls cell shape, spreading, and motility. Mol Biol Cell. 2011;22(20):3840–3852. | ||
Peng Y, Zhang P, Huang X, et al. Direct regulation of FOXK1 by C-jun promotes proliferation, invasion and metastasis in gastric cancer cells. Cell Death Dis. 2016;7(11):e2480. | ||
Wang J, Xue X, Hong H, et al. Upregulation of microRNA-524-5p enhances the cisplatin sensitivity of gastric cancer cells by modulating proliferation and metastasis via targeting SOX9. Oncotarget. 2017;8(1):574–582. | ||
Inamoto T, Azuma H, Sakamoto T, et al. Invasive ability of human renal cell carcinoma cell line Caki-2 is accelerated by gamma-aminobutyric acid, via sustained activation of ERK1/2 inducible matrix metalloproteinases. Cancer Invest. 2007;25(7):574–583. | ||
Kumar B, Koul S, Petersen J, et al. p38 mitogen-activated protein kinase-driven MAPKAPK2 regulates invasion of bladder cancer by modulation of MMP-2 and MMP-9 activity. Cancer Res. 2010;70(2):832–841. | ||
Langlois B, Perrot G, Schneider C, et al. LRP-1 promotes cancer cell invasion by supporting ERK and inhibiting JNK signaling pathways. PloS One. 2010;5(7):e11584. | ||
van Golen KL, Bao LW, Pan Q, Miller FR, Wu ZF, Merajver SD. Mitogen activated protein kinase pathway is involved in RhoC GTPase induced motility, invasion and angiogenesis in inflammatory breast cancer. Clin Exp Metastasis. 2002;19(4):301–311. | ||
Collisson EA, Kleer C, Wu M, et al. Atorvastatin prevents RhoC isoprenylation, invasion, and metastasis in human melanoma cells. Mol Cancer Ther. 2003;2(10):941–948. | ||
Nobes CD, Hall A. Rho GTPases control polarity, protrusion, and adhesion during cell movement. J Cell Biol. 1999;144(6):1235–1244. | ||
Jaffe AB, Hall A. Rho GTPases in transformation and metastasis. Adv Cancer Res. 2002;84:57–80. | ||
Ikoma T, Takahashi T, Nagano S, et al. A definitive role of RhoC in metastasis of orthotopic lung cancer in mice. Clin Cancer Res. 2004;10(3):1192–1200. | ||
Kai L, Wang J, Ivanovic M, et al. Targeting prostate cancer angiogenesis through metastasis-associated protein 1 (MTA1). Prostate. 2011;71(3):268–280. | ||
Zhu X, Guo Y, Li X, Ding Y, Chen L. Metastasis-associated protein 1 nuclear expression is associated with tumor progression and clinical outcome in patients with non-small cell lung cancer. J Thorac Oncol. 2010;5(8):1159–1166. | ||
Coussens LM, Fingleton B, Matrisian LM. Matrix metalloproteinase inhibitors and cancer: trials and tribulations. Science. 2002;295(5564):2387–2392. | ||
Iniesta P, Moran A, De Juan C, et al. Biological and clinical significance of MMP-2, MMP-9, TIMP-1 and TIMP-2 in non-small cell lung cancer. Oncol Rep. 2007;17(1):217–223. | ||
Poyer F, Coquerel B, Pegahi R, et al. Secretion of MMP-2 and MMP-9 induced by VEGF autocrine loop correlates with clinical features in childhood acute lymphoblastic leukemia. Leuk Res. 2009;33(3):407–417. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.