Back to Archived Journals » Orphan Drugs: Research and Reviews » Volume 4
Orphan drugs in glioblastoma multiforme: a review
Authors Lassen U, Mau-Sørensen M, Poulsen HS
Received 28 June 2014
Accepted for publication 31 July 2014
Published 14 November 2014 Volume 2014:4 Pages 83—91
DOI https://doi.org/10.2147/ODRR.S46018
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Lise Aagaard
Ulrik Lassen, Morten Mau-Sørensen, Hans Skovgaard Poulsen
Department of Oncology, Rigshospitalet, Copenhagen, Denmark
Abstract: Glioblastoma multiforme (GBM) is the most common and deadly brain tumor in adults. The incidence of GBM in the USA and Europe is 2–3 per 100,000. By definition, an orphan disease affects up to 200,000 persons in the USA (one in every 1,500). A search was made in the US Food and Drug Administration orphan drug listing. In addition, a PubMed search of orphan drugs designated for GBM or high-grade glioma was performed, followed by a search for clinical studies in GBM with orphan drugs designated for other indications. This included cytotoxic chemotherapy and targeted agents. Thirteen drugs with orphan designation for the treatment of glioblastoma, high-grade glioma, or primary malignant brain tumors were identified. In addition, 16 drugs with orphan designation for other indications were identified to have been evaluated in clinical studies of GBM. The efficacy data from the clinical studies is presented. A few agents have been approved by the US Food and Drug Administration for the treatment of high-grade gliomas following orphan drug designation, but most have failed to reach the market. However, a few patients may have benefited from receiving developmental agents within clinical trials. Biomarkers for selection of these patients may result in more success in the field of personalized medicine.
Keywords: orphan drugs, glioblastoma multiforme, brain tumor, targeted therapy, cytotoxic therapy
Introduction
Glioblastoma multiforme (GBM) is the most common and deadly malignant brain tumor in adults.1 Standard therapy for patients with good performance status (World Health Organization performance 0–2) consists of extensive surgery if surgery is possible followed by radiation therapy in combination with concomitant and adjuvant temozolomide. However, most patients experience relapse and the median survival is less than 15 months.2 A number of new tailored therapies have been tested, of which antiangiogenic therapy with bevacizumab in particular has shown promising results.3 However, the effect on survival has been very limited,3 and a demand for development of new therapeutic strategies therefore exists.
The incidence of GBM in the USA and Europe is 2–3 per 100,000. By definition, an orphan disease is one affecting <200,000 persons in the USA (one in every 1,500). In Europe, the definition is a bit narrower, with fewer than five in 10,000 (one in every 2,000) people affected.4 Accordingly, GBM is an orphan disease. Forty percent of orphan drugs are indicated to treat various cancers, and some top-selling oncology drugs indeed have orphan drug designation. These include Rituxan®, Herceptin®, and Avastin®. This paper describes the use of orphan drugs in GBM.
Materials and methods
A search was made in the US Food and Drug Administration (FDA) orphan drug list (http://www.accessdata.fda.gov/scripts/opdlisting/oopd/). This page searches the Orphan Drug Product designation database. Searches are run by entering the product name, orphan designation, or dates. The search was performed on May 6, 2014. A PubMed search of orphan drugs designated for GBM or high-grade gliomas (Table 1) was then performed. In addition, we searched PubMed for clinical studies in GBM with orphan drugs designated for other indications (Table 2). This included cytotoxic chemotherapy and targeted agents. The PubMed search used a broad search strategy to identify clinical studies with these agents, and reference lists of included studies were also reviewed. Thirteen drugs with orphan designation for the treatment of glioblastoma, high-grade glioma, or primary malignant brain tumors were identified (Table 1). In addition, 16 drugs with orphan designation for other indications were identified as having been evaluated in clinical studies of GBM (Table 2).
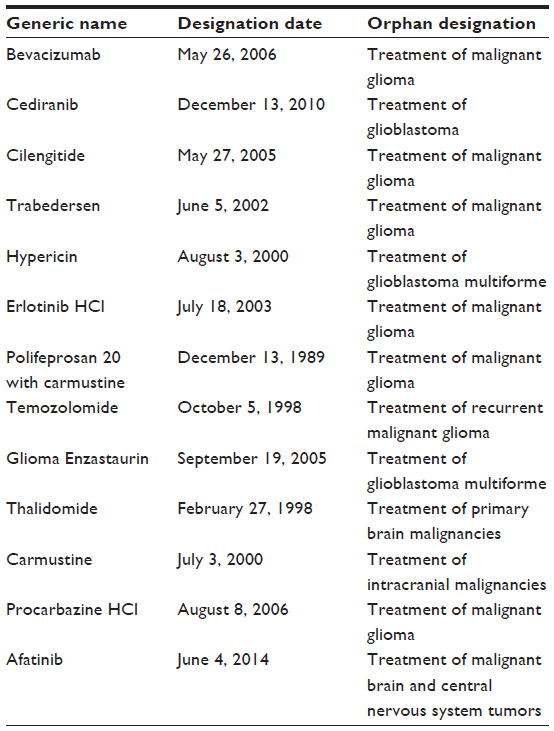 | Table 1 List of orphan drugs with designation for primary brain tumors |
 | Table 2 List of orphan drug with designation for indications other than primary brain tumors |
Orphan drugs designated for glioma, GBM, intracranial malignancies, or primary brain tumors
Antiangiogenic drugs
GBM is characterized by florid angiogenesis,5 and drugs targeting blood vessel formation have been developed.6
Bevacizumab
Bevacizumab, a humanized monoclonal antibody targeted to vascular endothelial growth factor (VEGF), received orphan drug designation for the treatment of glioma in 2006. Bevacizumab clears VEGF ligand in the extracellular space and therefore does not have to cross the blood–brain barrier to be active. Initial Phase II studies reported an impressive response rate of up to 42% and progression-free survival at 6 months (PFS6) of 46%.7,8 The led to the pivotal Phase II study, BRAIN (ClinicalTrials.gov identifier NCT00345163), that showed a benefit in patients with recurrent GBM.9 Several Phase II studies in GBM patients with recurrent disease have been performed, with prolongation of life and improved quality of life in responders.10–13 Recently, a randomized Phase II study comparing bevacizumab alone versus lomustine versus the combination of both agents was presented at the 2013 American Society of Clinical Oncology (ASCO) meeting. The study showed that the combination resulted in a significant increase in PFS6 from 18% (bevacizumab) and 11% (lomustine) to 41%.14 Confirmation by a Phase III study is awaited.
Two published, randomized Phase III studies, AVAGLIO (Avastin in Glioblastoma)15 and Radiation Therapy Oncology Group (RTOG) 0825,16 compared standard treatment versus standard treatment plus bevacizumab. Both studies included patients with newly diagnosed GBM and good performance status, and O6-methylguanine-DNA methyltransferase (MGMT)-methylated as well as non-methylated tumors. Known prognostic factors were comparable in the two studies except for surgery, in that more patients had debulking surgery in the RTOG study than in the AVAGLIO study. Both studies found a significant increase in median progression-free survival (from 6.2 months to 10.6 months [a 70% increase] in AVAGLIO and from 7.3 months to 10.7 months [a 47% increase] in RTOG 0825). However, no difference in overall survival was observed in any of the studies. A third randomized Phase III study that included non-methylated glioblastomas reported an increase in progression-free survival but not in overall survival.17
Thalidomide
Thalidomide received orphan drug designation for the treatment of primary brain malignancies in 1998. It was developed as a sedative in the late 1950s, but has antiangiogenic properties, so was evaluated in patients with vascular tumors. In a Phase II study of 39 patients with high-grade glioma, a response rate of 6% was observed, with a median time to progression of 10 weeks.18 Response rates of 5% and a median time to progression of 11 weeks have similarly been reported.19 Several studies have combined thalidomide with other agents, usually temozolomide, but these have been without evidence of additive effects.20
Inhibition of miscellaneous cell pathways
Cediranib
Cediranib is an orally available pan-VEGF receptor tyrosine kinase inhibitor with additional activity against platelet-derived growth factor-β and c-Kit. Cediranib received orphan drug designation for malignant glioma in 2010. An initial Phase II study showed a very encouraging radiographic response rate of 57% and PFS6 of 26%.21 The Phase III study, REGAL (ClinicalTrials.gov identifier NCT00777153), compared cediranib as monotherapy or in combination with lomustine versus lomustine alone in patients with recurrent GBM. However, this study failed to meet the primary endpoint of prolongation of progression-free survival with cediranib either as monotherapy or in combination with lomustine versus lomustine.22 Studies of first-line cediranib combined with radiotherapy and temozolomide are ongoing.
Cilengitide
Cilengitide is a specific inhibitor of the a vb 3 and vb 5 integrins. An orphan drug designation for malignant gliomas was recorded in 2005. According to preclinical studies, cilengitide has both antiangiogenic activity and anti-invasive activity. Modest activity at doses of 500–2,000 mg twice weekly has been reported,23 and a pharmacokinetic study has confirmed drug delivery and retention in tumor tissue in surgical specimens following three intravenous administrations. However, PFS6 was only 12% and median progression-free survival was 8 weeks, with no responses reported.24 In a joint European Organization for Research and Treatment of Cancer and National Cancer Institute of Canada (EORTC/NCIC) Phase III randomized study, cilengitide failed to prolong progression-free survival or overall survival in patients with newly diagnosed glioblastoma and the methylated MGMT gene promoter.25
Trabedersen
Trabedersen is a synthetic antisense phosphorothioate oligodeoxynucleotide complementary to the messenger RNA of the human transforming growth factor (TGF)-β2 gene, and was developed as a novel, targeted treatment for patients with high-grade glioma. The human TGF-β2 gene is overexpressed in more than 90% of high-grade gliomas.26 Orphan drug designation for malignant glioma was registered in 2002. A small hypothesis-generating randomized Phase II study comparing two doses of intratumoral trabedersen with temozolomide or PCV (procarbazine lomustine and vincristine) was negative and these combinations have not been studied since.27
Erlotinib
Primary GBM is characterized by overexpression and dysregulated activity of the epidermal growth factor receptor (EGFR), as a result of amplification and/or mutation. The most common EGFR mutation in GBM is the constitutive active deletion variant EGFRvIII, which has been correlated with increased malignant potential.28 Both EGFR and EGFRvIII are only found in GBM and not in the normal brain. Although EGFR likely plays an important role in GBM, inhibition of the receptor pathway alone has not proven as clinically effective as expected.
Several Phase II trials evaluating the benefit of EGFR inhibitors have been performed in patients with recurrent malignant glioma. Erlotinib was granted orphan designation for malignant glioma in 2003. Objective response rates range from 0% to 26%, and no apparent survival benefit has been observed. The two largest Phase II studies reported a PFS6 in recurrent GBM of only 3%29 and 20%,30 and response rates of and 0% and 6.3%, respectively.
A randomized Phase II study compared erlotinib with temozolomide or lomustine in recurrent GBM. PFS6 in the erlotinib arm was 11.4% (95% confidence interval 4.6–21.5), and 24% in the control arm. The response rate for erlotinib was only 3.7%, and the response was not correlated with EGFR expression or mutation.31 A recent Phase II study confirmed that erlotinib had little or no activity in patients with relapsed GBM expressing EGFRvIII and phosphatase and tensin homolog (PTEN).32
Afatinib
Afatinib, an irreversible blocker of the erbB family (including EGFRvIII), received orphan drug designation in April 2014 for the treatment of malignant brain tumors. Because mutated EGFRvIII is also targeted with this agent, activity may be seen in GBM. One Phase I–II study presented at the 2011 ASCO meeting compared afatinib monotherapy with afatinib in combination with temozolomide or temozolomide monotherapy in recurrent GBM. PFS6 was 3%, 17%, and 22%, respectively, and a response was seen in one, five, and six patients, respectively. It was concluded that afatinib has low single agent activity in recurrent GBM.33
Enzastaurin
Enzastaurin, a potent inhibitor of protein kinase C and the phosphatidylinositol-4,5-bisphosphate 3-kinase/v-Akt Murine Thymoma Viral Oncogene (AKT) pathway, was granted orphan designation in 2005 for the treatment of GBM. Encouraging response rates were reported in a Phase I–II study, with 21 of 84 patients achieving a radiological response (25%).34 However, a subsequent Phase III study comparing enzastaurin with lomustine was negative. The study was terminated for futility after a preplanned interim analysis showed a median progression-free survival of 1.5 months versus 1.6 months (hazard ratio 1.28) and a PFS6 of 11.1% versus 19% (P=0.03). Overall survival (6.6 months and 7.1 months, respectively; hazard ratio 1.2) did not differ significantly between the enzastaurin and lomustine arms. An objective response was observed in 2.9% and 4.3% of patients, respectively.35
Chemotherapeutic drugs
Temozolomide
Temozolomide, an orally administered imidazotetrazine derivative, received initial orphan drug designation in 1998 for the treatment of recurrent malignant glioma. The early Phase II studies in recurrent GBM defined the standard regimen as 200 mg/m2 for 5 days every 28 days, with a response rate of 8% and PFS6 of 18%.36 Temozolomide was granted FDA approval for the treatment of recurrent anaplastic astrocytoma in 1999, and later for recurrent GBM. In 2005, temozolomide was also approved for the treatment of newly diagnosed GBM concomitantly with radiotherapy and then as maintenance treatment. This was based on the EORTC/NCIC randomized Phase III study that assessed concomitant and adjuvant temozolomide with radiotherapy versus radiotherapy alone as primary therapy for histologically confirmed GBM in patients who had undergone biopsy or resection, but excluded patients over 70 years of age. Temozolomide resulted in increased survival (hazard ratio 0.60, 95% confidence interval 0.46–0.79; P=0.0003) when compared with radiation only.37 This study defined the new standard of care for GBM. Subsequently, a subgroup analysis revealed that temozolomide only improved the outcome in patients with methylation of MGMT and not in those without.38 As temozolomide is an alkylating agent, its therapeutic benefit depends on its ability to alkylate/methylate DNA. This methylation damages DNA and triggers the death of tumor cells. However, some tumor cells are able to repair this type of DNA damage by expressing a protein known as O6-alkylguanine DNA alkyltransferase (AGT), encoded in humans by the MGMT gene, which diminishes the therapeutic efficacy of temozolomide. Therefore, GBM can be divided into two groups, one harboring MGMT promoters that are methylated so the enzyme does not repair DNA, and another group harboring non-methylated MGMT promoters that repair DNA damage. Therefore, GBM patients with methylated MGMT respond better and live longer than those with non-methylated MGMT.38
In the recurrent setting, temozolomide improves progression-free survival and may have beneficial effects on quality of life, but does not improve overall survival.39 Other regimens with temozolomide have been evaluated, including two dose-dense regimens: one with temozolomide 100–150 mg/m2 for 21 of 28 days and the other with 150 mg/m2 for 7 of 14 days (7 days on/7 days off). A small Phase II study of the first regimen reported a response rate of 10% and PFS6 of 44%, and no correlation with MGMT status.40 The other dose-dense regimen, ie, the 21/28-day schedule, was evaluated in 18 patients with a response rate of 22%.41 However, in a randomized Phase II study, this regimen was found to be inferior to the standard regimen of 5 days every 28 days.42
Polifeprosan 20 with carmustine
The polifeprosan 20 with carmustine implant, also known as the Gliadel® wafer, was granted orphan designation in 1989. At that time, the primary treatment for GBM was surgery followed by radiotherapy alone. Gliadel wafer implantation at the time of surgery could simplify the management logistics when compared with systemic chemotherapy, which at that time was not the standard of care. During surgery, the wafer is implanted in order to release carmustine in the tumor bed over a period of approximately 5 days.43 Several Phase II studies have reported Gliadel to be safe in GBM.44 Initial placebo-controlled Phase III studies of Gliadel were positive in the first-line setting (hazard ratio 0.65, 95% confidence interval 0.48–0.86, P=0.003).45 However, in recurrent GBM, placebo-controlled Phase III studies have failed to demonstrate a survival benefit,46 and based on a recent Cochrane review, Gliadel is not recommended for recurrent GBM.47 Nevertheless, a recent review by Olson et al recommended Gliadel wafers in the management of progressive glioblastoma as a surgical adjunct when cytoreductive surgery is indicated, taking into account the associated toxicities seen with this modality.48
Carmustine
Carmustine, a nitrosurea, received orphan drug designation for the treatment of intracranial malignancies in 2000. Initial studies reported response rates of 15% and PFS6 of 17%,49 and a response of 6% with PFS6 of 13%.50 Carmustine has not been studied as an infusional drug in a Phase III trial, but has been extensively studied as the implantable Gliadel wafer.
Procarbazine
Procarbazine was granted orphan designation in 2006 for the treatment of malignant glioma. However, this agent had been approved by the FDA in 1969 for the treatment of Hodgkin’s lymphoma. Most studies of procarbazine in GBM have been in combination with a nitrosurea and vincristine (as the PCV regimen). Retrospective reports have shown modest response rates of 3%–11%, with PFS6 of 29%–38%.51 Only a few prospective Phase II studies have been performed in recurrent GBM. A study of 58 chemotherapy-naïve patients reported a response rate of 29%, including a 10% complete response rate and a PFS6 of 42%.52 The PCV combination is now approved by the FDA for the treatment of recurrent high-grade glioma and is often used as a control regimen in randomized Phase III studies. Still, the reported data only represent level III evidence.
Miscellaneous agents
Hypericin
Hypericin received orphan designation for the treatment of GBM in 2000. It is a natural compound found in the stems and petals of St John’s wort (Hypericum perforatum). A synthetic form is used in clinical trials, as a potent protein kinase C inhibitor.53 In a Phase II study of hypericin in recurrent high-grade glioma, only one partial response were seen in 35 patients with GBM, while a partial response was observed in one of seven patients with anaplastic astrocytoma.54 The PFS6 was not reported.
Orphan drugs designated for other indications in clinical trials for GBM
Table 3 describes the key features of the agents reported in Table 2.55–70 Of special interest are studies with imatinib and hydroxyurea, since initial Phase II studies indicated favorable activity, and this combination was therefore evaluated in a randomized Phase III trial. A total of 240 patients with recurrent GBM were randomized to treatment with hydroxyurea alone or with hydroxyurea and imatinib. No differences between the two regimens were observed, with a PFS6 of 5% and 7%, respectively, and response rates of 2% and 1%, respectively.71 In addition, various combinations of bevacizumab and some of these agents have been performed, including combinations of EGFR inhibitors, but the data have not indicated any additional effect compared with bevacizumab alone.
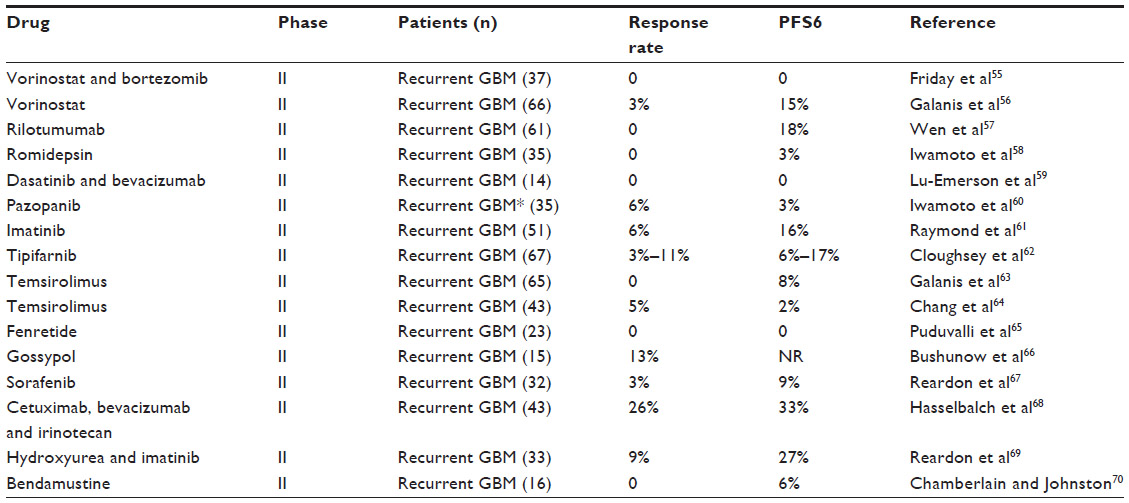 | Table 3 Efficacy data from reported clinical trials in GBM with orphan drug designated for other indications |
Discussion
Recently, it has been shown that progression-free survival is a reasonable surrogate endpoint for the activity of new drugs.10 In most of the studies mentioned above, this endpoint for activity has been recorded.
Based on that, only few agents with orphan drug designation in GBM have been approved. Temozolomide was initially approved for the treatment of recurrent GBM, but following the landmark study by Stupp et al in 2005,37 temozolomide was granted approval in newly diagnosed GBM concomitant with radiotherapy and followed by adjuvant temozolomide. A subgroup analysis revealed that temozolomide only improved the outcome in patients with MGMT methylation and not in those without.38
Gliadel wafers received FDA approval in 2003. However, the randomized pivotal Phase III study was underpowered, and according to the Cochrane review, Gliadel wafers are not recommended for use in recurrent GBM. Procarbazine is also approved by the FDA for recurrent GBM in combination with PCV. However, this combination does not result in significant response rates or better PFS6, and new agents are indeed warranted. Given that GBM is highly vascularized, many inhibitors of angiogenesis have been extensively evaluated, with special focus on identifying biomarkers to predict activity. Accordingly, the majority of the tested agents affect angiogenesis.
Addition of other agents to the “Stupp” regimen has failed to improve survival. Most recently, three randomized Phase III studies of bevacizumab failed to reach the primary endpoint of improved survival, although progression-free survival was improved.14–16 However, bevacizumab is approved by the FDA for the treatment of recurrent GBM, whereas the European Medicines Agency declined bevacizumab for this indication, since no survival benefit was documented.
Approximately one third of GBM patients experience beneficial effects from anti-VEGF treatment, but all inevitably progress. Classically, two main mechanisms of resistance to anti-VEGF have been suggested, ie, adaptive and intrinsic. Adaptive resistance refers to the ability of a tumor to display mechanisms (eg, as a result of upregulation of alternative angiogenic mechanisms) enabling it to evade anti-VEGF therapy, and intrinsic resistance refers to the ability of the tumor to be insensitive to anti-VEGF therapy as a result of genomic factors.72 The classical molecular subtype of GBM appears to respond more frequently to anti-VEGF, but in general this molecular resistance is not well understood and no molecular features have been identified that enable prediction of which patients should be treated.73–75 Upregulation of alternative angiogenic signaling pathways may be one resistance mechanism. Proangiogenic mechanisms might be stimulated by subpopulations of cancer cells and stromal cells (eg, pericytes and inflammatory cells) in the microenvironment, including fibroblast growth factor 2, delta-like ligand 4, stromal cell-derived factor 1, platelet-derived growth factor-α, angiopoietins, and placental growth factor.72,76,77 Characterization of these mechanisms has led to new drug combinations in order to improve efficacy of anti-VEGF treatments that are currently being tested in clinical trials.
Several other mechanisms have been suggested, and in vivo studies and gene expression microarray data have shown that upregulation and activation of the tyrosine kinase c-mesenchymal-epithelial transition factor promotes tumor cell invasion in order to escape the pressure of antiangiogenic therapy.78,79 Therefore, targeting this factor in combination with antiangiogenic therapy might be effective in order to overcome resistance, and results from clinical trials are awaited.
Very recently, autocrine growth stimulation has been suggested as an important reason for tumor proliferation. Many GBM cells express the VEGF receptor, and VEGF is produced by the tumor cells. It is therefore suggested that an autocrine loop exists and is responsible for tumor growth. Since bevacizumab does not cross the blood–brain barrier, autocrine growth stimulation will probably not be targeted.80
According to the definition of GBM as a rare disease, drugs developed for GBM may receive orphan drug designation. A few agents have been approved by the FDA for the treatment of high-grade gliomas following orphan drug designation, but many have failed to reach the market. However, some patients have benefited from receiving developmental agents within clinical trials. Biomarkers for selection of these may result in more success in the field in the new era of personalized medicine.
The number of orphan drug designations seems to keep increasing, and cancer drugs represent the majority as compared with drugs for more traditional rare diseases. This may be seen as an indicator of the success of the Orphan Drug Act, which was initiated in 1983. However, the overall huge expenses associated with most of these agents will require strict health planning and evidence-based guidelines.
Disclosure
The authors report no conflicts of interest in this work.
References
Ohgaki H, Kleihues P. Epidemiology and etiology of gliomas. Acta Neuropathol. 2005;109:93–108. | |
Stupp R, Hegi ME, Mason WP, et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009;10:459–466. | |
Wick W, Weller M, Weiler M, Batchelor T, Yung AW, Platten M. Pathway inhibition: emerging molecular targets for treating glioblastoma. Neuro Oncol. 2011;13:566–579. | |
Franco P. Orphan drugs: the regulatory environment. Drug Discov Today. 2013;18:163–172. | |
Jain RK, di Tomaso E, Duda DG, Loeffler JS, Sorensen AG, Batchelor TT. Angiogenesis in brain tumours. Nat Rev Neurosci. 2007; 8:610–622. | |
de Groot JF, Mandel JJ. Update on anti-angiogenic treatment for malignant gliomas. Curr Oncol Rep. 2014;16:380. | |
Vredenburgh JJ, Desjardins A, Herndon JE, et al. Phase II trial of bevacizumab and irinotecan in recurrent malignant glioma. Clin Cancer Res. 2007;13:1253–1259. | |
Vredenburgh JJ, Desjardins A, Herndon JE, et al. Bevacizumab plus irinotecan in recurrent glioblastoma multiforme. J Clin Oncol. 2007;25:4722–4729. | |
Friedman HS, Prados MD, Wen PY, et al. Bevacizumab alone and in combination with irinotecan in recurrent glioblastoma. J Clin Oncol. 2009;27:4733–4740. | |
Han K, Ren M, Wick W, et al. Progression-free survival as a surrogate endpoint for overall survival in glioblastoma: a literature-based meta-analysis from 91 trials. Neuro Oncol. 2014;16:696–706. | |
Henriksson R, Asklund T, Poulsen HS. Impact of therapy on quality of life, neurocognitive function and their correlates in glioblastoma multiforme: a review. J Neurooncol. 2011;104:639–646. | |
Jakobsen JN, Hasselbalch B, Stockhausen MT, Lassen U, Poulsen HS. Irinotecan and bevacizumab in recurrent glioblastoma multiforme. Expert Opin Pharmacother. 2011;12:825–833. | |
Olson JJ, Nayak L, Ormond DR, Wen PY, Kalkanis SN, Ryken TC; AANS/CNS Joint Guidelines Committee. The role of targeted therapies in the management of progressive glioblastoma: a systematic review and evidence-based clinical practice guideline. J Neurooncol. 2014;118:557–599. | |
Taal W, Oosterkamp HM, Walenkamp AME, et al. A randomized phase II study of bevacizumab versus bevacizumab plus lomustine versus lomustine single agent in recurrent glioblastoma: the Dutch BELOB study. J Clin Oncol. 2013;31 Suppl 15:Abstr 2001. | |
Chinot OL, Wick W, Mason W, et al. Bevacizumab plus radiotherapy-temozolomide for newly diagnosed glioblastoma. N Engl J Med. 2014;370:709–722. | |
Gilbert MR, Dignam JJ, Armstrong TS, et al. A randomized trial of bevacizumab for newly diagnosed glioblastoma. N Engl J Med. 2014;370:699–708. | |
Herrlinger U, Schäfer N, Steinbach JP, et al. Survival and quality of life in the randomized, multicenter GLARIUS trial investigating bevacizumab/irinotecan versus standard temozolomide in newly diagnosed, MGMT-non-methylated glioblastoma patients. J Clin Oncol. 2014;32(Suppl 5):Abstr 2042. | |
Fine HA, Wen PY, Maher EA, et al. Phase II trial of thalidomide and carmustine for patients with recurrent high-grade gliomas. J Clin Oncol. 2003;21:2299–2304. | |
Marx GM, Pavlakis N, McCowatt S, et al. Phase II study of thalidomide in the treatment of recurrent glioblastoma multiforme. J Neurooncol. 2001;54:31–38. | |
Riva M, Imbesi F, Beghi E, et al. Temozolomide and thalidomide in the treatment of glioblastoma multiforme. Anticancer Res. 2007;27:1067–1071. | |
Batchelor TT, Duda DG, di Tomaso E, et al. Phase II study of cediranib, an oral pan-vascular endothelial growth factor receptor tyrosine kinase inhibitor, in patients with recurrent glioblastoma. J Clin Oncol. 2010;28:2817–2823. | |
Batchelor TT, Mulholland P, Neyns B, et al. Phase III randomized trial comparing the efficacy of cediranib as monotherapy, and in combination with lomustine, versus lomustine alone in patients with recurrent glioblastoma. J Clin Oncol. 2013;31:3212–3218. | |
Reardon DA, Fink KL, Mikkelsen T, et al. Randomized phase II study of cilengitide, an integrin-targeting arginine-glycine-aspartic acid peptide, in recurrent glioblastoma multiforme. J Clin Oncol. 2008;26:5610–5617. | |
Gilbert MR, Kuhn J, Lamborn KR, et al. Cilengitide in patients with recurrent glioblastoma: the results of NABTC 03-02, a phase II trial with measures of treatment delivery. J Neurooncol. 2012;106:147–153. | |
Stupp R, Hegi M, Gorlia T, et al. Cilengitide combined with standard treatment for patients with newly diagnosed glioblastoma with O6-methylguanine-DNA methyltransferase (MGMT) promoter methylation: final results of the multicenter, randomized, open-label, controlled, phase III CENTRIC study. J Clin Oncol. 2013; 31(Suppl 15):BA2009. | |
Kjellman C, Olofsson SP, Hansson O, et al. Expression of TGF-beta isoforms, TGF-beta receptors, and SMAD molecules at different stages of human glioma. Int J Cancer. 2000;89:251–258. | |
Bogdahn U, Hau P, Stockhammer G, et al; Trabedersen Glioma Study Group. Targeted therapy for high-grade glioma with the TGF-β2 inhibitor trabedersen: results of a randomized and controlled phase IIb study. Neuro Oncol. 2011;13:132–142. | |
Smith JS, Tachibana I, Passe SM, et al. PTEN mutation, EGFR amplification, and outcome in patients with anaplastic astrocytoma and glioblastoma multiforme. J Natl Cancer Inst. 2001;93:1246–1256. | |
Raizer JJ, Abrey LE, Lassman AB, et al; North American Brain Tumor Consortium. A phase II trial of erlotinib in patients with recurrent malignant gliomas and nonprogressive glioblastoma multiforme postradiation therapy. Neuro Oncol. 2010;12:95–103. | |
Yung WK, Vredenburgh JJ, Cloughesy TF, et al. Safety and efficacy of erlotinib in first-relapse glioblastoma: a phase II open-label study. Neuro Oncol. 2010;12:1061–1070. | |
van den Bent MJ, Brandes AA, Rampling R, et al. Randomized phase II trial of erlotinib versus temozolomide or carmustine in recurrent glioblastoma: EORTC brain tumor group study 26034. J Clin Oncol. 2009;27:1268–1274. | |
Gallego O, Cuatrecasas M, Benavides M, et al. Efficacy of erlotinib in patients with relapsed gliobastoma multiforme who expressed EGFRVIII and PTEN determined by immunohistochemistry. J Neurooncol. 2014;116:413–419. | |
Eisenstat DD, Nabors LB, Mason WP, et al. A phase II study of daily afatinib (BIBW 2992) with or without temozolomide (21/28 days) in the treatment of patients with recurrent glioblastoma. J Clin Oncol. 2011;29 Suppl:Abstr 2010. | |
Kreisl TN, Kotliarova S, Butman JA, et al. A phase I/II trial of enzastaurin in patients with recurrent high-grade gliomas. Neuro Oncol. 2010;12:181–189. | |
Wick W, Puduvalli VK, Chamberlain MC, et al. Phase III study of enzastaurin compared with lomustine in the treatment of recurrent intracranial glioblastoma. J Clin Oncol. 2010;28:1168–1174. | |
Brada M, Hoang-Xuan K, Rampling R, et al. Multicenter phase II trial of temozolomide in patients with glioblastoma multiforme at first relapse. Ann Oncol. 2001;12:259–266. | |
Stupp R, Mason WP, van den Bent MJ, et al; European Organisation for Research and Treatment of Cancer Brain Tumor and Radiotherapy Groups; National Cancer Institute of Canada Clinical Trials Group. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352:987–996. | |
Hegi ME, Diserens AC, Gorlia T, et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med. 2005;352:997–1003. | |
Hart MG, Grant R, Garside R, Rogers G, Somerville M, Stein K. Temozolomide for high grade glioma. Cochrane Database Syst Rev. 2013;4:CD007415. | |
Wick A, Felsberg J, Steinbach JP, et al. Efficacy and tolerability of temozolomide in an alternating weekly regimen in patients with recurrent glioma. J Clin Oncol. 2007;25:3357–3361. | |
Strik HM, Buhk JH, Wrede A, et al. Rechallenge with temozolomide with different scheduling is effective in recurrent malignant gliomas. Mol Med Rep. 2008;1:863–867. | |
Clarke JL, Iwamoto FM, Sul J, et al. Randomized phase II trial of chemoradiotherapy followed by either dose-dense or metronomic temozolomide for newly diagnosed glioblastoma. J Clin Oncol. 2009;27:3861–3867. | |
Fleming AB, Saltzman WM. Pharmacokinetics of the carmustine implant. Clin Pharmacokinet. 2002;41:403–419. | |
La Rocca RV, Hodes J, Villanueva WG, et al. A Phase II study of radiation with concomitant and then sequential temozolomide (TMZ) in patients with newly diagnosed supratentorial high-grade malignant glioma who have undergone surgery with carmustine (BCNU) wafer insertion. Neuro Oncol. 2007;8:391–500. | |
Westphal M, Hilt DC, Bortey E, Delavault P, Olivares R, Warnke PC. A phase 3 trial of local chemotherapy with biodegradable carmustine (BCNU) wafers (Gliadel wafers) in patients with primary malignant glioma. Neuro Oncol. 2003;5:79–88. | |
Brem H, Piantadosi S, Burger PC. Placebo-controlled trial of safety and efficacy of intraoperative controlled delivery by biodegradable polymers of chemotherapy for recurrence. Lancet. 1995;345:1008–1012. | |
Hart MG, Grant R, Garside R, Rogers G, Somerville M, Stein K. Chemotherapeutic wafers for high grade glioma. Cochrane Database Syst Rev. 2011;3:CD007294. | |
Olson JJ, Nayak L, Ormond DR, Wen PY, Kalkanis SN. The role of cytotoxic chemotherapy in the management of progressive glioblastoma: a systematic review and evidence-based clinical practice guideline. J Neurooncol. 2014;118:501–555. | |
Brandes AA, Tosoni A, Amistà P, et al. How effective is BCNU in recurrent glioblastoma in the modern era? A phase II trial. Neurology. 2004;63:1281–1284. | |
Reithmeier T, Graf E, Piroth T, Trippel M, Pinsker MO, Nikkhah G. BCNU for recurrent glioblastoma multiforme: efficacy, toxicity and prognostic factors. BMC Cancer. 2010;10:30. | |
Kappelle AC, Postma TJ, Taphoorn MJ, et al. PCV chemotherapy for recurrent glioblastoma multiforme. Neurology. 2001;56:118–120. | |
Brandes AA, Turazzi S, Basso U, et al. A multidrug combination designed for reversing resistance to BCNU in glioblastoma multiforme. Neurology. 2002;58:1759–1764. | |
Couldwell WT, Gopalakrishna R, Hinton DR, et al. Hypericin: a potential antiglioma therapy. Neurosurgery. 1994;35:705–709. | |
Couldwell WT, Surnock AA, Tobia AJ, et al. A phase 1/2 study of orally administered synthetic hypericin for treatment of recurrent malignant gliomas. Cancer. 2011;117:4905–4915. | |
Friday BB, Anderson SK, Buckner J, et al. Phase II trial of vorinostat in combination with bortezomib in recurrent glioblastoma: a North Central Cancer Treatment Group study. Neuro Oncol. 2012;14: 215–221. | |
Galanis E, Jaeckle KA, Maurer MJ, et al. Phase II trial of vorinostat in recurrent glioblastoma multiforme: a North Central Cancer Treatment Group study. J Clin Oncol. 2009;27:2052–2058. | |
Wen PY, Schiff D, Cloughesy TF, et al. A phase II study evaluating the efficacy and safety of AMG 102 (rilotumumab) in patients with recurrent glioblastoma. Neuro Oncol. 2011;13:437–446. | |
Iwamoto FM, Lamborn KR, Kuhn JG, et al. A phase I/II trial of the histone deacetylase inhibitor romidepsin for adults with recurrent malignant glioma: North American Brain Tumor Consortium Study 03-03. Neuro Oncol. 2011;13:509–516. | |
Lu-Emerson C, Norden AD, Drappatz J, et al. Retrospective study of dasatinib for recurrent glioblastoma after bevacizumab failure. J Neurooncol. 2011;104:287–291. | |
Iwamoto FM, Lamborn KR, Robins HI, et al. Phase II trial of pazopanib (GW786034), an oral multi-targeted angiogenesis inhibitor, for adults with recurrent glioblastoma (North American Brain Tumor Consortium Study 06-02). Neuro Oncol. 2010;12:855–861. | |
Raymond E, Brandes AA, Dittrich C, et al; European Organisation for Research and Treatment of Cancer Brain Tumor Group Study. Phase II study of imatinib in patients with recurrent gliomas of various histologies: a European Organisation for Research and Treatment of Cancer Brain Tumor Group Study. J Clin Oncol. 2008;26(28):4659–4665. | |
Cloughesy TF, Wen PY, Robins HI, et al. Phase II trial of tipifarnib in patients with recurrent malignant glioma either receiving or not receiving enzyme-inducing antiepileptic drugs: a North American Brain Tumor Consortium Study. J Clin Oncol. 2006;24:3651–3656. | |
Galanis E, Buckner JC, Maurer MJ, et al; North Central Cancer Treatment Group. Phase II trial of temsirolimus (CCI-779) in recurrent glioblastoma multiforme: a North Central Cancer Treatment Group Study. J Clin Oncol. 2005;23:5294–5304. | |
Chang SM, Wen P, Cloughesy T, et al; North American Brain Tumor Consortium and the National Cancer Institute. Phase II study of CCI-779 in patients with recurrent glioblastoma multiforme. Invest New Drugs. 2005;23:357–361. | |
Puduvalli VK, Yung WK, Hess KR, et al; North American Brain Tumor Consortium. Phase II study of fenretinide (NSC 374551) in adults with recurrent malignant gliomas: a North American Brain Tumor Consortium study. J Clin Oncol. 2004;22:4282–4289. | |
Bushunow P, Reidenberg MM, Wasenko J, et al. Gossypol treatment of recurrent adult malignant gliomas. J Neurooncol. 1999;43:79–86. | |
Reardon DA, Vredenburgh JJ, Desjardins A, et al. Effect of CYP3A-inducing anti-epileptics on sorafenib exposure: results of a phase II study of sorafenib plus daily temozolomide in adults with recurrent glioblastoma. J Neurooncol. 2011;101:57–66. | |
Hasselbalch B, Lassen U, Hansen S, et al. Cetuximab, bevacizumab, and irinotecan for patients with primary glioblastoma and progression after radiation therapy and temozolomide: a phase II trial. Neuro Oncol. 2010;12:508–516. | |
Reardon DA, Egorin MJ, Quinn JA, et al. Phase II study of imatinib mesylate plus hydroxyurea in adults with recurrent glioblastoma multiforme. J Clin Oncol. 2005;23:9359–9368. | |
Chamberlain MC, Johnston SK. Salvage therapy with single agent bendamustine for recurrent glioblastoma. J Neurooncol. 2011;105:523–530. | |
Dresemann G, Weller M, Rosenthal MA, et al. Imatinib in combination with hydroxyurea versus hydroxyurea alone as oral therapy in patients with progressive pretreated glioblastoma resistant to standard dose temozolomide. J Neurooncol. 2010;96:393–402. | |
Bergers G, Hanahan D. Modes of resistance to anti-angiogenic therapy. Nat Rev Cancer. 2008;8:592–603. | |
Hasselbalch B, Eriksen JG, Broholm H, et al. Prospective evaluation of angiogenic, hypoxic and EGFR-related biomarkers in recurrent glioblastoma multiforme treated with cetuximab, bevacizumab and irinotecan. APMIS. 2010;118:585–594. | |
Laffaire J, Di Stefano AL, Chinot O, et al. An ANOCEF genomic and transcriptomic microarray study of the response to irinotecan and bevacizumab in recurrent glioblastomas. Biomed Res Int. 2014;2014:282815. | |
Sathornsumetee S, Cao Y, Marcello JE, et al. Tumor angiogenic and hypoxic profiles predict radiographic response and survival in malignant astrocytoma patients treated with bevacizumab and irinotecan. J Clin Oncol. 2008;26:271–278. | |
Jubb AM, Browning L, Campo L, et al. Expression of vascular Notch ligands Delta-like 4 and Jagged-1 in glioblastoma. Histopathology. 2012;60:740–747. | |
Lucio-Eterovic AK, Piao Y, de Groot JF. Mediators of glioblastoma resistance and invasion during antivascular endothelial growth factor therapy. Clin Cancer Res. 2009;15:4589–4599. | |
Eckerich C, Zapf S, Fillbrandt R, et al. Hypoxia can induce c-Met expression in glioma cells and enhance SF/HGF-induced cell migration. Int J Cancer. 2007;121:276–283. | |
Jahangiri A, De Lay M, Miller LM, et al. Gene expression profile identifies tyrosine kinase c-Met as a targeie, mediator of antiangiogenic therapy resistance. Clin Cancer Res. 2013;19:1773–1783. | |
Hamerlik P, Lathia JD, Rasmussen R, et al. Autocrine VEGF- VEGFR2-Neuropilin-1 signaling promotes glioma stem-like cell viability and tumor growth. J Exp Med. 2012;209:507–520. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.