Back to Journals » Neuropsychiatric Disease and Treatment » Volume 15
Orthostatic hypotension and dementia incidence: links and implications
Authors Robertson AD ![]() , Udow SJ, Espay AJ
, Udow SJ, Espay AJ ![]() , Merola A
, Merola A ![]() , Camicioli R
, Camicioli R ![]() , Lang AE, Masellis M
, Lang AE, Masellis M
Received 30 April 2019
Accepted for publication 9 July 2019
Published 2 August 2019 Volume 2019:15 Pages 2181—2194
DOI https://doi.org/10.2147/NDT.S182123
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Roger Pinder
Andrew D Robertson,1,2 Sean J Udow,3 Alberto J Espay,4 Aristide Merola,4 Richard Camicioli,5 Anthony E Lang,6–8 Mario Masellis2,6,9
1Schlegel-UW Research Institute for Aging, University of Waterloo, Waterloo, ON, Canada; 2Hurvitz Brain Sciences Program, Sunnybrook Research Institute, University of Toronto, Toronto, ON, Canada; 3Division of Internal Medicine, Rady Faculty of Health Sciences, University of Manitoba, Winnipeg, MB, Canada; 4Department of Neurology, James and Joan Gardner Family Center for Parkinson’s Disease and Movement Disorders, University of Cincinnati, Cincinnati, OH, USA; 5Department of Medicine (Neurology), University of Alberta, Edmonton, AB, Canada; 6Division of Neurology, Department of Medicine, University of Toronto, Toronto, ON, Canada; 7Edmond J. Safra Program in Parkinson’s Disease, Toronto Western Hospital, University Health Network, Toronto, ON, Canada; 8Morton and Gloria Shulman Movement Disorders Clinic, Toronto Western Hospital, University Health Network, Toronto, ON, Canada; 9Cognitive and Movement Disorders Clinic, Sunnybrook Health Sciences Centre, Toronto, ON, Canada
Abstract: Orthostatic hypotension (OH) is a common condition, particularly in patients with α-synucleinopathies such as Parkinson’s disease, and has a significant impact on activities of daily living and quality of life. Recent data suggest an association with cognitive impairment. Herein, we review the evidence that OH increases the odds of incident mild cognitive impairment and dementia. Potential mechanisms underlying the putative relationship are discussed, including cerebral hypoperfusion, supine hypertension, white matter hyperintensities, and neurodegeneration. Finally, we highlight the challenges with respect to treatment and the negative impact on the quality of life and long-term prognosis presented by the coexistence of OH and dementia. Large population-based studies have reported that OH is associated with about a 20% increased risk of dementia in the general population, while smaller cohort studies suggest an even greater effect in patients with α-synucleinopathies (3- to 7-fold higher than controls). Ultimately, OH exposure is difficult to quantify, predominantly limited to pressure regulation during a one-time orthostatic challenge, and the causative association with dementia may turn out to be bidirectional, especially in α-synucleinopathies. Early diagnosis and treatment of OH may improve long-term prognosis.
Keywords: α-synucleinopathies, cognitive impairment, dementia, orthostatic hypotension, review
Introduction
Orthostatic hypotension (OH) is a disorder in which a sustained drop in blood pressure (BP) is observed in upright posture compared to supine. This is a common disorder in older adults with a prevalence ranging from between 6% and 30% in community-living individuals1,2 to over 50% in those living in geriatric institutions.3 While OH in the general population has multiple etiologies, including underlying cardiac and vascular pathologies which contribute to arterial baroreflex failure, pump failure, or hypovolemia,4 a subset of OH known as neurogenic OH is largely attributed to dysfunction of the autonomic nervous system, including inadequate norepinephrine release in cardiac and peripheral tissues.5,6 Neurogenic OH is a common feature of a specific group of neurodegenerative disorders involving intracellular α-synuclein accumulation, known as α-synucleinopathies, including Parkinson’s disease (PD) with and without cognitive impairment/dementia and dementia with Lewy bodies (DLB). A pooled estimate of OH prevalence in PD from 25 studies is ~30%,7 ranging up to 65% in one study.8 In DLB, the prevalence has been reported as high as 69%.9
OH symptoms, which include lightheadedness, dizziness, blurred vision, weakness, fatigue, nausea, palpitations, tremulousness, headache, and neck pain, as well as a corresponding increase in the likelihood of syncope, falls, and injury, contribute a significant burden to quality of life.10,11 Further, OH is associated with an increased risk of cardiovascular and cerebrovascular morbidity,12–14 and all-cause mortality.14,15 Despite a growing amount of cross-sectional evidence suggesting an association between OH and cognitive impairment,16,17 the notion of a causal relationship remains controversial.18–20 Over the last decade, several studies have examined the longitudinal association of OH and cognitive decline. Herein, we review the literature to assess the evidence for incident mild cognitive impairment (MCI) and dementia in OH and discuss physiological links and implications. Given the higher prevalence of OH in α-synucleinopathies, described earlier, we placed an emphasis on this clinical cohort in which case the causes of OH are more likely to be neurogenic. Unfortunately, most studies assessing OH and cognition have not adequately detailed the orthostatic response (eg, the full temporal profile of BP or the corresponding noradrenergic response) or characterized the cohort of interest (eg, the presence of parkinsonism, rapid eye movement sleep behavior disorder, or other autonomic dysfunction) such that OH etiology can be deduced. Hence, we refer to OH throughout the paper whether it is from neurogenic or non-neurogenic causes and highlight findings from populations in which an impaired adrenergic response is the likely underlying cause. In addition, we review potential mechanisms associated with OH which may precipitate cognitive impairment, as well as the treatment and prognostic implications of the coexistence of OH and dementia.
Search criteria
In this narrative review, we searched the MEDLINE database for original research articles involving humans and published in English after October 2008. Search terms (truncated with *) included a combination of exposure (orthost*, postur*, hypotens*, syncop*, or falls), timing (inciden*, novel, risk, predict*, cohort, prospect*, or longitud*), and outcome (dementia, cognit*, or delirium). Articles related to stroke or other traumatic events were excluded. The review, however, was not systematic or exhaustive in nature.
OH definition and measurement
Upon transition to an upright posture, a brief increase in arterial BP – due to compression of the veins in the lower limbs and abdomen, and increased venous return – is immediately followed by a large drop in BP due to a reduction in systemic vascular resistance and redistribution of blood to the splanchnic and peripheral vascular beds. The usual response to this transient hypotension is a reflexive increase in adrenergic outflow from postganglionic nerves to the heart and vasculature, via the arterial baroreflex, increasing heart rate, cardiac contractility, and vascular tone to restore BP within 30 s of the postural transition.21 In OH, the reflexive increases in cardiac output and peripheral vasoconstriction are deficient and cannot restore BP.
Classical, initial, and delayed OH
The classical definition of OH is a sustained drop in systolic BP (SBP) of ≥20 mmHg (≥30 mmHg when coexisting with supine hypertension (SH)) or diastolic BP (DBP) of ≥10 mmHg (≥15 mmHg with SH) usually within 3 mins after moving from the supine to the upright position or in the context of a head-up tilt of at least 60°.22 In most OH studies, BP is measured at 1 and 3 mins after the postural transition, while additional measures at 5 and 10 mins are made infrequently. Advances in non-invasive beat-by-beat BP monitoring, however, have expanded our understanding beyond classic OH to include the temporal variants of initial OH (IOH), delayed-recovery OH (OH-30), and delayed-onset OH (DOH).21,23 IOH is characterized by an exaggerated fall in SBP and/or DBP (≥40 and ≥20 mmHg, respectively) immediately upon assuming the upright posture but full recovery by 30 s. In OH-30, often a subset of IOH, full recovery is achieved within 3 mins but is delayed beyond the usual 30 s. As BP recovers within 3 mins of standing, OH-30 is not considered classical OH but does reflect impaired BP regulation.21 DOH describes a condition in which the BP drop occurs beyond 3 mins, and as late as after 30 mins of standing.23 In the general population, DOH has been reported to contribute to ~6% of all OH cases;24 however, reported values likely underestimate the true prevalence of DOH as it would remain undiagnosed by discontinuation of testing after only 3 mins. DOH appears to represent an early form of autonomic dysfunction encompassing cardiovagal impairment and has been reported to evolve to classic OH within 10 years in nearly 50% of the cases.23 Moreover, there is a prospective link between DOH and α-synucleinopathy within 10 years.23 A measure of the peripheral impairment in adrenergic outflow that drives neurogenic OH in Lewy body disorders is low plasma norepinephrine.5,25 Although multiple system atrophy also falls under the umbrella of α-synucleinopathies, the neuropathology with respect to autonomic failure is distinct from PD and DLB and involves a disconnect between central inputs and peripheral fibers. The peripheral fibers themselves, as well as circulating norepinephrine, remain relatively intact.26
Symptomatic OH
Another factor attenuating the true prevalence of OH in the general population is that only about one-half of older adults with classic OH report experiencing symptoms.27 A similar occurrence of symptoms is reported by adults with PD.28,29 Although symptoms are more likely to be reported in more severe OH, symptomatic and asymptomatic OH have a similar deleterious impact on activities of daily living, fall rate, and quality of life.11,29 These latter findings, in addition to the likelihood that cognitive impairment may reduce the reporting of symptoms, speak to the importance of obtaining objective – ideally, beat-by-beat – measures of BP regulation in at-risk individuals.
OH and incident cognitive decline
OH and MCI
To date, there is little evidence to suggest that OH is directly related to incident MCI in the general population. Three population-based prospective studies based in Europe and Southeast Asia did not observe any association between OH and cognitive decline, using only the Mini-Mental State Exam (MMSE) as criterion (ie, a drop in MMSE≥3 or below 24). In 651 home-dwelling and institutionalized older adults in northern Finland, OH at baseline was not associated with cognitive decline after 2.5 years.30 Similarly, in the Singapore Longitudinal Aging Study involving 1,347 non-demented older adults31 and the Progetto Veneto Anziani Study of 1,408 older adults,32 OH was not associated with changes in cognition during follow-up periods of up to ~4.5 years. In one small case–control study, however, involving 38 OH patients and 76 controls with a 4-year follow-up period, OH was associated with a 4-fold increase in the odds of MMSE dropping below 24.33 Consensus criteria for dementia were not assessed. These studies relied strictly on the MMSE, which was designed for screening and lacks the sensitivity or specificity to detect a change in cognitive state.34,35 Further, the MMSE is heavily weighted on memory and language, whereas cross-sectional associations between OH and cognitive function are strongest for executive function, attention, and visuospatial processes.36,37 Still, studies which incorporated supplementary neuropsychological testing have reported similar findings. For example, the Irish Longitudinal Study on Ageing (TILDA), which followed 3,417 community-living non-stroke, non-demented, and non-Parkinsonian individuals over 2 years, did not find any associations between OH and change in MMSE, verbal fluency, or 10-word recall scores.38 In addition to the limits of the MMSE, these studies reported follow-up cognitive function after only 2–4 years and the duration of OH prior to enrolment was unknown. Hence, OH exposure may have been insufficient to impart a detrimental cognitive effect in the short term. The Atherosclerosis Risk in Communities (ARIC) Study, involving 10,572 participants, found that those with OH at baseline had 28% greater odds of a decline in psychomotor speed over a longer 6-year follow-up period39 – a finding that was confirmed recently at the 25-year follow-up.40 The associations from both the 6-year and 25-year follow-up data, however, were not significant after accounting for baseline demographic and cardiovascular risk factors. The Good Aging in Skåne study – a population study within the Swedish National Study on Aging and Care (GÅS-SNAC) – monitored 1,480 older adults over a follow-up of 6 years.24 While there was a tendency for OH at baseline to be associated with an increased odds of developing MCI at follow-up, the results were no longer significant after adjusting for age. In contrast, the self-reported presence of OH symptoms within the year leading up to enrolment was associated with a 2.3-fold increase in the age-adjusted odds of incident MCI.24 Inconsistencies across studies may be due to the reliance on the MMSE, the relatively short follow-up periods used in most studies, and the methods of evaluating OH. Only the TILDA study, which assessed orthostatic response with beat-by-beat monitoring, measured BP at more than 2 timepoints post-transition. In a collection of longer-term population-based studies discussed later, ranging from 6 to 28 years of follow-up, a stronger link between OH and incident cognitive impairment is evident.
OH and dementia
A group of well-described population-based studies has provided evidence that OH is associated with an increased risk of developing dementia (Table 1). For the most part, there is agreement between studies that SBP regulation has a stronger link to cognitive impairment than that of DBP and that more severe OH carries a proportionally greater risk. The GÅS-SNAC study reported that the odds of incident dementia were increased ~2-fold when OH was present at baseline and that OH as a function of the SBP drop with standing had a greater risk than OH as a function of the drop in DBP.24 The Rotterdam study followed 6,204 non-demented, stroke-free individuals over a median 15.3-year follow-up period.41 Classic OH was associated with a 15% increased incidence of dementia. Further, a continuous scale of orthostatic dysregulation – quantified as SBP variability – was associated with an increased risk of dementia; a finding that persisted even in a subgroup that excluded individuals with classic OH. In the French Three-City Study, 7,425 community-living individuals older than 65 years of age were tracked over a mean follow-up period of 7.5 years, during which time 760 cases of dementia were diagnosed.42 OH was associated with a ~20% increased risk of incident dementia; however, when a more stringent threshold for OH was used (ie, SBP drop>30 mmHg or DBP drop>15 mmHg), the risk of dementia was even greater (~57%). The findings of the Rotterdam and the Three-City Study provide evidence that the risk of dementia increases with the severity of OH impairment and that the risk is elevated with even mild drops in BP that do not meet the criteria for classic OH.
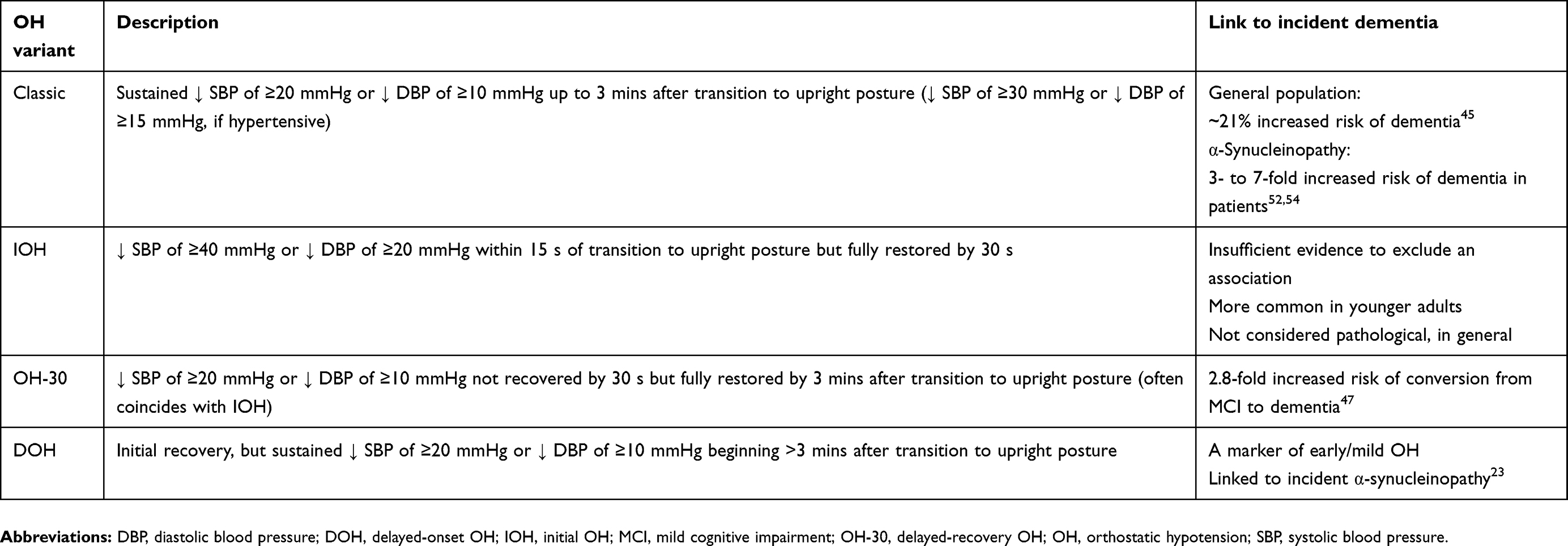 |
Table 1 OH variants and their link to dementia |
To date, two studies have data on the association of OH in midlife and incident dementia with long-term follow-up in excess of 20 years – the ARIC study40 and the Swedish Malmӧ Preventative Project.43 In the ARIC study, OH at baseline was associated with a 54% higher risk of developing dementia over ~25 years. Although this association remained significant after adjustment for demographic and clinical covariates, it was not significant in a sensitivity analysis using only a subgroup of participants without SH.40 The Malmӧ project is the longest prospective study of OH in the general population with a mean follow-up period of 28 years. While classic OH thresholds did not predict incident dementia, the magnitude of the DBP drop upon standing was associated with a 22% higher risk (per 10 mmHg) of dementia.43 This study stands out as the only population-based study not to observe an association between OH and dementia, and the only study to show a stronger link between the DBP drop and dementia compared to that of SBP. The results may be confounded by the fact that OH prevalence was lower (~2%) than is typically observed in the general population.
In a cohort of very old (ie, >80 years at baseline) 3,121 hypertensive adults from the international Hypertension in the Very Elderly Trial, OH was associated with a 36% increased risk of cognitive decline, using MMSE criteria, and a 34% increased risk of incident dementia, using consensus criteria, over a relatively short follow-up (mean 2 years).44 The dementia risk was even greater when considering only individuals who were symptomatic but did not experience a large orthostatic drop in BP. Of note, this study relied on bedside sit-to-stand criteria for OH – SBP/DBP thresholds of 15/7 mmHg45 – which have not yet been fully validated. Still, given that the association was apparent after such a short time period suggests that older hypertensive adults with OH may represent a cohort of exceptionally elevated risk. Peters and colleagues subsequently combined these results with other prospective literature in a meta-analysis and reported OH was associated with a 21% increased risk of dementia.44
Paradoxical evidence from both the GÅS-SNAC and ARIC studies indicates no relationship between OH and incident MCI but a direct relationship between OH and incident dementia.24,40 One reason for this apparent discrepancy might be floor effects. In both studies, OH was associated with poorer performance in some cognitive domains at baseline. Thus, it may follow that individuals with OH had less range to decline prior to crossing thresholds for dementia than those without OH. With differing cognitive baselines but similar changes in cognition over time, adults with OH would be more likely to develop dementia, and those without OH would be more likely to develop MCI. There is evidence to suggest that participants with MCI and OH are more likely to convert to dementia than those with MCI but not OH. In a follow-up study of 141 participants with MCI, 30% transitioned to dementia within 3 years.46 Although no baseline differences in the SBP drop on standing were observed between MCI-converters and non-converters, the rate of BP recovery following the initial drop was slower in those who converted to dementia. That is to say, individuals with OH-30 (ie, those who remained >30% below their supine SBP at 30 s after standing) were ~2.8 times more likely to convert to dementia during follow-up.46
At the population level, OH does not appear to be linked to a particular dementia type. In line with dementia prevalence in the general population, Alzheimer’s disease, vascular dementia, and mixed-type dementia were the most common in the population-based studies we reviewed, accounting for over 80% of incident dementia; DLB/PD dementia represented less than 10%.41,42 The Rotterdam study reported similar hazard ratios for both the Alzheimer’s disease and vascular dementia subgroups, but the incidence of other dementias was too low to assess risk.41 The lack of an association with Lewy body disorders is perhaps surprising, given neurogenic OH is more prevalent in PD with dementia compared to Alzheimer’s disease or vascular dementia.47 We speculate, however, that incident Lewy body disorders may be underdiagnosed in these studies given that the prevalence of Lewy bodies observed during autopsy is greater than that diagnosed clinically.48 Given the established association between autonomic dysfunction, including OH, and α-synucleinopathies, cohort studies involving this patient group separately from the general population have been pursued.
OH and dementia in α-synucleinopathies
As indicated earlier, the etiology of OH in the general population may be predominantly non-neurogenic.4 In α-synucleinopathies such as PD, however, autonomic dysfunction is more prevalent and OH is directly related to impaired adrenergic outflow. OH and other autonomic impairments may even be prodromal to PD and DLB, and more recently have been considered important biomarkers in the disease process.49 In a prospective study of the determinants of dementia in PD, 80 patients with PD were followed over a mean of 4.4 years with 27 (34%) developing dementia.50 For every 10 mmHg drop in SBP after standing, the odds of incident dementia increased 84% in models adjusted for age, sex, disease duration, and supine SBP. Using a less stringent definition of OH (ie, a drop in SBP>10 mmHg), the odds of developing dementia were increased 7-fold.50 When only considering the presence of orthostatic symptoms, the odds of incident dementia were 3-fold higher than those who did not report symptoms. This association of OH and incident dementia was later validated in two subsequent PD cohorts.51 Notably, the link between OH and dementia was stronger than between SH and dementia.50 This finding was replicated in a smaller cohort of 52 PD patients in which OH increased the odds of cognitive decline 5.5-fold over 3 years, after adjustment for age, sex education, and PD duration.52 Further, a retrospective study of PD patients without initial autonomic dysfunction reported the time to the development of global autonomic dysfunction, as well as time to develop OH in particular, was associated with an increased risk of cognitive impairment and reduced survival.53 Recently, a large international prospective study examined factors associated with the risk of parkinsonism and dementia in idiopathic rapid eye movement behavioral disorder.54 Both an SBP drop greater >10 mmHg within 3 mins of standing and orthostatic symptoms tended to be related to an elevated risk of parkinsonism or dementia, although the associations did not meet statistically significant thresholds.54 In contrast to PD and DLB, a link between cognitive impairment and multiple system atrophy has only recently gained interest55 and the mediating effect of OH has not been explored prospectively.
Impaired cardiac sympathetic innervation, which is associated with a muted cardiac response following a hypotensive stimulus,56 has also been linked to cognitive dysfunction in α-synucleinopathies. In a study of 93 de novo PD patients, postganglionic adrenergic denervation was assessed by cardiac iodine-123-meta-iodobenzylguanidine uptake.57 Low uptake at baseline was associated with a 3.5-fold increased risk of incident dementia over a mean 6.7-year follow-up period.57 This latter study provides specific evidence of a physiological pathway involved in the link between neurogenic OH and cognitive impairment.
Putative mechanisms linking OH and cognitive decline
The evidence reviewed above suggests an association between OH and incident cognitive impairment, with greater risk in α-synucleinopathies. While it remains plausible that OH is simply a biomarker that reflects underlying pathological processes leading to cognitive impairment, biological plausibility exists for mechanisms that may underlie a direct contribution of OH to dementia (Table 2).
 |
Table 2 Factors contributing to cognitive impairment related to OH |
Cerebral blood flow
Three observations suggest that repeated bouts of transient cerebral hypoperfusion may contribute to an increased risk of dementia with OH. First, OH has a stronger link with incident dementia than any other autonomic variable. OH but not bowel, urinary, or sexual dysfunction, as assessed by the Unified Multiple Systems Atrophy Scale, was linked to an increased risk of dementia in PD.50 This suggests that the ability to maintain cerebral perfusion pressure, specifically, rather than autonomic function, in general, has a key role in modulating dementia risk. Second, simply the presence of OH symptoms is sufficient to increase risk. The GÅS-SNAC study reported that OH symptoms during the preceding 12 months but not empirically measured OH were associated with an increased incidence of MCI over the course of a 6-year follow-up.24 In theory, symptoms reflect transient cerebral hypoperfusion that may occur even during mild BP drops that are below OH thresholds. This is supported by the observation that SBP variability, even in individuals who do not meet OH criteria, is associated with incident dementia.41 Further, the likelihood of reporting theoretically increases with symptom frequency such that individuals with repeated bouts over the preceding 12 months would be more likely to report. It is important to note, however, that the GÅS-SNAC study reported a link between symptoms and incident MCI but not dementia. The lack of an association of symptoms with dementia may be a consequence of either inaccurate symptom reporting in individuals with early cognitive dysfunction or longer-term exposure to OH in which case symptoms may be less likely to manifest. The third observation which suggests that cerebral hypoperfusion mediates the relationship between OH and cognition is that the level of risk of cognitive decline has been linked to the severity of OH.41–43,50 Intact cerebral autoregulatory mechanisms would help to mitigate the effects of mild-to-moderate OH by maintaining CBF relatively constant despite small orthostatic decreases in BP. More severe OH, however, could drop BP below autoregulatory thresholds, leading to cerebral hypoperfusion, which may contribute to the cognitive dysfunction.
The literature is mixed with regards to whether autoregulatory function is impaired in autonomic dysfunction.58–60 In a transcranial Doppler ultrasound study of non-PD adults with OH, 3 distinct responses of CBF velocity were observed during head-up tilt.59 Three-quarters of participants had either a flat response or an increase in velocity following a drop in BP, suggesting a maintained or expanded autoregulatory range. In the remaining cohort, velocity dropped in concert with BP reflecting impaired autoregulation.59 Although this was a small study of only 21 participants, the 3:1 ratio in favor of maintained or increased CBF velocity during upright posture is roughly in line with that of asymptomatic-to-symptomatic OH.28 The terms compensated and uncompensated OH have been introduced to reflect a preservation or a drop in CBF, respectively.61 The degree to which cerebral autoregulation is altered in neurodegenerative disease is still uncertain. In PD patients with OH, cerebral autoregulatory indices were similar to controls using transfer function analysis of data from supine rest.58 Briefly, this frequency-based method assesses cerebral autoregulation by measuring the latency of CBF velocity signal change following spontaneous low-frequency fluctuations in BP without requiring posture change or other external BP perturbations. Despite this maintained autoregulation, symptoms were directly related to the magnitude of the drop in CBF velocity during head-up tilt when the drop in BP passed a specific threshold. This threshold, which was ~80% of the supine mean BP, may represent when BP dropped below the autoregulatory range for CBF maintenance.58 Similarly, a single photon emission computed tomography study of CBF during supine and upright postures in a small group of non-PD OH patients and controls reported reduced frontal lobe CBF in the OH group when upright, in which 5 of 6 participants had an SBP drop of over 20%.62
In addition to periods of transient cerebral hypoperfusion that occur following postural transitions, multiple reports have shown that OH is associated with altered supine CBF regulation. A perfusion magnetic resonance imaging study of 9 symptomatic OH patients and controls reported that the OH was associated with elevated cerebral blood volume in white matter and elevated mean transit time in both white and grey matter.63 The authors also reported a trend for lower gray and white matter CBF in OH compared to controls and, paradoxically, a direct correlation between CBF and the extent of SBP drop during upright tilt (ie, a larger drop in SBP was associated with higher supine CBF).63 They posit that these findings are an indication of a more dilated cerebral vasculature. Cerebrovascular dilatation is in accordance with a left shift in cerebral autoregulation to protect against hypoperfusion during periodic hypotension;59 however, it simultaneously exposes the brain to elevated perfusion pressure during SH which is likely to be greater in those with more severe OH. While the study by Van Osch and colleagues purposefully excluded α-synucleinopathies, we have recently shown in a mixed group of patients with Lewy body disorders that the orthostatic drop in SBP and supine SBP were associated with distinct regional CBF patterns – namely, a greater orthostatic drop in SBP was associated with lower occipito-parietal CBF while higher supine SBP was associated with elevated CBF in the frontal cortices.37 CBF in the same occipito-parietal regions that were associated with orthostatic SBP was directly related to performance on neuropsychological tests of visuospatial function and attention. Twelve of the 15 participants in this mixed sample had already been diagnosed with dementia; however, so we cannot conclude that these distinct CBF patterns contribute to the risk of incident cognitive impairment. In the general population, lower CBF is associated with accelerated cognitive decline and a higher adjusted-risk of dementia over a mean 7-year follow-up period (31% increased risk per standard deviation drop in CBF),64 and occipito-parietal hypoperfusion, specifically, is relevant given that posterior cortical dysfunction (ie, visuospatial dysfunction) was associated with dementia risk in a prospective study of PD.65
Supine hypertension
In addition to the detrimental effects of repeated transient hypotensive episodes, it is common for those with OH to have SH (ie, ≥140/90 mmHg measured after at least 5 mins of supine rest).66 The TILDA cohort found that the prevalence of OH was 50% higher in hypertensive compared to normotensive adults.1 Importantly, SH is observed in up to 50% of the Parkinson’s patients with OH.67–69 There is ample evidence that high BP is a risk factor for future dementia,70 thus some cognitive changes in adults with OH may be attributed in part to the impact of chronic hypertension. Cross-sectional analysis from the TILDA cohort found that individuals with combined OH and SH had poorer performance on global and executive functioning cognitive tests, while OH without SH was not associated with cognitive performance.1 In contrast, a cohort study of Chinese older adults found that OH in hypertensives was associated with a reduced odds of cognitive impairment compared to OH in normotensives,31 although cognitive assessment in this study was limited to the MMSE and the study combined both uncontrolled and controlled hypertensive patients in the analysis. The duration and treatment efficacy of SH may confound the effect of OH on the incidence of cognitive impairment. While there is no prospective evidence linking the coexistence of OH and SH to dementia in α-synucleinopathies, PD patients with OH and SH (n=9) performed similarly to those with OH alone (n=14) on a battery of neuropsychological tasks71 and no difference in the prevalence of SH was observed in a cohort of PD with and without dementia.67 Managing OH in the context of coexisting SH, and vice versa, is a delicate balance68,72 and further prospective study of the coexistence of OH and SH in relation to dementia risk is needed.
White matter hyperintensities
The impact of OH and SH on the brain is particularly evident in examining the development of white matter hyperintensities (WMH).73,74 In PD specifically, two larger studies have reported that WMH burden is associated with the presence of OH,75,76 although this finding has not been universal.71 WMH are a known independent risk factor for cognitive impairment in older adults77 and in PD in particular.78–80 In de novo PD, the severity of WMH is independently associated with cognitive decline over a mean 4-year follow-up.81 Further, in PD patients with MCI, WMH were associated within an increased likelihood of conversion to PD dementia over 2 years.82 In considering both SH and WMH in the context of OH and the potential for periodic hypoperfusion, it is relevant to note that the link between cerebral hypoperfusion and dementia risk in the Rotterdam study was most profound in individuals with higher mean BP and more severe WMH burden.64
Parallel effects of neurodegeneration/protein aggregation
While there is little doubt that the independent effects of OH, along with SH, on CBF and the progression of WMH influence cognitive decline in the general population, diffuse pathological changes in the brain associated with neurodegeneration may have a complementary or synergistic role. The Health ABC study provides unique insight into the relationship between OH and cognitive impairment by way of examining OH and structural neuroimaging over the course of 15 years.83 OH was associated with a reduction in gray matter volume in multiple right-lateralized brain regions, including the dorsolateral prefrontal cortex, lingual gyrus, middle cingulate cortex, hippocampus, and parahippocampal gyri. Although no mediation analysis was performed, the relationship between OH and incident dementia remained significant after correcting for the presence of small vessel disease and gray matter atrophy which suggests that structural brain changes are not a direct interceding factor in the relationship between OH and cognition.83 Indeed, the possibility exists that central neuronal dysfunction independent of structural changes is an underlying factor contributing to both OH and cognitive dysfunction. The Rotterdam study reported an association between reduced heart rate response to postural change and dementia,41 which suggests that – even in a population-based study – the link between OH and dementia is, at least partly, neurogenic.84
Within the α-synuclein literature, one theory suggests the coexistence of OH and diffuse protein aggregation may contribute to more advanced disease states in the spectrum of Lewy body disorders.18 Within PD and DLB, progressive cortical and subcortical neurodegeneration associated with the presence of Lewy bodies results in not only dopaminergic impairments but also noradrenergic and cholinergic neurotransmitter dysfunction.85 In particular, degenerative changes in the brainstem and hypothalamus, as well as the anterior cingulate and insular cortices, impact how the central nervous system regulates the noradrenergic response to a hypotensive stimulus.86 Parallel dysfunction in the adrenergic and cholinergic innervation of pial and intracranial arteries and arterioles87 may contribute to regional cerebrovascular autoregulatory dysfunction and accentuate the impact of OH on cognition. As α-synuclein aggregates infiltrate wider cortical areas, distinct neuronal circuits are interrupted and, therefore, OH and cognitive dysfunction might present in parallel.88 Recent evidence in PD, however, has shown that the onset of autonomic dysfunction is unrelated to α-synuclein pathological burden.53 Indeed, the association between protein aggregates and clinical deterioration, as well as between protein aggregation and neurodegeneration, has recently come under question.89 A direct comparison of incident dementia in OH cohorts with and without α-synucleinopathies is necessary to disentangle the contributions of OH and parallel cortical dysfunction to dementia in diseases such as PD.
Challenges of coincident OH and cognitive impairment
Cognitive fluctuations
Cognitive fluctuations describe spontaneous changes in arousal, attention, and cognition. They are prevalent in Alzheimer’s disease, as well as DLB and PD with dementia,90,91 and have been shown to significantly impair activities of daily living and reduce quality of life in patients with dementia.92 Evidence that OH contributes significantly to posture-induced cognitive impairment, or the exacerbation of existing cognitive deficits, increases risk for patients during activities of daily living. In a cross-sectional within-group study of 37 non-demented individuals with mild-to-moderate idiopathic PD, cognitive deficits were more profound and broad while in an upright-tilted position compared to when supine in those with neurogenic OH vs those without OH.93 In particular, deficits in sustained attention, response inhibition, semantic fluency, and verbal memory (encoding and retention), which were apparent in the supine position in both PD-only and PD-OH groups, were exaggerated only in the PD-OH group when upright. Additional deficits in phonemic fluency, psychomotor speed, auditory working memory, and visuospatial function became apparent in the upright position.93 As further evidence, Peralta and colleagues reported that patients with PD dementia exhibited a larger SBP drop upon head-up tilt and a larger performance decrement in sustained attention tasks than PD patients without dementia.94 Importantly, treatment of OH has been shown to ameliorate cognitive fluctuations in geriatric patients with dementia and OH.95 A well-documented case of a patient with PD dementia just before, during, and after an episode of cognitive fluctuation demonstrated, without positional changes, normotension, hypotension, and partial restoration, respectively, suggesting a tight link between BP and cognitive fluctuations.96
Challenges related to treatment
Recognition of links between OH and dementia is important given that many pharmacological agents used to treat neurobehavioral, psychiatric, and motor symptoms have BP-lowering properties (eg, antipsychotics, cholinesterase inhibitors, anxiolytics, sedatives, and dopamine agonists).97 Furthermore, patients with cognitive impairment are often elderly, have multiple medical conditions, and are at risk of using potentially inappropriate medications.98 Medications that treat common medical problems like alpha-blockers for urinary dysfunction and antihypertensives for hypertension can worsen OH and should be reconsidered in the setting of OH or cognitive impairment. OH and cognitive impairment represent a dual threat of non-motor symptoms in α-synucleinopathies, so these conditions require special examination as treatment of the motor symptoms in PD may worsen non-motor symptoms. The iatrogenic effects of dopaminergic treatments include worsening of OH and cognitive impairment99 though these side effects are mediated through separate mechanisms. Recently, the treatment of dyskinesia and motor disturbances in patients with mild-to-moderate PD (Hoehn and Yahr median score: 2.5) using either deep brain stimulation of the subthalamic nucleus, intrajejunal levodopa infusion, or apomorphine infusion was shown to improve scores in the mood/cognition domain of the Non-Motor Symptom assessment scale for Parkinson’s Disease.100 The effect of deep brain stimulation on cognition may, in part, be attributed to a reduced levodopa equivalent daily dose at follow-up.
Consensus guidelines exist for treatment of OH in which the objectives are to minimize symptom burden, to improve quality of life, and to reduce morbidity and mortality.25 In patients with dementia, treatment of OH can reduce mental fluctuations, falls, lethargy, fatigue, and dizziness (see section “Cognitive fluctuations“),95 thereby improving quality of life. Given that all pressor therapies increase BP in both orthostatic and supine positions, the risk of iatrogenic SH and its potential contributions to cerebral small vessel disease must be considered. No current studies adequately assess the long-term complications of SH in those with comorbid OH, and current practice aims to balance the debilitating symptoms of OH against the potential negative consequences of elevated SH. As a result, correction of aggravating factors, including removal or titration of competing medications and investigation for anemia, and non-pharmacological treatment options, such as diet, salt- and fluid-loading, compression garments, and behavioral modifications to mitigate the risk of fainting or falling, take precedence over pharmacological treatment.25 A recent evidence-based panel commissioned by the International Parkinson and Movement Disorders Society concluded that, although insufficient evidence exists to rate any particular treatment for combatting OH in PD, sympathomimetic agents and prodrugs, including midodrine, fludrocortisone, and droxidopa are potentially useful, at least in the short term, given special monitoring for SH and pre-existing cardiac disorders.101 Domperidone may also be beneficial but evidence is weak.102 Combined approaches may be useful in individual patients.
Mortality
In addition to the impact exerted by the coexistence of OH and dementia on quality of life, these patients have a poorer long-term outlook. A 2014 meta-analysis of mortality risk for community-based OH reported a significant all-cause mortality adjusted-risk ratio of 1.4.103 In a more selected sample – the majority of whom had comorbid α-synucleinopathies, including PD or DLB, a median 53-month follow-up resulted in a 42% increase in mortality rate.15 Further, in a 10-year longitudinal study of DOH (n=48) and OH (n=42), mortality rates were 29% and 64%, respectively, and the co-existence of orthostatic abnormalities and α-synucleinopathies increased mortality further.23 Finally, a prospective study of PDD and DLB patients reported that the presence of OH was associated with a shorter survival time after adjusting for age, disease duration, and vascular risk factors.104
Conclusion
Herein, we have reviewed the evidence linking OH to incident dementia. Overall, evidence from large population-based cohorts suggests that OH increases the risk of dementia by up to ~50% in the general population, while smaller cohort studies of non-demented PD patients suggest that the risk of dementia is increased between 3- and 7-fold depending on how OH is characterized. In both the general population, and specifically in patients with α-synucleinopathies, an increased risk has been identified both with binary classification of OH and beat-by-beat measures of BP suggesting that there is a scaled association between BP regulation and cognitive decline, with a particular impact of SBP. Given the different levels of risk between the general population and Lewy body disorder cohorts, it is possible that incident cognitive impairment in α-synucleinopathies results from a synergistic interaction of cerebrovascular impairment and neuronal pathology. While some discrepancy between studies has been observed, this may be partially due to the method of assessment for OH and the ability to detect temporal variants of OH. The role of OH in the development and progression of cognitive impairment has distinct implications to quality of life. Clarification of the pathophysiological underpinnings of dementia associated with OH may be teased out through longitudinal, multimodal imaging studies using more precise identification of the cardiovascular and cerebrovascular responses to orthostatic challenges using beat-by-beat methods to characterize the temporal variants of OH. Ultimately, causality between OH and dementia may turn out to be bidirectional, especially in α-synucleinopathies. Early diagnosis and treatment of OH, however, may improve long-term prognosis.
Abbreviations
ARIC, Atherosclerosis Risk in Communities; BP, blood pressure; CBF, cerebral blood flow; DBP, diastolic blood pressure; DLB, dementia with Lewy Bodies; DOH, delayed onset orthostatic hypotension; GÅS-SNAC, Good Aging in Skåne study within the Swedish National Study on Aging and Care; IOH, initial orthostatic hypotension; MCI, mild cognitive impairment; MMSE, mini-mental state exam; OH, orthostatic hypotension; OH-30, delayed recovery orthostatic hypotension; PD, Parkinsons disease; SBP, systolic blood pressure; SH, supine hypertension; TILDA, the Irish Longitudinal Study on Aging; WMH, white matter hyperintensities.
Author contributions
ADR drafted the original article. All authors made substantial contributions to the conception of data; took part in critically revising the article; gave final approval of the version to be published; and agree to be accountable for all aspects of the work.
Disclosure
Alberto J Espay has received grant support from the NIH, Great Lakes Neurotechnologies and the Michael J Fox Foundation; personal compensation as a consultant/scientific advisory board member for AbbVie, Adamas, Acadia, Acorda, Neuroderm, Impax, Sunovion, Lundbeck, Osmotica Pharmaceutical, and USWorldMeds; publishing royalties from Lippincott Williams & Wilkins, Cambridge University Press, and Springer; and honoraria from USWorldMeds, Lundbeck, Acadia, Sunovion, the American Academy of Neurology, and the Movement Disorders Society. Aristide Merola has received grant support from Lundbeck, AbbVie, and Abbott; personal fees from Abbott, AbbVie, Theravance, and Medtronic; speaker honoraria from AbbVie, Lundbeck, Abbott, CSL Behring, and Theravance; and is supported by NIH (KL2 TR001426). Richard Camicioli has received grant support from the Canadian Institutes for Health Research CIHR (Canadian Consortium on Neurodegeneration in Aging), the Michael J Fox Foundation, and grant funding via subcontracts to the University of Calgary from Brain Canada/Parkinson Canada as a co-Investigator (Canadian Open Parkinson Network and Functional Assessment of Vascular Reactivity); he is on a gait advisory board for the Michael J Fox Foundation; and he is on editorial boards for Parkinsonism and Related Disorders, Frontiers in Neuroscience of Aging and Dementia and Geriatric Cognitive Disorders. Anthony E Lang has received support through employment at University Health Network, University of Toronto; grants from Brain Canada, Canadian Institutes of Health Research, Corticobasal Degeneration Solutions, Edmond J Safra Philanthropic Foundation, Michael J. Fox Foundation, the Ontario Brain Institute, National Parkinson Foundation, Parkinson Society Canada, and W. Garfield Weston Foundation; consultancies for AbbVie, Acorda, Biogen, Intracellular, Lundbeck, Sun Pharma, Kallyope, Retrophin, Paladin, Seelos, Theravance, Roche, and Corticobasal Degeneration Solutions; advisory board memberships with Jazz Pharma, PhotoPharmics, and Sunovion; honoraria from Sun Pharma, AbbVie and Sunovion; royalties from Elsevier, Saunders, Wiley-Blackwell, Johns Hopkins Press, and Cambridge University Press; personal fees from AbbVie, Accorda, Bristol-Myers Squibb, Biogen, Merck, Sun Pharma/SPARC, Corticoasal Solutions, Paladin, Medichem, Medtronic, Theravance, Jazz Pharma, Retrophin, Seelos, Syneos, Roche, Ono pharma, Intracellular, Jansen, Kallyope, Lundbeck, Lilly. Mario Masellis is supported by the Department of Medicine (Sunnybrook Health Sciences Centre and the University of Toronto), the Sunnybrook Foundation, the Hurvitz Brain Sciences Research Program and the Sunnybrook Research Institute; receives support as co-lead of the Ontario Neurodegenerative Disease Research Initiative funded by the Ontario Brain Institute; he has served as an advisor to Ionis Pharmaceuticals, Alector, Arkuda Therapeutics, and UCB; is an Associate Editor/Board Member for Current Pharmacogenomics and Personalized Medicine; received an investigator-initiated research grant from Teva; royalties from Henry Stewart Talks; personal fees from Arkuda Therapeutics, Ionis Pharmaceuticals, Alector, and Henry Stewart Talks; contract research support from Roche, Novartis, Eli Lilly and Axovant; reports grants from Parkinson Canada, Ontario Brain Institute/Ministry of Research Innovation and Science of Ontario, Ontario Ministry of Research, Innovation and Science, Canadian Institutes of Health Research, Alzheimer’s Drug Discovery Foundation (ADDF), Brain Canada, Weston Brain Institute, Roche, Washington University, Axovant, and Novartis. The authors report no other conflicts of interest in this work.
References
1. Frewen J, Finucane C, Savva GM, Boyle G, Kenny RA. Orthostatic hypotension is associated with lower cognitive performance in adults aged 50 plus with supine hypertension. J Gerontol Ser A Biol Sci Med Sci. 2014;69(7):878–885. doi:10.1093/gerona/glt171
2. Tilvis RS, Hakala SM, Valvanne J, Erkinjuntti T. Postural hypotension and dizziness in a general aged population: a four-year follow-up of the Helsinki Aging Study. J Am Geriatr Soc. 1996;44(7):809–814.
3. Vloet LC, Pel-Little RE, Jansen PA, Jansen RW. High prevalence of postprandial and orthostatic hypotension among geriatric patients admitted to Dutch hospitals. J Gerontol Ser A Biol Sci Med Sci. 2005;60:1271–1277. doi:10.1093/gerona/60.10.1271
4. Rutan GH, Hermanson B, Bild DE, Kittner SJ, LaBaw F, Tell GS. Orthostatic hypotension in older adults. The Cardiovascular Health Study. CHS Collaborative Research Group. Hypertens. 1992;19(6 Pt 1):508–519. doi:10.1161/01.HYP.19.6.508
5. Goldstein DS, Holmes CS, Dendi R, Bruce SR, Li S-T. Orthostatic hypotension from sympathetic denervation in Parkinson’s disease. Neurology. 2002;58(8):1247–1255. doi:10.1212/wnl.58.8.1247
6. Tipre DN, Goldstein DS. Cardiac and extracardiac sympathetic denervation in Parkinson’s disease with orthostatic hypotension and in pure autonomic failure. J Nucl Med. 2005;46(11):1775–1781.
7. Velseboer DC, de Haan RJ, Wieling W, Goldstein DS, de Bie RMA. Prevalence of orthostatic hypotension in Parkinson’s disease: a systematic review and meta-analysis. Parkinsonism Relat Disord. 2011;17(10):724–729. doi:10.1016/j.parkreldis.2011.04.016
8. Goldstein DS, Holmes C, Bentho O, et al. Biomarkers to detect central dopamine deficiency and distinguish Parkinson disease from multiple system atrophy. Parkinsonism Relat Disord. 2008;14(8):600–607. doi:10.1016/j.parkreldis.2008.01.010
9. Andersson M, Hansson O, Minthon L, Ballard CG, Londos E. The period of hypotension following orthostatic challenge is prolonged in dementia with Lewy bodies. Int J Geriatr Psychiatry. 2008;23(2):192–198. doi:10.1002/gps.1861
10. Antonini A, Barone P, Marconi R, et al. The progression of non-motor symptoms in Parkinson’s disease and their contribution to motor disability and quality of life. J Neurol. 2012;259:2621–2631. doi:10.1007/s00415-012-6557-8
11. Merola A, Romagnolo A, Rosso M, et al. Autonomic dysfunction in Parkinson’s disease: a prospective cohort study. Mov Disord. 2018;33(3):391–397. doi:10.1002/mds.27268
12. Juraschek SP, Daya N, Appel LJ, et al. Orthostatic hypotension and risk of clinical and subclinical cardiovascular disease in middle‐aged adults. J Am Heart Assoc. 2018;7(10):e008884. doi:10.1161/JAHA.118.008528
13. Milazzo V, Di Stefano C, Milan A, et al. Cardiovascular complications in patients with autonomic failure. Clin Auton Res. 2015;25(3):133–140. doi:10.1007/s10286-015-0275-0
14. Angelousi A, Girerd N, Benetos A, et al. Association between orthostatic hypotension and cardiovascular risk, cerebrovascular risk, cognitive decline and falls as well as overall mortality. J Hypertens. 2014;32(8):1562–1571. doi:10.1097/HJH.0000000000000235
15. Maule S, Milazzo V, Maule MM, Di Stefano C, Milan A, Veglio F. Mortality and prognosis in patients with neurogenic orthostatic hypotension. Funct Neurol. 2012;27(2):101–106.
16. Iseli R, Nguyen VTV, min S, Reijnierse EM, Lim WK, Maier AB. Orthostatic hypotension and cognition in older adults: a systematic review and meta-analysis. Exp Gerontol. 2019;120:40–49. doi:10.1016/j.exger.2019.02.017
17. Bocti C, Pépin F, Tétreault M, et al. Orthostatic hypotension associated with executive dysfunction in mild cognitive impairment. J Neurol Sci. 2017;382:79–83. doi:10.1016/j.jns.2017.09.028
18. Udow SJ, Robertson AD, MacIntosh BJ, et al. ‘Under pressure’: is there a link between orthostatic hypotension and cognitive impairment in α-synucleinopathies? J Neurol Neurosurg Psychiatry. 2016;87(12):1311–1321. doi:10.1136/jnnp-2016-314123
19. McDonald C, Newton JL, Burn DJ. Orthostatic hypotension and cognitive impairment in Parkinson’s disease: causation or association? Mov Disord. 2016;31(7):937–946. doi:10.1002/mds.26632
20. Sambati L, Calandra-Buonaura G, Poda R, Guaraldi P, Cortelli P. Orthostatic hypotension and cognitive impairment: a dangerous association? Neurol Sci. 2014;35(6):951–957. doi:10.1007/s10072-014-1686-8
21. van Wijnen VK, Finucane C, Harms MPM, et al. Noninvasive beat-to-beat finger arterial pressure monitoring during orthostasis: a comprehensive review of normal and abnormal responses at different ages. J Intern Med. 2017;282(6):468–483. doi:10.1111/joim.12636
22. Freeman R, Wieling W, Axelrod FB, et al. Consensus statement on the definition of orthostatic hypotension, neurally mediated syncope and the postural tachycardia syndrome. Clin Auton Res. 2011;21(2):69–72. doi:10.1007/s10286-011-0119-5
23. Gibbons CH, Freeman R. Clinical implications of delayed orthostatic hypotension. Neurology. 2015;85(16):1362–1367. doi:10.1212/WNL.0000000000002030
24. Elmståhl S, Widerström E. Orthostatic intolerance predicts mild cognitive impairment: incidence of mild cognitive impairment and dementia from the Swedish general population cohort Good Aging in Skåne. Clin Interv Aging. 2014;9:1993. doi:10.2147/CIA.S72316
25. Gibbons CH, Schmidt P, Biaggioni I, et al. The recommendations of a consensus panel for the screening, diagnosis, and treatment of neurogenic orthostatic hypotension and associated supine hypertension. J Neurol. 2017;264(8):1567–1582. doi:10.1007/s00415-016-8375-x
26. Jordan J, Shibao C, Biaggioni I. Multiple system atrophy: using clinical pharmacology to reveal pathophysiology. Clin Auton Res. 2015;25(1):53–59. doi:10.1007/s10286-015-0271-4
27. Arbogast SD, Alshekhlee A, Hussain Z, McNeeley K, Chelimsky TC. Hypotension unawareness in profound orthostatic hypotension. Am J Med. 2009;122(6):574–580. doi:10.1016/j.amjmed.2008.10.040
28. Palma JA, Gomez-Esteban JC, Norcliffe-Kaufmann L, et al. Orthostatic hypotension in Parkinson disease: how much you fall or how low you go? Mov Disord. 2015;30:639–645. doi:10.1002/mds.26079
29. Merola A, Romagnolo A, Rosso M, et al. Orthostatic hypotension in Parkinson’s disease: does it matter if asymptomatic? Parkinsonism Relat Disord. 2016;33:65–71. doi:10.1016/j.parkreldis.2016.09.013
30. Viramo P, Luukinen H, Koski K, Laippala P, Sulkava R, Kivelä SL. Orthostatic hypotension and cognitive decline in older people. J Am Geriatr Soc. 1999;47(5):600–604.
31. Yap PLK, Niti M, Yap KB, Ng TP. Orthostatic hypotension, hypotension and cognitive status: early comorbid markers of primary dementia? Dement Geriatr Cogn Disord. 2008;26(3):239–246. doi:10.1159/000160955
32. Curreri C, Giantin V, Veronese N, et al. Orthostatic changes in blood pressure and cognitive status in the elderly. Hypertension. 2016;68(2):427–435. doi:10.1161/HYPERTENSIONAHA.116.07334
33. Huang H, Zheng T, Liu F, Wu Z, Liang H, Wang S. Orthostatic hypotension predicts cognitive impairment in the elderly: findings from a cohort study. Front Neurol. 2017;8:121. doi:10.3389/fneur.2017.00121
34. Mitchell AJ. A meta-analysis of the accuracy of the mini-mental state examination in the detection of dementia and mild cognitive impairment. J Psychiatr Res. 2009;43(4):411–431. doi:10.1016/j.jpsychires.2008.04.014
35. Kaszás B, Kovács N, Balás I, et al. Sensitivity and specificity of Addenbrooke’s cognitive examination, mattis dementia rating scale, frontal assessment battery and mini mental state examination for diagnosing dementia in Parkinson’s disease. Parkinsonism Relat Disord. 2012;18(5):553–556. doi:10.1016/j.parkreldis.2012.02.010
36. Allcock LM, Kenny RA, Mosimann UP, et al. Orthostatic hypotension in Parkinson’s disease: association with cognitive decline? Int J Geriatr Psychiatry. 2006;21(8):778–783. doi:10.1002/gps.1562
37. Robertson AD, Messner MA, Shirzadi Z, et al. Orthostatic hypotension, cerebral hypoperfusion, and visuospatial deficits in Lewy body disorders. Parkinsonism Relat Disord. 2016;22:80–86. doi:10.1016/j.parkreldis.2015.11.019
38. Feeney J, O’Leary N, Kenny RA. Impaired orthostatic blood pressure recovery and cognitive performance at two-year follow up in older adults: the Irish Longitudinal Study on Ageing. Clin Auton Res. 2016;26(2):127–133. doi:10.1007/s10286-016-0340-3
39. Rose KM, Couper D, Eigenbrodt ML, Mosley TH, Sharrett AR, Gottesman RF. Orthostatic hypotension and cognitive function: the Atherosclerosis Risk in Communities Study. Neuroepidemiology. 2010;34(1):1–7. doi:10.1159/000255459
40. Rawlings AM, Juraschek SP, Heiss G, et al. Association of orthostatic hypotension with incident dementia, stroke, and cognitive decline. Neurology. 2018;91(8):e759–e768. doi:10.1212/WNL.0000000000006027
41. Wolters FJ, Mattace-Raso FUS, Koudstaal PJ, Hofman A, Ikram MA. Orthostatic hypotension and the long-term risk of dementia: a population-based study. PLoS Med. 2016;13(10):e1002143. doi:10.1371/journal.pmed.1002143
42. Cremer A, Soumaré A, Berr C, et al. Orthostatic hypotension and risk of incident dementia. Hypertension. 2017;70(1):44–49. doi:10.1161/HYPERTENSIONAHA.117.09048
43. Holm H, Nägga K, Nilsson ED, et al. Longitudinal and postural changes of blood pressure predict dementia: the Malmö Preventive Project. Eur J Epidemiol. 2017;32(4):327–336. doi:10.1007/s10654-017-0228-0
44. Peters R, Anstey KJ, Booth A, et al. Orthostatic hypotension and symptomatic subclinical orthostatic hypotension increase risk of cognitive impairment: an integrated evidence review and analysis of a large older adult hypertensive cohort. Eur Heart J. 2018;39(33):3135–3143. doi:10.1093/eurheartj/ehy418
45. Shaw BH, Garland EM, Black BK, et al. Optimal diagnostic thresholds for diagnosis of orthostatic hypotension with a ‘sit-to-stand test.’. J Hypertens. 2017;35(5):1019–1025. doi:10.1097/HJH.0000000000001265
46. Hayakawa T, McGarrigle CA, Coen RF, et al. Orthostatic blood pressure behavior in people with mild cognitive impairment predicts conversion to dementia. J Am Geriatr Soc. 2015;63(9):1868–1873. doi:10.1111/jgs.13596
47. Allan LM, Ballard CG, Allen J, et al. Autonomic dysfunction in dementia. J Neurol Neurosurg Psychiatry. 2006;78(7):671–677. doi:10.1136/jnnp.2006.102343
48. Brenowitz WD, Hubbard RA, Keene CD, et al. Mixed neuropathologies and estimated rates of clinical progression in a large autopsy sample. Alzheimer’s Dement. 2017;13(6):654–662. doi:10.1016/j.jalz.2016.09.015
49. Postuma RB, Gagnon JF, Pelletier A, Montplaisir J. Prodromal autonomic symptoms and signs in Parkinson’s disease and dementia with Lewy bodies. Mov Disord. 2013;28:597–604. doi:10.1002/mds.25445
50. Anang JB, Gagnon JF, Bertrand JA, et al. Predictors of dementia in Parkinson disease: a prospective cohort study. Neurology. 2014;83:1253–1260. doi:10.1212/WNL.0000000000000842
51. Anang JBM, Nomura T, Romenets SR, Nakashima K, Gagnon J-F, Postuma RB. Dementia predictors in Parkinson disease: a validation study. J Parkinsons Dis. 2017;7(1):159–162. doi:10.3233/JPD-160925
52. Hussain MW, Camicioli R. Nonmotor symptoms of Parkinson’s disease as predictors of dementia. Can J Neurol Sci. 2018;45(1):97–99. doi:10.1017/cjn.2017.239
53. De Pablo-Fernandez E, Tur C, Revesz T, Lees AJ, Holton JL, Warner TT. Association of autonomic dysfunction with disease progression and survival in Parkinson disease. JAMA Neurol. 2017;74(8):970. doi:10.1001/jamaneurol.2017.1125
54. Postuma RB, Iranzo A, Hu M, et al. Risk and predictors of dementia and parkinsonism in idiopathic REM sleep behaviour disorder: a multicentre study. Brain. 2019;142(3):744–759. doi:10.1093/brain/awz030
55. Stankovic I, Krismer F, Jesic A, et al. Cognitive impairment in multiple system atrophy: A position statement by the neuropsychology task force of the MDS multiple system atrophy (MODIMSA) study group. Mov Disord. 2014;29(7):857–867. doi:10.1002/mds.25880
56. Goldstein DS, Cheshire WP. Roles of cardiac sympathetic neuroimaging in autonomic medicine. Clin Auton Res. 2018;28(4):397–410. doi:10.1007/s10286-018-0547-6
57. Choi MH, Yoon JH, Yong SW. Cardiac sympathetic denervation and dementia in de novo Parkinson’s disease: a 7-year follow-up study. J Neurol Sci. 2017;381:291–295. doi:10.1016/j.jns.2017.09.010
58. Haubrich C, Pies K, Dafotakis M, Block F, Kloetzsch C, Diehl RR. Transcranial Doppler monitoring in Parkinson’s disease: cerebrovascular compensation of orthostatic hypotension. Ultrasound Med Biol. 2010;36:1581–1587. doi:10.1016/j.ultrasmedbio.2010.06.016
59. Novak V, Novak P, Spies JM, Low PA. Autoregulation of cerebral blood flow in orthostatic hypotension. Stroke. 1998;29(1):104–111.
60. Tsai SJ, Chen SC, Leu TM, et al. Impairment of cerebral hemodynamic response to the cold pressor test in patients with Parkinson’s disease. Parkinsonism Relat Disord. 2009;15:94–100. doi:10.1016/j.parkreldis.2008.03.007
61. Novak P. Cerebral blood flow, heart rate, and blood pressure patterns during the tilt test in common orthostatic syndromes. Neurosci J. 2016;2016:6127340.
62. Hayashida K, Nishiooeda Y, Hirose Y, Ishida Y, Nishimura T. Maladaptation of vascular response in frontal area of patients with orthostatic hypotension. J Nucl Med. 1996;37:1–4.
63. van Osch MJP, Jansen PAF, Vingerhoets RW. van der Grond J. Association between supine cerebral perfusion and symptomatic orthostatic hypotension. Neuroimage. 2005;27(4):789–794. doi:10.1016/j.neuroimage.2005.05.026
64. Wolters FJ, Zonneveld HI, Hofman A, et al. Cerebral perfusion and the risk of dementia. Circulation. 2017;136(8):719–728. doi:10.1161/CIRCULATIONAHA.117.027448
65. Williams-Gray CH, Foltynie T, Brayne CEG, Robbins TW, Barker RA. Evolution of cognitive dysfunction in an incident Parkinson’s disease cohort. Brain. 2007;130(7):1787–1798. doi:10.1093/brain/awm111
66. Fanciulli A, Jordan J, Biaggioni I, et al. Consensus statement on the definition of neurogenic supine hypertension in cardiovascular autonomic failure by the American Autonomic Society (AAS) and the European Federation of Autonomic Societies (EFAS). Clin Auton Res. 2018;28(4):355–362. doi:10.1007/s10286-018-0529-8
67. Fanciulli A, Göbel G, Ndayisaba JP, et al. Supine hypertension in Parkinson’s disease and multiple system atrophy. Clin Auton Res. 2016;26(2):97–105. doi:10.1007/s10286-015-0336-4
68. Espay AJ, LeWitt PA, Hauser RA, Merola A, Masellis M, Lang AE. Neurogenic orthostatic hypotension and supine hypertension in Parkinson’s disease and related synucleinopathies: prioritisation of treatment targets. Lancet Neurol. 2016;15(9):954–966. doi:10.1016/S1474-4422(16)30079-5
69. Goldstein DS, Pechnik S, Holmes C, Eldadah B, Sharabi Y. Association between supine hypertension and orthostatic hypotension in autonomic failure. Hypertension. 2003;42(2):136–142. doi:10.1161/01.HYP.0000081216.11623.C3
70. Skoog I, Lernfelt B, Landahl S, et al. 15-year longitudinal study of blood pressure and dementia. Lancet. 1996;347(9009):1141–1145. doi:10.1016/s0140-6736(96)90608-x
71. Pilleri M, Facchini S, Gasparoli E, et al. Cognitive and MRI correlates of orthostatic hypotension in Parkinson’s disease. J Neurol. 2013;260(1):253–259. doi:10.1007/s00415-012-6627-y
72. Jordan J, Fanciulli A, Tank J, et al. Management of supine hypertension in patients with neurogenic orthostatic hypotension. J Hypertens. 2019;37(8):1541–1546. doi:10.1097/HJH.0000000000002078
73. Ballard C, O’Brien J, Barber B, et al. Neurocardiovascular instability, hypotensive episodes, and MRI lesions in neurodegenerative dementia. Ann N Y Acad Sci. 2000;903:442–445. doi:10.1111/j.1749-6632.2000.tb06396.x
74. Colloby SJ, Vasudev A, O’Brien JT, Firbank MJ, Parry SW, Thomas AJ. Relationship of orthostatic blood pressure to white matter hyperintensities and subcortical volumes in late-life depression. Br J Psychiatry. 2011;199(5):404–410. doi:10.1192/bjp.bp.110.090423
75. Oh YS, Kim JS, Lee KS. Orthostatic and supine blood pressures are associated with white matter hyperintensities in Parkinson disease. J Mov Disord. 2013;6:23–27. doi:10.14802/jmd.13006
76. Ten Harmsen BL, van Rumund A, Aerts MB, et al. Clinical correlates of cerebral white matter abnormalities in patients with Parkinson’s disease. Parkinsonism Relat Disord. 2018;49:28–33. doi:10.1016/j.parkreldis.2017.12.029
77. Maillard P, Carmichael O, Fletcher E, Reed B, Mungas D, DeCarli C. Coevolution of white matter hyperintensities and cognition in the elderly. Neurology. 2012;79(5):442–448. doi:10.1212/WNL.0b013e3182617136
78. Lee S-J, Kim J-S, Yoo J-Y, et al. Influence of white matter hyperintensities on the cognition of patients with Parkinson disease. Alzheimer Dis Assoc Disord. 2010;24(3):227–233. doi:10.1097/WAD.0b013e3181d71a13
79. González-Redondo R, Toledo J, Clavero P, et al. The impact of silent vascular brain burden in cognitive impairment in Parkinson’s disease. Eur J Neurol. 2012;19(8):1100–1107. doi:10.1111/j.1468-1331.2012.03682.x
80. Kim J-S, Oh Y-S, Lee K-S, Kim Y-I, Yang D-W, Goldstein DS. Association of cognitive dysfunction with neurocirculatory abnormalities in early Parkinson disease. Neurology. 2012;79(13):1323–1331. doi:10.1212/WNL.0b013e31826c1acd
81. Dadar M, Zeighami Y, Yau Y, et al. White matter hyperintensities are linked to future cognitive decline in de novo Parkinson’s disease patients. NeuroImage Clin. 2018;20:892–900. doi:10.1016/j.nicl.2018.09.025
82. Sunwoo MK, Jeon S, Ham JH, et al. The burden of white matter hyperintensities is a predictor of progressive mild cognitive impairment in patients with Parkinson’s disease. Eur J Neurol. 2014;21(6):922–e50. doi:10.1111/ene.12412
83. O’Hare C, Kenny R-A, Aizenstein H, et al. Cognitive status, gray matter atrophy, and lower orthostatic blood pressure in older adults. J Alzheimer’s Dis. 2017;57(4):1239–1250. doi:10.3233/JAD-161228
84. Norcliffe-Kaufmann L, Kaufmann H, Palma J-A, et al. Orthostatic heart rate changes in patients with autonomic failure caused by neurodegenerative synucleinopathies. Ann Neurol. 2018;83(3):522–531. doi:10.1002/ana.25170
85. Kehagia AA, Barker RA, Robbins TW. Neuropsychological and clinical heterogeneity of cognitive impairment and dementia in patients with Parkinson’s disease. Lancet Neurol. 2010;9(12):1200–1213. doi:10.1016/S1474-4422(10)70212-X
86. Coon EA, Cutsforth-Gregory JK, Benarroch EE. Neuropathology of autonomic dysfunction in synucleinopathies. Mov Disord. 2018;33(3):349–358. doi:10.1002/mds.27186
87. Edvinsson L. Neurogenic mechanisms in the cerebrovascular bed. Autonomic nerves, amine receptors and their effects on cerebral blood flow. Acta Physiol Scand Suppl. 1975;427:1–35.
88. Del Tredici K, Braak H. Sporadic Parkinson’s disease: development and distribution of α-synuclein pathology. Neuropathol Appl Neurobiol. 2016;42(1):33–50. doi:10.1111/nan.12298
89. Espay AJ, Vizcarra JA, Marsili L, et al. Revisiting protein aggregation as pathogenic in sporadic Parkinson and Alzheimer diseases. Neurology. 2019;92(7):329–337. doi:10.1212/WNL.0000000000006926
90. Ballard CG, Aarsland D, McKeith I, et al. Fluctuations in attention: PD dementia vs DLB with parkinsonism. Neurology. 2002;59(11):1714–1720. doi:10.1212/01.wnl.0000036908.39696.fd
91. Escandon A, Al-Hammadi N, Galvin JE. Effect of cognitive fluctuation on neuropsychological performance in aging and dementia. Neurology. 2010;74(3):210–217. doi:10.1212/WNL.0b013e3181ca017d
92. Sun M, Mainland BJ, Ornstein TJ, et al. The association between cognitive fluctuations and activities of daily living and quality of life among institutionalized patients with dementia. Int J Geriatr Psychiatry. 2018;33(2):e280–e285. doi:10.1002/gps.4788
93. Centi J, Freeman R, Gibbons CH, Neargarder S, Canova AO, Cronin-Golomb A. Effects of orthostatic hypotension on cognition in Parkinson disease. Neurology. 2017;88(1):17–24. doi:10.1212/WNL.0000000000003452
94. Peralta C, Stampfer-Kountchev M, Karner E, et al. Orthostatic hypotension and attention in Parkinson’s disease with and without dementia. J Neural Transm. 2007;114(5):585–588. doi:10.1007/s00702-006-0615-2
95. Freidenberg DL, Shaffer LET, Macalester S, Fannin EA. Orthostatic hypotension in patients with dementia. Cogn Behav Neurol. 2013;26(3):105–120. doi:10.1097/WNN.0000000000000003
96. Riley DE, Espay AJ. Cognitive fluctuations in Parkinson’s disease dementia: blood pressure lability as an underlying mechanism. J Clin Mov Disord. 2018;5(1):1. doi:10.1186/s40734-018-0068-4
97. Press Y, Punchik B, Freud T. Orthostatic hypotension and drug therapy in patients at an outpatient comprehensive geriatric assessment unit. J Hypertens. 2016;34(2):351–358. doi:10.1097/HJH.0000000000000781
98. Redston MR, Hilmer SN, McLachlan AJ, Clough AJ, Gnjidic D. Prevalence of potentially inappropriate medication use in older inpatients with and without cognitive impairment: a systematic review. J Alzheimer’s Dis. 2018;61(4):1639–1652. doi:10.3233/JAD-170842
99. Kujawa K, Leurgans S, Raman R, Blasucci L, Goetz CG. Acute orthostatic hypotension when starting dopamine agonists in Parkinson’s disease. Arch Neurol. 2000;57(10):1461–1463.
100. Dafsari HS, Martinez‐Martin P, Rizos A, et al. EuroInf 2: subthalamic stimulation, apomorphine, and levodopa infusion in Parkinson’s disease. Mov Disord. 2019;34(3):353–365. doi:10.1002/mds.27626
101. Seppi K, Ray Chaudhuri K, Coelho M, et al. Update on treatments for nonmotor symptoms of Parkinson’s disease-an evidence-based medicine review. Mov Disord. 2019;34(2):180–198. doi:10.1002/mds.27602
102. Bacchi S, Chim I, Kramer P, Postuma RB. Domperidone for hypotension in Parkinson’s disease: a systematic review. J Parkinsons Dis. 2017;7(4):603–617. doi:10.3233/JPD-171209
103. Xin W, Lin Z, Mi S. Orthostatic hypotension and mortality risk: a meta-analysis of cohort studies. Heart. 2014;100(5):406–413. doi:10.1136/heartjnl-2013-304121
104. Stubendorff K, Aarsland D, Minthon L, Londos E. The impact of autonomic dysfunction on survival in patients with dementia with Lewy bodies and Parkinson’s disease with dementia. PLoS One. 2012;7(10):e45451. doi:10.1371/journal.pone.0045451
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.