Back to Journals » Diabetes, Metabolic Syndrome and Obesity » Volume 14
Osteoclasts May Affect Glucose Uptake-Related Insulin Resistance by Secreting Resistin
Authors Li X, Sun F, Lu J, Zhang J, Wang J, Zhu H, Gu M, Ma J
Received 6 May 2021
Accepted for publication 6 July 2021
Published 31 July 2021 Volume 2021:14 Pages 3461—3470
DOI https://doi.org/10.2147/DMSO.S316964
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Juei-Tang Cheng
Xiangqi Li,* Fei Sun,* Jiancan Lu, Jichen Zhang, Jingnan Wang, Hongling Zhu, Mingjun Gu, Junhua Ma
Department of Endocrinology, Shanghai Gongli Hospital, The Second Military Medical University, Shanghai, 200135, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Junhua Ma
Department of Endocrinology, Shanghai Gongli Hospital, The Second Military Medical University, Miaopu Road 219, Shanghai, 200135, People’s Republic of China
Tel +86 21 58858730
Email [email protected]
Objectives: Bone may play a role in the modulation of insulin sensitivity. Insulin resistance can be caused by increased resistin. However, whether osteoclasts affect the insulin resistance via resistin remains unclear. In the present study, we show the expression of resistin in osteoclasts and the possible underlying role of resistin on glucose uptake-related insulin resistance in vitro.
Methods: Conditioned mediums (CM) were collected from Raw264.7 cells treated without (CCM) or with RANKL (CM3, treated with RANKL for 3 days; CM5, treated with RANKL for 5 days) and transfected with control or resistin siRNA (CMsiRNA). The osteoclast formation was examined by tartrate resistant acid phosphatase (TRAP) staining. C2C12 myoblasts were cultured with the CM or CMsiRNA. Glucose uptake was evaluated by 2-NBDG fluorescence intensity. Resistin expression was evaluated by quantitative real-time polymerase chain reaction (qRT-PCR) and enzyme-linked immunosorbent assay. Statistical analysis was performed by an independent two sample t-test or one-way ANOVA.
Results: The 2-NBDG fluorescence intensity was higher in C2C12 cells treated with CCM compared to those that received CM3 and CM5 (p < 0.05). Resistin mRNA and protein expressions were both increased in RAW264.7 cells treated with RANKL for 3 days and 5 days compared with those cells without RANKL administration. The 2-NBDG fluorescence intensities in C2C12 cells treated with CMsiRNA and CM5+Anti-resistin antibody were significantly higher than those cultured with CM5 (p < 0.05).
Conclusion: Osteoclasts may promote glucose uptake-related insulin resistance by secreting resistin.
Keywords: bone, insulin resistance, osteoclast, resisin, diabetes mellitus
Introduction
Insulin resistance is a pathophysiological condition in which cells in muscles, fat, and liver do not respond well to normal doses of insulin and cannot use glucose from the blood for energy.1 Insulin resistance is one of the critical reasons for the pathogenesis of multiple metabolic syndrome, including type 2 diabetes mellitus and obesity.2
Recently, an increasing number of studies have shown that bone is one type of endocrine organ which can regulate the energy metabolism.3,4 Obesity, high blood sugar levels and insulin resistance occurred in osteocalcin (OC) knockout mice.3 Insulin can promote osteoblasts to produce OC, and the bone regulates carbohydrate metabolism via OC. Osteoclast is another type of cell in bone. However, the role of osteoclast on insulin resistance has not been totally clarified. A recent study indicated that denosumab, a monoclonal antibody that binds with high specificity to the human receptor activator of NF-kB ligand (RANKL), could significantly diminish the insulin resistance condition in the fourth week.5 Similar trends were observed in the 12th and 24th week, but without statistical significance. Interestingly, Passeri and colleagues showed that denosumab might positively affect hepatic insulin sensitivity.6 The in vivo study indicated that blockage of RANKL signaling in mouse models resulted in a marked improvement of hepatic insulin sensitivity.7 We speculate that osteoclast may play an important role in insulin resistance because RANKL is a critical cytokine for osteoclast formation. However, whether osteoclasts affect the insulin resistance and the possible underlying mechanisms are not completely understood.
Resistin is a type of adipocytokines which is produced by adipocytes.8 It acts on myocytes, hepatocytes, and adipocytes, reducing their sensitivity to insulin.9 It also takes part in the inflammatory response and increases the expression of some proinflammatory cytokines.10 Insulin resistance can be caused by resistin-related chronic inflammation. Resistin can induce insulin resistance by both AMPK-dependent and AMPK-independent pathways.11 It has been shown that resistin is expressed in human macrophages, placenta, and pancreas.12–14 Inflammatory cells such as macrophages are considered the predominant source of circulating resistin.13 A recent study showed that resistin independently and positively correlated with fasting glucose in elderly people with metabolic syndrome.15 Preosteoclasts and osteoclasts are both macrophages. Thommesen et al16 showed that the expression of resistin mRNA in RAW 264.7, a type of preosteoclastic cell, was increased during differentiation. Recently, resistin expression was also up-regulated in periodontal cells and tissues that had periodontitis.17 However, whether osteoclasts affect the insulin resistance via resistin remains unclear. Insulin resistance was defined as a reduced sensitivity to insulin by the body’s insulin-dependent processes, such as glucose uptake.1 In the present study, we aimed to show the effect of osteoclasts on insulin resistance from the aspect of glucose uptake and the possible underlying role of resistin.
Materials and Methods
Cell Culture
RAW264.7 cells and C2C12 cells were obtained from National Collection of Authenticated Cell Cultures (Shanghai, China). RAW264.7 cells were cultured in Dulbecco’s modified Eagle’s Medium (DMEM; 25 mM glucose; Gibco, USA) containing 10% FBS, 1% penicillin/streptomycin, and with 20 ng/mL RANKL at 37 °C in a humidified atmosphere of 95% air and 5% CO2. On the third or fifth day, the cells were cultured with RANKL-free and glucose-free medium for 12h. Then, the medium was collected for indirect co-culturing with C2C12 cells. The medium from RAW264.7 cells without RANKL induction was also collected as control.
C2C12 myoblasts were cultured in DMEM supplemented with 10% FBS (Gibco, USA), 100 U/mL penicillin, and 100 μg/mL streptomycin at 37 °C in a humidified atmosphere of 95% air and 5% CO2. On the second day, the medium was changed to DMEM with 2% horse serum (Gibco, USA) to induce differentiation for an additional 4 days. The medium was replaced every two days.
The differentiated C2C12 myoblasts (104 cells/well) were cultured with the conditioned mediums (CMs) containing 40% medium collected from RAW264.7 cells (non-RANKL-treated, CCM; RANKL-treated for 3 days and 5 days, CM3 and CM5) and 60 mM of glucose (n = 6) for 24 h in a 24-well plate.
RNA Interference
Resistin siRNA (sense 5ʹ-CCAAAUGCAAUAAAGAACAUUGGCU-3ʹ; antisense 5ʹ-AGCCAAUGUUCUUUAUUGCAUUUGG-3ʹ; each strand with a 3ʹ dTdT overhang) and negative control (NC) siRNA (sense 5ʹ-TTCTCCGAACGTGTCGACACGGAGAC −3ʹ; antisense 5ʹ-TAATGTACGTGCAGTTAGCCTGACGC −3ʹ) were obtained from Life Technologies. Transfection of siRNA was performed using Lipofectamine RNAiMAX and Opti-MEM (both from Life Technologies) according to the manufacturer’s instructions. Raw264.7 cells were cultured as previously described. On day 2, 50 nM of each siRNA was added in a maturation medium and cultured. On the third or fifth day, the cells were cultured with RANKL-free and glucose-free medium for 12h. Then, the medium (NC, CCMi, CM3i and CM5i) was collected for the indirect co-culturing with C2C12 cells.
Cell Viability
The cell viability of RAW264.7 cells transfected with NC and resistin siRNA was evaluated by a CCK8 kit. Briefly, RAW264.7 cells transfected with NC and resistin siRNA were seeded into 96-well plates for 24 h. Then, 10 μL of CCK8 solution was added to each well and the plate was incubated in the incubator for 4 hours. The absorbance of the cells was measured at 450 nm using a microplate reader. Similarly, the effects of CMs on C2C12 viability were also evaluated by the CCK8 kit.
Glucose Uptake Assay
C2C12 cells treated with the CMs were washed with PBS and starved in glucose-free DMEM containing 2% FA-free FBS for 3 h. They were subsequently incubated in the presence of insulin (100 nM) for 10 min, then washed with ice-cold Krebs buffer and 2-NBDG (Cayman, USA) (150 μg/mL) was added into the culture medium for 30 min at 37 °C. The 2-NBDG fluorescence intensity was measured using a microplate reader at an excitation wavelength of 480 nm and an emission wavelength of 540 nm.
In addition, 10 ng anti-resistin antibody (ab136877, Abcam, USA) was added into the obtained 20 mL CM5 (CM5-AntiR). Then, the C2C12 cells were cultured with the mixed medium. Glucose uptake was assessed using the 2-NBDG fluorescence intensity. The CMM value for the glucose uptake was set as 100%.
Osteoclast Formation
RAW264.7 cells could differentiate to osteoclasts in the presence of receptor activator of nuclear factor-κ B ligand (RANKL) induction. The osteoclast formation was evaluated by tartrate resistant acid phosphatase (TRAP) staining. Briefly, RAW264.7 cells were washed with cold PBS, fixed with 4% paraformaldehyde for 10 min, and stained for TRAP using a commercial kit (Sigma-Aldrich, USA). The TRAP-positive multinucleated cells containing three or more nuclei were counted as mature osteoclasts. The resistin mRNA expression during osteoclast formation (0, 3 and 5 days) was observed (n = 4).
Primary osteoclasts were also isolated from new-born Sprague–Dawley rats. The study protocol was approved by Animal Care Committee of Shanghai Gongli Hospital. All experiments were conducted according to Guiding Principles for Care and Use of Animals (National Research Council, 8th edition). Briefly, the long bones were placed in alpha-MEM and the soft tissues were removed. Then, the bones were curetted and the dispersed cell solution was seeded into a 6-well plate for 30 min. The osteoclasts were attached to the plate. The cells were used for RNA extraction and supernatants were used for the resistin protein assay at 24h and 72 h (n = 3).
RNA Extraction and Quantitation of mRNA Expression by Quantitative Real-Time Polymerase Chain Reaction (qRT-PCR)
Total RNA was isolated from osteoclasts by using Trizol reagent. Complementary DNA was synthesized by using a Quantscript RT kit (Tiangen Biotech Co., China) (0.5 μg total RNA). The individual cDNA species were amplified in a reaction mixture containing cDNA aliquot (50 ng), and the relevant sense and antisense primers (Resistin: forward 5ʹ-TCACTTTTCACCTCTGTGGATATGAT-’3, reverse 5ʹ-TGCCCCAGGTGGTGTAAA-’3). Then, real-time PCR was performed using an ABI PRISM 7500 Sequence Detection System (Life Technologies) and SYBR-Green. β-actin mRNA (forward 5ʹ-TTCCTTCTTGGGTATGGAAT-’3, reverse 5ʹ-GAGCAATGATCTTGATCTTC-’3) was used as an internal control. We also detected leptin (forward 5ʹ-GGATCAGGTTTTGTGGTGCT-’3, reverse 5ʹ-TTGTGGCCCATAAAGTCCTC-’3) and adiponectin mRNA (forward 5ʹ-CAGGTCTTCTTGGTCCTAAGGG-’3, reverse 5ʹ-GTCCACATTCTTTTCCTGATACTG-’3) expression. The reactions protocol was as follows: initiated at 94 °C for 5 min and PCR (94 °C for 5 s, 60 °C for 20 s) for 35 cycles. Relative mRNA expression was determined using the 2-ΔΔCt method.
Enzyme Linked Immunosorbent Assay (ELISA)
Resistin released from RAW264.7 cells was determined using the Mouse Resistin Quantikine ELISA Kit (R&D Systems, Minneapolis, MN, USA) according to the manufacturer’s instructions. The detection limit was 8 pg/mL. The CV of the intra-assay and inter-assay was lower than 7%. The recovery rate was 85–108%.
Statistical Analysis
All statistical analyses were performed by using SPSS 20 (IBM Corp., Armonk, NY, USA). Normal distribution was checked by Shapiro–Wilk test. The data are shown as mean ± SD (normal distribution) or median (interquartile range). Statistical analysis was performed by an independent two sample t-test or One-way ANOVA. p < 0.05 was considered to be statistically significant.
Results
Osteoclasts Affected Glucose Uptake in C2C12 Cells
Firstly, we determined the glucose uptake in C2C12 cells treated with different CMs. The glucose uptake was decreased in C2C12 cells treated with CM3 and CM5 compared to those that received CCM (p < 0.01). As shown in Figure 1, the 2-NBDG fluorescence intensity was significantly decreased in the CM3 and CM5 groups (Figure 1A and B). There were no significant differences in cell viability in C2C12 cells treated with CMs (Figure 1C).
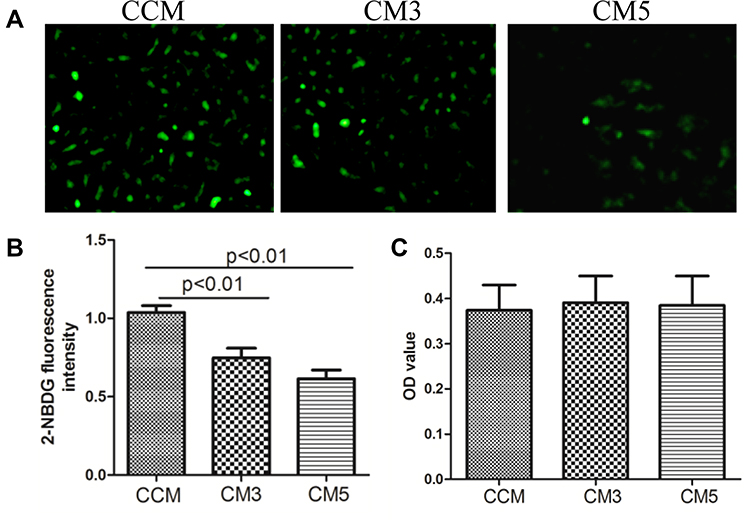 |
Figure 1 Osteoclast affected glucose uptake in C2C12 cell (A and B) and effects of conditioned mediums (CMs) on cell viability (C). The 2-NBDG fluorescence intensity were significantly decreased in CM3 and CM5 groups. No significant differences were observed in cell viability cultured with CMs. CCM: conditioned medium from RAW264.7 without RANKL induced; CM3: conditioned medium from RAW264.7 treated with RANKL for 3 days; CM5: conditioned medium from RAW264.7 treated with RANKL for 5 days. |
Resistin Expression During Osteoclast Formation
Subsequently, we determined the mRNA and protein expression of resistin during osteoclast formation (Figure 2). RAW264.7 differentiated into osteoclasts in the presence of RANKL induction (Figure 2A). Relative resistin mRNA (Figure 2B) and protein expressions (Figure 2C) were both increased in RAW264.7 cells treated with RANKL for 3 days and 5 days compared with those cells without RANKL administration. The resistin level was positively correlated with the osteoclast number (r = 0.63, p < 0.05) (Figure 2D). However, no significant differences were observed in leptin and adiponectin mRNA during during osteoclast formation.
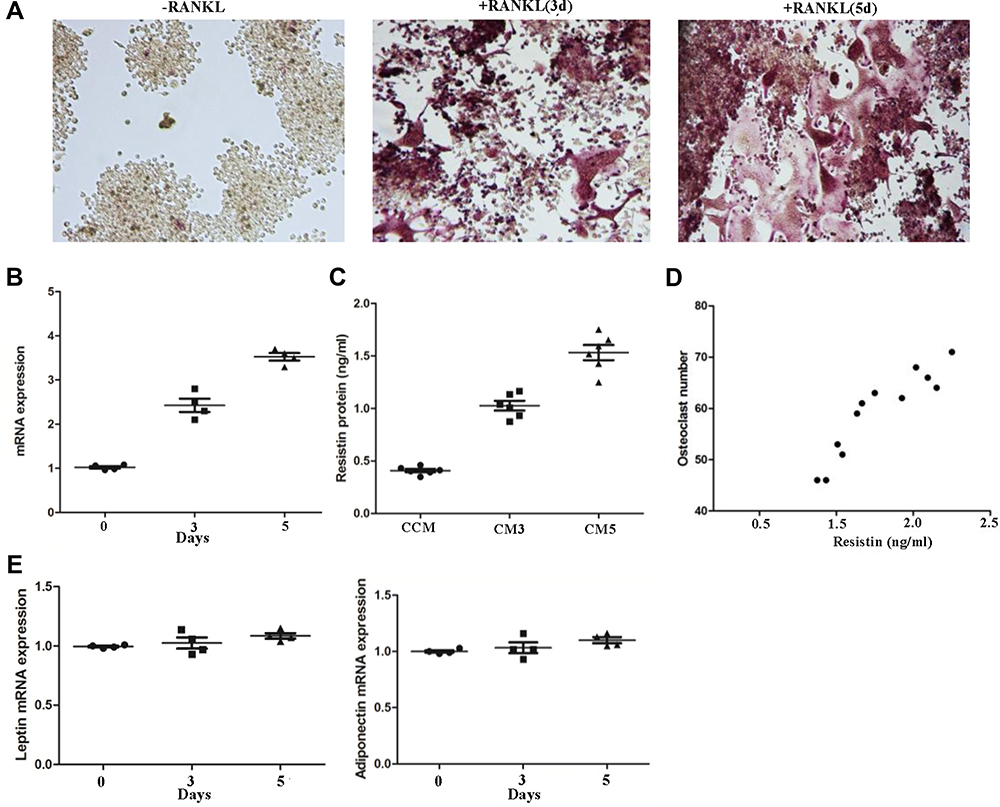 |
Figure 2 Resistin expression during osteoclast formation. RANKL promoted RAW264.7 cell differentiation to osteoclast (A). The relative mRNA (B) and protein (C) expression were both increased in RANKL treated cells (n = 3). The resistin protein level was positively correlated with osteoclast number (D). There were no significant changes in leptin and adiponectin mRNA during osteoclast formation (E). CCM: conditioned medium from RAW264.7 without RANKL induced; CM3: conditioned medium from RAW264.7 treated with RANKL for 3 days; CM5: conditioned medium from RAW264.7 treated with RANKL for 5 days. |
The number of osteoclasts at 72h was increased compared to that at 24 h (Figure 3A). The primary osteoclast model also showed that relative resistin mRNA (Figure 3B) and protein expressions (Figure 3C) were also increased with the increase in osteoclast formation.
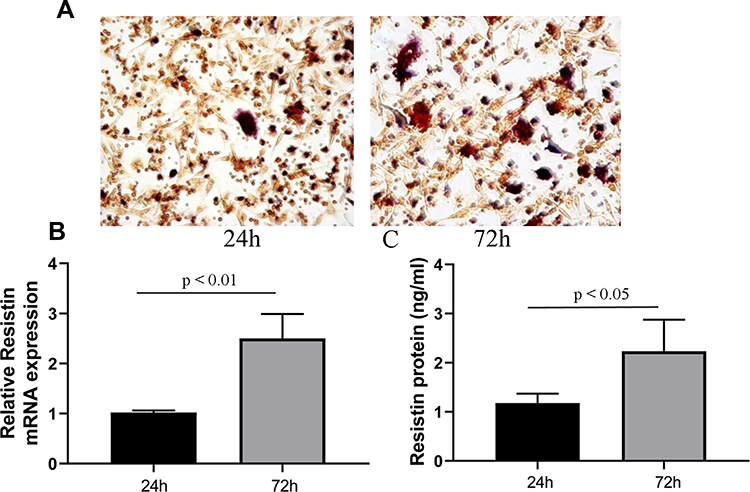 |
Figure 3 Resistin expression in primary osteoclast. Primary osteoclast was isolated from the long bone of new-born rat. The number of osteoclast at 72 h was larger than that at 24 h (A). The relative mRNA (B) and protein (C) expression at 72 h were also higher than those at 24 h (n = 3). |
Resistin Knockdown
The fluorescent images showed that resistin siRNA and negative control siRNA were transfected into the RAW264.7 cells (Figure 4A). The transfection did not show any obvious cytotoxic effects (Figure 4B). The relative resistin mRNA and protein expression in cells treated with resistin siRNA were significantly lower than those treated with negative control siRNA (Figure 4C and D). Relative resistin mRNA expression was decreased by approximately 65% of that of the negative control siRNA-transfected cells (Figure 4E). ELISA further showed that the resistin protein was also decreased in cells transfected with resistin siRNA (Figure 4F).
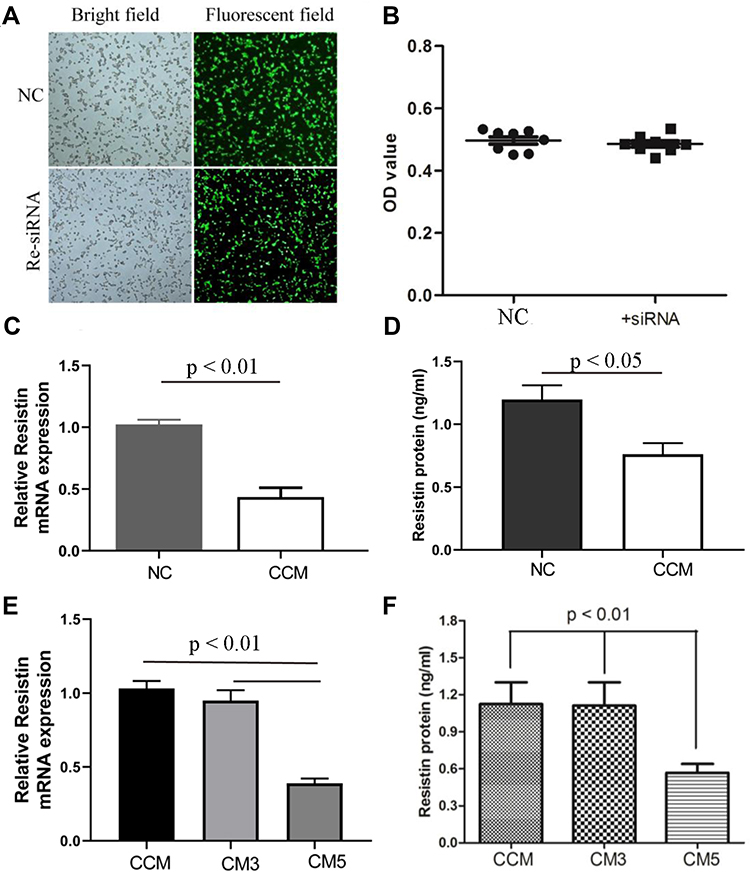 |
Figure 4 Resistin knockdown by siRNA. Resistin siRNA and negative control (NC) siRNA were transfected into the RAW264.7 cells (A). The transfection did not show obviously cytotoxic effects (B) (n = 8). Relative resistin mRNA and protein expression were decreased in resistin siRNA (CCM) compared NC group (C and D) (n = 4). In addition, relative resistin mRNA (C) and protein (D) expression at 3 days and 5 days of RANKL induction were also observed (E and F) (n = 4). NC: negative control siRNA; CCMi: conditioned medium from RAW264.7 transfected with resistin siRNA, but without RANKL induced; CM3i: conditioned medium from RAW264.7 transfected with resistin siRNA and treated with RANKL for 3 days; CM5i: conditioned medium from RAW264.7 transfected with resistin siRNA and treated with RANKL for 5 days. |
Glucose Uptake with Resistin Knockdown
The C2C12 cells were cultured with the CCMi and CM5i collected from the RANKL-induced RAW264.7 cells transfected with resistin siRNA. The 2-NBDG fluorescence intensity is shown in Figure 5. The intensity was significantly increased in C2C12 cells cultured with CM5i compared with the CM5 group (p < 0.05).
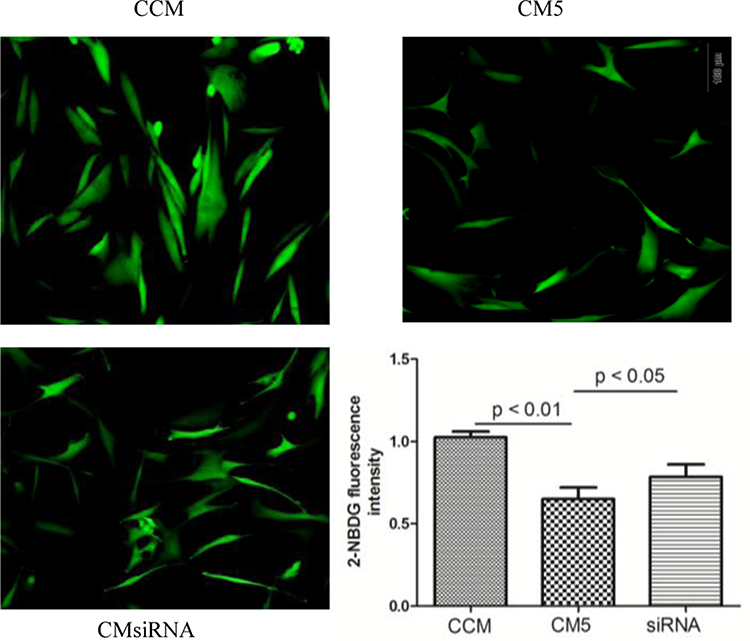 |
Figure 5 Glucose uptake in C2C12 cell treatment with different conditioned medium (CCM, CM5 and CMsiRNA). The 2-NBDG fluorescence intensity was significantly increased in C2C12 cells cultured with CMsiRNA compared with CM5 group (p < 0.05) (n = 4). CCM: conditioned medium from RAW264.7 without RANKL induced; CMsiRNA: conditioned medium from RAW264.7 transfected with resistin siRNA and treated with RANKL for 5 days; CM5: conditioned medium from RAW264.7 treated with RANKL for 5 days. |
Glucose Uptake in C2C12 Cells Cultured with CM5-AntiR
The 2-NBDG fluorescence intensity is shown in Figure 6. The intensity was significantly increased in C2C12 cells cultured with CM5-AntiR compared with the CM5 group (p < 0.05).
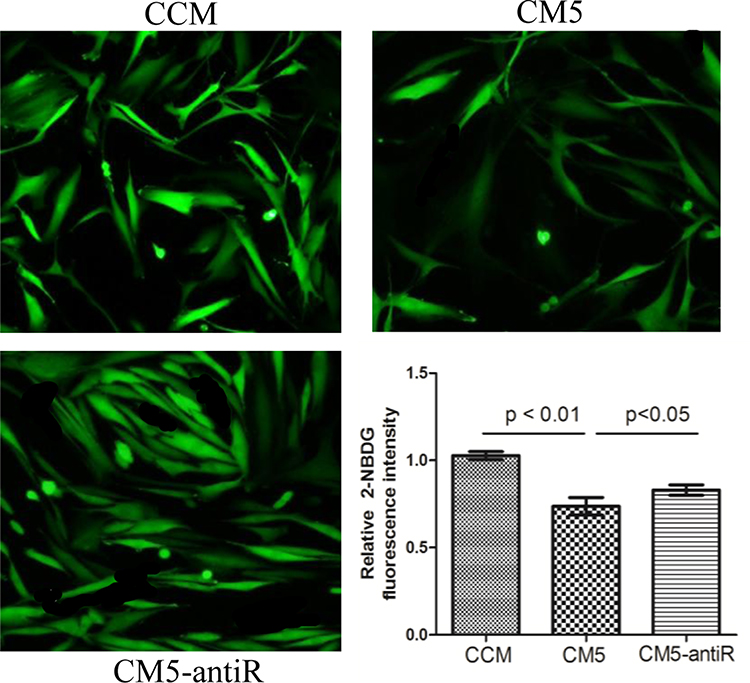 |
Figure 6 Glucose uptake in C2C12 cell treatment with different conditioned medium (CMM and CM5) and medium mixed with anti-resistin antibody (CM5-antiR). The 2-NBDG fluorescence intensity was significantly increased in C2C12 cells cultured with CM5-antiR compared with CM5 group (p < 0.05). CCM: conditioned medium from RAW264.7 without RANKL induced; CM5: conditioned medium from RAW264.7 treated with RANKL for 5 days. |
Discussion
Bone cells not only control bone remodeling, but also modulate adipose sensitivity and insulin secretion.18 Adipocytokines, such as adiponectin, resistin and vaspin, were suspected to participate in metabolic disorders and insulin resistance.19,20 Thommesen et al16 showed that the expression of resistin mRNA in preosteoclastic cells. However, few studies have shown the association between osteoclast differentiation and insulin resistance. Our data demonstrated that osteoclast differentiation may induce insulin resistance by secreting resistin. These results were further confirmed in a resistin knockdown cell model.
The association between osteoblasts and glucose homeostasis has been reported.18 Osteocalcin produced by the osteoblasts has been shown to affect adipocyte sensitivity to insulin and insulin secretion.3,18 However, few studies have investigated the effects of osteoclasts on glucose homeostasis or insulin resistance. Epidemiological data indicated that the concentration of soluble RANKL, a key cytokine in osteoclast differentiation, is an independent risk factor of type 2 diabetes mellitus.7 Interestingly, several reports show that inhibition of RANKL may improve insulin resistance in people and animal model.5,7 These studies indicated that osteoclast differentiation may contribute to insulin resistance. Our data evidenced that this hypothesis may be correct. The conditioned medium obtained during osteoclast differentiation induced glucose uptake-related insulin resistance in C2C12 cells.
Few data were found regarding the mechanism by which RANKL affected the glucose homeostasis. Kiechl et al7 showed that RANKL can affect the hepatic insulin resistance. They developed a hepatocyte-specific Rank (which could bind with RANKL) knockout (Rankko) mice and compared them with wild-type controls. The control mice developed insulin resistance after 4 weeks of a high-fat diet, but Rankko did not. Similar results were observed in mice that received a hydrodynamic injection of Rank shRNA (RANKi). They also found that specific deletion of Rank in the skeletal muscle or in pancreatic β-cells did not show improved insulin sensitivity. Therefore, they speculated that the role of RANKL on insulin resistance was due to its effects on hepatocytes. Osteoclasts are also target cells to RANKL. RANK is also expressed in preosteoclasts and mature osteoclasts. However, the role of osteoclasts in RANKL-related insulin resistance was not investigated in their study. Interestingly, preosteoclastic cells can express resistin,16 which is associated with metabolic impairments and insulin resistance.21,22 Our data showed that the resistin levels were increased during osteoclast differentiation. When the expression of resistin was downregulated by Resistin siRNA, C2C12 cells did not show abnormal glucose uptake. Our results indicated that osteoclasts may affect glucose uptake by resistin.
Many population and animal studies have shown positive correlation between resistin level and fasting blood glucose.15,23 Resistin may be involved in the regulation of glucose metabolism. However, no significant relationship between plasma levels of resistin and glucose metabolism was also reported in a number of population studies.23 This inconsistency may be due to the differences in study population, sample size or age/gender.
The molecular mechanism of resistin actions is still unclear. Several studies showed that resistin could inhibit insulin receptor substrate-1 and -2 phosphorylation and Akt activation24 and can also promote the production of inflammatory cytokine, including tumor necrosis factor α (TNF-α) and interleukin-6 (IL-6).10 Resistin can induce insulin resistance by both AMPK-dependent and AMPK-independent pathways.11 Toll-like-receptors (TLR) are considered a major factor in insulin resistance.8,25 The receptor or receptors of resistin especially in human is not fully understood. Recent investigations have shown that TLR4 is a potential candidate receptor for human resistin. The resistin/TLR4 signaling pathway may promote the onset of insulin resistance.8 Interestingly, TLR4 also plays an important role in C2C12 myoblasts, such as myogenic differentiation.26,27 These data all indicate that resistin may affect C2C12 cells in glucose uptake-related insulin resistance.
There are several limitations in this study. First, we only performed an in vitro study. Further studies are needed to test the role of osteoclasts on insulin resistance in animal models. Second, a RAW264.7 cell line and primary cell model were adopted in our study. Our results should be confirmed in more ostaoclastic cells model. Third, we did not investigate the mechanism of resistin on insulin resistance because many studies have shown the possible mechanisms. Moreover, the TLR4 pathway has been shown to play important role in the differentiation of C2C12 cells. Thus, our study could be an exploration.
Our study showed that the resistin expression was increased during osteoclast differentiation, and osteoclasts may affect glucose uptake in C2C12 cells. Osteoclasts may promote glucose uptake-related insulin resistance by secreting resistin. The inhibition on osteoclast formation may be a potential treatment strategy for insulin resistance or diabetes.
Abbreviations
CM, conditioned mediums; DMEM, dulbecco’s modified Eagle’s Medium; ELISA, Enzyme linked immunosorbent assay; IR, Insulin resistance; OC, osteocalcin; qRT-PCR, quantitative real-time polymerase chain reaction; TRAP, tartrate resistant acid phosphatase; RANKL, receptor activator of NF-kB ligand.
Data Sharing Statement
The data used and/or analyzed during the present study are available from the corresponding author on reasonable request.
Acknowledgments
This work was supported by grants from the Academic Leaders Training Program of Pudong Health Bureau of Shanghai (No. PWRd2019-12), the Talent Project of Shanghai Pudong New Area Gongli Hospital (No. GLRb2017-01), the Postdoctoral Cultivation Fund of Shanghai Pudong New Area Gongli Hospital (No. GLBH 2017005).
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Disclosure
The authors declare that they have no conflicts of interest.
References
1. DeFronzo RA, Tripathy D. Skeletal muscle insulin resistance is the primary defect in type 2 diabetes. Diabetes Care. 2009;32(Suppl 2):S157–S163. doi:10.2337/dc09-S302
2. Hojlund K. Metabolism and insulin signaling in common metabolic disorders and inherited insulin resistance. Dan Med J. 2014;61(7):B4890.
3. Lee NK, Sowa H, Hinoi E, et al. Endocrine regulation of energy metabolism by the skeleton. Cell. 2007;130(3):456–469. doi:10.1016/j.cell.2007.05.047
4. Komori T. Functions of osteocalcin in bone, pancreas, testis, and muscle. Int J Mol Sci. 2020;21(20):7513. doi:10.3390/ijms21207513
5. Lasco A, Morabito N, Basile G, et al. Denosumab inhibition of RANKL and insulin resistance in postmenopausal women with osteoporosis. Calcif Tissue Int. 2016;98(2):123–128. doi:10.1007/s00223-015-0075-5
6. Passeri E, Benedini S, Costa E, et al. A single 60 mg dose of denosumab might improve hepatic insulin sensitivity in postmenopausal nondiabetic severe osteoporotic women. Int J Endocrinol. 2015;2015:352858. doi:10.1155/2015/352858
7. Kiechl S, Wittmann J, Giaccari A, et al. Blockade of receptor activator of nuclear factor-κB (RANKL) signaling improves hepatic insulin resistance and prevents development of diabetes mellitus. Nat Med. 2013;19(3):358–363. doi:10.1038/nm.3084
8. Benomar Y, Taouis M. Molecular mechanisms underlying obesity-induced hypothalamic inflammation and insulin resistance: pivotal role of resistin/TLR4 pathways. Front Endocrinol. 2019;10:140. doi:10.3389/fendo.2019.00140
9. Shojima N, Sakoda H, Ogihara T, et al. Humoral regulation of resistin expression in 3T3-L1 and mouse adipose cells. Diabetes. 2002;51(6):1737–1744. doi:10.2337/diabetes.51.6.1737
10. Bokarewa M, Nagaev I, Dahlberg L, et al. Resistin, an adipokine with potent proinflammatory properties. J Immunol. 2005;174(9):5789–5795. doi:10.4049/jimmunol.174.9.5789
11. Luo Z, Zhang Y, Li F, et al. Resistin induces insulin resistance by both AMPK -dependent and AMPK-independent mechanisms in HepG2 cells. Endocrine. 2009;36(1):60–69. doi:10.1007/s12020-009-9198-7
12. Minn AH, Patterson NB, Pack S, et al. Resistin is expressed in pancreatic islets. Biochem Biophys Res Commun. 2003;310(2):641–645. doi:10.1016/j.bbrc.2003.09.061
13. Patel L, Buckels AC, Kinghorn IJ, et al. Resistin is expressed in human macrophages and directly regulated by PPAR gamma activators. Biochem Biophys Res Commun. 2003;300(2):472–476. doi:10.1016/S0006-291X(02)02841-3
14. Yura S, Sagawa N, Itoh H, et al. Resistin is expressed in the human placenta. J Clin Endocrinol Metab. 2003;88(3):1394–1397. doi:10.1210/jc.2002-011926
15. TD zięgielewska-Gęsiak S, Wyszomirska K, Fatyga E, et al. The role of oxidant-antioxidant markers and resistin in metabolic syndrome elderly individuals. Sci Prog. 2021;104(2):368504211006510.
16. Thommesen L, Stunes AK, Monjo M, et al. Expression and regulation of resistin in osteoblasts and osteoclasts indicate a role in bone metabolism. J Cell Biochem. 2006;99(3):824–834. doi:10.1002/jcb.20915
17. Nogueira AVB, Nokhbehsaim M, Tekin S, et al. Resistin is increased in periodontal cells and tissues: in vitro and in vivo studies. Mediators Inflamm. 2020;2020:9817095. doi:10.1155/2020/9817095
18. de Paula FJ, Horowitz MC, Rosen CJ. Novel insights into the relationship between diabetes and osteoporosis. Diabetes Metab Res Rev. 2010;26(8):622–630. doi:10.1002/dmrr.1135
19. Bahceci M, Gokalp D, Bahceci S, et al. The correlation between adiposity and adiponectin, tumor necrosis factor alpha, interleukin-6 and high sensitivity C-reactive protein levels. Is adipocyte size associated with inflammation in adults? J Endocrinol Invest. 2007;30(3):210–214. doi:10.1007/BF03347427
20. Choi SH, Hong ES, Lim S. Clinical implications of adipocytokines and newly emerging metabolic factors with relation to insulin resistance and cardiovascular health. Front Endocrinol. 2013;4:97. doi:10.3389/fendo.2013.00097
21. Hivert MF, Sullivan LM, Fox CS, et al. Associations of adiponectin, resistin, and tumor necrosis factor-alpha with insulin resistance. J Clin Endocrinol Metab. 2008;93(8):3165–3721. doi:10.1210/jc.2008-0425
22. Norata GD, Ongari M, Garlaschelli K, et al. Plasma resistin levels correlate with determinants of the metabolic syndrome. Eur J Endocrinol. 2007;156(2):279–284. doi:10.1530/eje.1.02338
23. Mostafazadeh M, Haiaty S, Rastqar A, et al. Correlation between resistin level and metabolic syndrome component: a review. Horm Metab Res. 2018;50(7):521–536. doi:10.1055/a-0637-1975
24. Satoh H, Nguyen MA, Miles PD, et al. Adenovirus-mediated chronic “hyper-resistinemia” leads to in vivo insulin resistance in normal rats. J Clin Invest. 2004;114(2):224. doi:10.1172/JCI20785
25. Rogero MM, Calder PC. Obesity, inflammation, toll-like receptor 4 and fatty acids. Nutrients. 2018;10(4):E432. doi:10.3390/nu10040432
26. Ono Y, Sakamoto K. Lipopolysaccharide inhibits myogenic differentiation of C2C12 myoblasts through the Toll-like receptor 4-nuclear factor-κB signaling pathway and myoblast-derived tumor necrosis factor-α. PLoS One. 2017;12(7):e0182040. doi:10.1371/journal.pone.0182040
27. Frost RA, Nystrom GJ, Lang CH. Lipopolysaccharide stimulates nitric oxide synthase-2 expression in murine skeletal muscle and C(2)C(12) myoblasts via Toll-like receptor-4 and c-Jun NH(2)-terminal kinase pathways. Am J Physiol Cell Physiol. 2004;287(6):C1605–15. doi:10.1152/ajpcell.00010.2004
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.