Back to Journals » Drug Design, Development and Therapy » Volume 14
Optimization of a Benzothiazole Indolene Scaffold Targeting Bacterial Cell Wall Assembly
Authors Chauhan J, Yu W ![]() , Cardinale S, Opperman TJ, MacKerell AD Jr
, Cardinale S, Opperman TJ, MacKerell AD Jr ![]() , Fletcher S, de Leeuw EPH
, Fletcher S, de Leeuw EPH
Received 7 August 2019
Accepted for publication 30 December 2019
Published 10 February 2020 Volume 2020:14 Pages 567—574
DOI https://doi.org/10.2147/DDDT.S226313
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Qiongyu Guo
Jay Chauhan,1 Wenbo Yu,2 Steven Cardinale,3 Timothy J Opperman,3 Alexander D MacKerell Jr,2,4 Steven Fletcher,1 Erik PH de Leeuw1
1Institute of Human Virology & Department of Biochemistry and Molecular Biology of the University of Maryland Baltimore School of Medicine, Baltimore, MD 21201, USA; 2Computer-Aided Drug Design Center, University of Maryland, School of Pharmacy, Baltimore, MD 21201, USA; 3Microbiotix, LLC., Worcester, MA 01605, USA; 4Department of Pharmaceutical Sciences, University of Maryland, School of Pharmacy, Baltimore, MD 21201, USA
Correspondence: Erik PH de Leeuw
Tel +1 410 706 3430
Email [email protected]
Background: The bacterial cell envelope is comprised of the cell membrane and the cell wall. The bacterial cell wall provides rigidity to the cell and protects the organism from potential harmful substances also. Cell wall biosynthesis is an important physiological process for bacterial survival and thus has been a primary target for the development of antibacterials. Antimicrobial peptides that target bacterial cell wall assembly are abundant and many bind to the essential cell wall precursor molecule Lipid II.
Methods: We describe the structure-to-activity (SAR) relationship of an antimicrobial peptide-derived small molecule 7771– 0701 that acts as a novel agent against cell wall biosynthesis. Derivatives of compound 7771– 0701 (2-[(1E)-3-[(2E)-5,6-dimethyl-3-(prop-2-en-1-yl)-1,3-benzothiazol-2-ylidene]prop-1-en-1-yl]-1,3,3-trimethylindol-1-ium) were generated by medicinal chemistry guided by Computer-Aided Drug Design and NMR. Derivatives were tested for antibacterial activity and Lipid II binding.
Results: Our results show that the N-alkyl moiety is subject to change without affecting functionality and further show the functional importance of the sulfur in the scaffold. The greatest potency against Gram-positive bacteria and Lipid II affinity was achieved by incorporation of a bromide at the R3 position of the benzothiazole ring.
Conclusion: We identify optimized small molecule benzothiazole indolene scaffolds that bind to Lipid II for further development as antibacterial therapeutics.
Keywords: Lipid II, antibiotics, drug development, cell wall
Cell wall biosynthesis is a complex process that occurs in three stages: the cytoplasmic stage, the membrane-associated stage and at the cell wall envelope.1,2 The main purpose of this process is the translocation of the peptidoglycan subunits N‐acetylglucosamine (GlcNAc) and N‐acetylmuramic acid (MurNAc) from the cytoplasm across the cellular membrane. These two amino sugars are coupled together by β‐1,4‐glycosidic bonding. Cross-linking of glycan chains occurs via amide to backbone bond formation of a pentapeptide moiety, which is attached to the MurNac sugar.2–5 Lipid II (GlcNAc-MurNAc(pentapeptide) phosphoryl undecaprenol) is an essential intermediate in cell wall biosynthesis. Since Lipid II is partly accessible to the extracellular compartment of the cytoplasmic membrane, it is a target for antibacterial compounds.2,5 These compounds include glycopeptides that have been in clinical use, such as vancomycin, as well as other classes of antibacterial peptides like lantibiotics, ramoplanins and defensins.2,6–9 Based on the interaction between Lipid II and the antimicrobial peptide Human Neutrophil Peptide −1 (HNP-1),9 we identified low molecular weight synthetic compounds that target Lipid II with high specificity and affinity.10 In this study, we report on the structural and functional relationships of derivatives of lead compound 7771–0701.
Materials and Methods
CADD Modeling and MD Simulations
Molecular Dynamics (MD) simulations were performed with the program CHARMM11 using the CHARMM36 lipid12 and protein force field13,14 for Lipid II, the CHARMM TIP3P model15 for water, along with the CHARMM General force field16–18 for the ligand. Based on the previously published model from MD simulation for the BAS00127538-Lipid II complex system in aqueous solution,10 the lead compound 7771–0701 was docked onto Lipid II by aligning its indolene group with that of BAS00127538. The system was then subjected to a short energy minimization following which a 100 ps MD simulation with a time step of 0.5 fs was carried out to further equilibrate the system. The system was then subjected to a 20 ns MD simulation run with a time step of 1 fs. Simulations were carried out in the NPT ensemble at 300 K and 1 atm with SHAKE of covalent bonds involving hydrogens, and there were no restraints in the simulations. Free energies of binding, ΔG, were estimated using the linear interaction energy (LIE) method,19 where
(Eq.1)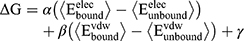
in which α = 0.5, β = 0.16, γ cancels out as we only considered the relative free energies ΔΔG, and the unbound interaction energies were computed from 5 ns MD simulations of the compound alone in water. This involved manually placing one of the inhibitor benzene rings and MurNac ring in Lipid II adjacent to each other. Harmonic restraints, k(r-r0)2, were placed between the geometric centers of the above groups, where k=50 kcal/(mol Å2), r0=3 Å and r is the distance between those geometric centers. The system was then subjected to a 2000 step SD energy minimization followed by a 1 ns gas phase Langevin simulation in the presence of the restraints followed by an additional 1 ns gas phase Langevin simulation in the absence of the restraints. The resulting complex was then solvated in a 48*48*48 Å3 pre-equilibrated (45) water box for condensed phase simulations. All water molecules within 2.8 Å of the non-hydrogen atoms of the complex are removed, and two sodium ions were added to neutralize the system, which contained 10385 atoms. While all nonbonded interactions were evaluated for gas phase simulations, nonbonded interactions were truncated at 12 Å for condensed phase simulations, with a force switch smoothing from 10 to 12 Å. Simulations were performed using periodic boundary conditions with the particle mesh Ewald summation method (46) used to treat the electrostatic interactions with a real space cutoff of 12 Å. The system was minimized for 2000 SD steps and subjected to an isobaric, isothermal (NPT) MD simulation at 300 K and 1 atm. Simulations were extended for 2 ns during which the inhibitor remains in close contact with Lipid II.
3-Lipid II Purification
Short-chain water-soluble Lipid II containing a lipid tail of three isoprene units (3-Lipid II or farnesyl-Lipid II) was generated and purified essentially as described.20 In short, M. flavus vesicles (40–80 nmol lipid-Pi) were incubated together with 200 nmol UDP-GlcNAc, 100 nmol UDP-MurNAC-pentapeptide, and 150 nmol farnesyl phosphate (3-P) in 100 mm Tris-HCl, pH 8.0, 5 mm MgCl2. The incubation lasted two hours at room temperature for 3-P. The synthesis 3-Lipid II was followed using reversed phase TLC (Merck). 3-Lipid II with RP-8 was developed in 75% methanol. For purification, the membranes were removed by centrifugation at 40,000 × g, and the supernatant was collected and loaded on a C18 HPLC column. The short chain Lipid II variant was eluted with a linear gradient from 50 mm ammonium bicarbonate to 100% methanol in 30 mins. Farnesyl-Lipid II (3-Lipid II) eluted at ∼60% methanol. The identity of the Lipid II species was confirmed by mass spectroscopy.
Surface Plasmon Resonance
Surface Plasmon Resonance binding experiments were carried out on a BIAcore T100 system (BIAcore Inc., Piscataway, NY) at 25°C. The assay buffer was 10 mM HEPES, 150 mM NaCl, 0.05% surfactant P20, pH 7.4 (± 3 mM EDTA) supplemented with 10% DMSO. 3-Lipid II (50 RUs) was immobilized on CM5 sensor chips using the amine-coupling chemistry recommended by the manufacturer. For initial determination of binding, compounds were introduced into the flow-cells (30 µL/min) in the running buffer at 10 µM. Resonance signals were corrected for nonspecific binding by subtracting the background of the control flow-cell. After each analysis, the sensor chip surfaces were regenerated with 50 mM NaOH for 30 s at a flow rate 100 μL/min, and equilibrated with the buffer prior to next injection. For binding kinetics studies, binding isotherms were analyzed with manufacturer-supplied software for BIAcore T100.
Mechanism-of-Action Studies
Macromolecular synthesis inhibition by 7771–0701 was investigated using S. aureus MMX100 (ATCC 29213). Cells were grown at 35oC overnight on Tryptic Soy Agar Broth (Remel, Lenexa, KS), and growth from the plate was used to inoculate 15 mL of Mueller Hinton Broth. The culture was grown to early exponential growth phase (OD600 = 0.2 to 0.3) while incubating in a shaker at 35°C and 150 rpm. For each macromolecular assay, the test agent was added at either 0, 0.25, 0.5, 1, 2, or 4-fold their respective MIC values for S. aureus ATCC 29213. As positive control drugs, the following antibiotics were added at 8X MIC in order to validate each assay: Vancomycin (cell wall synthesis); ciprofloxacin (DNA synthesis), rifampin (RNA synthesis), cerulenin (lipid synthesis), and linezolid (protein synthesis).
For DNA and protein synthesis, 100 μL of cell culture reaching early exponential phase was added to triplicate wells containing various concentrations of test compound or control antibiotics (2.5 μL) at 40X the final concentration in 100% DMSO (0.1% methanol in water for Rifampicin). A 2.5% DMSO treated culture served as the “no drug” control for all experiments. Cells were added in 1.25X strength MHB to account for the volume of drug added to each reaction, or in M9 minimal medium for protein synthesis reactions. Following a 5-mins incubation at room temperature either [3H]Thymidine (DNA synthesis) or [3H]Leucine (protein synthesis) was added at 0.5–1.0 µCi per reaction, depending on the experiment. Reactions were allowed to proceed at room temperature for 15–40 min and then stopped by adding 12 µL of cold 5% trichloroacetic acid (TCA) or 5% TCA/2% casamino acids (protein synthesis). Reactions were incubated on ice for 30 min and the TCA precipitated material was collected on a 25 mm GF/1.2 µm PES 96-well filter plate (Corning). After washing five times with 200 µL per well of cold 5% TCA, the filters were allowed to dry, and then counted using a Packard Top Count microplate scintillation counter.
For cell wall synthesis, bacterial cells in early exponential growth phase were transferred to M9 minimal medium and added to 1.5 mL eppendorf tubes (100 μL/tube) containing various concentrations of test compound or control antibiotics (2.5 µL) at 40X the final concentration in 100% DMSO as described above. Following a 5-mins incubation at 37°C, [14C] N-acetyl-glucosamine (0.4 μCi/reaction) was added to each tube and incubated for 45 min in a 37oC heating block. Reactions were stopped through the addition of 100 µL of 8% SDS to each tube. Reactions were then heated at 95oC for 30 min in a heating block, cooled, briefly centrifuged, and spotted onto pre-wet HA filters (0.45 μM). After washing three times with 5 mL of 0.1% SDS, the filters were rinsed two times with 5 mL of deionized water, allowed to dry, and then counted using a Beckman LS3801 liquid scintillation counter.
For lipid synthesis, bacterial cells were grown to early exponential growth phase in MHB and 100 µL was added to 1.5 mL Eppendorf tubes (in triplicate) containing various concentrations of test compound or control antibiotics as described above. Following a 5-mins incubation at room temp., [3H] glycerol was added at 0.5 µCi per reaction. Reactions were allowed to proceed at room temperature for 40 min and then stopped through the addition of 375 µL of chloroform/methanol (1:2) followed by vortexing for 20 sec after. Chloroform (125 μL) was then added to each reaction and vortexed, followed by the addition of 125 µL dH2O and vortexing. Reactions were centrifuged at 13,000 rpm for 10 min, and then 150 µL of the organic phase was transferred to a scintillation vial and allowed to dry in a fume hood for at least 1 hr. Samples were then counted via liquid scintillation counting.
Antibacterial Activity Assay
The antibacterial activity of 7771–0701 and analogs against Staphylococcus aureus ATCC 29213 and Escherichia coli 25922 was carried out in a 96-well turbidimetric assay essentially as described previously with the following modifications: bacteria were exposed for 30 min to compounds in 10 mM phosphate buffer containing 5% DMSO prior to addition of 2 x Muller-Hinton medium. Bacterial growth was monitored for 12 hrs and data were analyzed as described (26). Determination of the Minimal Inhibitory Concentrations (MIC) by dilution was carried out by broth dilution according to CLSI standards.21
Results
We used mechanism-of-action studies to assess the mode of bacterial killing by the lead 7771–0701 compound (Figure 1). 7771–0701 most potently inhibited cell wall synthesis (IC50 of 0.29 μg/mL vs 0.64, 0.53 and 0.85 μg/mL for DNA, protein or lipid synthesis, respectively).
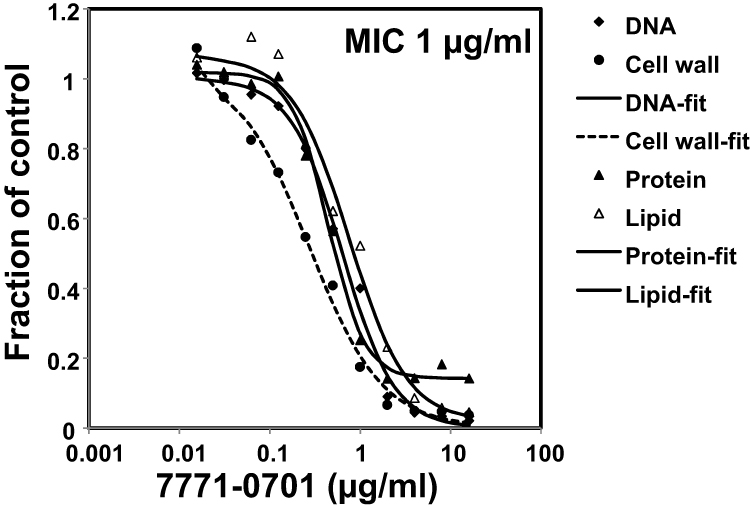 |
Figure 1 Mechanism of action studies of 7771–0701. Exponentially growing S. aureus 29213 cells were exposed to compound and comparators in triplicate using 2.5% DMSO as “no drug” control. Cells were added to Mueller-Hinton Broth or M9 medium for protein synthesis and further incubated in the presence of 13CN-acetyl glucosamine (cell wall), 3Hglycerol (lipid), 3HThymidine (DNA), or 3HLeucine (protein). Following incubation, reactions were stopped by adding TCA (DNA, protein), 8% SDS (cell wall) or chloroform/methanol (lipid) and analyzed by scintillation counting. |
We next used NMR spectroscopy to verify the interaction between Lipid II and this compound, 7771–0701. The NMR analysis (not shown), indicated a binding mode similar to that of the small molecule Lipid II binder we reported on, BAS00127538.10 Accordingly, a CADD model of 7771–0701 in complex with soluble Lipid II was generated and subjected to 20 ns MD simulations. A stable binding mode was found in the simulations with ΔΔG values relative to BAS0012753810 of −1.0 kcal/mol. Together, these data show that compound 7771–0701 inhibits cell wall biosynthesis by binding to Lipid II.
The complex structure from MD simulation as shown in Figure 2 suggests compound 7771–0701 binds with lipid II in a similar way as BAS00127538.10 The indolene moiety in 7771–0701 wraps around the aliphatic tail of lipid II and methylated phenyl ring in the benzothiazole group faces sugar moiety in lipid II. Positive charged nitrogen in benzothiazole group interacts with lipid II phosphate group.
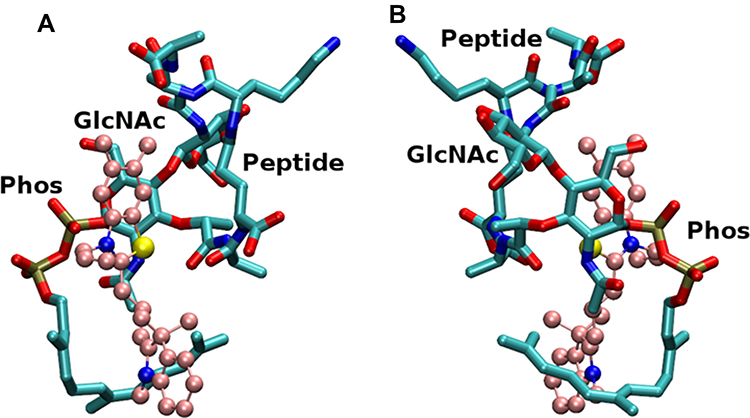 |
Figure 2 Model of lead compound 7771–0701 (CPK representation with carbons in pink color) in complex with lipid II (licorice representation with carbons in cyan color), based on the NMR data. The phosphate (Phos), sugar (GlcNAc) and pentapeptide (Peptide) of Lipid II are labeled. (A) and (B) panels are approximately 180 degree rotation of the complex. |
To identify optimized lipid II-binding analogs and establish SAR, we synthesized 26 derivatives of the 7771–0701 scaffold (see Scheme 1 and Supporting information), based on the CADD modeling. The first generation of molecules synthesized were to simplify the 7771–0701 scaffold that would allow access to a wide array of functionalized analogs (Scheme 1).
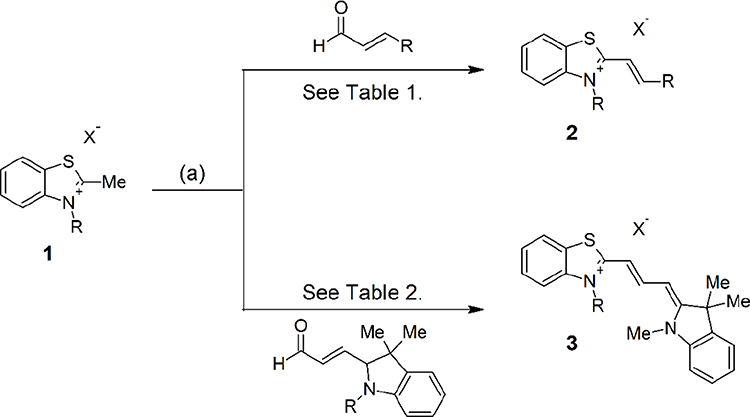 |
Scheme 1 General synthetic route to 7771 analogs. Note: (a) Aldehyde (1 eq.), acetic anhydride, 100 deg. C, 5 h. |
2-methylbenzothiazole salts 1, where R = Me, were reacted with aromatic aldehydes in acetic acid to afford alkenyl salt compounds 2 after filtration. These six compounds were tested for activity against S. aureus, E. faecium and were examined for Lipid II binding by surface plasmon resonance (Table 1). Reaction with dimethylamino benzaldehyde gave compound 7jc65-1 with MIC of 8, 16 and kd of 1.0 x 10–6 for S. aureus, E. faecium and Lipid II, respectively. Extending the alkene linker, 7jc65-1, gave a modest increase in activity. Enlarging the steric bulk around the 4-amino position (7jc66-2, 7jc67-1 and 7jc67-2) decreased potency drastically for all three compounds. The most potent compound of the first generation series was 7jc66-1, which gave Lipid II binding affinity of 9.0 x 10–7. Based on these observations, we conclude that simplifying the scaffold did not enhance the functional parameters compared to lead scaffold.
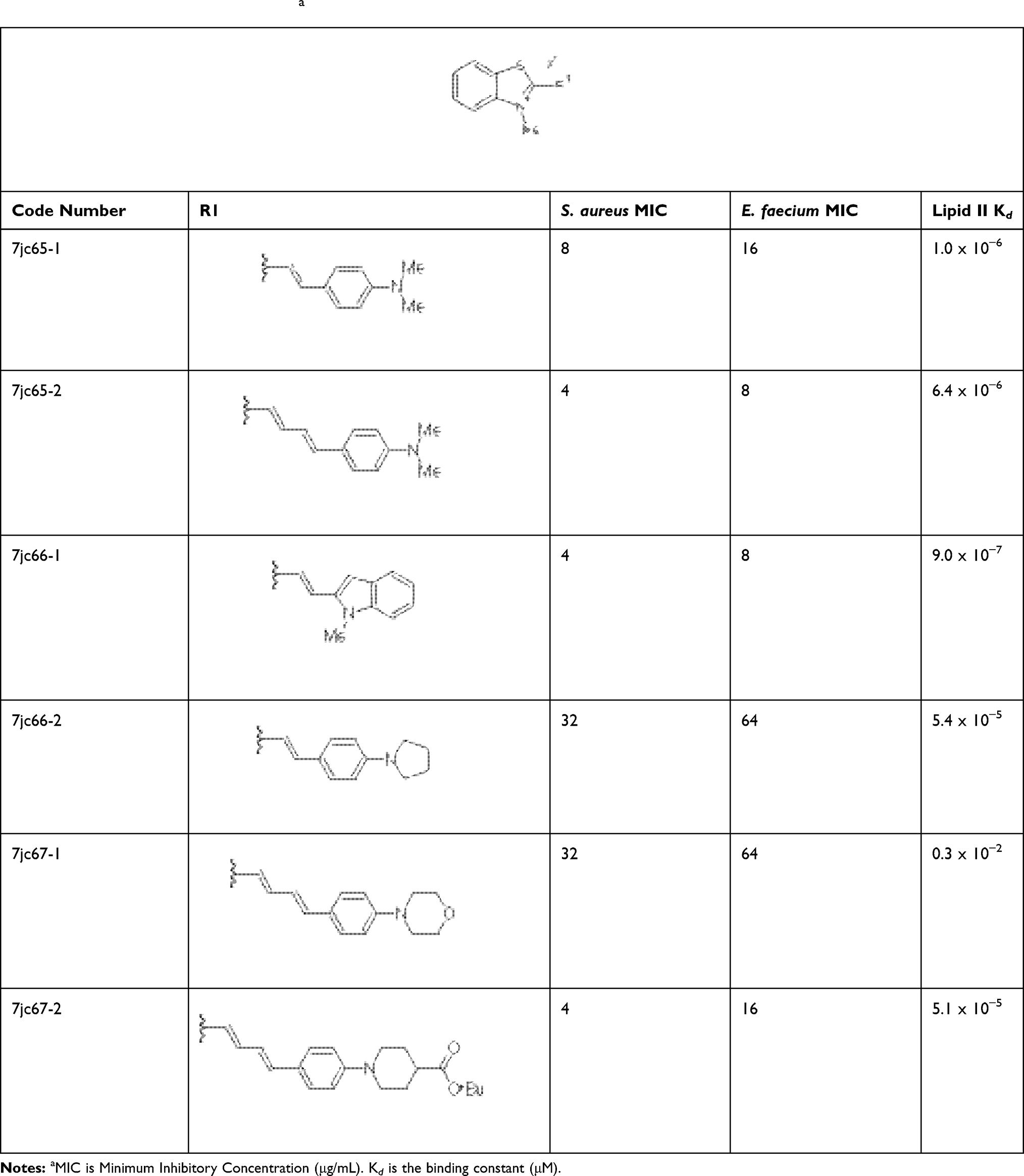 |
Table 1 First-Generation Compoundsa |
We next generated a series of compounds which more closely resembled the lead compound and tested potency against both Gram-positive bacteria as well as determined Lipid II binding affinity. The one-step general procedure is outlined in Scheme 1, where N-alkylated benzothiazoles 1 were condensed with indolene-acetaldehyde to afford compounds 3 after filtration. All 20 compounds gave superior results compared to the first generation series (Tables 1 and 2). 7jc63 where R1 = Me gave MIC of 1, 4 and kd of 7.0 x 10–7 for S. aureus, E. faecium and Lipid II, respectively. Substituting the methyl group to allyl provided 7jc71-1 (R3 = H) which gave comparable results to lead compound 7771 (R3 = OMe).10 Exchanging the R2 aromatic proton to halogens gave no significant increase in potency (7jc71-2 to 7jc71-4), although substituting to an acetate group (7jc73) diminished potency considerably. The importance of sulfur in the aromatic ring is demonstrated by the replacement with oxygen (7jc75-1) and drastically reduced Lipid II affinity, Kd = >64. Incorporating a chlorine in the R4 position (7jc83-1 to 7jc85-2) produced greater potency and comparable binding when compared to 7jc71-1. The introduction of a nitro group at this position (7jc91-1 to 7jc96-1) reduced the antibacterial activity 4- to 8-fold. While the N-allyl group can be replaced by ethyl (7jc81-1) or but-2-ene (7jc81-2), no significant change was observed in the activity when compared to 7jc63 or 7jc71-1. This demonstrates that the N-alkyl group is flexible to changes without affecting functionality. Finally, the greatest potency against Gram-positive bacteria and Lipid II affinity were achieved when a bromide was incorporated at the R3 position of the benzothiazole ring affording compound 7jc75-2 with MIC of 1 against S. aureus, 0.5 against E. faecium and high-affinity Lipid II binding (Kd 5.5 x 10−9).
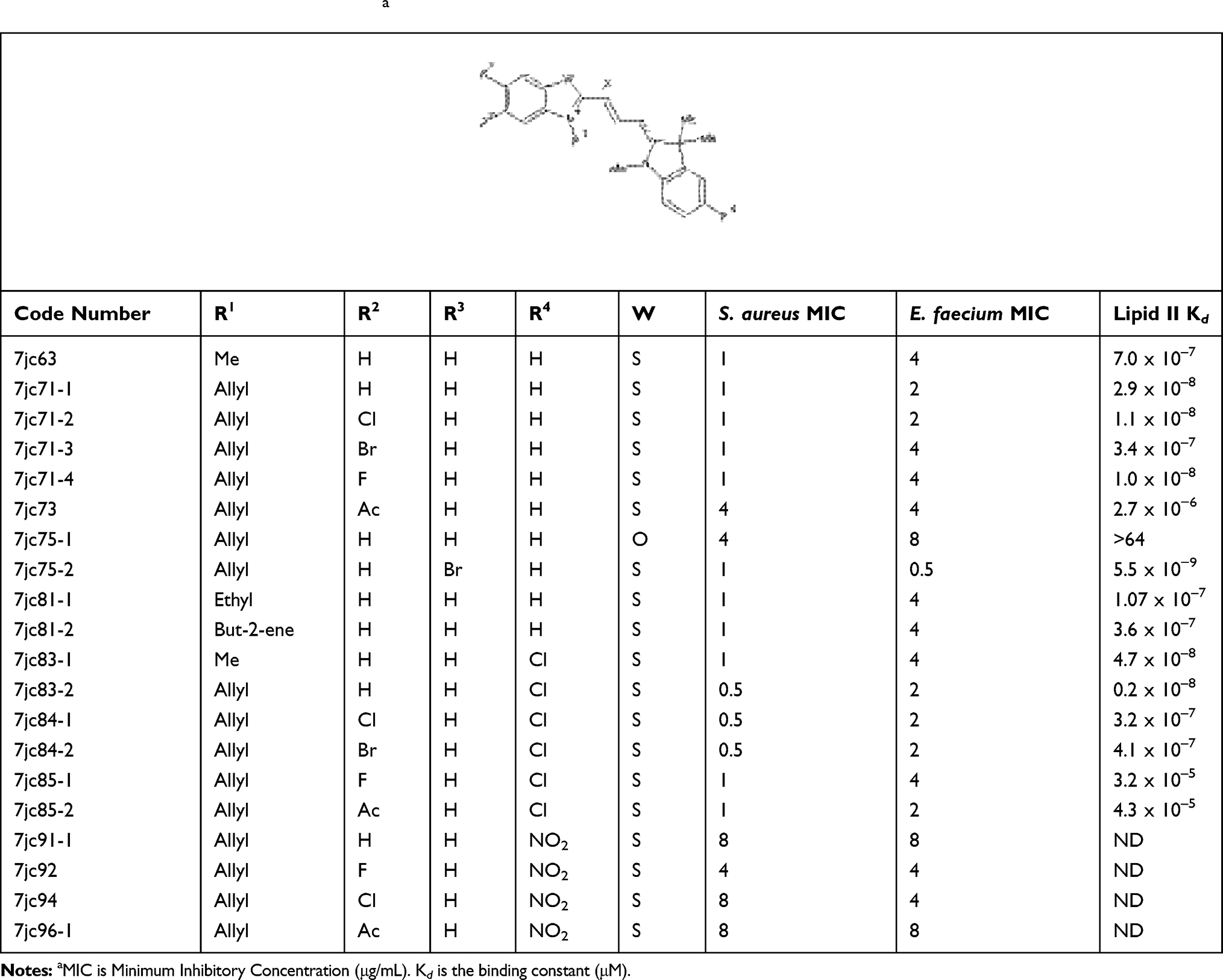 |
Table 2 Second-Generation Compoundsa |
Conclusion
Fully synthetic compounds that bind to Lipid II have not yet been developed. Using CADD, we have identified and optimized a novel scaffold that efficiently binds to this essential bacterial component of cell wall assembly. Subsequent experimental characterization of these compounds showed preferential activity against Gram-positive organisms. 7771–0701 binds to aminosugar moiety of Lipid II, thus making cross-resistance with vancomycin unlikely, since vancomycin binds to the penta-peptide. Studies like these on small molecule Lipid II-binding agents will provide insight for future development, design and synthesis of such compounds as promising therapeutic leads.
Abbreviations
S. aureus, Staphylococcus aureus; E. faecium, Enterococcus faecium; MIC, Minimal Inhibitory Concentration; MurNAc, N-acetyl muramic acid.
Author Contributions
All authors contributed to data analysis, drafting and revising the article, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Funding
This work was supported by Center for Biomolecular Therapeutics (PR155EDL1; UMB), the UMB Computer-Aided Drug Design Center, a UM Ventures Seed grant and Center for Maryland Advanced Ventures grant to EdL.
Disclosure
AM Jr and EdL are inventors on US patents # 8,796,323 and # 9,351,963 to University of Maryland, Baltimore; JC, SF, AM Jr and EdL are inventors on PCT/US11/59432. AM Jr is a co-founder and CSO for SilcsBio LLC. The authors report no other conflicts of interest in this work.
References
1. Walsh C. Where will new antibiotics come from? Nat Rev Microbiol. 2003;1(1):65–70. doi:10.1038/nrmicro727
2. Breukink E, de Kruijff B. Lipid II as a target for antibiotics. Nat Rev Drug Discov. 2006;5(4):321–332. doi:10.1038/nrd2004
3. Koch AL. Bacterial wall as target for attack: past, present, and future research. Clin Microbiol Rev. 2003;16(4):673–687. doi:10.1128/CMR.16.4.673-687.2003
4. Schneider T, Sahl HG. Lipid II and other bactoprenol-bound cell wall precursors as drug targets. Curr Opin Investig Drugs. 2010;11(2):157–164.
5. van Heijenoort J. Lipid intermediates in the biosynthesis of bacterial peptidoglycan. Microbiol Mol Biol Rev. 2007;71(4):620–635. doi:10.1128/MMBR.00016-07
6. Hsu ST, Breukink E, Tischenko E, et al. The nisin-lipid II complex reveals a pyrophosphate cage that provides a blueprint for novel antibiotics. Nat Struct Mol Biol. 2004;11(10):963–967. doi:10.1038/nsmb830
7. Muller A, Ulm H, Reder-Christ K, Sahl HG, Schneider T. Interaction of type A lantibiotics with undecaprenol-bound cell envelope precursors. Microb Drug Resist. 2012;18(3):261–270. doi:10.1089/mdr.2011.0242
8. Schneider T, Kruse T, Wimmer R, et al. Plectasin, a fungal defensin, targets the bacterial cell wall precursor Lipid II. Science. 2010;328(5982):1168–1172. doi:10.1126/science.1185723
9. de Leeuw E, Li C, Zeng P, et al. Functional interaction of human neutrophil peptide-1 with the cell wall precursor lipid II. FEBS Lett. 2010;584(8):1543–1548. doi:10.1016/j.febslet.2010.03.004
10. Varney KM, Bonvin AM, Pazgier M, et al. Turning defense into offense: defensin mimetics as novel antibiotics targeting lipid II. PLoS Pathog. 2013;9(11):e1003732. doi:10.1371/journal.ppat.1003732
11. Brooks BR, Brooks CL
12. Klauda JB, Venable RM, Freites JA, et al. Update of the CHARMM all-atom additive force field for lipids: validation on six lipid types. J Phys Chem B. 2010;114(23):7830–7843. doi:10.1021/jp101759q
13. MacKerell AD
14. Best RB, Zhu X, Shim J, et al. Optimization of the additive CHARMM all-atom protein force field targeting improved sampling of the backbone φ, ψ and side-chain χ1 and χ2 dihedral angles. J Chem Theory Comp. 2012;8:3257–3273. doi:10.1021/ct300400x
15. Jorgensen WL. Transferable intermolecular potential functions for waters, alcohols, and ethers. application to liquid water. J Am Chem Soc. 1981;103:335. doi:10.1021/ja00392a016
16. Vanommeslaeghe K, Hatcher E, Acharya C, et al. CHARMM general force field: a force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J Comp Chem. 2010;31(4):671–690.
17. Vanommeslaeghe K, Mackerell AD
18. Vanommeslaeghe K, Raman EP, MacKerell AD
19. Åqvist J, Medina C, Samuelsson JE. A new method for predicting binding affinity in computer-aided drug design. Protein Eng. 1994;7:385–391. doi:10.1093/protein/7.3.385
20. Breukink E, van Heusden HE, Vollmerhaus PJ, et al. Lipid II is an intrinsic component of the pore induced by nisin in bacterial membranes. J Biol Chem. 2003;278(22):19898–19903. doi:10.1074/jbc.M301463200
21. CLSI. Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria That Grow Aerobically; Approved Standard—Eighth Edition. 2009.
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.