Back to Journals » Breast Cancer: Targets and Therapy » Volume 6
Oseltamivir phosphate monotherapy ablates tumor neovascularization, growth, and metastasis in mouse model of human triple-negative breast adenocarcinoma
Authors Haxho F, Allison S, Alghamdi F, Brodhagen L, Kuta V, Abdulkhalek S, Neufeld RJ, Szewczuk M ![]()
Received 23 September 2014
Accepted for publication 10 October 2014
Published 9 December 2014 Volume 2014:6 Pages 191—203
DOI https://doi.org/10.2147/BCTT.S74663
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Pranela Rameshwar
Fiona Haxho,1 Stephanie Allison,2 Farah Alghamdi,1,3 Lacey Brodhagen,1 Victoria EL Kuta,1 Samar Abdulkhalek,1,4 Ronald J Neufeld,2 Myron R Szewczuk1
1Department of Biomedical and Molecular Sciences, 2Department of Chemical Engineering, Queen’s University, Kingston, ON, Canada; 3The King Fahd Armed Forces Hospital, Serology, Jeddah, Saudi Arabia; 4Department of Molecular Genetics, Lerner Research Institute, Cleveland Clinic, Cleveland, OH, USA
Background: Triple-negative breast cancers (TNBCs) lack the estrogen, progesterone, and epidermal growth factor (EGF) receptor-2 (HER2/neu) receptors. Patients with TNBC have typical high grading, more frequent relapses, and exhibit poorer outcomes or prognosis compared with the other subtypes of breast cancers. Currently, there are no targeted therapies that are effective for TNBC. Preclinical antitumor activity of oseltamivir phosphate (OP) therapy was investigated to identify its role in tumor neovascularization, growth, invasiveness, and long-term survival in a mouse model of human TNBC.
Methods: Live cell sialidase, water soluble tetrazolium, WST-1 cell viability, and immunohistochemistry assays were used to evaluate sialidase activity, cell survival, and the expression levels of tumor E-cadherin, N-cadherin, and host endothelial CD31+/PECAM-1 cells in archived paraffin-embedded TNBC MDA-MB-231 tumors grown in RAGxCγ double mutant mice.
Results: OP, anti-Neu1 antibodies, and matrix metalloproteinase-9-specific inhibitor blocked Neu1 activity associated with EGF-stimulated TNBC MDA-MB-231 cells. OP treatment of MDA-MB-231 and MCF-7 cells and their long-term tamoxifen-resistant clones reproducibly and dose-dependently reduced the sialidase activity associated with EGF-stimulated live cells and the cell viability after 72 hours of incubation. Combination of 1 µM cisplatin, 5-FU, paclitaxel, gemcitabine, or tamoxifen with OP dosages ≥300 µg/mL significantly reduced cell viability at 24, 48, and 72 hours when compared to the chemodrug alone. Heterotopic xenografts of MDA-MB-231 tumors developed robust and bloody tumor vascularization in RAG2xCγ double mutant mice. OP treatment at 30 mg/kg daily intraperitoneally reduced tumor vascularization and growth rate as well as significantly reduced tumor weight and spread to the lungs compared with the untreated cohorts. OP treatment at 50 mg/kg completely ablated tumor vascularization, tumor growth and spread to the lungs, with significant long-term survival at day 180 postimplantation, tumor shrinking, and no relapses after 56 days off-drug. OP 30 mg/kg cohort tumors expressed significantly reduced levels of human N-cadherins and host CD31+ endothelial cells with concomitant significant expression of E-cadherins compared to the untreated cohorts.
Conclusion: OP monotherapy may be the effective treatment therapy for TNBC.
Keywords: triple-negative breast cancer, chemoresistance
Introduction
Breast cancers that are basal-like tumors constitute 75% of triple-negative breast cancer (TNBC) classification, with unique clinical (eg, premenopausal age onset, high parity, prevalence in African-American and Hispanic women, visceral and brain metastases) and histological features (eg, grade 3, high proliferative rate, apoptotic cells, central necrosis, lymphocytic response, and glomeruloid microvascular proliferation).1 Unfortunately, this classification of TNBC includes ~25% of breast cancer tumors that are not basal-like. Thus, patients incorrectly labeled as having estrogen receptor (ER) and progesterone receptor negative invasive disease are at risk of an incomplete diagnosis for an effective and possible life-saving therapy. Clinically, the therapy for TNBC is to use standard chemotherapeutics, but these treatments are associated with a high rate of relapse at local and systemic regions. This “good chemosensitivity–bad prognosis”, often referred to as the triple-negative paradox, is the clinical feature of this type of breast cancer.1 To date, there are no targeted therapies that are effective for TNBC. The current state of the art for TNBC is eloquently reviewed by Rastelli et al.1
In the clinical setting, targeted therapies based on single agents like specific antibodies or small molecule inhibitors against receptor tyrosine kinase have failed because they do not meet the primary endpoint of improvement for overall survival of the patients.2 For TNBCs, there are no current data that can predict a significant correlation between the receptor tyrosine kinase status and the response rates to tyrosine kinase inhibitors. For example, targeted therapies include: 1) zaltrap (aflibercept) as an angiogenesis inhibitor for prostate cancer; 2) torisel (temsirolimus or CCI-779) as a rapamycin (mTOR) inhibitor for first-line renal cell carcinoma in combination with avastin (bevacizumab) and a second-line renal cell carcinoma following therapy with sutent (sunitinib); and 3) avastin (bevacizumab) as an angiogenesis inhibitor in addition to chemotherapy following surgery for triple-negative adjuvant breast cancer.
Here, we describe an alternate therapeutic approach for breast cancers classified as TNBC, which is based on our newly discovered signaling platform that regulates epidermal growth factor (EGF) receptors (EGFRs).3 EGF binding to its receptor induces a G-protein coupled receptor signaling process to activate matrix metalloproteinase-9 (MMP-9) and Neu1 sialidase. The report discloses a novel complex of neuromedin B G-protein coupled receptor, MMP-9, and Neu1 tethered to EGFR at the ectodomain of the receptor on the cell surface. Activated Neu1 specifically hydrolyzes α-2,3-sialyl residues of the receptors, which facilitates the removal of steric hindrance between receptors, allowing subsequent kinase activation and cellular signaling. We have previously reported that an identical signaling paradigm is used by nerve growth factor TrkA receptors,4 insulin receptors,5 cell surface TOLL-like receptor-4,6–10 and intracellular TOLL-like receptor-7 and -9;11 all of these receptors are known to play major roles in tumorigenesis. In addition, we have found that oseltamivir phosphate (OP) targeted and inhibited the induction of Neu1 activity associated with these receptor activations.4,10,11 Furthermore, we have reported a new therapeutic approach using OP as an anticancer agent.3 Since Neu1 is involved as one of the key central players for EGFR signaling, preclinical molecular-targeting studies provide the proof-of-evidence for an effective OP therapy in the treatment of human pancreatic cancer growth and metastatic spread in heterotopic xenograft of tumors growing in RAGxCγ double mutant mice.3 In addition, OP overcame the chemoresistance of pancreatic PANC1 cancer cells to cisplatin and gemcitabine alone or in combination in a dose-dependent manner, and disabled the cancer cell survival mechanism(s) against the chemotherapeutic drugs.12 Interestingly, the data in the report also provided additional confirmation for OP therapy in reversing epithelial–mesenchymal transition (EMT) associated with resistance to standard, clinical chemotherapeutics. Recently, we reported that silencing transcriptional factor snail in ovarian carcinoma cells ablated the abnormal robust and bloody tumor vascularization in RAGxCγ double mutant mice with a concomitant abolishment of tumor growth and metastatic spread to the lungs.13 Collectively, the data suggest another molecular level of a novel organizational signaling platform connecting the snail–MMP-9 signaling axis in amplifying the Neu1 sialidase and MMP-9 cross-talk in regulating EGFRs, tumor neovascularization, growth, and invasiveness.
The present report describes an alternate therapeutic approach for the treatment of TNBC. OP monotherapy ablates in a dose-dependent manner tumor neovascularization, growth, and metastasis in mouse model of human triple-negative breast adenocarcinoma.
Materials and methods
Cell lines
MCF-7 (ATCC® HTB-22™) and MDA-MB-231 (ATCC® HTB-26™) are adherent epithelial adenocarcinomas obtained from the mammary gland, breast, which are derived from the metastatic pleural effusion site. MDA-MB-231 is a triple-negative, basal B subtype, mesenchymal stem-like, invasive ductal carcinoma with BRAF, CDKN2A, KRAS, NF2, TP53, and PDGFRA mutations.14 MCF-7 is a non-triple negative human breast adenocarcinoma cell line. The cells were grown in culture media containing 1× Dulbecco’s Modified Eagle’s Medium (DMEM; Gibco, Rockville, MD, USA), conditioned medium, supplemented with 10% fetal calf serum (FCS; HyClone, Logan, UT, USA), and 5 μg/mL plasmocin™ (InvivoGen, San Diego, CA, USA) in a 5% CO2 incubator at 37°C. At ~80% confluence, the cells were passaged at least five times before use in the experiments.
MCF-7 and MDA-MB-231 cell lines resistant against 5 μM and 10 μM tamoxifen were established in culture to gradual increases in concentrations of the indicated drugs in 1× DMEM conditioned medium. After removing dead cells, the viable cells were maintained in culture at the indicated chemodrug concentration. At ~80% confluence, cells were passaged in the same concentration of the chemotherapeutic agent for over 1 year. Stable MCF-7 and MDA-MB-231 resistant clones against 5 μM and 10 μM tamoxifen were used for the in vitro experiments.
Reagents
EGF (Sigma-Aldrich, St Louis, MO, USA), the natural ligand of the EGFR, was reconstituted in sterile 1× phosphate-buffered saline (PBS) at a stock concentration of 1 mg/mL and stored at −20°C. EGF concentrations to stimulate cells were 30–100 ng/mL. Incubation times varied between experiments and thus are indicated.
cis-Diamineplatinum(II) dichloride (P4394; Sigma-Aldrich) was reconstituted in dimethyl sulfoxide (DMSO) to make a 27.7 mM stock solution. Gemcitabine hydrochloride (G6423; Sigma-Aldrich) was reconstituted in 1× PBS to make a 133.5 mM stock solution. 5-Fluorouracil (5-FU) (F6627; Sigma-Aldrich) was reconstituted in a mixture of 1 mL DMSO and 9 mL 1× PBS to make 2.31 mM 5-FU stock. Paclitaxel from Taxus brevifolia, (T7402, Sigma-Aldrich), was reconstituted in DMSO to make 1.17 mM stock. These stocks were further diluted in 1× DMEM conditioned medium to make various dosages of the chemotherapeutic agents to be used in in vitro experiments.
Inhibitors
OP 75 mg capsules were reconstituted in sterile 1× PBS and centrifuged at 1,000 rpm for 10 minutes to obtain OP in the supernatant as previously reported.12 The stock-extracted OP solution had a concentration of 15 mg/mL. OP (~98% purity) was obtained from Hangzhou DayangChem Co, Ltd (Hangzhou City, People’s Republic of China). Cell culture in 1× DMEM conditioned medium containing different concentrations of OP (200–800 μg/mL) were used for the in vitro and in vivo experiments. MMP-9 inhibitor (CAS1177749-58-4) was obtained from Calbiochem-EMD Chemicals Inc. (half maximal inhibitory concentration =5 nM).
Primary antibodies
Neutralizing antibodies were used to inhibit sialidase function: rabbit anti-human Neu1 immunoglobulin G (IgG) antibody (Santa Cruz Biotechnology, Santa Cruz, CA, USA). Rabbit monoclonal anti-human E-cadherin antibody (Cell Signaling Technology, Inc., Danvers, MA, USA) was used at 1:400 dilution for immunohistochemistry according to the manufacturer’s instructions. Rabbit monoclonal anti-human N-cadherin (Cell Signaling Technology, Inc.) was used at 1:200 dilution according to the manufacturer’s instructions.
DyLight™ 488 donkey anti-rabbit IgG secondary antibody (Santa Cruz Biotechnology, Inc.) was used at 40 μg/mL to detect primary antibodies against human E- and N-cadherins in archived paraffin-embedded xenogafts of human MDA-MB-231 tumors. DyLight™ 488 rat monoclonal anti-mouse CD31/PECAM-1 antibody (Novus Biologicals Canada ULC, Oakville, ON, Canada) was used at a 1:300 dilution according to the manufacturer’s instructions to detect mouse CD31+/PECAM-1 endothelial cells in the archived paraffin-embedded xenogafts of MDA-MB-231 tumors.
Sialidase assay in live cells
Cells were grown on 12 mm circular glass slides in 1× DMEM conditioned medium in sterile 24-well tissue plates for 24 hours or until they reached ~70% confluence as previously optimized in the live cell sialidase assay.8,10 After removing medium, 2′;-(4-methylumbelliferyl)-α-D-N-acetylneuraminic acid (4-MUNANA; Biosynth Intl.) substrate at 0.318 mM in Tris-buffered saline, pH 7.4, was added to each well alone (control), with EGF (30 ng/mL), or in combinations of EGF and inhibitors or neutralizing antibodies at the indicated concentrations. 4-MUNANA substrate is hydrolyzed by sialidase activity to give free 4-methylumbelliferone at fluorescence emission of 450 nm, followed with an excitation at 365 nm. Fluorescent images were taken after 1–2 minutes using Zeiss M2 Imager epifluorescent microscopy (EC PLnN 20×/0.5 DICII objective, 200× total magnification). The mean fluorescence surrounding the cells was quantified using the Image J program.
Mouse model of human TNBC
A mouse model of human TNBC that has a double mutation in the combining recombinase activating gene (RAG1) and common cytokine receptor γ chain (Cγ) (RAG1xCy) was used as previously described for human pancreatic cancer.3 The RAG1xCγ double mutant mice on a non-obese diabetic (NOD.Cg-Rag1tm1Mom Il2rgtm1Wjl/SzJ; The Jackson Laboratory) genetic background are completely deficient in T-, B-, and natural killer-cells; show no spontaneous tumor formation; and exhibit normal hematopoietic parameters.15 A breeding colony of mice were generated by intercrossing and were maintained in specific pathogen-free isolators in the Animal Care Facility, Queen’s University, Kingston, ON, Canada. All mice were kept under sterile conditions in microisolators equipped with autoclaved food and water. Experimental protocols using mice in these studies received approval from the Animal Care Committee, Queen’s University. Mice between 6 and 8 weeks of age were used.
Cancer cell implantation in RAGxCγ double mutant xenograft mice
MDA-MB-231 cells grown in 75 cm2 cell culture flasks at 80% confluence were resuspended into solution using TrypLE Express (Gibco) and washed in sterile saline. The cells in suspension were centrifuged for 3 minutes at 900 rpm, and the cell pellet suspended in sterile saline at a concentration of 5–10×106 cells/mL for 0.5×106 cell implantation cutaneously into the right back flank of the mouse. There were four mice in each cohort of untreated, 30 mg/kg OP, and 50 mg/kg OP. Treatment with 30 mg/kg and 50 mg/kg of soluble OP in sterile saline were injected daily intraperitoneally starting at day 10 postimplantation, when the tumor volumes reached 10–20 mm3. For the 50 mg/kg OP cohort, the daily treatment regimen continued to day 111, then once a week until day 124, when the mice were taken off the drug treatment. The treatment regimen with OP was based on previously reported results on pancreatic3 and ovarian13 cancer cells. Tumor volumes were measured twice a week and determined as
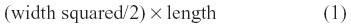
At the endpoints at day 180 or earlier due to skin lesions, mice were weighed and euthanized by cervical dislocation, and live necropsy tumor, liver, lung, pancreas, heart, and spleen were weighed. Archived paraffin-embedded tumor, liver, and lung necropsy tissues were used for hematoxylin and eosin staining and immunostaining for CD31+/PECAM-1 cells in tumor tissues, followed by microscopic analysis using a Zeiss Imager M2 fluorescence microscope.
Immunohistochemistry
The presence of the characteristic human epithelial marker, E-cadherin, and human mesenchymal marker, N-cadherin, in human MDA-MB-231 tumors was detected using immunohistochemistry and specific antibodies. Processed tumors were embedded in paraffin blocks following necropsy in each experiment. Microtome sections of tumor at 5 μm thickness were deparaffinized, heated for 8 minutes in citrate buffer, and rinsed three times in 1× PBS, followed by blocking with 1% bovine serum albumin (Thermo Fisher Scientific) in 1× PBS overnight at 4°C. Deparaffinized and processed tumor sections (5 μm) were immunostained with primary antibody for either 2 μg/mL rabbit anti-E- or anti-N-cadherin followed by Alexa Fluor® 488 secondary donkey anti-rabbit IgG antibody. The stained cells were covered with Entellan® rapid mounting medium, and images were observed using a Zeiss Imager M2 fluorescence microscope at 200× magnification. Additional 5 μm microtome sections of tumors were analyzed for mouse endothelial marker CD31/PECAM-1 using a 1:300 dilution of a 1 mg/mL stock of rat monoclonal antibody conjugated with DyLight-488 fluorochrome against mouse CD31/PECAM-1 (Novus Biologicals Canada ULC) and a Zeiss Imager M2 fluorescence microscope.
WST-1 assay
The WST-1 assay is a standard measure of cell viability based on the reduction of a tetrazolium compound to a soluble derivative.16 The absorbance of the reaction at 420 nm is proportional to the number of living cells in culture. Cells at 80%–90% confluence were added to 96-well microtiter plates at a density of 5,000 cells/well for 24 hours. They were treated with different concentrations of OP, chemodrugs, or left untreated as controls for 24, 48, and 72 hours. Absorbance readings were taken at 0, 24, 48, and 72 hours by adding 10% WST-1 reagent (Roche Diagnostics, Laval-des-Rapides, QC, Canada) in 1× PBS to each well, followed by an incubation at 37°C for 2 hours before taking readings at the indicated time points. Cell viability as a percentage of control was illustrated as a bar graph using GraphPad Prism software (GraphPad Software, La Jolla, CA, USA). The following formula was used to determine cell viability as a percent of control for each time point after drug treatments:

The “no treatment” WST-1 assay values were calculated for 24, 48, and 72 hours without drug treatment:

The 0 hours refers to the sum of plated cells and the WST-1 assay reagent and readings taken after 2 hours.
Statistical analysis
Statistical analysis of data used GraphPad Prism software. Comparison of results were made by one-way ANOVA at 95% confidence using Bonferroni’s multiple comparison test or the unpaired t-test.
Results
Neu1 sialidase activity is associated with EGF stimulation of TNBC MDA-MB-231 cells
Since the coexpression of EGFR and MMP-9 has prognostic value in cancer,17,18 and Neu1 and MMP-9 cross-talk regulates EGFRs in pancreatic3 and ovarian13 cancer cells, we investigated the molecular targeting potential of the Neu1–MMP-9 cross-talk platform in a TNBC MDA-MB-231 cell line in vitro. We initially performed the sialidase assay on EGF-stimulated live cells as previously described by our group.8,10,19 It is well known and characterized that MDA-MB-231 and MCF-7 breast cancer cells express the EGFRs.20,21 As shown in Figure 1A, EGF stimulation of live MDA-MB-231 cells induced sialidase activity as revealed in the periphery surrounding the cells using the fluorogenic sialidase-specific substrate, 4-MUNANA, which fluoresces at 450 nm due to the emission of 4-methylumbelliferone. The mean fluorescence of 50 multipoint replicates was quantified using the Image J software and is depicted in the bar graph. We also used specific neutralizing antibodies against the human Neu1 as well as the neuraminidase inhibitor OP and a specific inhibitor of MMP-9, all of which blocked the sialidase activity associated with EGF-treated live MDA-MB-231 cells comparable to the levels of no EGF-treated controls. The anti-Neu1 antibody used is specific for the epitope corresponding to amino acids 116–415 mapping at the C-terminus of Neu1 of human origin. It also detects Neu1 of mouse, rat, and human origin. The data depicted in Figure 1A are consistent with our previous reports for EGFRs in pancreatic3 and ovarian13 cancer cells.
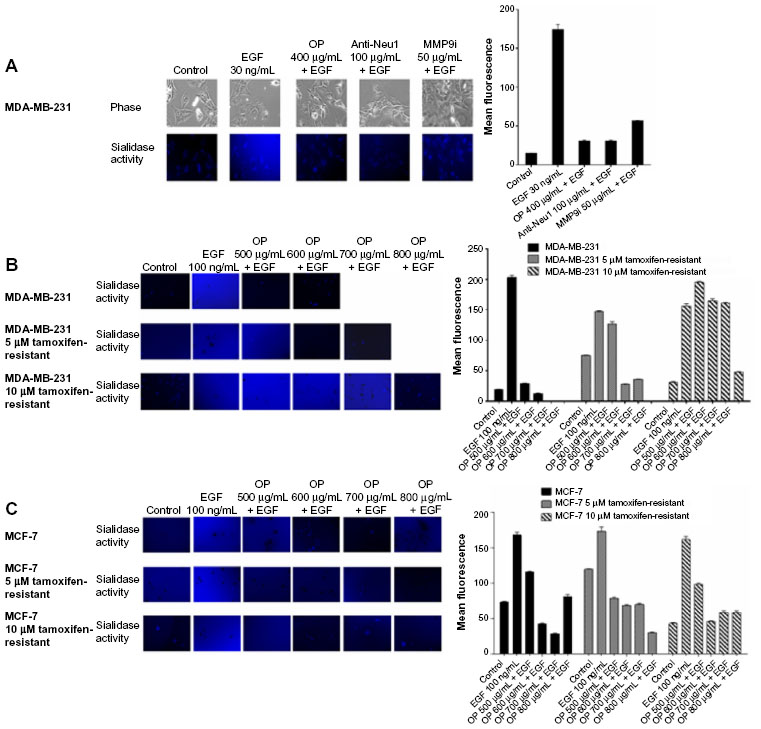 | Figure 1 EGF induces sialidase activity in live (A) TNBC MDA-MB-231 cells, (B) MDA-MB-231 cells and its resistant clones against 5 μM and 10 μM tamoxifen, and (C) MCF-7 cells and its resistant clones against 5 μM and 10 μM tamoxifen. |
In addition, we generated long-term (1 year) resistant clones of MDA-MB-231 and MCF-7 against 5 μM and 10 μM tamoxifen. With TNBC survival mechanisms against chemotherapeutics, the effects may be in large part manifested by the compensatory action of other downstream signaling molecules as described for the AXL and EGFR inhibitors.22 Martin et al have described in detail the current state of multidrug resistance in breast cancer, including the molecular mechanisms that contribute to tamoxifen resistance.23 Here, we hypothesized that tamoxifen-resistant MDA-MB-231 and MCF-7 clones at 5 μM and 10 μM may have a compensatory action(s) on other signaling pathways. In the presence of OP, the cancer cell survival mechanism(s) against the chemotherapeutic agents may be disabled and become more sensitive to the chemodrugs, as we have previously reported.12 As shown in Figure 1B and C, EGF stimulation of live tamoxifen-resistant MDA-MB-231 and MCF-7 cells induced sialidase activity. OP monotherapy dose-dependently blocked the sialidase activity associated with EGF-treated live cells comparable to the levels of no EGF-treated controls. It is noteworthy that the 10 μM tamoxifen-resistant MDA-MB-231 cells required a higher dosage of OP to block sialidase activity associated with EGF-stimulated cells compared to the 5 μM tamoxifen and to the parental cells. To explain these latter data, it is proposed that long-term resistance against tamoxifen may have upregulated in a stoichiometric manner several other signaling pathways to compensate for the level of tamoxifen in the growth medium.
Viability of MCF-7 and MDA-MB-231 and their tamoxifen-resistant clones when treated with OP at different doses using the WST-1 assay
To test the in vitro effects of OP treatment on the cell viability of MCF-7 and MDA-MB-231 and their corresponding tamoxifen-resistant clones, we performed the WST-1 cell proliferation assay16 using various concentrations of tamoxifen or OP for 24, 48, and 72 hours as optimally predetermined. Cell viability as a percentage of control ± standard error of the mean of triplicate values was determined based on the reduction of a tetrazolium compound to the soluble form. The data shown in Figure 2A and B indicate that the treatment of MCF-7 and MDA-MB-231 cells with tamoxifen dose-dependently decreased the cell viability (as a percentage of untreated control), with a lethal dose to kill 50% of viable cells of 9 μM for MCF-7 (Figure 2A) and 15 μM for MDA-MB-231 (Figure 2C) after 72 hours of incubation. We also tested the in vitro effects of OP treatment on cell viability using the tamoxifen-resistant MCF-7 and MDA-MB-231 cell clones. The data shown in Figure 2B and D indicate that OP treatment reproducibly and dose-dependently, markedly decreased the cell viability (as a percentage of untreated control) for the tamoxifen-resistant clones when compared to the parental MCF-7 and MDA-MB-231 cell lines.
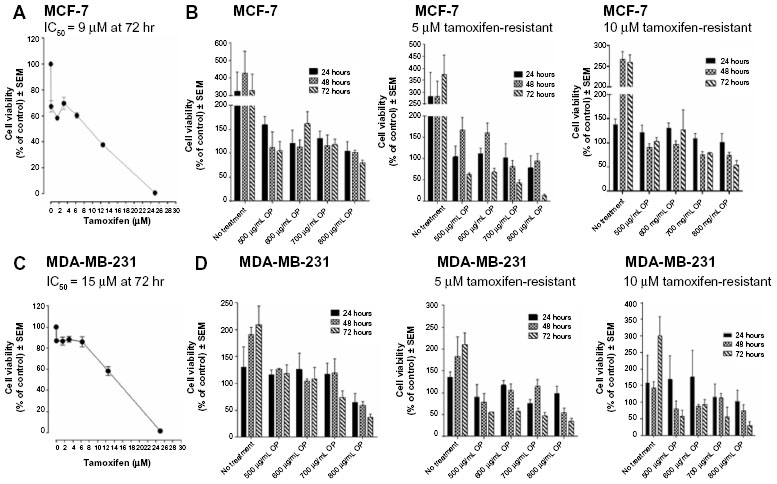 | Figure 2 Cell viability of (A) MCF-7 and (C) MDA-MB-231 cells treated with tamoxifen at indicated doses, (B) MCF-7 and (D) MDA-MB-231 cells and their resistant clones against 5 μM and 10 μM tamoxifen treated with OP at indicated doses using the WST-1 assay. |
The viability of MDA-MB-231 cells following treatment with different dosages of OP in combination with 1 μM of either cisplatin, 5-FU, paclitaxel, gemcitabine, or tamoxifen was compared with that of the chemodrug alone. The data in Figure 3 show that for the combination of cisplatin, 5-FU, paclitaxel, gemcitabine with OP dosages ≥300 μg/mL significantly reduced cell viability at 24, 48, and 72 hours when compared to the cell viability after single chemodrug treatment. In contrast, the combination of tamoxifen with OP at 800 μg/mL significantly reduced cell viability at 24, 48, and 72 hours.
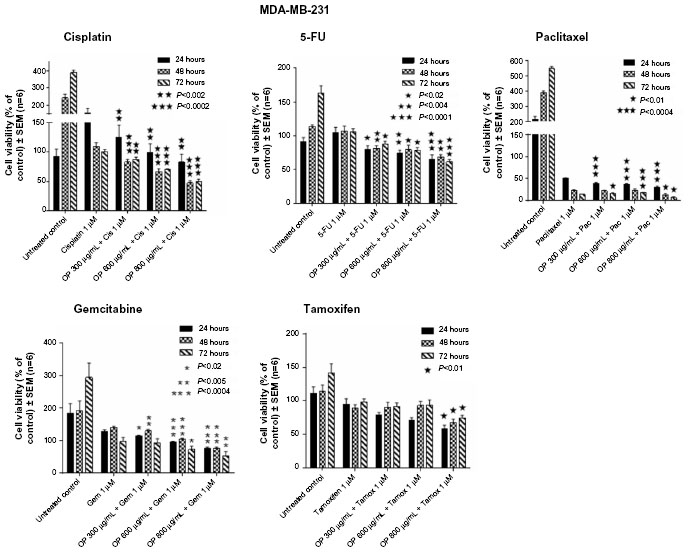 | Figure 3 Cell viability of MDA-MB-231 cells treated with OP at indicated doses in combination with 1 μM of either cisplatin, 5-FU, paclitaxel, gemcitabine, or tamoxifen using the WST-1 assay. |
OP monotherapy dose-dependently ablates tumor vascularization, growth, and invasiveness in heterotopic xenografts of TNBC MDA-MB-231 cells in RAGxCγ double mutant mice
The preclinical in vivo antitumor activity of OP was investigated in the RAGxCγ double mutant mouse model of human TNBC MDA-MB-231 cells. The RAGxCγ double mutant mouse lacks mature T- and B-cells, functional natural killer-cells, and is deficient in cytokine signaling, leading to better engraftment of human cells than any other published mouse strain.3 As previously demonstrated for pancreatic epithelial carcinoma cell line,3 the hypothesis is that OP monotherapy prevents the in vivo growth and spread of tumors in the heterotopic xenograft mouse model of human TNBC. MDA-MB-231 cells at 0.5×106 cells in 0.2 mL were implanted cutaneously in the right back flank of these mice. Twice a week, following implantation of the cancer cells, each mouse was monitored for tumor volume growth at the site of implantation, and body weight and condition were scored. All of the experimental mice showed no loss of body weight, and body condition scoring was normal until they developed skin lesions. Scoring the body condition of rodents is a noninvasive method for assessing health and establishing endpoints for adults where body weight is not a viable monitoring tool. For heterotopic implantation of MDA-MB-231 cells, mice developed skin lesions due to the abnormal tumor vascularization, which was the end point for body condition scoring. The data in Figure 4A show live tumors with skin lesions for each mouse (labeled A1, A2, and A4 for untreated control, B1–4 for OP 30 mg/kg, and C1 and C3 for OP 50 mg/kg) and live necropsy tumors with an unexpected, abnormal, robust, and bloody tumor vasculature at day 42 or earlier postimplantation of MDA-MB-231 cells. Due to the abnormal tumor vasculature, we also immunostained paraffin-embedded tumors from each mouse for host endothelial cell marker CD31/PECAM-1. There was an extensive migration of host CD31+ cells to the tumors for the untreated cohort but markedly reduced levels for the 30 mg/kg OP-treated cohort (Figure 4B). For the 50 mg/kg OP cohort, there were no tumors at day 180 postimplantation. As expected, the necropsy tumors from the 30 mg/kg OP-treated cohort revealed significantly high levels of tumor E-cadherin with concomitant significantly reduced N-cadherin levels (Figure 4B).
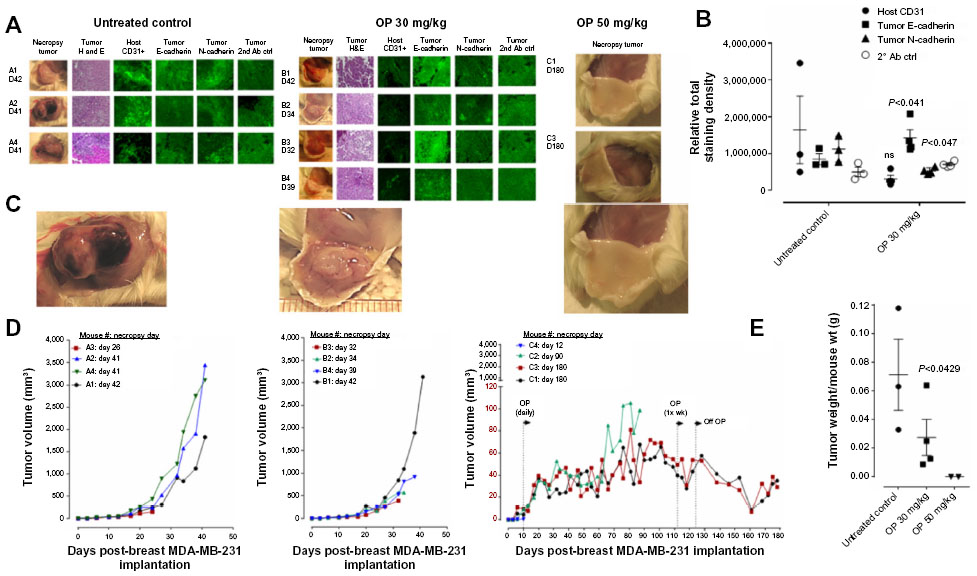 | Figure 4 OP treatment of RAGxCγ double mutant mice bearing heterotopic xenograft of MDA-MB-231 tumors. |
Treatment with 30 mg/kg and 50 mg/kg of soluble OP in sterile saline with daily injections intraperitoneally at day 10 postimplantation when the tumor volume reached 10–20 mm3 attenuated the aggressive tumor vascularization with skin lesions (Figure 4C) and the tumor growth (Figure 4D). The treatment regimen with OP was based on previously reported results on pancreatic3 and ovarian13 cancer cells. It is proposed that OP treatment regimen for cancers with robust tumor vascularization requires higher doses for attenuating tumor growth.
For the 50 mg/kg OP cohort, daily injections of OP continued to day 111 postimplantation, followed by 1× weekly injections, and off OP treatment at day 124 until the end point of the experiment at day 180. The data revealed tumor shrinking with no relapse. Live tumor weight per mouse body weight indicated a significant reduction for the OP 30 mg/kg cohort at days 32–42 (Figure 4E) compared to the untreated cohort. There were no visible tumors for the 50 mg/kg OP cohort at day 180. In addition, the survival rate of the 50 mg/kg OP-treated cohort was significant compared to the untreated control group (Figure 5A). The daily dosage of OP injections had no effect on the weights of the pancreas, heart, and liver per mouse body weight, but there was a marked decrease in the weight of the spleen per mouse body weight for the 50 mg/kg OP treatment (Figure 5B). These latter data are consistent with other reported results with patients with breast cancer where a reduction in spleen size was prognostic indicator.24–27 It is noteworthy that women in the weight category of less than 60 kg at diagnosis compared with those having the highest category of 79 kg revealed a 2.5-fold increased risk of dying from breast cancer (hazard ratio, 2.54 [95% confidence interval, 1.08–6.00]; trend P=0.02).28
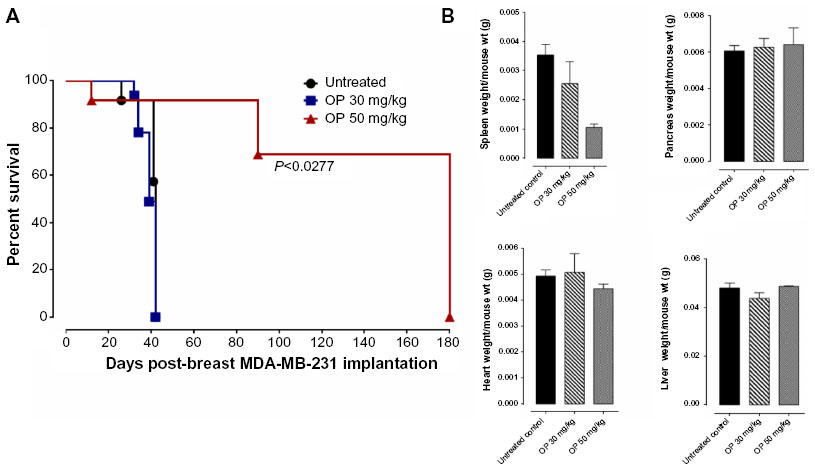 | Figure 5 (A) Percent survival of indicated cohorts of mice taken from Figure 4D and (B) mean ± SEM of live necropsy spleen, pancreas, heart, and liver weight per mouse body weight for each of the cohorts in Figure 4. |
Live necropsy lungs (Figure 6A) showed no visible tumor nodules. There were no tumor nodules nor metastatic clusters of MDA-MB-231 cancer cells in the liver for each of the cohorts (data not shown). The daily dosage of 30 mg/kg and 50 mg/kg OP treatment intraperitoneally, significantly attenuated the metastatic spread of MDA-MB-231 breast cancer cells to the lungs (Figure 6B) compared to the extensive metastatic clusters of cancer cells in the lungs for the untreated cohort. OP treatment had no adverse effects on the lungs as indicated by lung weight per mouse body weight (Figure 6C). This lack of metastatic spread of the MDA-MB-231 cancer cells to the lung following OP treatment is dose-dependent on the treatment regime. The lack of invasiveness due to OP treatment may be due in part to a disruptive tumor vasculature development (eg, reduced host CD31+ endothelial cell migration) or a decreased expression of N-cadherin with a concomitant increase in E-cadherin (Figure 4).
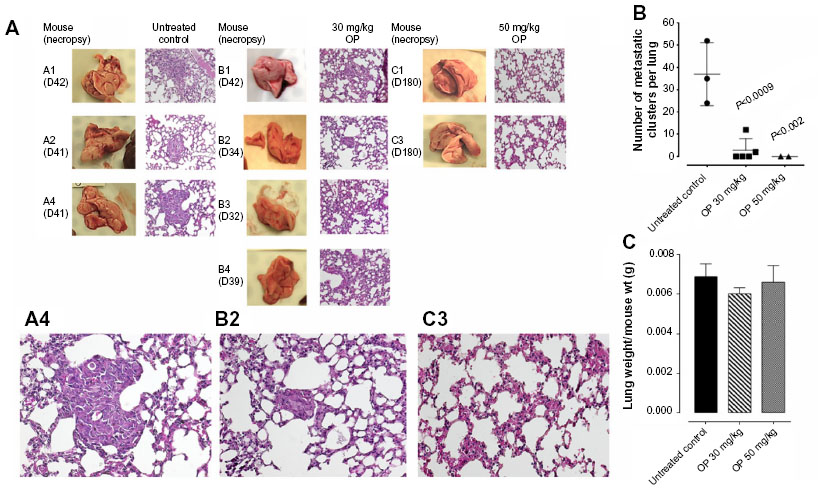 | Figure 6 (A) H&E staining of necropsy lung for number of metastatic clusters per lung (B) in paraffin-embedded tissues taken from xenograft A2780 tumor-bearing RAGxCγ double mutant mice. |
Discussion
Clinically, TNBC is insensitive to most of the effective therapies available for breast cancer treatments.29,30 For example, they include the HER2-targeted therapy such as Herceptin (trastuzumab) as well as tamoxifen or the aromatase inhibitors. These findings suggest that the growth of the cancer is not supported by the hormones estrogen and progesterone, nor by the presence of HER2 receptors. Combination chemotherapy using a metronomic dosage schedule is the standard therapy for early-stage TNBC.29 A prospective analysis of 1,118 patients who received neoadjuvant chemotherapy, of whom 255 (23%) had TNBC, indicated that patients with TNBC had higher pathologic complete response rates compared with non-TNBC patients (22% versus 11%; P=0.034).30
In our studies, the data indicate that both MDA-MB-231 and non-TNBC MCF-7 cells developed resistance against 5 μM and 10 μM of tamoxifen for over 1 year. At least for the non-TNBC cells, it has been reported that cross-talk between ER, HER-2, p38, and ERK may contribute to tamoxifen resistance.31 In another report, tamoxifen was shown to behave as an estrogen agonist in breast cancer cells that express high levels of AIB1 (amplified in breast cancer-1) and HER2, resulting in de novo resistance against tamoxifen.32 In addition, the study had demonstrated that gefitinib, an inhibitor of EGFR tyrosine kinase, by binding to the adenosine triphosphate-binding site of the enzyme, eliminated tamoxifen’s agonist effects and restored its antitumor activity both in vitro and in vivo in MCF-7/HER2-18 cells.32 Unfortunately, in another study, 25%–30% of patients with EGFR-activating mutations acquired resistance to gefitinib.33 The data in the report also showed that hepatocyte growth factor, a ligand of MET oncoprotein, facilitated the process of gefitinib resistance of lung adenocarcinoma cells, with EGFR-activating mutations, by restoring the phosphatidylinositol 3-kinase/Akt signaling pathway via phosphorylation of MET, but not with EGFR or ErbB3. Hepatocyte growth factor was also shown to induce resistance to the anti-EGFR antibody cetuximab in lung cancer cells, regardless of EGFR gene status.34
Insight into the mechanism of EGF-induced receptor activation came from our recent report on Neu1 sialidase and MMP-9 cross-talk in regulating the EGFR.3 The findings in the report on EGFRs also propose an alternative therapeutic approach using OP as a new anticancer agent targeting Neu1 as the key central enzyme regulating this Neu1–MMP-9 cross-talk within the EGFR signaling platform.3 Preclinical molecular targeting studies provide the proof-of-mechanism for OP as an effective treatment to inhibit growth and spread of human pancreatic cancer in heterotopic tumor xenografts growing in RAG2xCγ double mutant mice.3 It is noteworthy that alternate agents without cross-resistance to platinum or taxanes has been shown to improve the prognosis of platinum-resistant patients.35,36 Despite improvements using carboplatin/paclitaxel-based chemotherapy, 30% of patients with ovarian cancer fail to respond to primary therapy, with a high percentage of the responders relapsing within 1–2 years from the end of primary treatment, and dying of the disease within 5 years from their initial diagnosis.37 Gemcitabine, on the other hand, has been shown to be an active agent, single and/or in combination with carboplatin and paclitaxel, in the treatment of patients with recurrent ovarian cancer.37 Unfortunately, other reports have observed complications to the chemotherapeutics, including hypersensitivity pneumonitis-like patterns in gemcitabine-induced cases38 and hypersensitivity reactions to oxaliplatin.39 Based on our present results, it is proposed that treatment regimens for triple-negative breast adenocarcinoma could follow a combination therapy of OP at a high dosage with low doses of either cisplatin, 5-FU, paclitaxel, gemcitabine, or tamoxifen. The data in Figure 3 show that for the combination with paclitaxel, OP dosage ≥300 μg/mL caused ~95% reduction in cell viability compared to combination with the other chemodrugs. Based on the fact that hypersensitivity reactions to standard clinical chemotherapeutics are noted complications in cancer patients,38,39 it is proposed from our studies that combination therapy of OP and standard clinical chemotherapeutics may have a beneficial effect. Indeed, we have reported that OP significantly inhibited endotoxin lipopolysaccharide-induced NFκB activation and the production of nitric oxide and proinflammatory IL-6 and TNFα cytokines in primary and macrophage cell lines.10 In another report, we have shown that OP disables the chemoresistance of PANC1 pancreatic cancer cells against cisplatin and gemcitabine alone or in combination in a dose-dependent manner.12 The report also showed that OP treatment of PANC1 cells reversed the EMT associated with resistance to drug therapy.
The findings in the current report also reveal an unusual robust and bloody tumor vascularization in heterotopic xenografts of MDA-MB-231 TNBC tumors in RAGxCγ double mutant mice (Figure 4C). We recently reported that when snail small hairpin RNA (shRNA) A2780 ovarian cancer cells were injected cutaneously, the xenografts of tumors showed no robust bloody tumor vascularization compared with A2780 and A2780 shRNA slug tumors, and the mice were completely devoid of tumor growth and spread to the lungs.13 These observations together with another report have shown that the knockdown of snail expression not only suppresses ovarian cancer metastasis but also inhibits primary tumor growth.40 Snail not only plays a dual role in controlling the growth and metastasis of ovarian cancer but also functions as a mediator of tumor neovascularization.13
It is noteworthy that the transcriptional factor slug is expressed at high levels in normal breast, which may play a role in tubulogenesis.41 As an antiapoptotic agent, it could also protect epithelial cells from programmed cell death during ductal lumen formation and breast involution. In breast carcinomas, snail has been linked to tumor progression and invasiveness,41 the mechanism(s) which may cause repression of the E-cadherin gene. However, it is unclear how this transcriptional activity by these factors is regulated in vivo. For tumor neovascularization, we have reported that silencing slug in ovarian A2780 cells did not prevent the robust vascularization in RAGxCγ double mutant mice, whereas silencing snail in the A2780 cohort had a profound, complete inhibitory effect.13 Collectively, the signaling factors in the pathways that regulate the expression of the snail and slug proteins during different developmental processes have been reported to be tissue- and organism-dependent.42 Other reports have shown that snail is associated with cancer invasion and prognosis,43 and the overexpression of snail in epithelial Madin–Darby canine kidney (MDCK) cells promotes the EMT toward the invasive phenotype.44 Snail expression in MDCK cells had also induced the promoter activity and expression of MMP-9 through the PI3K/MAPK signaling pathway. For A2780 ovarian and TNBC MDA-MB-231 cells, it is proposed in our studies that snail may play an essential role in tumor neovascularization, the mechanism(s) which may involve the snail–MMP-9 signaling axis in facilitating the signaling paradigm for the activation of growth factor tyrosine kinase receptors to induce Neu1.11,13 If MMP-9 is playing a major role in the activation of Neu1 in complex with EGFR as our data suggest, logistically, snail in inducing MMP-9 in either ovarian A278013 or TNBC MDA-MB-231 cells in this report may be the molecular mechanism(s) by which the snail–MMP-9 signaling axis functions in tumor neovascularization. The data have shown that OP, anti-Neu1 antibody, and specific MMP-9 inhibitor treatments of the MDA-MB-231 breast cancer cell line blocked Neu1 activity associated with EGF-stimulation of the live cells. Based on these observations, we propose here that Neu1 might be an intermediate candidate connecting the snail–MMP-9 signaling axis in tumor neovascularization and in promoting the growth and invasiveness of human TNBC.
Conclusion
Collectively, the snail–MMP-9 signaling axis involvement in tumor neovascularization suggests that the modification of glycosylation on growth factor receptors involves the activation of Neu1. It follows that the molecular-targeting with OP treatment of Neu1 tethered to these receptors would be critically dose-dependent. Given the ability of OP to increase E-cadherin expression and decrease N-cadherin expression in TNBC MDA-MB-231 tumors and in ovarian tumors,12 TNBC treated with OP may become more adherent to the surrounding tissue and not metastasize as our data indicate (Figure 6). OP treatment strategies are proposed here to take the form of a horizontal approach, of which different oncogenic signaling pathways as well as macrophage-mediated tumorigenesis are targeted with promising therapeutic intent.
Acknowledgments
This work was supported in part by grants to MR Szewczuk and RJ Neufeld from the Natural Sciences and Engineering Research Council of Canada and private sector cancer funding from the Josefowitz Family to MR Szewczuk. The cancer contents of this report are covered by a Patent Cooperation Treaty international patent application number, PCT/CA2011/050690 (filing November 4, 2011).
We thank Lacey Brodhagen and Alexandra Morris, veterinarian technologists, for their expertise and assistance with the animal work.
Author contributions
F Alghamdi and S Allison performed the sialidase assay; L Brodhagen and S Allison performed the WST-1 assay; F Haxho performed the immunohistochemistry and the immunostaining for host CD31+ cells in tumor tissues and the liver and lung metastatic tumor analyses; S Abdulkhalek and V Kuta generated and maintained chemoresistant breast cancer cell lines; All authors read and critically revised the manuscript. F Haxho and MR Szewczuk wrote the paper. MR Szewczuk supervised the research design and MR Szewczuk and RJ Neufeld co-supervised F Haxho who prepared the first draft of the manuscript.
Disclosure
S Abdulkhalek is a recipient of the RS McLaughlin scholarship, the Ontario Graduate scholarship, and the Canadian Institutes of Health Research Doctoral award (Frederick Banting and Charles Best Canada Graduate scholarship). F Alghamdi was the recipient of the King Abdullah Scholarship from the Ministry of Higher Education, Saudi Arabia. The authors report no conflicts of interest in this work.
References
Rastelli F, Biancanelli S, Falzetta A, et al. Triple-negative breast cancer: current state of the art. Tumori. 2010;96(6):875–888. | |
Top 10 clinical trial failures of 2012 [webpage on the Internet]. Genetic Engineering and Biotechnology News; 2013. Available from: http://genengnews.com/insight-and-intelligence/top-10-clinical-trial-failures-of-2012/77899765/. Accessed February 3, 2013. | |
Gilmour AM, Abdulkhalek S, Cheng TS, et al. A novel epidermal growth factor receptor-signaling platform and its targeted translation in pancreatic cancer. Cell Signal. 2013;25(12):2587–2603. | |
Jayanth P, Amith SR, Gee K, Szewczuk MR. Neu1 sialidase and matrix metalloproteinase-9 cross-talk is essential for neurotrophin activation of Trk receptors and cellular signaling. Cell Signal. 2010;22(8): 1193–1205. | |
Alghamdi F, Guo M, Abdulkhalek S, Crawford N, Amith SR, Szewczuk MR. A novel insulin receptor-signaling platform and its link to insulin resistance and type 2 diabetes. Cell Signal. 2014;26(6): 1355–1368. | |
Abdulkhalek S, Amith SR, Franchuk SL, et al. Neu1 sialidase and matrix metalloproteinase-9 cross-talk is essential for Toll-like receptor activation and cellular signaling. J Biol Chem. 2011;286(42):36532–36549. | |
Abdulkhalek S, Guo M, Amith SR, Jayanth P, Szewczuk MR. G-protein coupled receptor agonists mediate Neu1 sialidase and matrix metalloproteinase-9 cross-talk to induce transactivation of TOLL-like receptors and cellular signaling. Cell Signal. 2012;24(11):2035–2042. | |
Amith SR, Jayanth P, Finlay T, et al. Detection of Neu1 sialidase activity in regulating Toll-like receptor activation. J Vis Exp. 2010(43). | |
Amith SR, Jayanth P, Franchuk S, et al. Neu1 desialylation of sialyl alpha-2,3-linked beta-galactosyl residues of TOLL-like receptor 4 is essential for receptor activation and cellular signaling. Cell Signal. 2010;22(2):314–324. | |
Amith SR, Jayanth P, Franchuk S, et al. Dependence of pathogen molecule-induced toll-like receptor activation and cell function on Neu1 sialidase. Glycoconj J. 2009;26(9):1197–1212. | |
Abdulkhalek S, Hrynyk M, Szewczuk MR. A novel G-protein-coupled receptor-signaling platform and its targeted translation in human disease. Res Reports in Biochemistry. 2013;3:17–30. | |
O’Shea LK, Abdulkhalek S, Allison S, Neufeld RJ, Szewczuk MR. Therapeutic targeting of Neu1 sialidase with oseltamivir phosphate (Tamiflu®) disables cancer cell survival in human pancreatic cancer with acquired chemoresistance. Onco Targets Ther. 2014;7:117–134. | |
Abdulkhalek S, Geen OD, Brodhagen L, et al. Transcriptional factor snail controls tumor neovascularization, growth and metastasis in mouse model of human ovarian carcinoma Clin Transl Med. 2014;3:28. | |
Lehmann BD, Bauer JA, Chen X, et al. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J Clin Invest. 2011;121(7):2750–2767. | |
Mazurier F, Fontanellas A, Salesse S, et al. A novel immunodeficient mouse model – RAG2 x common cytokine receptor gamma chain double mutants – requiring exogenous cytokine administration for human hematopoietic stem cell engraftment. J Interferon Cytokine Res. 1999;19(5):533–541. | |
Tan AS, Berridge MV. Superoxide produced by activated neutrophils efficiently reduces the tetrazolium salt, WST-1 to produce a soluble formazan: a simple colorimetric assay for measuring respiratory burst activation and for screening anti-inflammatory agents. J Immunol Methods. 2000;238(1–2):59–68. | |
Cox G, Jones JL, O’Byrne KJ. Matrix metalloproteinase 9 and the epidermal growth factor signal pathway in operable non-small cell lung cancer. Clin Cancer Res. 2000;6(6):2349–2355. | |
Swinson DE, Cox G, O’Byrne KJ. Coexpression of epidermal growth factor receptor with related factors is associated with a poor prognosis in non-small-cell lung cancer. Br J Cancer. 2004;91(7):1301–1307. | |
Woronowicz A, Amith SR, De Vusser K, et al. Dependence of neurotrophic factor activation of Trk tyrosine kinase receptors on cellular sialidase. Glycobiology. 2007;17(1):10–24. | |
Subik K, Lee JF, Baxter L, et al. The expression patterns of ER, PR, HER2, CK5/6, EGFR, Ki-67 and AR by immunohistochemical analysis in breast cancer cell lines. Breast Cancer (Auckl). 2010;4:35–41. | |
Masuda H, Zhang D, Bartholomeusz C, Doihara H, Hortobagyi GN, Ueno NT. Role of epidermal growth factor receptor in breast cancer. Breast Cancer Res Treat. 2012;136(2):331–345. | |
Meyer AS, Miller MA, Gertler FB, Lauffenburger DA. The receptor AXL diversifies EGFR signaling and limits the response to EGFR-targeted inhibitors in triple-negative breast cancer cells. Sci Signal. 2013;6(287):ra66. | |
Martin HL, Smith L, Tomlinson DC. Multidrug-resistant breast cancer: current perspectives. Breast Cancer (Dove Med Press). 2014;6:1–13. | |
Roberts JG, Chare MJ, Fortt RW, Leach KG, Baum M. Tumour histology and spleen size in women with breast cancer. Clin Oncol. 1980;6(1): 33–38. | |
Roberts JG, Chare MJ, Leach KG, Baum M. Spleen size in women with breast cancer. Clin Oncol. 1979;5(4):317–323. | |
Roberts JG, Wisbey M, Leach KG, Baum M. Proceedings: Spleen size in patients with breast cancer. Br J Cancer. 1975;31(2):262. | |
Roberts JG, Mander AM, Fortt RW, Leach KG, Bligh AS, Baum M. Proceedings: The relationship between spleen size, clinical staging and histological features in breast cancer. Br J Surg. 1975;62(2):159–160. | |
Enger SM, Greif JM, Polikoff J, Press M. Body weight correlates with mortality in early-stage breast cancer. Arch Surg. 2004;139(9):954–958; discussion 958–960. | |
Mehta RS. Dose-dense and/or metronomic schedules of specific chemotherapies consolidate the chemosensitivity of triple-negative breast cancer: a step toward reversing triple-negative paradox. J Clin Oncol. 2008;26(19):3286–3288; author reply 3288. | |
Liedtke C, Mazouni C, Hess KR, et al. Response to neoadjuvant therapy and long-term survival in patients with triple-negative breast cancer. J Clin Oncol. 2008;26(8):1275–1281. | |
Gutierrez MC, Detre S, Johnston S, et al. Molecular changes in tamoxifen-resistant breast cancer: relationship between estrogen receptor, HER-2, and p38 mitogen-activated protein kinase. J Clin Oncol. 2005;23(11):2469–2476. | |
Shou J, Massarweh S, Osborne CK, et al. Mechanisms of tamoxifen resistance: increased estrogen receptor-HER2/neu cross-talk in ER/HER2-positive breast cancer. J Natl Cancer Inst. 2004;96(12): 926–935. | |
Yano S, Wang W, Li Q, et al. Hepatocyte growth factor induces gefitinib resistance of lung adenocarcinoma with epidermal growth factor receptor-activating mutations. Cancer Res. 2008;68(22): 9479–9487. | |
Yamada T, Takeuchi S, Kita K, et al. Hepatocyte growth factor induces resistance to anti-epidermal growth factor receptor antibody in lung cancer. J Thorac Oncol. 2012;7(2):272–280. | |
Bamias A, Bamia C, Zagouri F, et al. Improved survival trends in platinum-resistant patients with advanced ovarian, fallopian or peritoneal cancer treated with first-line paclitaxel/platinum chemotherapy: the impact of novel agents. Oncology. 2013;84(3):158–165. | |
Kelly MG, Mor G, Husband A, et al. Phase II evaluation of phenoxodiol in combination with cisplatin or paclitaxel in women with platinum/taxane-refractory/resistant epithelial ovarian, fallopian tube, or primary peritoneal cancers. Int J Gynecol Cancer. 2011;21(4):633–639. | |
Fruscella E, Gallo D, Ferrandina G, D’Agostino G, Scambia G. Gemcitabine: current role and future options in the treatment of ovarian cancer. Crit Rev Oncol Hematol. 2003;48(1):81–88. | |
Tamura M, Saraya T, Fujiwara M, et al. High-resolution computed tomography findings for patients with drug-induced pulmonary toxicity, with special reference to hypersensitivity pneumonitis-like patterns in gemcitabine-induced cases. Oncologist. 2013;18(4):454–459. | |
Kim MY, Kang SY, Lee SY, et al. Hypersensitivity reactions to oxaliplatin: clinical features and risk factors in Koreans. Asian Pac J Cancer Prev. 2012;13(4):1209–1215. | |
Jin H, Yu Y, Zhang T, et al. Snail is critical for tumor growth and metastasis of ovarian carcinoma. Int J Cancer. 2010;126(9):2102–2111. | |
Côme C, Arnoux V, Bibeau F, Savagner P. Roles of the transcription factors snail and slug during mammary morphogenesis and breast carcinoma progression. J Mammary Gland Biol Neoplasia. 2004;9(2):183–193. | |
De Craene B, van Roy F, Berx G. Unraveling signalling cascades for the Snail family of transcription factors. Cell Signal. 2005;17(5): 535–547. | |
Mikami S, Katsube K, Oya M, et al. Expression of Snail and Slug in renal cell carcinoma: E-cadherin repressor Snail is associated with cancer invasion and prognosis. Lab Invest. 2011;91(10):1443–1458. | |
Jordà M, Olmeda D, Vinyals A, et al. Upregulation of MMP-9 in MDCK epithelial cell line in response to expression of the Snail transcription factor. J Cell Sci. 2005;118(Pt 15):3371–3385. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.