")
Back to Journals » International Journal of Nanomedicine » Volume 9 » Issue 1
Optimized synthesis of glycyrrhetinic acid-modified chitosan 5-fluorouracil nanoparticles and their characteristics
Authors Cheng M, Chen H, Wang Y, Xu H, He B, Han J, Zhang Z
Received 2 October 2013
Accepted for publication 6 December 2013
Published 24 January 2014 Volume 2014:9(1) Pages 695—710
DOI https://doi.org/10.2147/IJN.S55255
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Mingrong Cheng,1,2,* Houxiang Chen,3,* Yong Wang,4,* Hongzhi Xu,5 Bing He,5 Jiang Han,1 Zhiping Zhang1
1Department of General Surgery, 2Department of Endoscopy, Pudong New Area District Zhoupu Hospital, Shanghai, People's Republic of China; 3Zhejiang Huafon Fiber Research Institute, Zhejiang Huafon Spandex Co, Ltd, Wenzhou, People's Republic of China; 4School of Materials Science and Engineering, Wuhan University of Technology, Wuhan, People’s Republic of China; 5Department of General Surgery, Shanghai Fifth People’s Hospital, Fudan University, Shanghai, People’s Republic of China
*These authors contributed equally to this work
Abstract: The nanoparticle drug delivery system, which uses natural or synthetic polymeric material as a carrier to deliver drugs to targeted tissues, has a broad prospect for clinical application for its targeting, slow-release, and biodegradable properties. Here, we used chitosan (CTS) and hepatoma cell-specific binding molecule glycyrrhetinic acid to synthesize glycyrrhetinic acid-modified chitosan (GA-CTS). The synthetic product was confirmed by infrared (IR) spectra and hydrogen-1 nuclear magnetic resonance. The GA-CTS/5-fluorouracil (5-FU) nanoparticles were synthesized by combining GA-CTS and 5-FU and conjugating 5-FU onto the GA-CTS nanomaterial. The central composite design was performed to optimize the preparation process as CTS:tripolyphosphate sodium (TPP) weight ratio =5:1, 5-FU:CTS weight ratio =1:1, TPP concentration =0.05% (w/v), and cross-link time =50 minutes. GA-CTS/5-FU nanoparticles had a mean particle size of 193.7 nm, a polydispersity index of 0.003, a zeta potential of +27.4 mV, and a drug loading of 1.56%. The GA-CTS/5-FU nanoparticle had a protective effect on the drug against plasma degrading enzyme, and provided a sustained release system comprising three distinct phases of quick, steady, and slow release. Our study showed that the peak time, half-life time, mean residence time and area under the curve of GA-CTS/5-FU were longer or more than those of the 5-FU group, but the maximum concentration (Cmax) was lower. We demonstrated that the nanoparticles accumulated in the liver and have significantly inhibited tumor growth in an orthotropic liver cancer mouse model.
Keywords: liver cancer, targeted therapy, chemotherapy, pharmacokinetics efficacy
Introduction
Drug delivery systems carry drugs to the targeted cells by exploiting the different physiological and biochemical characteristics of tumor and normal cells.1 These systems can be used to reduce the distribution and metabolism of 5-fluorouracil (5-FU) in nontarget organs and tissues. They also improve the drug efficacy and reduce side effects as a result of the lower doses that are administered. The natural polymer chitosan (CTS) and its analogues have been widely studied as drug vectors, based on their lack of toxicity, biodegradability, good biocompatibility, and absorption.2–4 Small-molecule drugs, such as 5-FU and paclitaxel, are first-line anticancer drugs that inhibit tumor cell proliferation by interfering with the synthesis of nucleic acid. However, their efficacy is affected by low lipophilicity and low bioavailability.5,6 In addition, their clinical use is limited by unwanted side effects such as gastrointestinal reactions, myelosuppression, alopecia, and ataxia, and by their narrow therapeutic index (the therapeutic dose is close to the toxic dose).7 If 5-FU were carried by CTS or its derivatives, the result would be extended release, improved bioavailability, and reduced side effects.8–10 Carrier agents, such as CTS, also have adhesion and biodegradability properties that give them the potential to improve drug efficacy.
Targeted drug delivery has been shown to concentrate the drugs at the site of diseased tissue, thereby greatly reducing side effects in normal tissues and improving the biodegradability and drug efficacy.11,12 Glycyrrhetinic acid (GA) has been shown to specifically bind to receptors on the liver cell membrane as there are more GA binding sites in hepatoma cells than in other cells.13 Therefore, nanomaterials combined with GA will tend to accumulate in hepatoma cells, leading to improved growth inhibition.14,15 In this study, we prepared GA-modified chitosan (GA-CTS), which was used to synthetize a GA-CTS/5-FU nanoparticle. The central composite design (CCD) was applied for optimizing the preparation process of GA-CTS/5-FU nanoparticles, which efficiently targeted the drug to liver, and its slow-release properties were observed.
Materials and methods
Mice and cell lines
The human hepatocellular carcinoma cell line (SMMC-7721) and normal liver cells (LO2) were obtained from the Committee on Type Culture Collection of Chinese Academy of Sciences (Shanghai, People’s Republic of China). The human colon cancer cell line (SW480) was purchased from the American Type Culture Collection (Manassas, VA, USA). Female BALB/c mice, 7 weeks old and weighing about 20 g, were obtained from the Science Department of Experimental Animals of the Fudan University (Shanghai, People’s Republic of China). All mice were housed in a specific pathogen-free level B animal facility.
GA-CTS synthesis
1-ethyl-3-(3-dimethyl aminopropyl) (carbodiimide hydrochloride; Sigma-Aldrich, St Louis, MO, USA) and N-hydroxysuccinimide (Sigma-Aldrich) were added to GA solution (1 g; Xi’an Fujie Pharmaceutical Co, Ltd, Sanyuan County, Shaanxi Province, People’s Republic of China) in dimethylformamide (Amresco, LLC, Solon, OH, USA). The solution was mixed with 2% of chitosan in acetic acid and stirred at room temperature. After 48 hours, the mixture was precipitated with acetone and the precipitate was washed with 60% ethanol and ether. The final product was obtained after vacuum drying.
Fourier transform infrared (FT-IR) spectroscopy
Eighty-five percent deacetylated CTS powder (Sigma-Aldrich) and GA-CTS were pressed into a potassium bromide pellet. The composites were analyzed by FT-IR (NEXUS, Nicolet/Natus Medical Incorporated, San Carlos, CA, USA ) in the range 4,000–400 cm−1.
1H-NMR experiments
To verify the structure of CTS and GA-CTS, samples were dissolved in a solution of deuterium chloride and deuterium oxide. Hydrogen-1 nuclear magnetic resonance (1H-NMR) spectras were recorded using a Varian NMR System 600 machine (Varian, Inc./Agilent Technologies, Santa Clara, CA, USA) at a resonance frequency of 600 MHz. Tetramethylsilane was used as a reference compound.
Preparation of GA-CTS/5-FU nanoparticles
The GA-CTS/5-FU nanoparticles were prepared following the procedure described. 5-FU was added into 1% (w/v) GA-CTS solution (dissolved in 0.1 M acetic acid and pH was adjusted to appropriate value with 0.5 M sodium hydroxide [NaOH]). Then, an aqueous tripolyphosphate sodium (TPP) was added dropwise into the mixture and stirred. After a time, GA-CTS/5-FU nanoparticles were formed by cross-linking. The suspensions of nanoparticles were centrifuged at 10,000 revolutions per minute (rpm) for 30 minutes. Purified nanoparticles were lyophilized and stored at 4°C. The CTS/5-Fu nanoparticles were prepared in the same way. The zeta potential of nanoparticles was determined using Zetasizer Nano-Z (Malvern Instruments, Malvern, UK) and the average particle size was detected by scanning electron microscopy (SEM). The analysis was performed at 25.0°C±0.2°C using sample solutions in deionized distilled water or in 10% fetal bovine serum. The results of the zeta potential and average particle size were average values of three independent measurements.
Optimal design of GA-CTS/5-FU nanoparticles preparation
CCD is one of the most commonly used design types in response surface methodology.16 This design uses a quadratic regression fitting for both factors and responses to find the optimal parameters for a process and to solve multivariate problems through regression analysis.17 Recently, CCD has been widely used in optimizing experimental conditions because it can provide the most accurate experimental information with the smallest number of experiments. In order to study the physicochemical properties of GA-CTS/5-FU nanoparticles, we used CCD to optimize the preparation process of GA-CTS/5-FU nanoparticles, and we prepared the nanoparticles using these optimized parameters.
Factor selection and experimental design
Based on previous studies and on the preparation process of CTS nanoparticles, we selected four factors that could affect the formation of CTS/5-FU nanoparticles, which were: 1) CTS/TPP weight ratio (X1), which was set between 3.5:1 and 5.5:1; 2) 5-FU/CTS weight ratio (X2), which was set between 0.2:1 and 1:1; 3) TPP concentration (%, w/v [X3]), which was set between 0.04 and 0.16; and 4) cross linking time (X4), which was set between 10 and 50 minutes. In order to facilitate statistical calculation, the variable Xi was compiled according to the formula below, and the results are listed in Table 1. Mean particle size and drug loading were used as indicators for the CTS drug carrying nanoparticles.
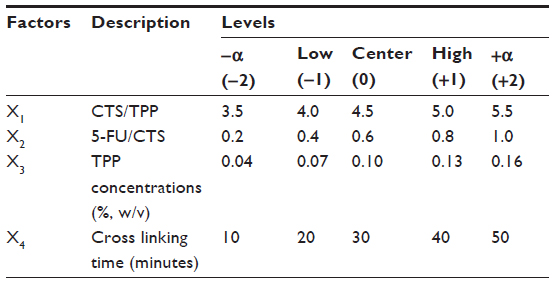 | Table 1 Independent variables and their coded levels investigated in CCD |
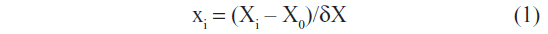
In this formula, X0 indicates the Xi at the center of nanoparticles, and δX stands for the tolerance of variance for each factor.
For tests with a single indicator, determinations could be made directly according to experimental results, but for tests with multiple indicators, a condition favorable for one indicator may not be favorable for another due to the interactions between indicators. In order to achieve the best balance between particle size and drug loading, we quantified the responses of each indicator as “importance”, and introduced the total desirability function (D). Each indicator was normalized to a “normalized value” between 0 and 1, and D was the geometric mean value of all indicators calculated according to the formula below.

In this formula, n indicates the number of responses, d indicates the response value, and rj indicates the importance of each indicator. We set the importance of drug loading as 2, and the importance of particle size as 1.
For an indicator determined as better when the response value is smaller (such as particle size), we normalized such indicator according to the formula below:

In this formula, Ymax and Ymin stand for the acceptable response values of the variable, and Yi stands for the experimental value.
For an indicator determined as better when the response value is larger (such as drug loading), we normalized such indicator according to the formula below:

In this formula, Ymax and Ymin stand for the acceptable response values of the variable, and Yi stands for the experimental value.
We then used a second-order model to fit the dependent variables (indicators) and the independent variables (factors). The formula for this model was based on the equation below:

In this formula, b0, bi, and bij stand for the intercept, coefficients of the linear, and quadratic regression, respectively.
The software Design-Expert 8 (Version 8.0.2, Stat-Ease Inc., Minneapolis, MN). were used for statistical analysis. Analysis of variance (ANOVA), response surface plots, and numerical optimization were used to evaluate the influences of factors on response value.
Chromatographic equipment and conditions
The high performance liquid chromatography (HPLC) system (Shimadzu, Kyoto, Japan) consists of a series III pump, a SPD-10A ultraviolet detector, and a LabAlliance AS-3000 autosampler. Data collection and processing were performed with ANASTAR chromatography workstation (Autoscience Instrument Co. Ltd., Tianjin, People’s Republic of China). Chromatographic separation was performed on a Diamonsil C18 reversed-phase column (250 mm × 4.6 mm, ID 5 μm; Dikma Technologies Inc, Lake Forest, CA, USA).
The mobile phase composed of acetonitrile and water (50:50, v/v) was used throughout the analysis at a flow rate of 1.0 mL/minute. The column temperature was maintained at 30°C by column oven. The ultraviolet detector was set at 254 nm. The injection volume was 20 μL.
In vitro release of GA-CTS/5-FU nanoparticles
To prepare a standard curve, a series of working solutions of 5-FU (0.1 mg/L, 0.2 mg/L, 0.5 mg/L, 1.0 mg/L, 5.0 mg/L, 10 mg/L, and 20 mg/L) were obtained by diluting 5-FU standard stock solution with 0.5 mL human plasma. A calibration curve was generated by linear regression of the peak area ratio of the 5-FU concentration.
GA-CTS/5-FU nanoparticles and 5-FU were placed into dialysis bags and immersed in 30 mL of simulated body fluid (SBF; 0.11 mol/L sodium chloride [NaCl], 0.005 mol/L potassium chloride [KCl], 0.0025 mol/L calcium chloride [CaCl2], 0.0012 mol/L monopotassium phosphate [KH2PO4], 0.0024 mol/L magnesium sulfate [MgSO4], 0.0017 mol/L sodium bicarbonate [NaHCO3], 0.0013 mol/L disodium hydrogen phosphate [Na2HPO4]) at pH 7.4. The particles were incubated at 37°C and shaken at a constant speed of 60 rpm. The medium was replaced with fresh SBF at 20 and 40 minutes, 1 and 6 hours, and then daily for 10 days. Drug concentration in each sample was assayed in triplicate by measuring absorbance at 265 nm. Drug release and loading capacity were calculated by the following formulas:

Preliminary pharmacokinetics of GA-CTS/5-FU
One-hundred-and-twenty mice were randomly divided into two groups with 60 mice in each group; one group for administration with 5-FU solution (1.857 mg/mL) at a dose of 0.371 mg for every mouse and the other group for administration with GA-CTS/5-FU nanoparticles at an equivalent drug dose to the 5-FU group. At 5 minutes, 30 minutes, 1 hour, 2 hours, 4 hours, 8 hours, 12 hours, 16 hours, 24 hours, and 48 hours after administration through tail vein injection, the blood sample was drawn from the neck vein of 12 mice (six mice from each group). All blood samples were collected in heparinized tubes and were immediately centrifuged (3,000 rpm for 10 minutes). Plasma samples were obtained and stored in a 4°C freezer.
Targeting GA-CTS/5-FU nanoparticles in hepatocellular carcinoma in vivo
Following tumor implantation, the mice were randomly divided into three groups (n=5 per group) and treated with 200 μL of 5-FU, CTS/5-FU, or GA-CTS/5-FU (5-FU: 0.371 mg). The mice were sacrificed after 30 minutes. Liver tumor, liver, spleen, kidney, lung, muscle, heart, and small intestine were harvested, washed in saline solution, and dried in filter papers. The tissue (0.5–1.0 g) was homogenized and the concentration of 5-FU in 0.5 mL of the homogenates was determined.
Targeting of GA-CTS nanoparticles in hepatic cancer in vitro
SMMC-7721 and LO2 cells were plated into 6-well plates and incubated for 24 hours. The cells were exposed to fresh medium containing CTS or GA-CTS nanoparticles labeled with different fluorescein isothiocyanate (FITC) concentrations for 4 hour. The cells were fixed in 4% paraformaldehyde at room temperature for 20 minutes and stained with Hoechst 33258 nuclear dye (Sigma-Aldrich). After washing three times in 0.01 M phosphate buffered saline, coverslips were added and the slides were observed under a fluorescence microscope using a 488 nm excitation filter for FITC and a 405 nm filter for Hoechst 33258. The images were analyzed using NIS-Elements software. (A1R, Nikon Corp., Tokyo, Japan).
Biochemical analysis of the blood
Blood samples were collected from mice treated with the nanoparticles for 10 days. Serum alanine aminotransferase (ALT), aspartate aminotransferase (AST) and creatinine were determined using a DA 3500 Discrete Analyzer (Fuji Medical System Co, Ltd, Tokyo, Japan). White blood cell, red blood cell, hemoglobin, and platelet counts were analyzed using a Sysmex XS-800 automatic blood cell analyzer (Sysmex Shanghai Ltd, Shanghai, People’s Republic of China).
Data analysis
All data are expressed as mean ± standard deviation. One-way analysis of variance and the least significant difference test were used for comparisons between groups. Kaplan Meier survival plots were used for survival analysis. A P-value <0.05 was considered significant.
Results
IR spectra and 1H-NMR of GA-CTS
Figure 1A shows the IR spectra of GA, CTS, and GA-CTS. IR spectra of GA existed as an oleanane-type pentacyclic triterpene skeleton. The characteristic absorption peak was at the bending vibration absorption of the C-H in-plane group of the gem-dimethyl moiety connected to C4. The characteristic absorption peak of p-π conjugated carbonyl group of C11 was at 1,664 cm−1. It existed as a C30 carboxyl group with an absorption peak at 1,706 cm−1. The peak at 3,440 cm−1 represented the stretch vibration absorption peak of C3. IR spectra of CTS showed three characteristic amide absorption peaks: an amide band I at 1,655 cm−1; an amide band II at 1,600 cm−1, and amide band III at 1,323 cm−1. The relatively weak absorption peak of amide band I and the relatively strong absorption peak of amide band II indicates that there was a high degree of deacetylation of CTS. Characteristic absorption peaks of carbohydrates were observed at 1,155 cm−1 for the asymmetric stretching vibration absorption peak of C-O-C, and at 1,078 cm−1 and 1,025 cm−1 for the skeleton stretching vibration absorption peak of C-O. The stretching vibration absorption peak of O-H and N-H was represented by the wave at 3,440 cm−1 and by the stretching vibration peak at 2,877 cm−1. Absorption bands shifts for GA/CTS were seen at 1,655 cm−1 of amide band I, 1,600 cm−1 of amide band II, and 1,323 cm−1 of amide band III in CTS, and at 1,654 cm−1, 1,560 cm−1 and 1,314 cm−1, respectively, in the GA/CTS nanoparticles. The intensity of amide band I increased, while the intensity of amide band II decreased. The absorption band of carboxyl group in GA (1,706 cm−1) disappeared. These changes were attributed to the formation of amide bond between GA carboxyl group and CTS amine. Figure 1B shows 1H-NMR results of CTS and GA-CTS. Chemical shifts of 1.410 parts per million and strong characteristic absorption peaks at 0.566 parts per million were observed for CTS, which were attributed to the protons in the -CH3, -CH2, and -CH groups of GA. There being only quaternary carbons around the tertiary carbon in GA residue of GA-CTS, the -CH group (filled circle) of GA residue exhibited a characteristic single peak at 2.06 parts per million. These data indicate the successful connection of GA to the amino group of CTS. The degree of substitution of GA in CTS was calculated by comparing the peak areas of the -CH group (filled circle) and the -CH3 group (filled box) as shown in Figure 1B. The degree of substitution was estimated to be 30.67 mol%.
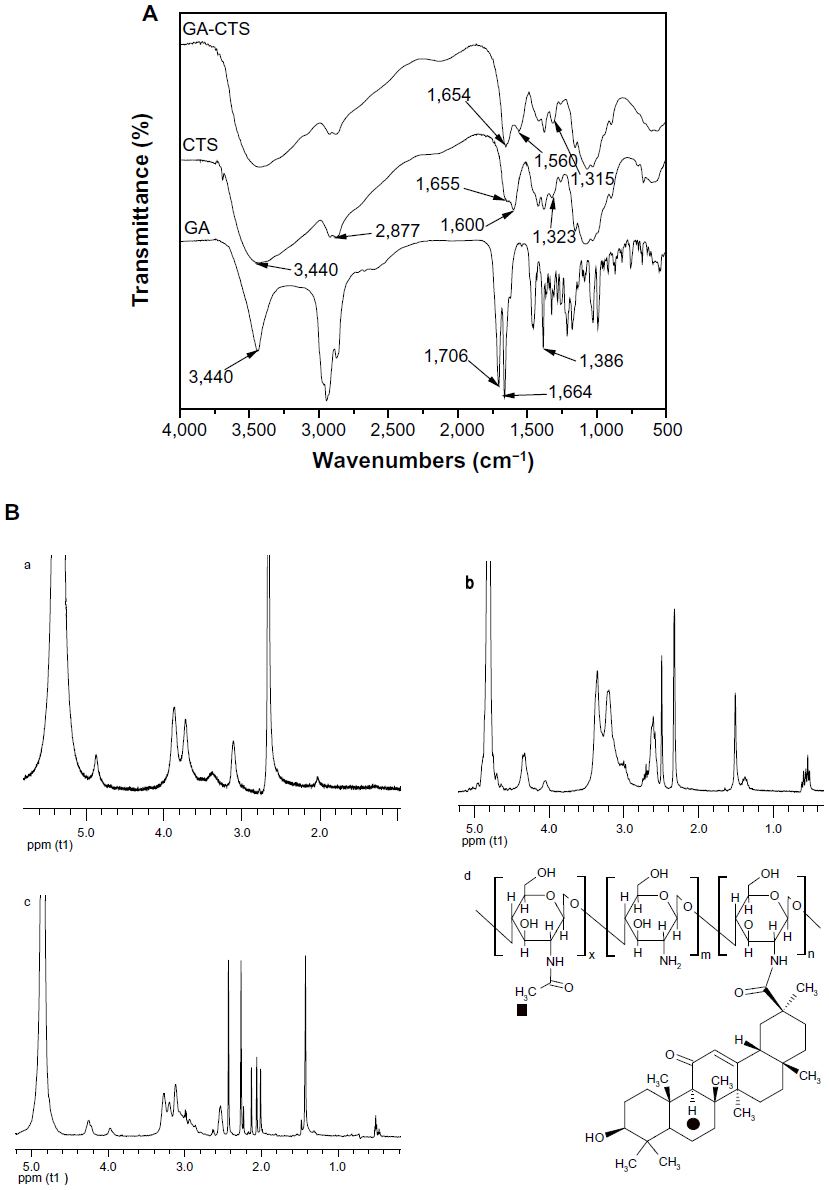 | Figure 1 IR spectra and 1H-NMR of GA-CTS. |
CCD analysis
CCD was performed using CTS and 5-FU. The CCD table consisted of 2k factorial designs (after compilation, ± stands for 1), 2k coordinate axes (±α, 0, 0 … 0), (0, ±α, 0 … 0) … (0, 0, 0 … ±α), and n center duplicate points (0, 0 … 0) (k stands for the number of experimental factors). When all data points are equidistant from the center point (α=2k/4), the CCD is rotatable as all experimental points are located on the spherical surface. Since the variances of predicted value for each regression equation are equal, it is easier to determine the optimum area, and the error interference was not required to be considered.18 This rotatable CCD consisted of 31 experiments, including 16 (24) factorial experiments, eight (2×4) axis points, and seven repeat experiments. Experimental design and results are shown in Table 2.
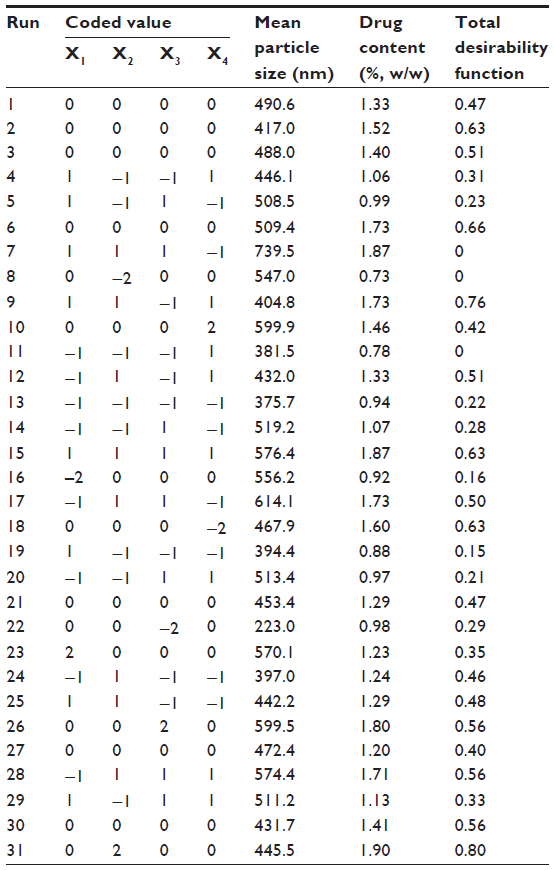 | Table 2 CCD matrix with coded level and measured responses |
Mean particle size of CTS nanoparticles
The mean sizes of nanoparticles prepared using different conditions are listed in Table 2. Our results showed that the mean particle size ranged from 223.0 to 739.5 nm, and that the maximum particle size was 3.3 times the minimum particle size, indicating that particle size was sensitive to the experimental factors we selected. In order to describe the association between factors and response (mean particle size, Y1) we used a quadratic model to fit the experimental data:

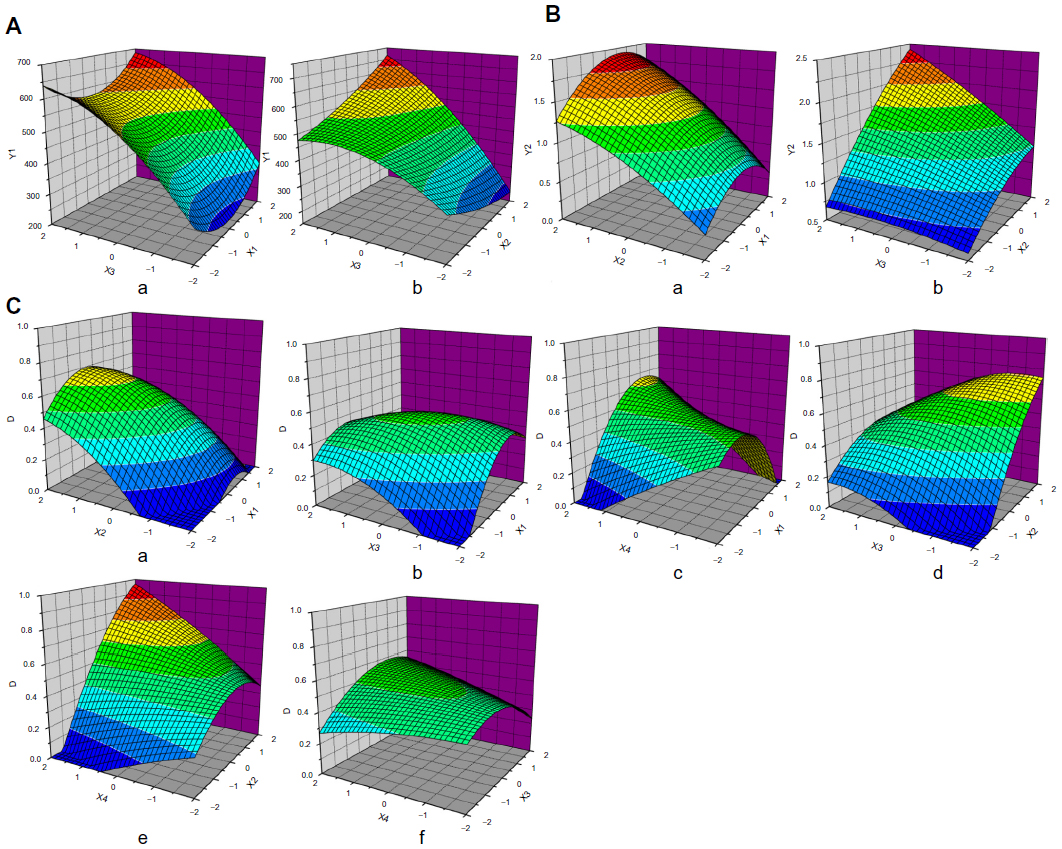
This quadratic model had an F-value of 5.79 and a P-value <0.01, suggesting that the regression equation could be used as a model for the optimal prediction of responses due to its high significance and good fitting. Response surface analysis was performed to variables that significantly affected mean particle size, and the 3-dimensional surface map was obtained using the Origin software (Figure 2A). TPP concentration had a considerable impact on particle size. An increase in TPP concentration resulted in a significant increase in the mean nanoparticle size. The nanoparticle size was the lowest when the CTS/TPP weight ratio was 5:1; higher or lower weight ratio led to increased particle size. Figure 2A(b) indicates that appropriate selection of TPP concentration and 5-FU/CTS weight ratio could optimize the diameter of nanoparticles due to the interactions between TPP concentrations and 5-FU/CTS ratio.
 | Figure 2 Response surface plots of CCD. |
Drug loading of CTS nanoparticles
The loading capacities of CTS nanoparticles prepared using different conditions ranged from 0.73% to 1.90% (Table 2), suggesting that drug loading was sensitive to the experimental factors we selected. A quadratic model was used to fit the response of drug loading, and the equation is:

This quadratic model satisfactorily described the association between drug loading and the factors we selected, and the equation was significant (P<0.0001) and had a good applicability (R=0.9226). We then obtained a 3-dimensional surface map (Figure 2B). In order to investigate the possibility of taking CTS nanoparticles as drug carriers, we used 5-FU as model drug. Though there was no significant interaction (P=0.056) between TPP concentration and 5-FU/CTS, elevating TPP concentration could significantly increase the drug loading of CTS nanoparticles.
Total desirability function (D)
When evaluating several responses, a total desirability value was introduced to achieve the best balance between different responses. For simplicity, the function value was set between 0 and 1, but for such a composite function, the interpretation of the D-value is complex. According to the experimental data, we performed multivariate nonlinear regression on the D-value using other selected factors, and obtained a binomial equation:

ANOVA showed that this quadratic model was significant (P<0.001) and had good fitting effect. Thus, it can be used to describe the association between responses and other factors. In terms of selected factors, X2 (5-FU/CTS weight ratio) had the most significant effect on the D-value (P<0.001), while X12, X1X4, and X2X4 had significant effect on the D-value (P<0.05). The 3-dimensional surface map was obtained using the Origin software, according to the equation (Figure 2C), and response surface analysis suggested that the parameters in Figure 2C(c) and 2C(e) had the greatest impact on the D-value.
Optimization
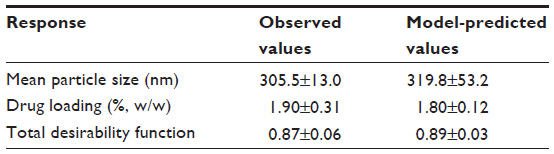
Based on the ANOVA results, we performed predictive analysis and numerical optimization using the Design-Expert software. Based on the impact of the selected factors on particle size and drug loading, we selected one point at the optimal area (with smaller particle size, higher drug loading, and good composite indicators) and conducted verifications. The four factors were: CTS/TPP =5 (compilation =+1); 5-FU/CTS =1 (compilation =+2); TPP concentration =0.05% (w/v, compilation =–1.67); and cross linking time =50 minutes (compilation =+2). Predictive and experimental values (average values for three repeated experiments) are listed in Table 3. Experimental data satisfactorily fitted the predictive model, indicating that the CCD-established model for preparation process of the CTS/5-FU nanoparticles had a good predictability.
 | Table 3 Observed and model-predicted values at optimum conditions |
Preparation of the GA-CTS/5-FU nanoparticles
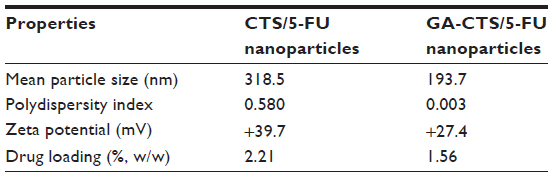
The CCD-optimized conditions for CTS/5-FU nanoparticle preparation were: CTS/TPP weight ratio =5:1; 5-FU/CTS weight ratio =1:1; TPP concentration =0.05% (w/v); and cross linking time = 50 minutes. Based on the conditions for CTS/5-FU nanoparticles preparation, we modified the conditions for GA-CTS/5-FU nanoparticles as: GA-CTS/TPP weight ratio =5:1; 5-FU/GA-CTS =1:1; TPP concentration =0.05% (w/v); and cross linking time =50 minutes. The results of three different drug delivery systems are shown in Table 4 and Figure 3A and B.
 | Table 4 Results of CTS and GA-CTS/5-FU nanoparticles at optimum conditions |
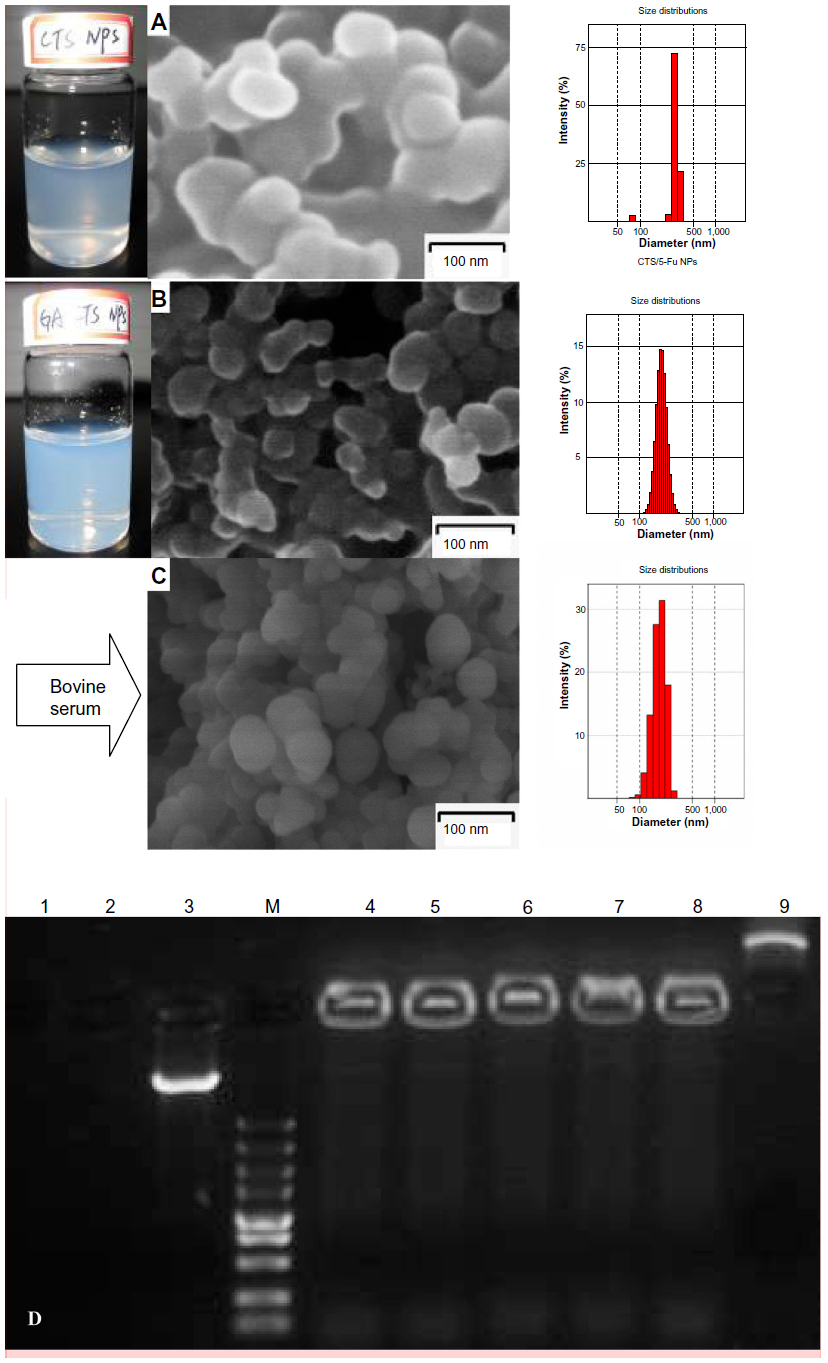 | Figure 3 Scanning electron microscopy of nanoparticles; their protective effect on drugs in vitro. |
Characterization of GA-CTS/5-FU nanoparticles
SEM of GA-CTS/5-FU showed the presence of spherical nanoparticles (size 193.7 nm) with smooth surfaces (Figure 3A). The polydispersity index (PI) was 0.003, indicating narrow nanoparticle distribution (Figure 3B). The zeta potential was +27.4 mV and drug loading efficiency was 1.56%. Figure 3C shows GA-CTS/5-FU nanoparticle with bovine serum; the size of GA-CTS/5-FU nanoparticles (206.0 nm) was increased a little and the charge (+14.3 mV) was decreased obviously, and the PI was 0.135. Figure 3D shows the naked plasmid DNA (pIRES-GM-CSF-IL21 was synthesized in our laboratory and maintained in previous experiments) with DNase I, in which bovine serum was completely digested, while the GA-CTS/plasmid DNA, either with DNase I or mixed with plasma, was still well preserved from 30 minutes to eight hours. There were light bands in the image of gel electrophoresis; it is indicated that GA-CTS nanoparticles can protect the plasmid DNA against DNase I enzymes or plasma degrading enzyme (Figure 3D).
To study the release of GA-CTS/5-FU nanoparticles, an in vitro release curve was generated using SBF. Linear regression of the peak area ratio of the 5-FU concentration (0.1 to 20 mg/L) curve was calculated as y = 2.5721x+0.685 (R=0.9956). Rapid release was observed for 5-FU (SBF) within a cumulative release percentage of 95.7% within 1 hour. Nanoparticle release occurred in three stages (Figure 4A). Rapid release was observed between 0 to 6 hours, which was followed by a cumulative release of 20.6%, due to the diffusion of surface 5-FU into the SBF solution. A smooth slow-release process occurred between 6 hours and day 7, resulting in a cumulative release of 91.4%. During day 7 to 10, the release reached a plateau, with a cumulative release percentage of 95.6%, and additional release of only 4.2% at day 10.
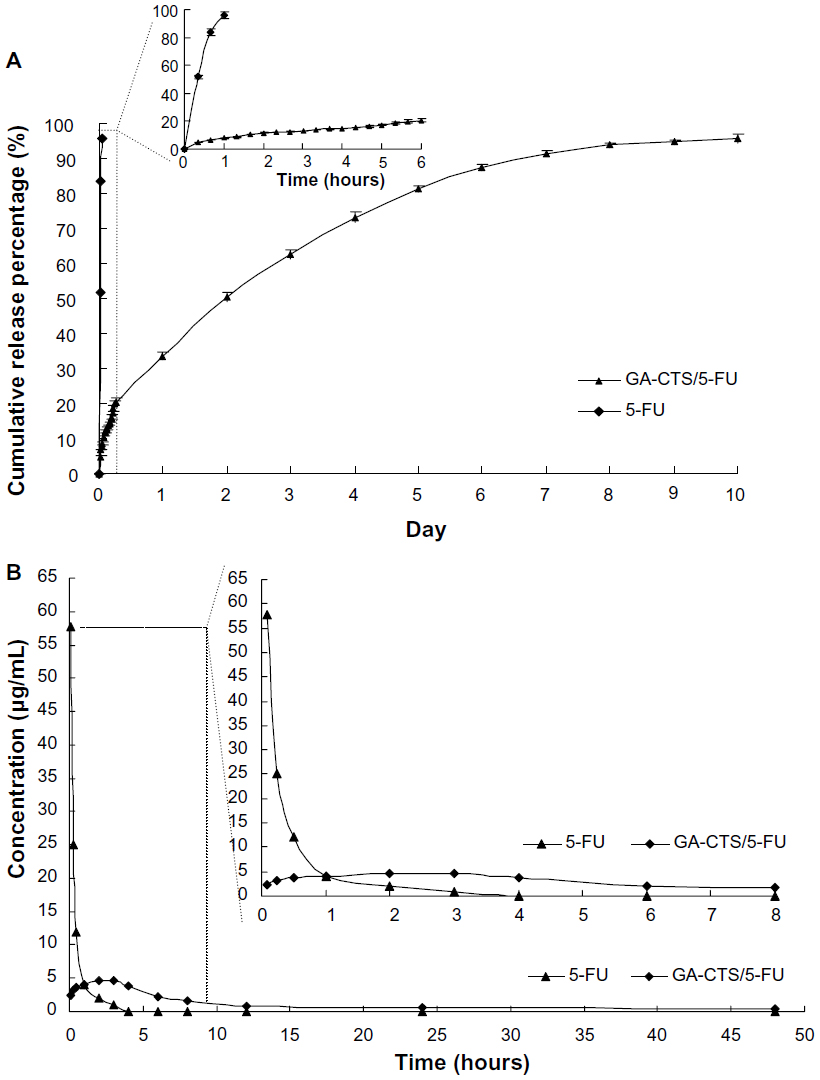 | Figure 4 The releasing curve of GA-CTS/5-FU nanoparticles in vitro and in vivo. |
Preliminary pharmacokinetics of GA-CTS/FU in mice
The maximum concentration (Cmax) of 5-FU group appeared within 5 minutes, and then the concentration stepped down quickly. The concentration was near 0 after 4–6 hours. The concentration of GA-CTS/5-FU was slowly increased and reached Cmax in about 3 hours, then the concentration stepped down slowly and maintained for 30–60 hours. GA-CTS/5-FU group’s Cmax of 5-FU in plasma was lower than the 5-FU group, but the half-life time was obviously longer than that of the 5-FU group (Figure 4B). Table 5 showed that, besides prolonging of half-life, the Time to maximum concentration (Tmax) and mean residence time (MRT) in the GA-CTS/5-FU group were longer than that in the 5-FU group. Meanwhile, the area under the curve (AUC) reflecting the drug absorption or release into the blood in GA-CTS/5-FU group increased significantly and reached 2.37-fold that of the 5-FU group. Taken from the above data, we confirmed that GA-CTS/5-FU is a sustained release and effective drug that has longer half-time, MRT, and AUC.
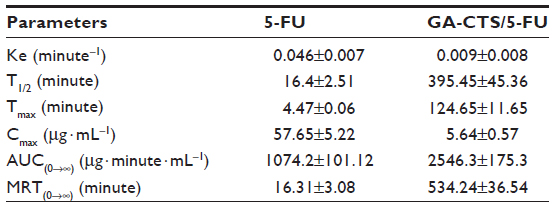 | Table 5 Pharmacokinetic parameters of 5-FU and GA-CTS/5-FU |
Determination of in vitro and in vivo liver-targeting of GA-CTS/5-FU
We used an orthotropic liver cancer mouse model to determine the in vivo targeting of GA-CTS/5-FU. Concentrations of 5-FU were measured in different tissues 30 minutes after injection of 5-FU, CTS/5-FU, and GA-CTS/5-FU. Significant differences were observed for the concentrations of 5-FU after each treatment (Figure 5A, P<0.01). The concentration of 5-FU in hepatic cancer cells, after treatment with GA-CTS/5-FU, was 2.81 and 5.81 times higher than in mice treated with CTS/5-FU or 5-FU, respectively. The lowest concentration was seen with 5-FU. In other tissues, the levels of 5-FU were lower following treatment with GA-CTS/5-FU compared to the other groups. Following administration of GA-CTS/5-FU, the concentration of 5-FU in hepatic cancer cells was 7.29, 12.26, 41.99, and 44.71 times higher than in normal liver, kidney, heart, and blood, respectively (Figure 5B). These results indicate targeting of GA-CTS in liver.
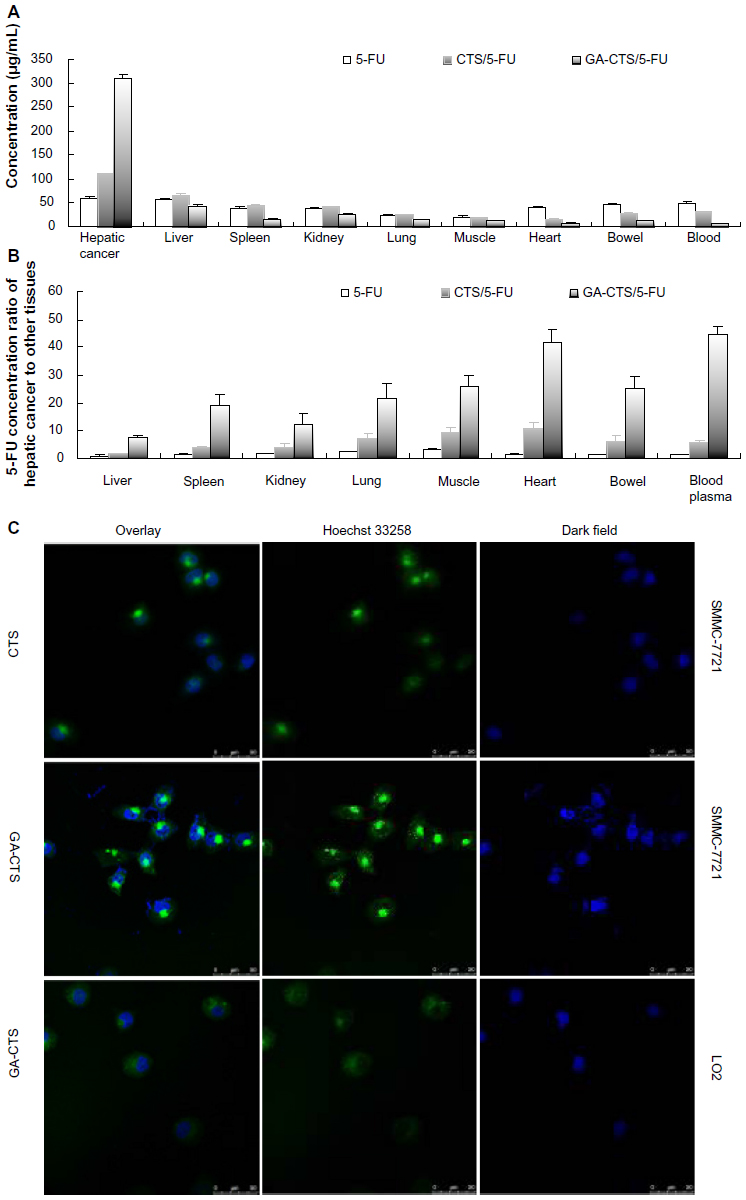 | Figure 5 GA-CTS/5-FU liver targeting in vitro and in vivo. |
Endocytosis of GA-CTS nanoparticles by normal liver LO2 cells and by hepatic cancer SMMC-7721 cells was analyzed under confocal microscopy. As shown in Figure 5C, strong green fluorescence was observed in SMMC-7721 cells following exposure to GA-CTS nanoparticles, indicating endocytosis of a large number of GA-CTS particles. SMMC-7721 cells exposed to CTS nanoparticles showed moderate intensity fluorescence. However, only weak intensity fluorescence was observed in LO2 cells exposed to GA-CTS nanoparticles. These data indicate that GA-CTS nanoparticles target hepatic cancer cells and entered the cells through receptor-mediated endocytosis.
In vivo antitumor efficacy of GA-CTS/5-FU
As shown in Figure 6A, tumor weights were significantly lower in the GA-CTS/5-FU group (0.52±0.11 g) than in GA-CTS (1.62±0.15 g) or control groups (1.64±0.21 g; P<0.01). In the 5-FU group, tumor weight was significantly less than in the control group (P<0.01), but no differences were seen between the GA-CTS group and controls (P>0.05). We also showed that GA-CTS/5-FU attenuated liver injury and leukemia caused by 5-FU (Table 6). Figure 6B shows that the highest cell density, cell proliferation, and division are obvious in the control group and GA-CTS group, and there is decreasing tendency for cell density, cell proliferation, and division from the 5-FU and GA-CTS/5-FU groups in turn; the tumor cell necrosis demonstrated opposite trends, and the control and GA-CTS group presented less necrosis, while there was obvious necrosis in the GA-CTS/5-FU group.
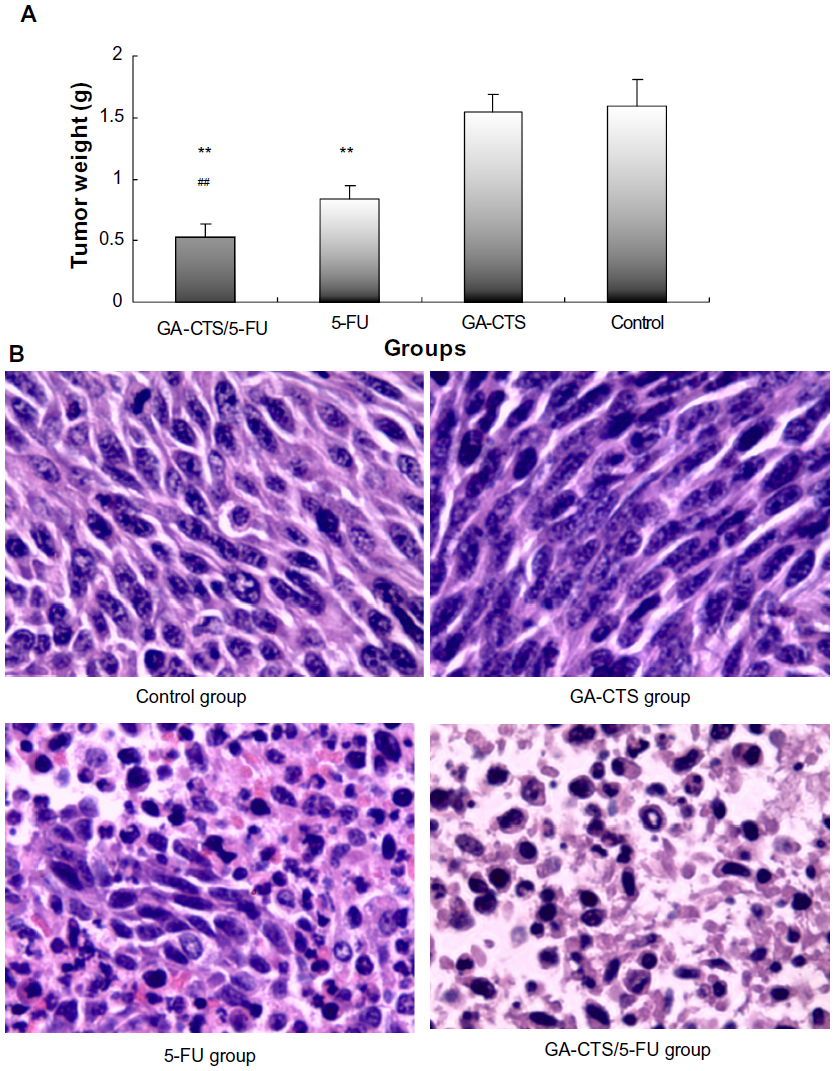 | Figure 6 Inhibition by GA-CTS/5-FU nanoparticles in orthotopic liver transplantation mouse model. |
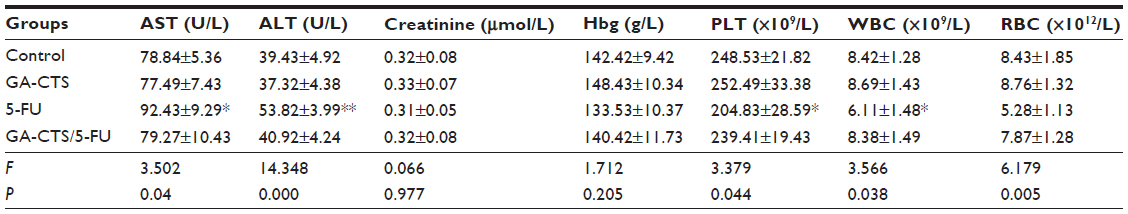 | Table 6 Serum levels of blood biochemical parameters in different groups by day 10 |
Discussion
Targeted drug therapy can specifically deliver drugs to tumor tissues, resulting in increased local concentrations and reduced toxicity. GA, an aglycone of glycyrrhizin, acts as an antioxidant and detoxifying agent. It has been shown to increase apoptosis in hepatoma cells.19,20 It also has the ability to target the liver21,22 and has been shown to specifically bind to receptors on the liver cell membrane.23 These properties make GA a suitable candidate for use in the development of novel drugs that target liver disease. In this study, GA-CTS, identified by IR and 1H-NMR, was used to investigate the feasibility of using GA-CTS as a drug carrier to the liver.
Nanoparticle size and drug loading are two important parameters for a drug delivery system. The pore size of normal vascular endothelial cells is about 2 nm, and the pore size of postcapillary venules is about 6 nm. Liver endothelial cells have pore sizes ranging from 106 to 175 nm, while hepatoma cells have pore sizes from 380 to 580 nm.24–26 Therefore, an ideal nanoparticle size should be larger than the largest pore size of normal liver cells and smaller than the smallest pore size of hepatoma cells. Thus, the nanoparticle size should be between 175 and 380 nm. Drug loading is another important parameter for a drug delivery system. The more drugs a nanoparticle can carry, the higher drug loading it will possess. Thus, many studies18,27,28 have been done to find nanoparticles with high drug loading through rational experimental design and optimization. In this study, we used CCD to optimize the preparation process of nanoparticles, and we obtained optimized nanoparticles with relatively small particle size and high drug loading.
We used CTS nanomaterial and found that the mean particle size increased with TPP concentration; while the CTS/TPP weight ratio reached 5:1, mean particle size reached the lowest. This may be because TPP is a multivalent anion containing 5 negative charges, and in acidic solution the amino group (–NH2) on CTS molecules could form the protonated amino group (–NH3+). During nanoparticle formation, TPP could freely diffuse into CTS droplets to form CTS/TPP cross linked particles with –NH3+ in the CTS. The increase of TPP concentrations will lead to increased particle size due to partial cation saturation in the CTS chain. When CTS/TPP weight ratio reached 5:1, the most effective cross linking could occur between CTS and TPP (Figure 2B); thus, the newly formed nanoparticles had the most compact structure and the smallest diameter. As shown in Figure 2A(a), when the CTS/TPP weight ratio exceeded 5:1, many –NH3+ on CTS chains could not be neutralized and CTS could not be effectively cross linked, resulting in increased nanoparticle size. When the CTS/TPP weight ratio was lower than 5:1, partial cross link between CTS and TPP also resulted in increased particle size and even elevated pH value.
Figure 2B shows that increased TPP concentrations significantly elevated the drug loading of CTS nanoparticles. This may be because the higher efficiency of counter ion improved the cross link density of CTS matrix. Increased 5-FU/CTS weight ratio also elevated nanoparticles’ drug loading due to the enhanced interaction between 5-FU and CTS. When the CTS/TPP weight ratio reached 5:1, the cross link between CTS, TPP, and 5-FU was the most effective and the drug loading was the highest. In the 31 experiments we carried out during CCD, the highest drug loading of CTS/5-FU nanoparticles reached 1.9%. This low drug loading may be because 5-FU is a hydrophilic small molecule that can be easily dissolved in water and it may diffuse into the solution during nanoparticle formation. Another explanation could be that the relatively small particle size limits the nanoparticles’ drug loading. Also, hydrogen bond breaking during particle degradation leads to increased solubility, which in turn reduced the drug loading of nanoparticles.
Increasing 5-FU/CTS weight ratio and cross linking time could increase the total desirability, because the cross link between CTS and TPP could be finished in 60 minutes, and the prolonged cross linking time could help to increase drug loading. Total desirability also increased significantly as the 5-FU concentration was elevated (positive parameter X2X4, P<0.05). This was because high 5-FU concentrations increased drug loading of the nanoparticles, thus improving total desirability. On the other hand, as shown in Figures 2C(a) and 2C(d) (the corresponding coefficients are negative), increases in 5-FU/CTS weight ratio increased the nanoparticle size and decreased total desirability. However, the impact was not very significant (X1X2, P>0.05; X2X3, 0.1>P>0.05). Figure 2C(c) indicates that there was a significant interaction between CTS/TPP weight ratio and crosslinking time. The positive coefficients for X1 indicated that increased CTS/TPP ratio could contribute to the improvement of total desirability. A proper ratio between CTS and TPP could result in effective crosslink and compact particle structure, but the CTS/TPP should not be too large due to the interaction effect. In addition, Figure 2C(b) proves that out-of-range CTS/TPP ratio negatively contributed to total desirability (negative parameter X1X3, 0.1>P>0.05), and that such contribution became more significant as the TPP concentration was increased. Considering the impact of the selected factors on particle size and drug loading, we chose one point from the optimum area for verification, and the parameters were: CTS/TPP ratio =5:1; 5-FU/CTS ratio =1:1 TPP concentration =0.05%; and cross link time =50 minutes. Experimental data were well fitted to the predicted data, and the CCD-optimized CTS/5-FU preparation process had satisfactory predictability.
After CCD optimization, the mean CTS/5-FU nanoparticles size was 318.0 nm, which was larger than the GA-CTS/5-FU nanoparticles (193.7 nm), because there were lots of -GA hydrophobic groups in the -GA-CTS/5-FU nanoparticles and the size was that of a hydrated particle. The GA-CTS/5-FU nanoparticles had significantly decreased zeta potential compared with CTS/5-FU nanoparticles, because the introduction of GA groups neutralized free amino groups on CTS. The GA-CTS/5-FU nanoparticles had low drug loading compared with CTS/5-FU nanoparticles because the GA hydrophobic groups into CTS and 5-FU decreased the adsorption of 5-FU on GA-CTS. PI is an important indicator for the suspending stability of nanoparticles. A smaller PI value means a better long-term suspending stability of nanoparticles. A PI between 0.1 and 0.25 suggests a very good uniformity of nanoparticles, while a PI >0.5 means a poor uniformity and nonnormal distribution of nanoparticles.29 The three different nanoparticles (all PI <0.1) had a spherical shape and showed agglomeration, which occurred during SEM sample preparation, as the dispersion of nanoparticles was incomplete after centrifugation.
5-FU is a widely used anticancer drug that is toxic to normal cells and has a short plasma half-life of 15 to 20 minutes. Improving the bioavailability and site-specific delivery of 5-FU and reducing its side effects may provide obvious therapeutic advantages. In this study, we prepared GA-CTS/5-FU nanoparticles with an average diameter of 193.7 nm. SEM imaging showed that GA-CTS/5-FU nanoparticles were spherical, smooth surface structures that adhered to one another and were not fully dispersed after centrifugation. Prior to centrifugation, the GA-CTS/5-FU nanoparticles used in our study had PI value of 0.003, with a single peak, indicating good uniformity. There was a slight increase in particle size and the potential was decreased when GA-CTS/5-FU nanoparticles were mixed with bovine serum. Its PI was 0.135. It is indicated that GA-CTS/5-FU nanoparticles still have a uniform dispersion with bovine serum, and it was found that nanoparticles have a protective effect of the drug against plasma degrading enzyme. The release of nanoparticles in SBF occurred in stages, characterized by sustained release, which was similar to previous studies.30 Rapid release was observed between 0 and 6 hours, with a cumulative release of 20.6%, resulting from diffusion of surface 5-FU into the SBF solution. Smooth slow-release occurred after between 6 hours and 7 days. Following the gradual degradation of insoluble hydrophobic material, the drug diffused through the membrane, resulting in a cumulative release of 60.8%. Between day 7 and 10, a residual drug release of 4.2%. Then, we further observed the change of pharmacokinetic parameters of GA-CTS/5-FU in vivo, such as half-life time, the maximum of concentration, MRT, etc. In this experiment, pharmacokinetic parameters of 5-FU and its GA-CTS nanoparticles in mice showed that GA-CTS/5-FU nanoparticle groups’ Cmax of 5-FU in plasma were lower than the 5-FU group, the half-life times and MRT were prolonged, and the AUCs were higher. It indicated that 5-FU that embed to GA-CTS/5-FU nanoparticles slowly released to blood while the carrying nanoparticles slowly degraded. The absorption and disposition in the body of nanoparticles is different from macrobead matter because of its small architecture. Perhaps it would solve the problem of short plasma half-life and/or severe side effects with high doses. To explore the liver targeting efficiency of GA-CTS/5-FU nanoparticles, in vivo distribution was evaluated. Absorption in hepatoma cells was analyzed using confocal microscopy. The results showed that the concentration of 5-FU in liver cancer from GA-CTS/5-FU was 2.81, 5.81, and 7.29 times higher than that achieved with CTS/5-FU, 5-FU, and control, respectively. Confocal microscopy provided evidence of a stronger green fluorescence in hepatoma cells exposed to GA-CTS nanoparticles than in those exposed to CTS. Hepatoma cells exposed to GA-CTS nanoparticles also had a stronger green fluorescence than normal liver cells. These results provide evidence of enrichment of GA-CTS nanoparticles on the surface of hepatoma cells, which facilitates the binding of GA to GA receptors or other binding sites. This is followed by endocytosis of 5-FU at optimal local concentrations.14
Our results also demonstrated that tumor weight in a mouse orthotopic liver transplantation model was significantly lower in animals who had received GA-CTS/5-FU than in other groups, and the necrosis of the GA-CTS/5-FU group is more obvious than that of the 5-FU groups, ie, there was less necrosis in the other groups. When compared with the 5-FU and GA-CTS/5-FU nanoparticles, the GA-CTS nanoparticles induced a more important endocytosis of 5-FU, resulting in higher 5-FU concentration and a lower tumor weight in GA-CTS/5-FU-injected mice. This suggests that GA binding might result in a higher concentration of 5-FU in the hepatoma cells.31 GA-CTS/5-FU also attenuated the toxicity caused by 5-FU.
Conclusion
In conclusion, we have successfully used CCD to optimize the synthesis process of liver-targeting GA-CTS/5-FU nanoparticles. GA-CTS/5-FU nanoparticles have a protective effect of the drug against plasma degrading enzymes our study showed that the peak time, half-life time, MRT, and AUC of GA-CTS/5-FU were longer or more than those of the 5-Fu group; however, the Cmax was lower. GA-CTS/5-FU nanoparticles have good slow-release properties, are targeted to the liver, and have significantly inhibited tumor growth in an orthotropic liver cancer mouse model.
Acknowledgments
This work was supported by Natural Science Foundation of Shanghai (12nm0502202 and 114119a4700), Pudong New Area Science and Technology Development Fund (PKJ2012-Y24), and the Pudong New Area Health System discipline lead development program (PWRd2013–10).
Disclosure
The authors report no conflicts of interest in this work.
References
Oh IH, Min HS, Li L, et al. Cancer cell-specific photoactivity of pheophorbide a-glycol chitosan nanoparticles for photodynamic therapy in tumor-bearing mice. Biomaterials. 2013;34(27):6454–6463. | |
Hamman JH. Chitosan based polyelectrolyte complexes as potential carrier materials in drug delivery systems. Mar Drugs. 2010;8:1305–1322. | |
Arulmozhi V, Pandian K, Mirunalini S. Ellagic acid encapsulated chitosan nanoparticles for drug delivery system in human oral cancer cell line (KB). Colloids Surf B Biointerfaces. 2013;110:313–320. | |
Javid A, Ahmadian S, Saboury AA, Kalantar SM, Rezaei-Zarchi S. Chitosan coated superparamagnetic iron oxide nanoparticles for doxorubicin delivery: synthesis and anticancer effect against human ovarian cancer cells. Chem Biol Drug Des. 2013;82(3):296–306. | |
Toriumi F, Kubota T, Saikawa Y, et al. Thymidylate synthetase (TS) genotype and TS/dihydropyrimidine dehydrogenase mRNA level as an indicator in determining chemosensitivity to 5-fluorouracil in advanced gastric carcinoma. Anticancer Res. 2004;24:2455–2463. | |
Johnson KR, Wang L, Miller MC 3rd, Willingham MC, Fan W. 5-Fluorouracil interferes with paclitaxel cytotoxicity against human solid tumor cells. Clin Cancer Res. 1997;3:1739–1745. | |
Fang JY, Liu PF, Huang CM. Decreasing systemic toxicity via transdermal delivery of anticancer drugs. Curr Drug Metab. 2008;9:592–597. | |
Huang X, Wang Y, Cai JP, et al. Sustained release of 5-fluorouracil from chitosan nanoparticles surface modified intra ocular lens to prevent posterior capsule opacification: an in vitro and in vivo study. J Ocul Pharmacol Ther. 2013;29:208–215. | |
Xu J, Ma L, Liu Y, Xu F, Nie J, Ma G. Design and characterization of antitumor drug paclitaxel-loaded chitosan nanoparticles by W/O emulsions. Int J Biol Macromol. 2012;50:438–443. | |
Yan S, Zhu J, Wang Z, Yin J, Zheng Y, Chen X. Layer-by-layer assembly of poly(L-glutamic acid)/chitosan microcapsules for high loading and sustained release of 5-fluorouracil. Eur J Pharm Biopharm. 2011;78:336–345. | |
Park JH, Saravanakumar G, Kim K, Kwon IC. Targeted delivery of low molecular drugs using chitosan and its derivatives. Adv Drug Deliv Rev. 2010;62:28–41. | |
Yu JM, Li YJ, Qiu LY, Jin Y. Polymeric nanoparticles of cholesterol-modified glycol chitosan for doxorubicin delivery: preparation and in-vitro and in-vivo characterization. J Pharm Pharmacol. 2009;61:713–719. | |
He ZY, Zheng X, Wu XH, et al. Development of glycyrrhetinic acid-modified stealth cationic liposomes for gene delivery. Int J Pharm. 2010;397:147–154. | |
Tian Q, Zhang CN, Wang XH, et al. Glycyrrhetinic acid-modified chitosan/poly(ethylene glycol) nanoparticles for liver-targeted delivery. Biomaterials. 2010;31:4748–4756. | |
Huang W, Wang W, Wang P, et al. Glycyrrhetinic acid-modified poly(ethylene glycol)-b-poly(gamma-benzyl l-glutamate) micelles for liver targeting therapy. Acta Biomater. 2010;6:3927–3935. | |
Bei YY, Zhou XF, You BG, et al. Application of the central composite design to optimize the preparation of novel micelles of harmine. Int J Nanomedicine. 2013;8:1795–1808. | |
Zou TB, Jia Q, Li HW, Wang CX, Wu HF. Response Surface Methodology for Ultrasound-Assisted Extraction of Astaxanthin from Haematococcus pluvialis. Mar Drugs. 2013;11:1644–1655. | |
Kollipara S, Bende G, Movva S, Saha R. Application of rotatable central composite design in the preparation and optimization of poly(lactic-co-glycolic acid) nanoparticles for controlled delivery of paclitaxel. Drug Dev Ind Pharm. 2010;36:1377–1387. | |
Jeong HG, You HJ, Park SJ, et al. Hepatoprotective effects of 18beta-glycyrrhetinic acid on carbon tetrachloride-induced liver injury: inhibition of cytochrome P450 2E1 expression. Pharmacol Res. 2002;46:221–227. | |
Lallemand B, Gelbcke M, Dubois J, Prévost M, Jabin I, Kiss R. Structure-activity relationship analyses of glycyrrhetinic acid derivatives as anticancer agents. Mini Rev Med Chem. 2011;11:881–887. | |
Tian Q, Wang XH, Wang W, Zhang CN, Wang P, Yuan Z. Self-assembly and liver targeting of sulfated chitosan nanoparticles functionalized with glycyrrhetinic acid. Nanomedicine. 2012;8:870–879. | |
Huang W, Wang W, Wang P, et al. Glycyrrhetinic acid-functionalized degradable micelles as liver-targeted drug carrier. J Mater Sci Mater Med. 2011;22:853–863. | |
Negishi M, Irie A, Nagata N, Ichikawa A. Specific binding of glycyrrhetinic acid to the rat liver membrane. Biochim Biophys Acta. 1991;1066:77–82. | |
Marcucci F, Lefoulon F. Active targeting with particulate drug carriers in tumor therapy: fundamentals and recent progress. Drug Discov Today. 2004;9:219–228. | |
Hobbs SK, Monsky WL, Yuan F, et al. Regulation of transport pathways in tumor vessels: role of tumor type and microenvironment. Proc Natl Acad Sci U S A. 1998;95:4607–4612. | |
Ballet F. Hepatic circulation: potential for therapeutic intervention. Pharmacol Ther. 1990;47:281–328. | |
Hassan EE, Parish RC, Gallo JM. Optimized formulation of magnetic chitosan microspheres containing the anticancer agent, oxantrazole. Pharm Res. 1992;9:390–397. | |
Singh B, Bhatowa R, Tripathi CB, Kapil R. Developing micro-/nanoparticulate drug delivery systems using “design of experiments”. Int J Pharm Investig. 2011;1:75–87. | |
Patravale VB, Date AA, Kulkarni RM. Nanosuspensions: a promising drug delivery strategy. J Pharm Pharmacol. 2004;56:827–840. | |
Zhu L, Ma J, Jia N, Zhao Y, Shen H. Chitosan-coated magnetic nanoparticles as carriers of 5-fluorouracil: preparation, characterization and cytotoxicity studies. Colloids Surf B Biointerfaces. 2009;68:1–6. | |
Hibasami H, Iwase H, Yoshioka K, Takahashi H. Glycyrrhetic acid (a metabolic substance and aglycon of glycyrrhizin) induces apoptosis in human hepatoma, promyelotic leukemia and stomach cancer cells. Int J Mol Med. 2006;17:215–219. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.