Back to Journals » Neuropsychiatric Disease and Treatment » Volume 10
Optimal management of seizures associated with tuberous sclerosis complex: current and emerging options
Received 22 June 2014
Accepted for publication 21 July 2014
Published 23 October 2014 Volume 2014:10 Pages 2021—2030
DOI https://doi.org/10.2147/NDT.S51789
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Shelly Wang,1 Aria Fallah2,3
1Department of Neurosurgery, University of Toronto, Toronto, ON, Canada; 2Department of Neurosurgery, Miami Children’s Hospital, Miami, FL, USA; 3Department of Clinical Epidemiology and Biostatistics, McMaster University, Hamilton, ON, Canada
Abstract: Seizures are clinically significant manifestations associated with 79%–90% of patients with tuberous sclerosis complex. Often occurring within the first year of life in the form of infantile spasms, seizures interfere with neuropsychiatric, social, and cognitive development and carry significant individual and societal consequences. Prompt identification and treatment of seizures is an important focus in the overall management of tuberous sclerosis complex patients. Medical management, either after seizure onset or prophylactically in infants with electroencephalographic abnormalities, is considered first-line therapy. Vigabatrin and adrenocorticotropic hormone have emerged over the past few decades as mainstay pharmacologic modalities. Furthermore, emerging research on mammalian target of rapamycin inhibitors demonstrated promise for the management of seizures and subependymal giant cell astrocytoma. For appropriate surgical candidates with an epileptogenic zone associated with one or more glioneuronal hamartomas, ideally in noneloquent cortex, resective surgery can be considered, which provides a cure in 56% of patients. For medically refractory patients who do not meet criteria for curative surgery, palliative surgical approaches focused on reducing seizure burden, in the form of corpus callosotomy and vagus nerve stimulation, are alternative management options. Lastly, the ketogenic diet, a reemerging therapy based on the anticonvulsant effects of ketone bodies, can be utilized independently or in conjunction with other treatment modalities for the management of difficult-to-treat seizures.
Keywords: epilepsy, adrenocorticotropic hormone, vigabatrin, mammalian target of rapamycin, ketogenic diet, vagus nerve stimulation
Introduction
Tuberous sclerosis complex (TSC) is an autosomal dominant neurocutaneous disorder characterized by the formation of hamartomas in multiple organ systems. TSC is estimated to affect 25,000–40,000 individuals in the United States and 1–2 million individuals worldwide, with a prevalence of 1 in 6,000 live births.1
TSC1 or TSC2 mutations are found in over 85% of patients with TSC, either through autosomal dominant inheritance, de novo mutations, or rarely, gonadal mosaicism.2 De novo mutations account for approximately 80% of TSC cases.3 The prevalence of TSC2 mutations is four times as common as TSC1 mutations among de novo cases and is approximately equal among familial TSC cases.4
TSC1, located on chromosome 9, codes for the hamartin protein, which is widely expressed in normal tissues. TSC2, located on chromosome 16, codes for the tuberin protein, which is expressed ubiquitously in all tissues. These two proteins interact in the Golgi complex, and the hamartin–tuberin complex acts as a tumor-suppressor protein through the Ras homologue enriched in brain protein to limit activation of the mammalian target of rapamycin (mTOR) complex 1. In TSC, inadequate suppression of the mTOR complex 1 leads to abnormal cellular growth, proliferation, and protein synthesis.5–7 The severity and phenotype of TSC among individuals varies due to mosaicism, differences between TSC1 and TSC2 genes, and a variety of mutation types found in each gene.
The central nervous system is one of the most commonly affected systems in TSC. Brain pathology includes subependymal nodules or subependymal giant cell astrocytoma (SEGA), glioneuronal hamartomas (cortical tubers), and white matter heterotopia. These findings are present in approximately 90% of children with TSC and are associated with abnormal cognitive and seizure development.8,9 Due to the associations between epilepsy, development, and neuropsychiatric outcome, prompt evaluation and management of seizures is a clear focus of interest. In this article, the medical, surgical, and emerging management options will be discussed.
Seizures in TSC
Seizures are the most common presenting features of TSC, affecting 73%–90% of patients in population-based studies.8,10,11 The majority (63%) of TSC patients experience seizure onset within the first year of life, although adults without seizure history continue to be at risk, with 12% of adults in this subgroup later developing epilepsy.8,12 Epilepsy develops in 96%–99% of TSC patients with a single seizure.11,12 Infantile spasm (IS) is the most common initial seizure subtype, although 54% of TSC patients develop multiple seizure types, including simple partial, complex partial, and secondary generalized seizures.12,13 IS, characterized by hypsarrhythmia on electroencephalography (EEG) and Lennox-Gastaut syndrome, characterized by frequent seizure activity of different morphology, are significant risk factors for the development of medically refractory epilepsy.
In patients with TSC, seizures are closely intertwined with global development. Specifically, cognitive disability is associated with a history of IS, refractory seizures, and to a lesser extent, the number or volume of glioneuronal hamartomas. A newly recognized entity of TSC-associated neuropsychiatric disorders describes the interrelated clinical manifestations of cerebral dysfunction, including aggressive behaviors, autism spectrum disorders, intellectual disabilities, psychiatric disorders, neuropsychological deficits as well as school and occupational difficulties.14 In a population-based study of TSC patients with early-onset epilepsy, refractory seizures (55%), intellectual disability (80%), and autism (30%) were prevalent.15 It is evident that a complex relationship exists between TSC, epilepsy, treatment of epilepsy, and neuropsychiatric outcomes.
In addition to interference with neuropsychiatric development, children with epileptic encephalopathy experience significant sleep disturbance patterns. This was manifested in altered sleep-related polysomnography parameters including reduced total sleep time and sleep percentage, increased number of awakenings, increased rate in stage shifting, and higher rapid eye movement latency.16
Due to the importance of early recognition and management of seizures, all TSC patients should undergo a baseline EEG evaluation. If it is abnormal, or in the case of patients exhibiting TSC-associated neuropsychiatric disorders, a 24-hour video EEG should be considered to assess for unrecognized or subclinical seizure activity.17 Furthermore, all patients with suspected TSC, regardless of age, should undergo brain magnetic resonance (MR) imaging at diagnosis and every 1–3 years until age 25 for surveillance of cerebral pathology.18
The characteristic brain lesion in TSC is SEGA, which is a World Health Organization grade 1, slow-growing tumor that usually arises in the periventricular area, sometimes from subependymal nodules.19 Due to its propensity toward growth near the foramen of Monro, it carries a clinically significant risk of morbidity and mortality from acute hydrocephalus, which is directly proportional to the SEGA volume.20 Secondly, cortical glioneuronal hamartomas are composed histologically of enlarged atypical and disorganized neuronal and glial elements with astrocytosis. Because glioneuronal hamartomas form during embryogenesis, disruption of normal cortical development and function occurs early in gestation, leading to a predisposition to seizures and cognitive deficits. The number and volume of tubers, as demonstrated by the tuber/brain proportion, have been linked to poor intelligence and cognitive indices.21,22 Additionally, a subgroup of cyst-like tubers that show low signal intensity on fluid-attenuated inversion recovery and T1-weighted sequences and increased signal on T2-weighted sequences are associated with increased frequency of IS, epilepsy, and refractory epilepsy, and may represent a more epileptogenic focus and aggressive seizure phenotype.23 Lastly, white matter heterotopia are radial lines extending from the ventricles to the cortex, often in relation with subependymal nodules, and are thought to represent demyelination, dysmyelination, or hypomyelination from a migrational disorder.24
Early management of seizures is important in preventing subsequent epileptic encephalopathy and in reducing the cognitive and neuropsychiatric consequences.25,26 In one study of long-term neurologic outcome of TSC children with early-onset epilepsy, a shorter duration between seizure onset and start of antiepileptic treatment led to reduced risk of epileptic encephalopathy (including autism and intellectual disability).15 In an open-label randomized trial, early implementation of antiepileptic drugs in TSC infants with multifocal EEG abnormalities, before the onset of seizures, resulted in a higher ratio of seizure-free patients, lower incidence of drug-resistant epilepsy, and lower number of patients requiring polytherapy at 24 months of age.27 Although no standards exist, some clinicians advocate for prophylactic treatment of seizures based on this data. Comprehensive assessment in a center familiar with TSC management, involvement of a multidisciplinary team, and early initiation of treatment are considered key principles in the management of seizures in the TSC population.
Medical management
Adrenocorticotropic hormone (ACTH) and vigabatrin (VGB) are considered mainstay therapies for the treatment of seizures in TSC patients. Over the past few decades, emerging clinical evidence has supported the use of VGB as a first-line agent for TSC patients with IS. However, the adverse event of retinal toxicity remains a clinically relevant consideration.
Other medications can be considered for other seizure types, including carbamazepine for partial or complex partial seizures and rufinamide for focal or atonic seizures. Furthermore, there is emerging evidence supporting the use of mTOR inhibitors for the treatment of seizures in TSC patients with SEGA.
ACTH
ACTH, in addition to steroids (hydrocortisone, prednisone, or dexamethasone), has been used since the 1950s for the treatment of IS and Lennox-Gastaut syndrome. ACTH has demonstrated superior efficacy in comparison to steroids and is considered one of the first-line therapies for TSC-related seizures. In addition to stimulating the release of adrenal glucocorticoids, ACTH also suppresses the production of the stress-activated neuropeptide corticotropin-releasing hormone (CRH) through steroid-independent activation of central melanocortin receptors.28
In prospective trials of ACTH therapy for IS, cessation of seizures occurred in 42%–87% of infants, with increased efficacy in patients with cryptogenic IS compared to symptomatic IS (Table 1). The time from initiation of treatment to cessation of seizures averaged from 7 to 12 days. In some cases, the effects of ACTH were found to persist after therapy discontinuation. Two prospective randomized controlled trials demonstrated that the efficacy of low dose ACTH therapy (either 0.2 IU/kg/day or 20–30 IU/day) was comparable to higher dose therapy and resulted in decreased adverse effects.29,30 Although there is insufficient evidence to precisely define the optimum ACTH dose and duration, clinicians can consider a 2-week course of ACTH at 20–30 IU/day, followed by a careful taper to discontinuation.31,32
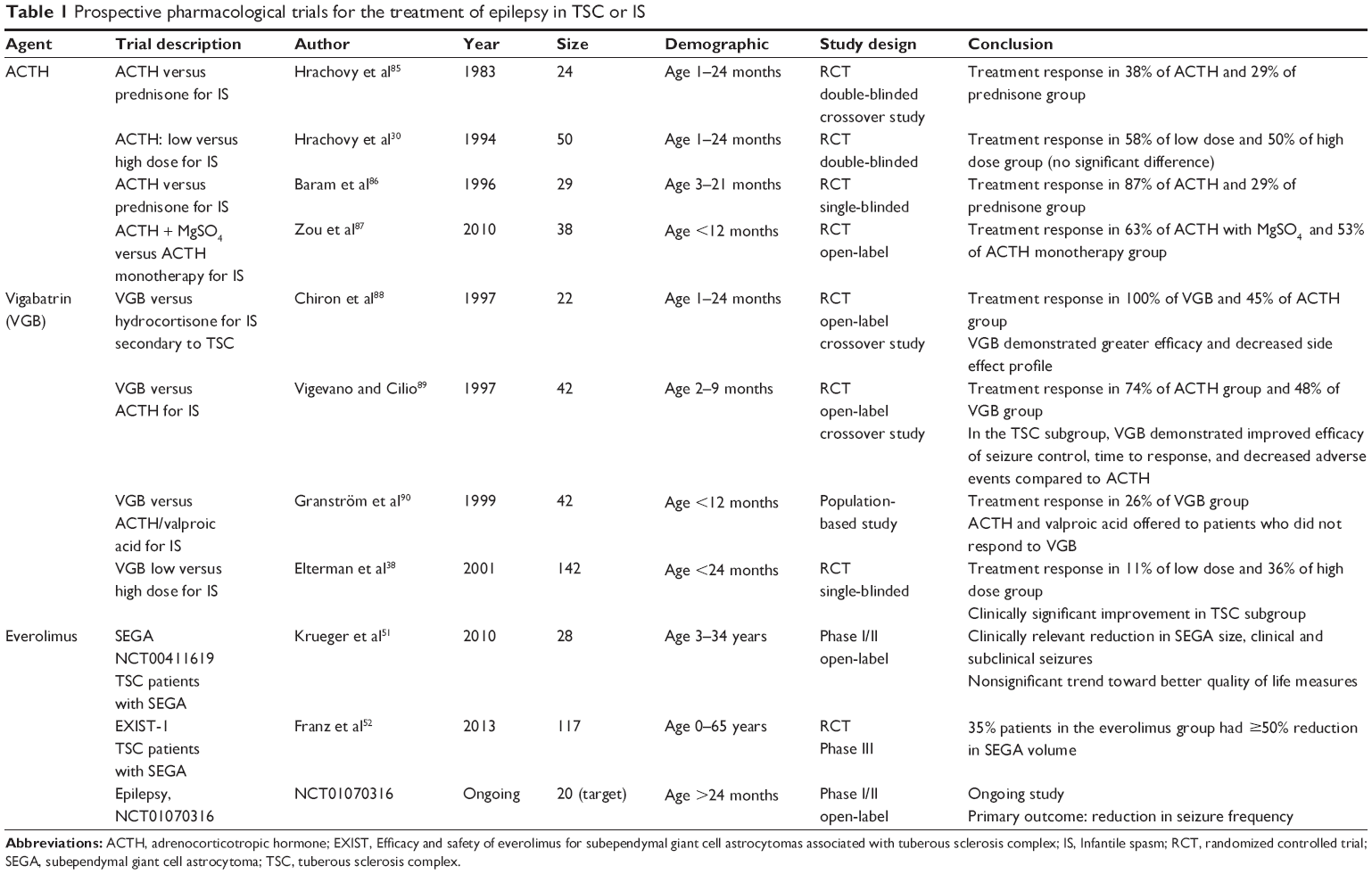 | Table 1 prospective pharmacological trials for the treatment of epilepsy in TSC or IS |
Adverse effects occurred in 37%–85% of treated patients, most commonly being hypertension, immune suppression and infection, electrolyte imbalance, gastrointestinal disturbances, ocular opacities, hypertrophic cardiomyopathy, cerebral atrophy, and growth impairment.33,34 A transient cushingoid appearance, hirsutism, irritability, and sleep disorders were also observed. Patients receiving ACTH treatment should be monitored closely for hypertension, electrolyte abnormalities, and infections.
VGB
VGB is an antiepileptic drug that inhibits the catabolism of gamma-aminobutyric acid (GABA) by irreversibly inhibiting GABA transaminase. VGB was approved by the US Food and Drug Administration (FDA) in 2009 as an adjunctive therapy for adult patients with refractory partial complex seizures and as monotherapy for pediatric patients aged 1 month to 2 years with IS. It has emerged as a first-line treatment for TSC patients with IS.
VGB has shown efficacy in the treatment of TSC patients with IS and partial seizures in a number of retrospective and prospective studies, with a response rate of 73%–96%.35 In a retrospective review of partial seizures, 25% of patients became seizure-free or experienced a >90% decrease in seizure frequency with the addition of VGB to their regimens.36 Children with epilepsy onset after 1 year of age were most likely to demonstrate a good response to VGB treatment. Even in the case of treatment-resistant children (to carbamazepine, benzodiazepines, and sodium valproate) with IS, the addition of VGB led to seizure reduction in the majority of children, while 43% showed complete suppression of spasms, the best short- and long-term outcomes observed in those with TSC.37 Prospective trials comparing VGB and ACTH also demonstrated its superiority in seizure control, time to control of symptoms, and in some cases favorable side effect profile (Table 1). Therefore, VGB has been identified by many countries as the first-line medical therapy for the treatment of TSC-related seizures.
The optimal dosage of VGB is unclear, although a range of 18–200 mg/kg/day has been used in literature.32 Two randomized trials demonstrated that higher (100–148 mg/kg/day) doses of VGB led to increased seizure cessation and shorter time to response compared to lower doses of therapy.38,39 The US consensus report on the management of IS recommends initial commencement of VGB at a dose of 50 mg/kg/day, escalating to 100–150 mg/kg/day as required for clinical response.31
Adverse events of VGB include somnolence, hypotonia, weight gain, excitation, and insomnia. The most significant adverse effect is peripheral visual field defect from retinal cone and amacrine cell dysfunction. The prevalence varies according to patient age and extent of exposure to VGB and is approximately 25%–50% in adults, 15% in children, and 15%–31% in infants.40 For patients initiated on VGB treatment, age-appropriate visual field testing is required at baseline and then repeated at intervals in patients who continue therapy. VGB has also been reported to produce MR imaging abnormalities in infants treated for IS.41,42 These abnormalities, including hyperintense lesions on T2-weighted images with restricted diffusion on diffusion-weighted imaging in the basal ganglia, thalamus, brainstem, and dentate nucleus appear to be a subclinical, dose-related phenomenon that resolves with discontinuation of VGB.
The time from initiation of therapy to cessation of spasms ranged from 12 to 35 days; during this time, the risk of peripheral visual field defect development is low.32 VGB should be discontinued if no clinical improvement is observed within 12 weeks. If patients have a meaningful reduction in seizures or achieve seizure freedom, then the clinician and patient or caregiver must determine the benefits and risks of VGB therapy continuation. Mixed evidence exist for discontinuation of VGB therapy following a period of clinical benefit. Some data suggest that VGB can be withdrawn in children who have been seizure-free for 6 months,43 although other literature revealed spasm relapse after therapy discontinuation.44
mTOR inhibitors
mTOR is a protein kinase that controls cell growth, proliferation, and survival. mTOR inhibitors bind to a domain separate from the catalytic site and inhibit a subset of mTOR functions. Sirolimus (rapamycin) and everolimus have been shown to be effective in the treatment of upregulation in cancers, and preliminary data for its use in the TSC population are promising.45,46 Everolimus was recently approved by the FDA in 2010 to treat SEGA in TSC following encouraging animal and human clinical data.
Case studies of TSC patients with medically refractory epilepsy and SEGA demonstrated dramatic reduction of seizure frequency and SEGA size with the administration of sirolimus and everolimus.47–49 A study involving five TSC patients with either SEGA or pilocytic astrocytoma who were treated with oral sirolimus demonstrated either lesional regression or necrosis. One of these patients had regrowth of a SEGA after having received sirolimus treatment that was interrupted for 4 months.50 A Phase II trial has demonstrated clinically relevant reductions of SEGA volume, including mean reductions of ≥30% from baseline in 75% of patients and ≥50% from baseline in 32% of patients.51 The overall frequency of clinical seizures and electrographic seizures, as assessed by 24-hour video EEG, was also significantly decreased at 6 months. No patients developed new lesions, worsening hydrocephalus, or evidence of increased intracranial pressure, and no patients required surgical resection or other SEGA treatment. In a Phase III double-blind randomized controlled trial of 177 patients, 42% of patients had reductions in total SEGA volume by ≥50%. Most importantly, the reduction in SEGA volume was maintained in patients who received everolimus for ≥3 years, and a reduction of ≥30% in the volume of the primary SEGA lesion was maintained for a median duration of 24 months.52 Furthermore, diffusion tensor imaging showed improvements from baseline in the integrity of normal-looking white matter at 12 and 18 months.
Adverse effects of mTOR inhibitors include immunosuppression. In the Phase III trial, 49% of patients receiving everolimus had adverse events requiring dose reduction or temporary interruption of treatment. Most were grade 1–2 adverse effects consisting of gastritis, mouth ulceration, pyrexia, and pneumonia. No adverse events led to discontinuation from the study, and no patients died during the study.52 mTOR inhibitors have emerged as a promising treatment modality that may play a large role in the treatment paradigm of TSC patients.
Surgical management
Medical management of TSC-associated epilepsy is considered first-line therapy although some patients remain medically refractory. In one population-based study, 37% of patients had drug-resistant epilepsy.11 In this subset of patients, there may be a role for curative resective strategies or palliative disconnective or neuromodulatory strategies.
Resective strategies
If an epileptogenic zone (EZ) associated with one or more glioneuronal hamartomas, ideally in noneloquent cortex, can be localized, resective surgery may be offered as a cure. Resective surgery may be offered in the form of tuberectomy(ies), lobectomy, multilobar resection, and hemispherectomy. Determination of surgical candidacy, technique, and specific risks of the operation is a complex process requiring multidisciplinary collaboration. The groups’ recommendation is presented to the child and/or parent, who must ultimately be willing to accept the risks in hopes of attaining a good seizure outcome. Although similar principles of epilepsy surgery are utilized, the evaluation and ultimate recommendation for each patient is variable from institution to institution and are influenced by personal expertise and local experience with biotechnology (positron emission tomography, single-photon emission computed tomography, magnetoencephalography, and invasive EEG) available for localization of the EZ.
If resective surgery is an option, 56% of these patients may achieve seizure freedom following an operation.53 Other benefits include the possibility of decreasing or discontinuing antiepileptic drugs, ability to obtain/retain employment, ability to drive, improved independent functioning, and improved social relationships. Resective strategies still leave a large proportion of children (>40%) with ongoing seizures. The major surgical morbidity rate is approximately 3%, which includes early postoperative death (secondary to hemorrhage, infection, and hydrocephalus) and late postoperative death (unexplained or related to seizures).
Given the invasiveness and irreversibility of surgical resection, it is of utmost importance to develop strategies to identify appropriate candidates. An individual participant data meta-analysis was performed to identify such predictors. Predictors of favorable seizure outcomes (Engel classification 1 or 2; Table 2) following resective epilepsy surgery include absence of generalized seizure semiology, mild or no developmental delay, unifocal ictal scalp EEG abnormality, and EEG/MR imaging concordance.53 However, there are several important challenges in identification of predictors of seizure outcome in these children, the two most important being the rare incidence of disease and variability between centers in determining epilepsy surgery candidacy.
 | Table 2 Engel classification of seizure outcomes following surgical resection |
Palliative strategies
The goal of palliative surgical approaches toward epilepsy is to control the symptoms or reduce seizure burden and subsequently increase the quality of life. Palliative techniques include disconnective (corpus callosotomy [CC]) and neuromodulatory (vagus nerve stimulation [VNS]) procedures for the relief of seizure frequency.
CC
CC is a disconnective procedure characterized by division of the corpus callosum to limit the spread of ictal discharges and reduce the occurrence of generalized epileptic symptomatology. The indications for CC are primarily frontal and bilaterally generated seizures. TSC patients with atonic seizures were the most responsive to CC.54 Seizure remission was achieved in 90% of TSC patients who underwent complete CC and 67% who underwent partial CC.54 Prior to offering CC, a thorough evaluation is required to ensure that there are no clearly defined focal EZ that could be amenable to a resective strategy. The benefits of CC over VNS include its availability, lower cost, and freedom from hardware-related complications. The limitations include its invasiveness and irreversibility.
Specific surgical complications from a CC include injury to the superior sagittal sinus and bridging veins, pericallosal artery ischemia, cingulate contusions, and bilateral mesiofrontal damage. CC is associated with a disconnection syndrome, which includes the inability to name objectives presented to the left hemifield, left hemi alexia, left hemianopia, unilateral tactile anomia, and unilateral left agraphia. The overall morbidity is 5% while the mortality is 1% from this procedure.54 To minimize morbidity incurred from a disconnection syndrome, some physicians may elect to perform an anterior two-thirds CC prior to consideration of a full CC, if the seizures continue to be intractable. However, recent observational studies have supported a one-stage complete CC procedure, especially in severely affected or lower-functioning children, for improved seizure control.55–57
VNS
An alternative palliative strategy includes surgery for neuromodulation in the form of VNS. Children with TSC and medically intractable epilepsy who are not considered candidates for a resective procedure may be candidates for VNS implantation. In 1997, the VNS Therapy System™ was approved by the FDA as adjunctive therapy for adults and adolescents over 12 years of age whose partial-onset seizures were refractory to antiepileptic drugs. The main indications of VNS include nonlocalizable medically intractable epilepsy, those that fail surgical resection, or who have bilateral cortical localization. Similar to CC, VNS plays an important role in the treatment of atonic seizures.
The vagus nerve comprises approximately 20% efferent fibers and 80% afferent fibers, which serve as the target of VNS.58 Afferent vagal pathways lead to bilateral synapses on the nucleus of the solitary tract and to various cerebral regions involved in seizure generation. Vagus nerve pathways of seizure modulation of significance include projections to the pontine parabrachial nucleus, the parabrachial nucleus, and the locus ceruleus. Animal studies have revealed antiepileptogenic properties of VNS through a variety of mechanisms, including abortion of ongoing seizures, acute seizure prophylaxis, chronic seizure prophylaxis, and inhibition of epileptogenesis in animal models of seizure kindling.59–65
The procedure involves an anterior cervical incision where the electrode leads are wrapped along the left vagus nerve in the carotid sheath. A separate subclavicular incision is made where the pulse generator is inserted into a subcutaneous pocket. Adverse effects include voice alteration, hoarseness, and coughing during stimulation. Following insertion, the VNS requires programming and lifetime follow-up with an epileptologist and intermittent battery change, the frequency of which is determined by the stimulation parameters. The patient is discharged from hospital with a magnet that can be held over the pulse generator to abort seizures.
VNS is generally efficacious, with limited adverse events. Four reviews with a combined total of 38 children demonstrated VNS as an effective and well-tolerated adjunctive therapy for refractory seizures. Overall, 72% reported at least 50% seizure reduction.66–69 Furthermore, a significant improvement in quality of life and neuropsychological outcome (attention, cognition, behavior, mood) was recognized.70,71
Ketogenic diet
The ketogenic diet (KD) is a high fat, adequate protein, and low carbohydrate diet that results in a state of ketosis. Initially developed in the early 1920s,72 the KD had fallen into disuse during the 1970s with the rapid development of new antiepileptic drugs. In the past decade, resurgence of its therapeutic use had a major impact on the treatment of refractory seizures.
The KD leads to changes in plasma levels of ketones, insulin, glucose, glucagon, and free fatty acids. Formation of ketone bodies in the liver from long and medium-chain fatty acids have a direct anticonvulsant effect when crossing the blood–brain barrier. Increased mitochondrial biogenesis, oxidative phosphorylation, enhanced GABA levels, reduced neuronal excitability and firing, and stabilized synaptic function have been shown to occur in patients on the KD.73,74 Additionally, the roles of carbohydrate reduction, activation of adenosine triphosphate-sensitive potassium channels by mitochondrial metabolism, inhibition of the mTOR pathway, and inhibition of glutamatergic excitatory synaptic transmission have been described.75,76
Animal studies and human clinical studies of the KD have both demonstrated efficacy in the treatment of medically refractory seizures. In one study of 150 medically refractory children (on an average of 6.2 medications), improved seizure control was observed immediately after initiation of the KD and during a 3–6 year follow-up.13,14 Twelve months after initiating the diet, 7% of the children were seizure free and another 20% had ≥90% decrease in seizures; 3–6 years later, 27% of these same children had fewer or no seizures. Several recent meta-analyses of the KD for seizure control revealed complete seizure cessation achieved in 10%–16%, ≥90% reduction in seizures in 32%, and ≥50% reduction in seizures in 33%–56%.77–80 In one small study of 12 TSC patients, 92% of children had a ≥50% reduction in seizures at 6 months, and 67% had a ≥90% reduction at 5 months.81 The specific effects of the KD was described in a study of three boys with TSC and refractory partial seizures, who started the diet at 3.5–6 years of age.82 The KD effectively and completely controlled seizures in two patients and significantly decreased the frequency of atonic seizures in the third patient while he was between multistage surgical procedures. The only randomized controlled trial for the role of the KD in medically refractory epilepsy demonstrated statistically significant seizure control: at 3 months, 38% had ≥50% reduction in seizures and 7% experienced ≥90% reduction.83
Early-onset adverse effects associated with the KD include acidosis, hypoglycemia, gastrointestinal distress, dehydration, and lethargy. They are typically transient and easily managed and are minimized if patients are not fasted. Later adverse effects include dyslipidemia, kidney stones, acidosis, growth retardation, and decreased bone density.84 Common reasons for diet intolerability or discontinuation included <50% seizure reduction (47%), diet restrictiveness (16%), and incurrent illness or diet side effects (13%).80
Conclusion
Seizures associated with TSC carry significant individual and societal impact. Often arising early in life, they are difficult to treat and interfere with normal cognitive and neuropsychiatric development. This review focused on the medical, surgical, and emerging management modalities for seizures in the TSC population. Mainstay pharmacologic treatments of ACTH and VGB have emerged as first-line therapies for TSC patients with IS. Emerging evidence for mTOR inhibitors have supported its role for the treatment of epilepsy and SEGA in TSC patients. For individuals with a localized EZ, resective strategies can be considered for cure. Other surgical strategies, including CC and VNS, can be used for palliative purposes to decrease seizure frequency. Lastly, the KD is a reemerging therapy for difficult-to-control seizures of all types. Although the treatment of seizures in TSC patients is complex and often multimodal, groundwork over the past few decades has advanced this field significantly. Emerging research in both medical and surgical modalities continues to hold promise for improved medical and developmental outcomes in this population.
Disclosure
The authors report no conflicts of interest in this work.
References
National Institute of Neurological Disorders and Stroke. Tuberous sclerosis fact sheet. NIH publication number 07-1846 2014 [webpage on the Internet]. Bethesda, MD: National Institutes of Health; 2014 [updated April 16, 2014]. Available from: http://www.ninds.nih.gov/disorders/tuberous_sclerosis/detail_tuberous_sclerosis.htm | ||
Crino PB, Nathanson KL, Henske EP. The tuberous sclerosis complex. N Engl J Med. 2006;355(13):1345–1356. | ||
Roach ES, Sparagana SP. Diagnosis of tuberous sclerosis complex. J Child Neurol. 2004;19(9):643–649. | ||
Au KS, Williams AT, Roach ES, et al. Genotype/phenotype correlation in 325 individuals referred for a diagnosis of tuberous sclerosis complex in the United States. Genet Med. 2007;9(2):88–100. | ||
Curatolo P, Bombardieri R, Jozwiak S. Tuberous sclerosis. Lancet. 2008;372(9639):657–668. | ||
Huang J, Manning BD. The TSC1-TSC2 complex: a molecular switchboard controlling cell growth. Biochem J. 2008;412(2):179–190. | ||
Chan JA, Zhang H, Roberts PS, et al. Pathogenesis of tuberous sclerosis subependymal giant cell astrocytomas: biallelic inactivation of TSC1 or TSC2 leads to mTOR activation. J Neuropathol Exp Neurol. 2004;63(12):1236–1242. | ||
Yates JR, Maclean C, Higgins JN, et al; Tuberous Sclerosis 2000 Study Group. The Tuberous Sclerosis 2000 Study: presentation, initial assessments and implications for diagnosis and management. Arch Dis Child. 2011;96(11):1020–1025. | ||
Inoue Y, Nemoto Y, Murata R, et al. CT and MR imaging of cerebral tuberous sclerosis. Brain Dev. 1998;20(4):209–221. | ||
Joinson C, O’Callaghan FJ, Osborne JP, Martyn C, Harris T, Bolton PF. Learning disability and epilepsy in an epidemiological sample of individuals with tuberous sclerosis complex. Psychol Med. 2003;33(2):335–344. | ||
Vignoli A, La Briola F, Turner K, et al. Epilepsy in TSC: certain etiology does not mean certain prognosis. Epilepsia. 2013;54(12):2134–2142. | ||
Chu-Shore CJ, Major P, Camposano S, Muzykewicz D, Thiele EA. The natural history of epilepsy in tuberous sclerosis complex. Epilepsia. 2010;51(7):1236–1241. | ||
Riikonen R, Simell O. Tuberous sclerosis and infantile spasms. Dev Med Child Neurol. 1990;32(3):203–209. | ||
de Vries P. Neurodevelopmental, psychiatric and cognitive aspects of tuberous sclerosis complex. In: Kwiatkowsi DJ, Whittemore VH, Thiele EA, editors. Tuberous Sclerosis Complex: Genes, Clinical Features. Weinheim: Wiley-Blackwell. 2010:229–267. | ||
Cusmai R, Moavero R, Bombardieri R, Vigevano F, Curatolo P. Long-term neurological outcome in children with early-onset epilepsy associated with tuberous sclerosis. Epilepsy Behav. 2011;22(4):735–739. | ||
Carotenuto M, Parisi P, Esposito M, Cortese S, Elia M. Sleep alterations in children with refractory epileptic encephalopathies: a polysomnographic study. Epilepsy Behav. 2014;35:50–53. | ||
Curatolo P, Jóźwiak S, Nabbout R; TSC Consensus Meeting for SEGA and Epilepsy Management. Management of epilepsy associated with tuberous sclerosis complex (TSC): clinical recommendations. Eur J Paediatr Neurol. 2012;16(6):582–586. | ||
Krueger DA, Northrup H; International Tuberous Sclerosis Complex Consensus Group. Tuberous sclerosis complex surveillance and management: recommendations of the 2012 International Tuberous Sclerosis Complex Consensus Conference. Pediatr Neurol. 2013;49(4):255–265. | ||
Goh S, Butler W, Thiele EA. Subependymal giant cell tumors in tuberous sclerosis complex. Neurology. 2004;63(8):1457–1461. | ||
de Ribaupierre S, Dorfmüller G, Bulteau C, et al. Subependymal giant-cell astrocytomas in pediatric tuberous sclerosis disease: when should we operate? Neurosurgery. 2007;60(1):83–89; discussion 89–90. | ||
Goodman M, Lamm SH, Engel A, Shepherd CW, Houser OW, Gomez MR. Cortical tuber count: a biomarker indicating neurologic severity of tuberous sclerosis complex. J Child Neurol. 1997;12(2):85–90. | ||
Jansen FE, Vincken KL, Algra A, et al. Cognitive impairment in tuberous sclerosis complex is a multifactorial condition. Neurology. 2008;70(12):916–923. | ||
Chu-Shore CJ, Major P, Montenegro M, Thiele E. Cyst-like tubers are associated with TSC2 and epilepsy in tuberous sclerosis complex. Neurology. 2009;72(13):1165–1169. | ||
Iwasaki S, Nakagawa H, Kichikawa K, et al. MR and CT of tuberous sclerosis: linear abnormalities in the cerebral white matter. AJNR Am J Neuroradiol. 1990;11(5):1029–1034. | ||
Bombardieri R, Pinci M, Moavero R, Cerminara C, Curatolo P. Early control of seizures improves long-term outcome in children with tuberous sclerosis complex. Eur J Paediatr Neurol. 2010;14(2):146–149. | ||
Camposano SE, Major P, Halpern E, Thiele EA. Vigabatrin in the treatment of childhood epilepsy: a retrospective chart review of efficacy and safety profile. Epilepsia. 2008;49(7):1186–1191. | ||
Jóźwiak S, Kotulska K, Domańska-Pakieła D, et al. Antiepileptic treatment before the onset of seizures reduces epilepsy severity and risk of mental retardation in infants with tuberous sclerosis complex. Eur J Paediatr Neurol. 2011;15(5):424–431. | ||
Brunson KL, Avishai-Eliner S, Baram TZ. ACTH treatment of infantile spasms: mechanisms of its effects in modulation of neuronal excitability. Int Rev Neurobiol. 2002;49:185–197. | ||
Yanagaki S, Oguni H, Hayashi K, et al. A comparative study of high-dose and low-dose ACTH therapy for West syndrome. Brain Dev. 1999;21(7):461–467. | ||
Hrachovy RA, Frost JD, Glaze DG. High-dose, long-duration versus low-dose, short-duration corticotropin therapy for infantile spasms. J Pediatr. 1994;124(5 Pt 1):803–806. | ||
Pellock JM, Hrachovy R, Shinnar S, et al. Infantile spasms: a U.S. consensus report. Epilepsia. 2010;51(10):2175–2189. | ||
Mackay MT, Weiss SK, Adams-Webber T, et al; American Academy of Neurology; Child Neurology Society. Practice parameter: medical treatment of infantile spasms: report of the American Academy of Neurology and the Child Neurology Society. Neurology. 2004;62(10):1668–1681. | ||
Riikonen R, Donner M. ACTH therapy in infantile spasms: side effects. Arch Dis Child. 1980;55(9):664–672. | ||
Lombroso CT. A prospective study of infantile spasms: clinical and therapeutic correlations. Epilepsia. 1983;24(2):135–158. | ||
Aicardi J, Mumford JP, Dumas C, Wood S. Vigabatrin as initial therapy for infantile spasms: a European retrospective survey. Sabril IS Investigator and Peer Review Groups. Epilepsia. 1996;37(7):638–642. | ||
Friedman D, Bogner M, Parker-Menzer K, Devinsky O. Vigabatrin for partial-onset seizure treatment in patients with tuberous sclerosis complex. Epilepsy Behav. 2013;27(1):118–120. | ||
Chiron C, Dulac O, Beaumont D, Palacios L, Pajot N, Mumford J. Therapeutic trial of vigabatrin in refractory infantile spasms. J Child Neurol. 1991;Suppl 2:S52–S59. | ||
Elterman RD, Shields WD, Mansfield KA, Nakagawa J; US Infantile Spasms Vigabatrin Study Group. Randomized trial of vigabatrin in patients with infantile spasms. Neurology. 2001;57(8):1416–1421. | ||
Elterman RD, Shields WD, Bittman RM, Torri SA, Sagar SM, Collins SD. Vigabatrin for the treatment of infantile spasms: final report of a randomized trial. J Child Neurol. 2010;25(11):1340–1347. | ||
Willmore LJ, Abelson MB, Ben-Menachem E, Pellock JM, Shields WD. Vigabatrin: 2008 update. Epilepsia. 2009;50(2):163–173. | ||
Pearl PL, Vezina LG, Saneto RP, et al. Cerebral MRI abnormalities associated with vigabatrin therapy. Epilepsia. 2009;50(2):184–194. | ||
Wheless JW, Carmant L, Bebin M, et al. Magnetic resonance imaging abnormalities associated with vigabatrin in patients with epilepsy. Epilepsia. 2009;50(2):195–205. | ||
Guideline for prescribing vigabatrin in children has been revised. Vigabatrin Paediatric Advisory Group. BMJ. 2000;320(7246):1404–1405. | ||
Kröll-Seger J, Kaminska A, Moutard ML, et al. Severe relapse of epilepsy after vigabatrin withdrawal: for how long should we treat symptomatic infantile spasms? Epilepsia. 2007;48(3):612–613. | ||
Zeng LH, Xu L, Gutmann DH, Wong M. Rapamycin prevents epilepsy in a mouse model of tuberous sclerosis complex. Ann Neurol. 2008;63(4):444–453. | ||
Ehninger D, Han S, Shilyansky C, et al. Reversal of learning deficits in a Tsc2+/− mouse model of tuberous sclerosis. Nat Med. 2008;14(8):843–848. | ||
Muncy J, Butler IJ, Koenig MK. Rapamycin reduces seizure frequency in tuberous sclerosis complex. J Child Neurol. 2009;24(4):477. | ||
Perek-Polnik M, Jóźwiak S, Jurkiewicz E, Perek D, Kotulska K. Effective everolimus treatment of inoperable, life-threatening subependymal giant cell astrocytoma and intractable epilepsy in a patient with tuberous sclerosis complex. Eur J Paediatr Neurol. 2012;16(1):83–85. | ||
Koenig MK, Butler IJ, Northrup H. Regression of subependymal giant cell astrocytoma with rapamycin in tuberous sclerosis complex. J Child Neurol. 2008;23(10):1238–1239. | ||
Franz DN, Leonard J, Tudor C, et al. Rapamycin causes regression of astrocytomas in tuberous sclerosis complex. Ann Neurol. 2006;59(3):490–498. | ||
Krueger DA, Care MM, Holland K, et al. Everolimus for subependymal giant-cell astrocytomas in tuberous sclerosis. N Engl J Med. 2010;363(19):1801–1811. | ||
Franz DN, Belousova E, Sparagana S, et al. Efficacy and safety of everolimus for subependymal giant cell astrocytomas associated with tuberous sclerosis complex (EXIST-1): a multicentre, randomised, placebo-controlled phase 3 trial. Lancet. 2013;381(9861):125–132. | ||
Fallah A, Guyatt GH, Snead OC, et al. Predictors of seizure outcomes in children with tuberous sclerosis complex and intractable epilepsy undergoing resective epilepsy surgery: an individual participant data meta-analysis. PLoS One. 2013;8(2):e53565. | ||
Shimizu H. Our experience with pediatric epilepsy surgery focusing on corpus callosotomy and hemispherotomy. Epilepsia. 2005;46(Suppl 1):30–31. | ||
Jalilian L, Limbrick DD, Steger-May K, Johnston J, Powers AK, Smyth MD. Complete versus anterior two-thirds corpus callosotomy in children: analysis of outcome. J Neurosurg Pediatr. 2010;6(3):257–266. | ||
Kasasbeh AS, Smyth MD, Steger-May K, Jalilian L, Bertrand M, Limbrick DD. Outcomes after anterior or complete corpus callosotomy in children. Neurosurgery. 2014;74(1):17–28. | ||
Shim KW, Lee YM, Kim HD, Lee JS, Choi JU, Kim DS. Changing the paradigm of 1-stage total callosotomy for the treatment of pediatric generalized epilepsy. J Neurosurg Pediatr. 2008;2(1):29–36. | ||
Henry TR. Therapeutic mechanisms of vagus nerve stimulation. Neurology. 2002;59(6 Suppl 4):S3–S14. | ||
McLachlan RS. Suppression of interictal spikes and seizures by stimulation of the vagus nerve. Epilepsia. 1993;34(5):918–923. | ||
Woodbury DM, Woodbury JW. Effects of vagal stimulation on experimentally induced seizures in rats. Epilepsia. 1990;31(Suppl 2):S7–S19. | ||
Zabara J. Inhibition of experimental seizures in canines by repetitive vagal stimulation. Epilepsia. 1992;33(6):1005–1012. | ||
Lockard JS, Congdon WC, DuCharme LL. Feasibility and safety of vagal stimulation in monkey model. Epilepsia. 1990;31(Suppl 2):S20–S26. | ||
Takaya M, Terry WJ, Naritoku DK. Vagus nerve stimulation induces a sustained anticonvulsant effect. Epilepsia. 1996;37(11):1111–1116. | ||
Fernández-Guardiola A, Martínez A, Valdés-Cruz A, Magdaleno-Madrigal VM, Martínez D, Fernández-Mas R. Vagus nerve prolonged stimulation in cats: effects on epileptogenesis (amygdala electrical kindling): behavioral and electrographic changes. Epilepsia. 1999;40(7):822–829. | ||
Magdaleno-Madrigal VM, Valdés-Cruz A, Martínez-Vargas D, et al. Effect of electrical stimulation of the nucleus of the solitary tract on the development of electrical amygdaloid kindling in the cat. Epilepsia. 2002;43(9):964–969. | ||
Elliott RE, Carlson C, Kalhorn SP, et al. Refractory epilepsy in tuberous sclerosis: vagus nerve stimulation with or without subsequent resective surgery. Epilepsy Behav. 2009;16(3):454–460. | ||
Major P, Thiele EA. Vagus nerve stimulation for intractable epilepsy in tuberous sclerosis complex. Epilepsy Behav. 2008;13(2):357–360. | ||
Parain D, Penniello MJ, Berquen P, Delangre T, Billard C, Murphy JV. Vagal nerve stimulation in tuberous sclerosis complex patients. Pediatr Neurol. 2001;25(3):213–216. | ||
Zamponi N, Petrelli C, Passamonti C, Moavero R, Curatolo P. Vagus nerve stimulation for refractory epilepsy in tuberous sclerosis. Pediatr Neurol. 2010;43(1):29–34. | ||
Hallböök T, Lundgren J, Stjernqvist K, Blennow G, Strömblad LG, Rosén I. Vagus nerve stimulation in 15 children with therapy resistant epilepsy; its impact on cognition, quality of life, behaviour and mood. Seizure. 2005;14(7):504–513. | ||
Helmers SL, Wheless JW, Frost M, et al. Vagus nerve stimulation therapy in pediatric patients with refractory epilepsy: retrospective study. J Child Neurol. 2001;16(11):843–848. | ||
Bough KJ, Rho JM. Anticonvulsant mechanisms of the ketogenic diet. Epilepsia. 2007;48(1):43–58. | ||
Bough KJ, Wetherington J, Hassel B, et al. Mitochondrial biogenesis in the anticonvulsant mechanism of the ketogenic diet. Ann Neurol. 2006;60(2):223–235. | ||
Danial NN, Hartman AL, Stafstrom CE, Thio LL. How does the ketogenic diet work? Four potential mechanisms. J Child Neurol. 2013;28(8):1027–1033. | ||
Wilder RM. The effects of ketonemia on the course of epilepsy. Mayo Clin Proc. 1921;2:307-308. | ||
McDaniel SS, Rensing NR, Thio LL, Yamada KA, Wong M. The ketogenic diet inhibits the mammalian target of rapamycin (mTOR) pathway. Epilepsia. 2011;52(3):e7–e11. | ||
Levy RG, Cooper PN, Giri P. Ketogenic diet and other dietary treatments for epilepsy. Cochrane Database Syst Rev. 2012;3:CD001903. | ||
Lefevre F, Aronson N. Ketogenic diet for the treatment of refractory epilepsy in children: A systematic review of efficacy. Pediatrics. 2000;105(4):E46. | ||
Keene DL. A systematic review of the use of the ketogenic diet in childhood epilepsy. Pediatr Neurol. 2006;35(1):1–5. | ||
Henderson CB, Filloux FM, Alder SC, Lyon JL, Caplin DA. Efficacy of the ketogenic diet as a treatment option for epilepsy: meta-analysis. J Child Neurol. 2006;21(3):193–198. | ||
Kossoff EH, Thiele EA, Pfeifer HH, McGrogan JR, Freeman JM. Tuberous sclerosis complex and the ketogenic diet. Epilepsia. 2005;46(10):1684–1686. | ||
Coppola G, Klepper J, Ammendola E, et al. The effects of the ketogenic diet in refractory partial seizures with reference to tuberous sclerosis. Eur J Paediatr Neurol. 2006;10(3):148–151. | ||
Neal EG, Chaffe H, Schwartz RH, et al. The ketogenic diet for the treatment of childhood epilepsy: a randomised controlled trial. Lancet Neurol. 2008;7(6):500–506. | ||
Freeman JM, Kossoff EH, Hartman AL. The ketogenic diet: one decade later. Pediatrics. 2007;119(3):535–543. | ||
Hrachovy RA, Frost JD, Kellaway P, Zion TE. Double-blind study of ACTH vs prednisone therapy in infantile spasms. J Pediatr. 1983;103(4):641–645. | ||
Baram TZ, Mitchell WG, Tournay A, Snead OC, Hanson RA, Horton EJ. High-dose corticotropin (ACTH) versus prednisone for infantile spasms: a prospective, randomized, blinded study. Pediatrics. 1996;97(3):375–379. | ||
Zou LP, Wang X, Dong CH, Chen CH, Zhao W, Zhao RY. Three-week combination treatment with ACTH + magnesium sulfate versus ACTH monotherapy for infantile spasms: a 24-week, randomized, open-label, follow-up study in China. Clin Ther. 2010;32(4):692–700. | ||
Chiron C, Dumas C, Jambaqué I, Mumford J, Dulac O. Randomized trial comparing vigabatrin and hydrocortisone in infantile spasms due to tuberous sclerosis. Epilepsy Res. 1997;26(2):389–395. | ||
Vigevano F, Cilio MR. Vigabatrin versus ACTH as first-line treatment for infantile spasms: a randomized, prospective study. Epilepsia. 1997;38(12):1270–1274. | ||
Granström ML, Gaily E, Liukkonen E. Treatment of infantile spasms: results of a population-based study with vigabatrin as the first drug for spasms. Epilepsia. 1999;40(7):950–957. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.