")
Back to Journals » Therapeutics and Clinical Risk Management » Volume 10
Optimal management of pulmonary arterial hypertension: prognostic indicators to determine treatment course
Authors Baldi F, Fuso L, Arrighi E, Valente S
Received 30 May 2014
Accepted for publication 1 July 2014
Published 7 October 2014 Volume 2014:10 Pages 825—839
DOI https://doi.org/10.2147/TCRM.S48920
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Fabiana Baldi, Leonello Fuso, Eugenio Arrighi, Salvatore Valente
Pulmonary Medicine Unit, Catholic University, Rome, Italy
Abstract: Pulmonary arterial hypertension (PAH) is a rapidly progressive pulmonary vascular disease with a multifactorial etiopathogenesis that can result in right-sided heart failure and death. A number of studies indicate that an early therapeutic intervention yields better results on disease progression as compared to delayed treatment. In this review, we will analyze treatment strategies that may be used for monitoring disease progression and for guiding treatment decisions. Several factors (ie, symptoms, functional class, exercise capacity as assessed by a walking test and cardiopulmonary stress testing, hemodynamic parameters, cardiac magnetic resonance imaging, and plasma levels of biochemical markers) have been prognostic of survival. These indicators may be used both at the time of diagnosis and during treatment follow-up. No resolutive therapy is currently available for PAH; however, in the last decade, the advent of specific pharmacological treatments has given new hope to patients suffering from this debilitating disease with a poor prognosis. Combination drug therapies offer increased benefits over monotherapy, and current guidelines recommend a sequential “add on” design approach for patients in functional class II–IV. The goal-oriented “treat to target” therapy sets the timing for treatment escalation in case of inadequate response to currently known prognostic indicators. To date, further longitudinal studies should be urgently conducted to identify new goals that may improve therapeutic strategies in order to optimize personalized treatment in PAH patients.
Keywords: pulmonary hypertension, prognostic indicators, specific drug therapy, disease progression
Introduction
Pulmonary arterial hypertension (PAH) is a rapidly progressive multifactorial condition involving various biochemical pathways and different cell types.1 According to the most recent guidelines published jointly by the European Society of Cardiology (ESC) and the European Respiratory Society (ERS), pulmonary hypertension is a hemodynamic and pathophysiological state defined as an increase in mean pulmonary arterial pressure (PAPm) ≥25 mmHg at rest, as assessed by right heart catheterization (RHC).2 According to the clinical classification provided in the ESC/ERS 2009 guidelines,2 and which was recently updated,3 PAH is classified in group 1, which includes several subgroups (Table 1).
 | Table 1 Clinical classification of PH |
Irrespective of the underlying etiology, the most relevant pathogenic mechanism of PAH is a pulmonary vascular condition characterized by intimal proliferation, median hypertrophy, and the development of plexiform lesions.1 When associated with vasoconstriction and in situ thrombosis, these changes in the pulmonary vascular bed result in pulmonary vascular remodeling that may lead to raised pulmonary vascular resistance (PVR) and consequent elevated pulmonary arterial pressure (PAP), with increased right ventricular after-load. The right ventricle initially compensates for the increase in after-load through adaptive hypertrophy and, secondarily, via dilatation of the right side of the heart. Such effects of increased PAP levels on the right ventricle constitute a major determinant of symptoms and patient survival, as right-sided heart failure is the specific main cause of death in PAH patients.4 In this regard, echocardiographic and hemodynamic monitoring for the assessment of right ventricular adaptation and decreased function is a crucial factor for prognosis and the prediction of survival.5,6
The right ventricular function in PAH
In the asymptomatic phase of PAH, increased PAP and PVR do not alter cardiac output. However, as patients become symptomatic over time and move to World Health Organization (WHO) functional class (WHO-FC) II, PVR significantly increases, suggesting advanced vascular remodeling.
Under these circumstances, the heart initially compensates for the increase in after-load through hypertrophy and right ventricular remodeling. However, should cardiac overload persist over an extended period of time, right ventricular dilatation and failure may occur. Impaired cardiac function is associated with both systolic and diastolic failure, measured at RHC as elevated right atrial pressure (RAP) and decreased cardiac index (CI).7
In this regard, it is worth noting that PVR is a key indicator for hemodynamic abnormalities caused by pulmonary hypertension; however, PVR alone does not have any prognostic significance. On the other hand, both RAP and CI are valuable predictors of survival.2,8
In order to accurately stage the disease and adopt an adequate therapeutic strategy, it is useful to study right ventricular function using both direct parameters (echocardiographic and hemodynamic measurements) and indirect parameters such as symptoms, WHO-FC, exercise testing (ie, the 6-minute walking test [6MWT] and cardiopulmonary exercise test), and biochemical markers (ie, brain natriuretic peptide [BNP], N-terminal fragment of probrain natriuretic peptide [NT-proBNP], troponin T, uric acid, and platelet count).
The most recent guidelines published in 2009 recommend that the severity of the disease, as well as the response to therapy, should be evaluated by parameters of established prognostic value.2 Symptoms, as assessed through the WHO-FC scale, may be considered as the most significant prognostic indicators, as they are indices of the progression of right-sided heart failure caused by the pulmonary hypertension. Similarly, physical exercise capability and other hemodynamic parameters should be considered as additional prognostic indicators.
Prognostic indicators
Identifying the parameters directly associated with the risk of clinical deterioration and mortality is the key to select the therapeutic goals in a potentially fatal disease such as PAH. The goals of treatment currently used in clinical trials and those recommended in the ESC/ERS 2009 guidelines are based on the measurement of these prognostic parameters at basal conditions and during their follow-up reassessment (Table 2).
 | Table 2 Parameters with established importance for assessing disease severity, stability, and prognosis |
Namely, the following prognostic indicators are used: parameters obtained from clinical assessment; parameters derived from exercise tests; biochemical markers; and echocardiographic and hemodynamic parameters measuring the severity, prognosis, and stability of the disease.9,10 On this basis, the patient’s clinical conditions may be classified as “stable and satisfactory” (fulfilling the majority of criteria that are indicative of better prognosis), “stable but not satisfactory” (fulfilling only some of the criteria that are indicative of a better prognosis), and “unstable and deteriorating” (fulfilling the majority of the criteria that are indicative of a worse prognosis).2
Clinical evidence of right ventricular failure
The onset of peripheral edema, angina, and/or syncopal episodes is considered a sign of deterioration in right ventricular function. In particular, the presence of syncope is a negative prognostic factor and represents an independent predictor of mortality.11
WHO functional class
WHO-FC correlates the patient’s symptoms (dyspnea, fatigue, chest pain, and syncope) with daily routine activities. These are classified as classes: class I, no limitation of daily activity; class II, moderate activity leads to the onset of symptoms; class III, slight activity leads to the onset of symptoms; and class IV, an inability to carry out any physical activity without symptoms, or the symptoms may be present even at rest (Table 3).
 | Table 3 Functional classification of pulmonary hypertension according to the World Health Organization |
Functional class is one of the major factors that should be taken into consideration to determine the severity of pulmonary hypertension at diagnosis, irrespective of its etiology, as well as a therapeutic target during follow-up,12,13 and it is considered the best predictor for survival. Particularly, patients classified as WHO-FC IV at diagnosis have a significantly worse prognosis when compared with patients in class II or III.6,12–14 This has also been confirmed by the Registry to Evaluate Early and Long-Term PAH Disease Management (REVEAL) with respect to the 1-year survival rate.5 Survival estimates calculated at baseline for patients in WHO-FC III and class IV enrolled in long-term studies on epoprostenol were found to be more favorable for patients in class IV than for those in class III (47% versus 71%–81% at 3 years, respectively).13,14
The available data also highlight the prognostic significance of improvements in functional class, as they are associated with a downgrade to a lower class when administering a specific drug therapy. As a matter of fact, a few studies have shown that idiopathic PAH patients treated with epoprostenol improving from WHO-FC IV to class II during follow-up have a more favorable prognosis when compared to those who do not improve their functional class.13,14
Moreover, moving up from one functional class to another after a treatment period has prognostic value, even in etiologically different pulmonary hypertension (for example, PAH secondary to scleroderma). A study conducted on these patients showed that the survival rate of subjects classified as WHO-FC I–II after 4 months of treatment was 100%, 100%, and 86% at 1 year, 2 years, and 3 years, respectively, versus 78%, 38%, and 38% for patients who remained in WHO-FC III–IV.15,16 These data suggest that improving to WHO-FC II is a realistic therapeutic target for all PAH patients, as it is consistently associated with improved survival.
Exercise capacity test
In most clinical trials of approved therapies,2 change in 6MWT distance is used as a primary endpoint, and it is simple to use and easily reproducible.17,18 Notably, its prognostic significance lies in the distance (measured in meters) walked during the test: a negative prognostic value has been associated with a walk distance <300 m, and a better prognostic value to a distance >500 m.2
The absolute value of the walk distance achieved during treatment is prognostic of survival;13 however, this does not hold true for the relative change recorded during follow-up.13 As a matter of fact, patients suffering from severe PAH with a low walk distance at baseline need to recover more consistently in order to improve their prognosis if compared to subjects with more satisfactory exercise capabilities.
The cardiopulmonary exercise test is also a valid prognostic tool for monitoring right heart function. This test provides a continuous measurement of expired gas and ventilation during a maximal physical exercise. The prognostic parameters measured during the test are peak oxygen consumption (VO2 peak) (a value <12 mL/minute/kg is a negative prognostic indicator and a value >15 mL/minute/kg is a better prognostic indicator),2 and peak systolic arterial pressure (a value <120 mmHg is a negative prognostic indicator).19 Another parameter that can be considered is the ventilatory equivalent for carbon dioxide (EqCO2), which is less age-dependent than VO2 peak; values <45 and >55 L/minute/L/minute would seem like appropriate goals and warning signs, respectively, for the evaluation of exercise response in relation to treatment and prognosis.20
Assessment of right ventricular dysfunction
RHC has been recognized as the gold standard for the diagnosis of PAH, and it may be used to assess both the severity of the disease and the treatment response.9,21 Only CI (worse prognosis with values <2 L/minute/m2)14,22,23 and RAP (worse prognosis with values >15 mmHg)24 have proved to be of prognostic value among the parameters measured using this procedure. Surprisingly, PAPm, PVR, and mixed venous oxygen saturation do not play a significant prognostic role.
Echocardiography is an additional noninvasive test used to estimate over time any change in the right heart caused by the elevation of PAP. Notably, this examination provides the following prognostic information: tricuspid annular plane systolic excursion (worse prognosis with values <1.5 cm); myocardial performance index (Tei index) (worse prognosis for values >0.88); and left ventricular end-diastolic eccentricity index (worse prognosis with values >1.7). Enlargement of the right atrium and the presence of pericardial effusion also correlates with poor prognosis.25–32
In recent years, there has been increasing interest in cardiac magnetic resonance imaging as it provides a more accurate assessment of right ventricle size and function, and a noninvasive estimation of cardiac output and stroke volume as compared to echocardiography.33,34 A recent study assessed the value of cardiac magnetic resonance imaging measurements as a prognostic predictor of idiopathic PAH before starting intravenous prostacyclin therapy.35 The multivariate analyses suggested that the right ventricular end-diastolic volume indices, together with the WHO-FC, were independent predictors of both hospitalization for right-sided heart failure and mortality.
Biochemical markers and other prognostic indicators
Elevated biochemical markers such as blood BNP, NT-proBNP, troponin,36 and uric acid levels seem to correlate with poor prognosis. Particularly, BNP values >180 pg/mL have been shown to have a negative prognostic significance.22,23,37,38 Similarly, increased NT-proBNP plasma levels on follow-up have been associated with worse prognosis,39 whereas uric acid levels <6.4 mg/mL are associated with a more favorable prognosis.40
Recent clinical trials identified new prognostic factors. For instance, a study analyzing the trends of a variety of humoral and hemodynamic prognostic variables in patients with idiopathic PAH receiving combination therapy with bosentan, sildenafil, and epoprostenol,41 has demonstrated a significant inverse correlation between platelet levels at basal conditions and PAPm values. In particular, this study revealed that low platelet levels (<20×104/μL) correlate with low survival rates.41
Recent research indicates the role of proinflammatory cytokines in the underlying vascular remodeling process observed in PAH, and in the prognosis of these patients.42 Indeed, proinflammatory cytokines have been found to be independently associated with mortality in patients with PAH.43 Moreover, it has been shown that elevated blood levels of interleukin-6 (>4.7 pg/mL), particularly in idiopathic PAH patients with low BNP levels (<180 pg/mL), have a negative prognostic value as a significant independent predictor of mortality.44
Arterial pressure of carbon dioxide (PaCO2) measured by arterial blood gas analysis, systolic blood pressure at rest, heart rate (HR) at rest, and diffusing capacity of the lung for carbon monoxide (DLCO) are additional variables of potential prognostic significance. Levels of PaCO2 <4.25 kPa, systolic blood pressure <110 mmHg, HR >92 bpm, and DLCO <32% predicted have been shown to be significant independent predictors of mortality.5
Validation of the prognostic indicators
The validation of the aforementioned prognostic indicators is based on data from the REVEAL registry.5,45 This registry provides prospective data obtained from 2,716 patients with a recent diagnosis of group 1 PAH. The aim was to validate a variety of predictive parameters for short- and long-term survival during treatment, and to develop a simplified clinical risk calculator. This is based on a prognostic equation that results in a risk stratification scoring system that may guide therapeutic decisions, as shown in Table 4.46 The risk calculator has a minimum risk score of 0, a maximum score of 22, and is designed for clinical use. The predicted 1-year survival for patients in the validation cohort (number [n] =504) was 95%–100% in the low-risk group (score: 1–7), 90% to <95% in the average-risk group (score: 8), 85% to <90% in the moderately high-risk group (score: 9), 70% to <85% in the high-risk group (score: 10–11), and <70% in the very high-risk group (score ≥12).46
 | Table 4 REVEAL registry PAH risk score calculator |
Data from the REVEAL registry confirmed the prognostic value of parameters such as WHO-FC and the 6MWT distance, and the prognostic relevance of hemodynamic parameters such as CI and RAP, as well as lung function parameters, such as DLCO. Similarly, this registry has shown the prognostic relevance of biochemical markers (ie, BNP), vital signs (ie, HR and artery systolic pressure), as well as of comorbidities (ie, the presence of renal impairment) and demographic data (ie, age and race).45
Another registry, the French Pulmonary Arterial Hypertension Network, has also focused on survival and important prognostic indicators in patients with PAH.47 A total of 674 consecutive adult patients (121 incident and 553 prevalent cases) were prospectively enrolled in 2002–2003. In the entire cohort, 1-, 2-, and 3-year survival rates were 87%, 76%, and 67%, respectively. In the subset with prevalent idiopathic, familial, and anorexigen-associated PAH who were diagnosed <3 years prior to study entry (n=134), 1-, 2-, and 3-year survival rates were significantly higher than in incident patients (n=56). In the combined cohort of patients with idiopathic, familial, and anorexigen-associated PAH, multivariate analysis showed that survival could be estimated by means of a novel risk-prediction equation using the patient’s sex, walk distance, and cardiac output at the time of diagnosis.47
Recently, Nickel et al22 reported a systemic evaluation of prognostic markers at baseline and follow-up in a series of patients with idiopathic PAH. The follow-up assessments were found to be better outcome predictors than the baseline evaluations. Thus, both baseline and follow-up assessments are important when assessing the risk of patients with PAH. The baseline evaluation will determine disease severity at presentation, providing important information for choosing the initial therapy. Follow-up assessments are crucial for assessing the response to treatment, and they seem to provide a more reliable prognostic estimation than the baseline evaluation. Table 5 summarizes the variables used in clinical practice to determine the response to therapy and progress in patients with PAH.20
 | Table 5 Variables used in clinical practice to determine response to therapy and prognosis in patients with PAH |
General therapeutic strategies
Over the last decade, the therapeutic management of PAH patients has rapidly advanced. The introduction of specific treatments resulted in substantially improved prognosis and outcomes for patients suffering from this disease. Therefore, in the last few years, the expected survival of patients diagnosed with PAH has undoubtedly improved when compared with that reported in the early 1990s. However, poor prognosis and rapid clinical deterioration are still observed in some patients.6,12,48
At present, the main goal is to improve patient outcomes through currently available therapies, and the early diagnosis of PAH followed by treatment with first-line monotherapy is, above all, the best strategy for achieving this result.2,49 However, increasing emphasis is currently being placed on the importance of therapeutic escalation in patients who do not satisfactorily respond to the initial treatment. Historically, the decision of whether or not to use an escalation therapy was largely based on the presence of clinical deterioration.50,51 However, an approach has recently been adopted that allows for the implementation of an intervention before clinical and functional worsening is observed, since waiting for deterioration is not considered an acceptable option in a rapidly progressing disease like PAH.
Escalation therapy depends on the availability of different classes of specific drugs that use different modes of action, offering the possibility to combine several therapies. It is known that currently used drugs exert their effects on three main endogenous pathways: 1) the prostacyclin pathway; 2) the endothelin pathway; and 3) the nitric oxide (NO) pathway (Figure 1).
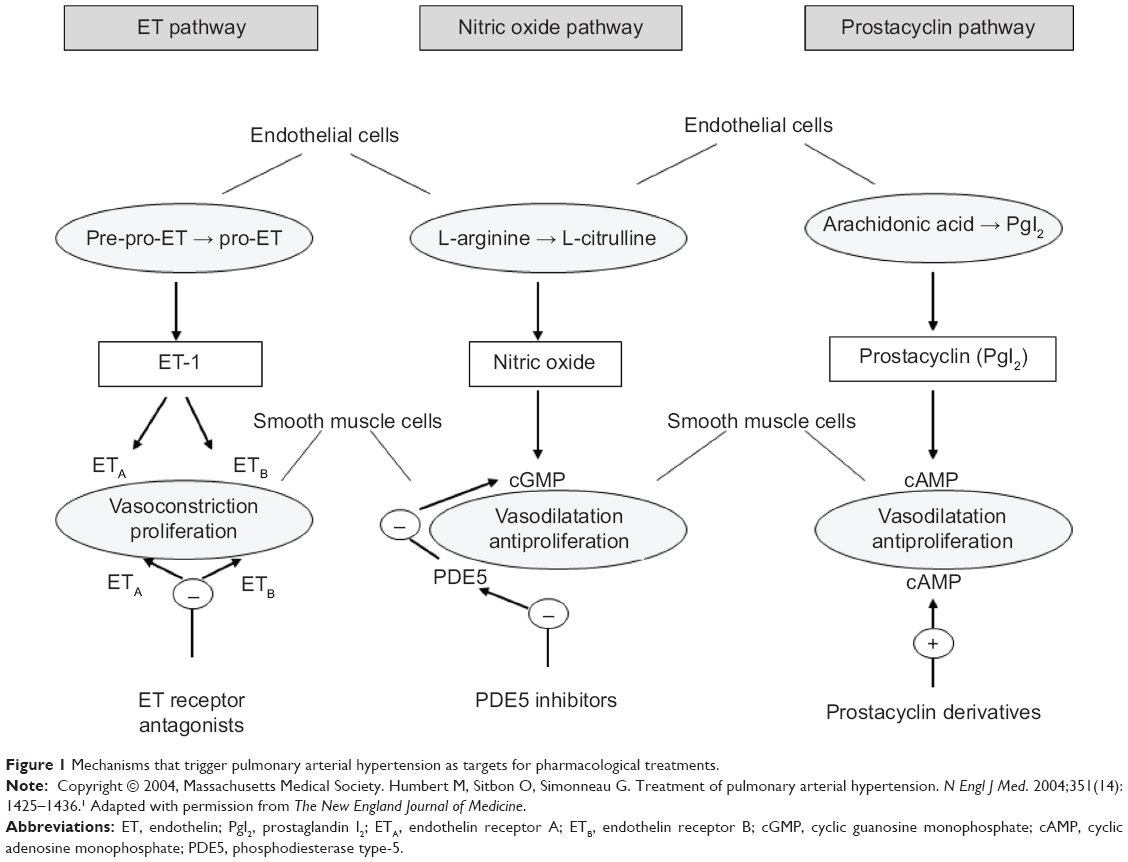 | Figure 1 Mechanisms that trigger pulmonary arterial hypertension as targets for pharmacological treatments. |
Prostacyclin has been shown to play a role in inducing pulmonary vasodilatation and in inhibiting smooth muscle cell growth.52 This pathway is targeted by epoprostenol, the first drug to be approved for the treatment of PAH, and by two other prostacyclin analogs (ie, treprostinil and iloprost). Treprostinil, available in both oral and inhaled forms, is approved in the United States, but not in Europe.
Circulating endothelin induces smooth muscle cell proliferation, fibrosis, inflammation, and vasoconstriction in the pulmonary vascular tree.53 Bosentan was the first oral medication specifically approved for the treatment of PAH, and it is a dual endothelin receptor (ET-A and ET-B) antagonist. Later on, selective type A endothelin receptor antagonists such as ambrisentan and sitaxsentan have been developed. However, sitaxsentan, which was approved in Europe but not in the US, was withdrawn worldwide from the market in December 2010 due to liver toxicity.
The NO pathway is enhanced by the phosphodiesterase type-5 inhibitors (sildenafil and tadalafil), and it is known to be involved in pulmonary vasodilatation and to inhibit the proliferation of smooth muscle cells.54 Sildenafil is approved in Europe, but not in the US, and it is also approved for pediatric PAH.
Randomized clinical trials conducted over the past decade have shown that monotherapy with these treatments is associated with improved survival,55,56 increased physical exercise capacity, improved hemodynamic parameters,55,57 and delayed clinical deterioration.57–61 Conversely, trials on combination therapy have not provided convincing results, but there is increasing evidence supporting its use and efficacy.62–64
Notably, there are three different ways to apply combination therapy to the management of PAH: the sequential “add-on” approach; as a “first-line” combination therapy; and as a “step-down” approach. The sequential “add-on” approach consists in adding a drug, typically with a different mechanism of action, for those patients who do not respond adequately to a monotherapy. The treatment algorithm from the ERS/ESC guidelines recommends a sequential approach for combination therapy.2 According to this approach, patients should be treated with a monotherapy and followed up regularly. If an inadequate clinical response to monotherapy is observed, adding a drug with a different mechanism of action may be considered.
The “first-line” combination therapy is another potential approach, and it consists of an initial therapy with more than one drug. Although such an aggressive approach to PAH could prevent clinical deterioration, little evidence exists supporting its efficacy; therefore, this option should only be considered when treating those severely affected patients in advanced functional classes such as WHO-FC IV at the time of diagnosis. A recent pilot study65 provided preliminary evidence of the long-term benefits of a “first-line” triple combination therapy (epoprostenol intravenous, bosentan, and sildenafil) in 18 patients with severe PAH. Significant improvements in 6MWT distance and hemodynamics were observed after 4 months and at the final evaluation (32±19 months). All patients were still alive after a mean follow-up of 41.2±13.4 months.65
Conversely, according to the “step-down” approach, it may be possible to gradually reduce the number of drugs and then step-down treatment to monotherapy in patients on combination therapy showing clinical improvement. However, this is a controversial proposal that has not been widely accepted.
There is now an increasing interest in another therapeutic approach: the up-front therapy approach, which is a treatment strategy that may be used in patients with severe PAH in WHO-FC III or IV at diagnosis.66 According to this approach, patients should be started on a combined therapy with epoprostenol intravenous, which is associated with a specific oral treatment such as an endothelin receptor antagonist or a phosphodiesterase type-5 inhibitor. At present, the results show improved functional class, better hemodynamic parameters, and increased physical exercise capacity after some months of treatment.
Surgical treatments
To date, surgical interventions such as balloon atrial septostomy or, in the worst cases, lung transplantation, should be undertaken in patients persistently in WHO-FC IV who are not expected to improve their prognostic goals, and who are not achieving treatment optimization with a combination therapy.67
Atrial septostomy
The creation of a right-to-left shunt by atrial septostomy can decompress the right heart chambers and increase left ventricular preload. The goal is to increase systemic blood flow by bypassing the pulmonary vascular obstruction. The consequent arterial desaturation that follows the procedure is offset by increased CO and the augmentation of systemic oxygen delivery.68 Atrial septostomy may be considered in individuals with refractory severe PAH and right-sided heart failure, despite aggressive specific therapy and maximal diuretic therapy.69 It may also be considered in patients who have signs of impaired systemic blood flow (such as syncope) due to reduced left heart filling. Stepwise balloon dilatation is the procedure of choice.70 However, procedure-related mortality may be as high as 15%–20%,68,71 and it is difficult to predict which patients will benefit and which will deteriorate after this therapy.
Transplantation
The advent of disease-specific therapy for severe PAH has reduced patient referral for lung transplant programs.70 However, the long-term outcomes of medically treated patients remains uncertain, and transplantation should remain an important option for those who fail on such therapies. Studies indicate that up to 25% of patients with idiopathic PAH may fail to improve on disease-specific therapy, and the prognosis of patients who remain in WHO-FC III or IV is poor.72,73 Thus, transplantation is considered by some to be the final effective treatment for selected patients with idiopathic PAH. Bilateral lung or heart–lung transplantation is the procedure of choice.70 The 3-year survival of patients who had a lung or heart–lung transplant for idiopathic PAH is approximately 50%.69,74 The timing of transplantation is critical. Guidelines for when to refer a patient for transplant evaluation are as follows:75 WHO-FC III or IV; mean RAP >10 mmHg; PAPm >50 mmHg; CI <2.5 L/minute/m2; failure to improve functionally despite medical therapy; and rapidly progressive disease. Transplant organizations and organ allocation policies are influenced by medical, ethical, geographical, and political factors, and systems vary from country to country. In the US, since May 2005, the order of patients on the waiting list for lung transplantation has been based on a Lung Allocation Score (LAS) that was developed to address high waiting list mortality rates, and resulting in progressively earlier placement of patients on the waiting list. The three main objectives of the LAS include: 1) reducing the number of deaths on the lung transplant waiting list; 2) increasing the transplant benefit for lung recipients; and 3) ensuring the efficient and equitable allocation of lungs to active transplant candidates. The LAS system works by assigning a score ranging from 0–100 to all candidates older than the age of 12 years. It is a weighted combination of the predicted risk of death during the following year on the waiting list and the predicted likelihood of survival during the first year following transplantation.76 Higher scores represent higher urgency and greater potential transplant benefit. In the 2 years following the implementation of the LAS system, waitlist times have decreased and the mean LAS score of transplant recipients has increased, which is consistent with a greater urgency for transplantation; the total number of patients transplanted has also increased.77
Treat to target therapy
Current treatment goals correlate with baseline values of prognostic indicators, leading to a goal-oriented therapeutic strategy. This “treat to target” approach is recommended by the ESC/ERS 2009 guidelines.2 Therefore, the prognostic assessment of PAH patients at baseline or during follow-up is remarkably helpful in guiding therapeutic decisions according to the current “treat to target” strategy, which primarily aims to improve the functional class and to maintain patients in WHO-FC II.78
The aim of treatment in PAH patients is to delay the time to clinical worsening and to extend the patients’ survival. The “goal-oriented therapy”, a treatment strategy that uses known prognostic indicators as treatment targets, can achieve this result by providing early intervention and therapeutic escalation before the clinical status of patients deteriorates. The key difference between “goal-oriented therapy” and previous approaches is that clinically stabilized patients, or even those who improve slightly, can undergo therapeutic escalation if predetermined goals are not met.
Hoeper et al79 described a “goal-oriented therapy” algorithm in 2005, as shown in Figure 2. According to this algorithm, PAH patients in WHO-FC III or IV, who were negative on the vasoreactivity test, received a first-line bosentan monotherapy and three treatment goals were assessed at baseline (6MWT distance >380 m, VO2 peak >10.4 mL/minute/kg, and peak systolic blood pressure >120 mmHg during exercise). If patients met all these three therapeutic goals in follow-up visits, bosentan monotherapy was continued. Alternatively, if one goal was not met, an additional therapy was added, respectively, in the following order: sildenafil; inhaled iloprost; intravenous iloprost; and then, eventually, lung transplantation was proposed. After a 3-year follow-up period, the adoption of this treatment strategy improved survival estimates when compared to historical controls (80% versus 63% survival at 3 years, respectively).79
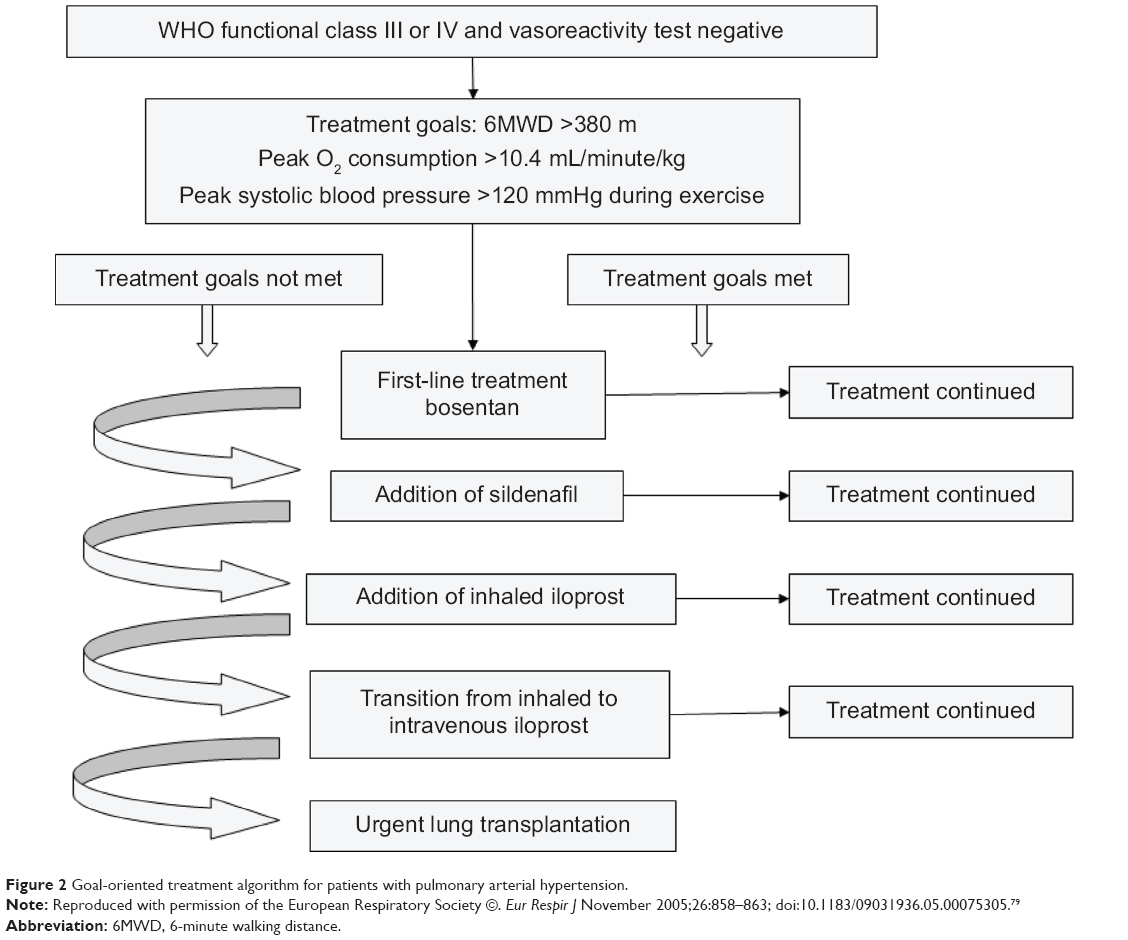 | Figure 2 Goal-oriented treatment algorithm for patients with pulmonary arterial hypertension. |
New drugs according to different pathways
Recent advances in the knowledge of the pathogenesis and progression of PAH encouraged the development of novel and specific drugs that may improve the prognosis of these patients. New specific treatments targeting the three known endogenous pathways are waiting for approval.
Prostacyclin pathway
The prostacyclin pathway is thought to play an established role in the pathogenesis of PAH.1,80 PAH patients have reduced levels of endogenous prostacyclin and reduced expression of prostacyclin synthase in the lung.81,82 Several specific therapies targeting the prostacyclin pathway have been developed over the last few years. However, disadvantages associated with these therapies include inconvenient routes of administration (by continuous intravenous infusion, subcutaneous injection, or inhalation), a short half-life, and nonspecific receptor interactions.83,84
A variety of oral prostacyclin or prostacyclin analogs are currently being tested in clinical trials. One of these, oral treprostinil, has been studied in a Phase III clinical trial both as a monotherapy and as part of a combination therapy (studies FREEDOM-C).85,86 The primary endpoint for these trials was an improvement in 6MWT distance at 12 weeks and 16 weeks of treatment, and the secondary endpoints included dyspnea score according to Borg scale. Both endpoints have been achieved, although treprostinil does not exert any effects in terms of improving functional class or the time to clinical worsening.85,86 Another study, the FREEDOM-M trial,87 showed that oral treprostinil improved exercise capacity in PAH patients not receiving other treatments; this drug was generally well tolerated and could provide a convenient, first-line prostacyclin treatment option for patients not requiring more intensive therapy. Based on this trial, oral treprostinil was approved in the US in December 2013.
Beraprost is another orally active prostacyclin analog. The limitations of this drug include a very short half-life (1 hour) and only modest, transient clinical benefits.88 A recent Phase III trial with beraprost failed to meet its primary endpoint and was suspended.89,90 Beraprost is, at present, approved only in Japan and South Korea.
More encouraging results were announced by Actelion on June 16, 2014 in an ongoing Phase III trial (GRIPHON study) on selexipag, a potent, orally active molecule that is rapidly hydrolyzed to an active metabolite, ACT-333679. Both selexipag and its metabolite are highly selective for the IP prostacyclin receptor compared with other prostanoid receptors such as EP, DP, FP, and TP.1,80,83,84 Such selectivity for the IP receptor may potentially improve tolerability of selexipag in terms of side effects (for example, nausea and vomiting), which might result from the activation of other prostanoid receptors.91 In addition, selexipag has a 7.9-hour half-life that allows for oral dosing twice daily.
The results reported in another study on selexipag have demonstrated a significant 30% reduction in PVR at 17 weeks of therapy and a significantly increased CI value.92 It is worth noting that the primary endpoint for this study is the time to the first morbidity/mortality event, which can provide crucial information on the long-term effects of selexipag in patients with PAH.
Endothelin pathway
It is widely known that endothelin is a key mediator of PAH, as it drives the changes that induce pulmonary vascular remodeling.53,93 The effects of endothelin are mediated by two receptor subtypes (ET-A and ET-B) to which it binds with high affinity.94,95 ET-A receptors are located in smooth muscle cells, whereas ET-B receptors are found in both endothelial and smooth muscle cells. When released from the endothelium, endothelin exerts its effects primarily on smooth muscle cells, leading to vasoconstriction and proliferation; in addition, endothelin acts on fibroblasts to induce proliferation and fibrosis, and it acts on the endothelium itself to generate vasoconstriction.
In addition to the already known endothelin receptor antagonists (bosentan and ambrisentan), a Phase III trial (SERAPHIN study)96 has demonstrated the safety and tolerability of a novel aspecific endothelin receptor antagonist, macitentan. This drug, approved in both Europe and the US, has shown superior efficacy, improved receptor binding affinity, and enhanced tissue penetration when compared with the currently available receptor blockers. It is worth noting that the primary endpoint for the SERAPHIN study is the time to first morbidity and/or mortality event, which is unlike other clinical trials that employ 6MWT distance as the primary endpoint.96
Toxicity data and results from the clinical trial have proved decreased hepatotoxicity and, therefore, it is less likely expected to cause elevations in liver enzymes than bosentan. Pharmacokinetic data also demonstrated that daily dosing can be taken irrespective of food intake, and no interaction has been reported with sildenafil, warfarin, ketoconazole, and cyclosporine for this molecule.97,98
Nitric oxide pathway
PAH is associated with reduced production of NO, an endothelium-derived vasodilator.10 In healthy individuals, NO acts on smooth muscle cells to induce vasodilatation and it inhibits proliferation through the activation of soluble guanylate cyclase, leading to an increased production of the secondary messenger cyclic guanosine monophosphate (cGMP).99,100 The two existing drugs working in this pathway (sildenafil and tadalafil) exert their effects by inhibiting phosphodiesterase type-5 (ie, the enzyme that catalyzes the conversion of cGMP to GMP).101 On the contrary, riociguat, a novel drug belonging to this class, acts directly on soluble guanylate cyclase, stimulating the enzyme and increasing its sensitivity to low levels of NO.102 The efficacy and safety of this new treatment have been demonstrated in Phase III clinical trials in idiopathic PAH (PATENT1–PATENT2 study) and in chronic post-thromboembolic PAH patients (CHEST1–CHEST2 study), respectively.103,104 In both studies, riociguat significantly improved the 6MWT distance, and it also improved secondary endpoints such as PVR, NT-proBNP, WHO-FC, time to clinical worsening, and the Borg dyspnea score.103,104 Riociguat is approved in both Europe and the US.
Platelet-derived growth factor pathway
The platelet-derived growth factor (PDGF) is one of the most promising targets in PAH.105,106 It is well known that PDGF plays a role in endothelial dysfunction and cell proliferation, and in the migration of smooth muscle cells. Studies on animal models of pulmonary hypertension demonstrate a regression of pulmonary vascular remodeling following the administration of imatinib mesylate, a tyrosine kinase inhibitor approved for the treatment of chronic myelogenous leukemia.107 Imatinib seems to play a role as a PDGF receptor antagonist, suppressing PDGF-induced vascular remodeling.108 A 24-week Phase III clinical trial (IMPRES study)109 has investigated the benefits generated by imatinib in 202 PAH patients receiving at least two specific treatments, using a double-blind, randomized, controlled study design. A significant improvement was observed for the primary endpoint of 6MWT distance (+32 m with imatinib versus placebo) and for the secondary endpoint of hemodynamic parameters.109
However, imatinib is not approved for use in PAH. Despite the approval of imatinib for numerous oncology conditions, its use in PAH is not recommended due to several cases of subdural hematoma and due to the long-term cardiac side effects reported for this drug.110,111
Therapy according to functional class
A treatment algorithm for PAH patients is shown in Figure 3. It is based on severity of the disease and includes new specific drugs.112
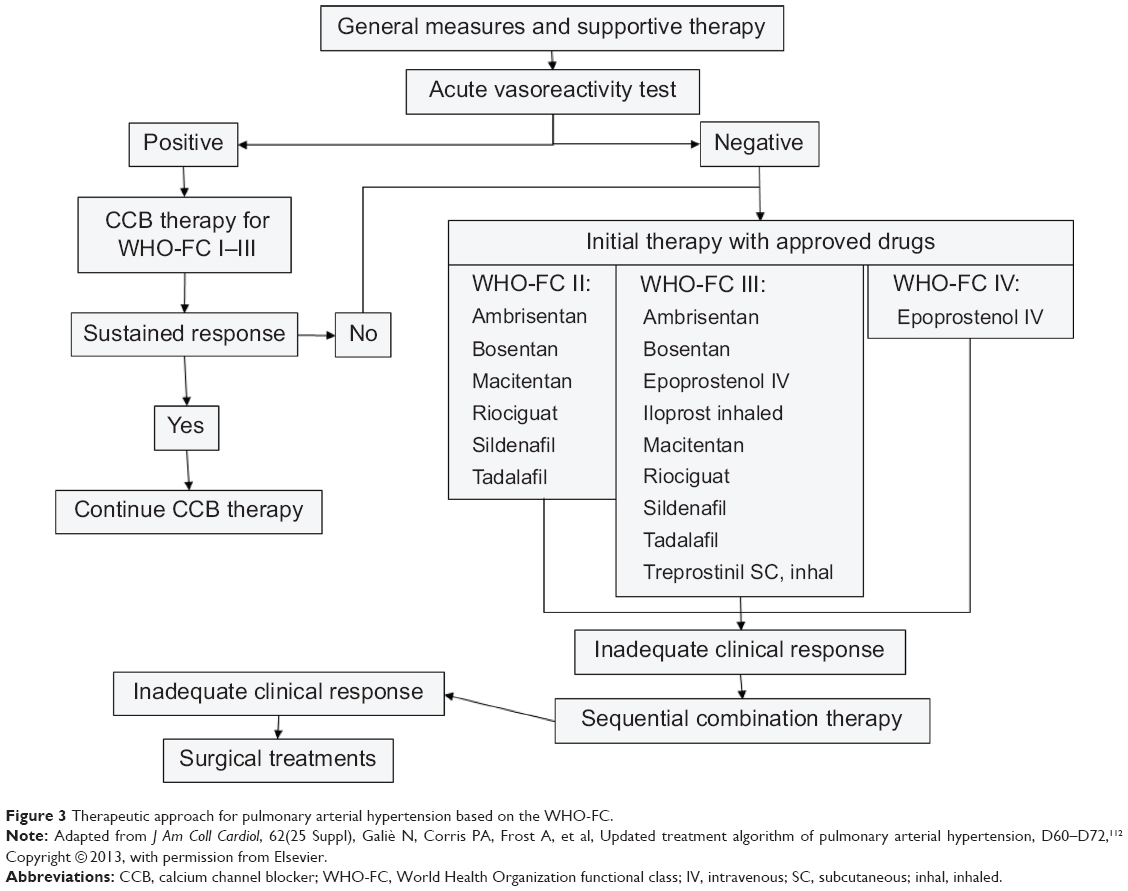 | Figure 3 Therapeutic approach for pulmonary arterial hypertension based on the WHO-FC. |
When considering mild functional limitations, there is not a clear indication for treatment in patients of WHO-FC I, except for primary therapy if there is an associated disease causing PAH. In patients with idiopathic PAH and a positive response to the vasoreactivity test, calcium channel blockers (CCB) may be effective.
Patients in WHO-FC II–IV should to be evaluated for a specific therapy. In addition, the need for oxygen, diuretic, and anticoagulant therapy should be assessed. All patients selected for a specific therapy should undergo RHC and a vasoreactivity test. For patients who have a positive vasoreactivity test, a trial of CCB therapy is recommended. Patients who respond to such therapy should be reassessed after 3–6 months of treatment. For patients who have a negative vasoreactivity test or who fail CCB therapy, specific therapy with prostanoids, endothelin receptor antagonists, and phosphodiesterase type-5 inhibitors is indicated. The preferred agent is related to the functional severity of the disease, and the choice should also be made according to the physician’s experience, a drug’s availability, and the patient’s preference. An inadequate clinical response to a single agent represents an indication for combination therapy using two or more classes of drugs that are used simultaneously. Finally, atrial septostomy and lung transplantation are reserved for patients who are refractory to medical therapy.
Challenges for the future
Although the studies conducted thus far provide valuable data that can help in the selection of therapeutic targets, further large cohort, prospective studies are still needed. The aim of these studies should be to determine the most appropriate treatment goals associated with the best outcomes for each subgroup of patients, and to investigate the difference in disease progression between incidence and prevalence cases. Moreover, these trials should combine variables with more extended survival time and include PAH patients with different etiologies. As a matter of fact, only PAH patients in group 1 have been enrolled in clinical trials so far, and this represents a major limitation.
Conclusion
PAH is a progressive disease that requires an early diagnosis to ensure a better prognosis, as well as regular monitoring to guide therapeutic escalation if treatment goals with initial monotherapy are not met. To date, the “add-on” combination therapy appears to be the best option to achieve treatment goals. At present, available data indicate that the most relevant goal is the improvement of patients to lower functional classes, at least to WHO-FC II, as well as improved hemodynamic and functional parameters. Treatment goals have remarkably evolved over the last decade and are becoming increasingly sophisticated, but additional data are required in order to identify new parameters of prognostic value and to improve treatment strategies leading to the optimized and personalized clinical and therapeutic management of PAH patients.
Disclosure
The authors report no conflicts of interest in this work.
References
Humbert M, Sitbon O, Simonneau G. Treatment of pulmonary arterial hypertension. N Engl J Med. 2004;351(14):1425–1436. | ||
Galiè N, Hoeper MM, Humbert M, et al; ESC Committee for Practice Guidelines (CPG). Guidelines for the diagnosis and treatment of pulmonary hypertension: the Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS), endorsed by the International Society of Heart and Lung Transplantation (ISHLT). Eur Heart J. 2009;30(20):2493–2537. | ||
Simonneau G, Gatzoulis MA, Adatia I, et al. Updated clinical classification of pulmonary hypertension. J Am Coll Cardiol. 2013; 62(25 Suppl):D34–D41. | ||
Chin KM, Kim NH, Rubin LJ. The right ventricle in pulmonary hypertension. Coron Artery Dis. 2005;16(1):13–18. | ||
Benza RL, Miller DP, Gomberg-Maitland M, et al. Predicting survival in pulmonary arterial hypertension: insights from the Registry to Evaluate Early and Long-Term Pulmonary Arterial Hypertension Disease Management (REVEAL). Circulation. 2010;122(2):164–172. | ||
D’Alonzo GE, Barst RJ, Ayres SM, et al. Survival in patients with primary pulmonary hypertension. Results from a national prospective registry. Ann Intern Med. 1991;115(5):343–349. | ||
Bogaard HJ, Abe K, Vonk Noordegraaf A, Voelkel NF. The right ventricle under pressure: cellular and molecular mechanisms of right-heart failure in pulmonary hypertension. Chest. 2009;135(3):794–804. | ||
Champion HC, Michelakis ED, Hassoun PM. Comprehensive invasive and noninvasive approach to the right ventricle-pulmonary circulation unit: state of the art and clinical and research implications. Circulation. 2009;120(11):992–1007. | ||
Galiè N, Hoeper MM, Humbert M, et al; Task Force for Diagnosis and Treatment of Pulmonary Hypertension of European Society of Cardiology (ESC); European Respiratory Society (ERS); International Society of Heart and Lung Transplantation (ISHLT). Guidelines for the diagnosis and treatment of pulmonary hypertension. Eur Respir J. 2009;34(6):1219–1263. | ||
McLaughlin VV, McGoon MD. Pulmonary arterial hypertension. Circulation. 2006;114(13):1417–1431. | ||
Le RJ, Fenstad ER, Maradit-Kremers H, et al. Syncope in adults with pulmonary arterial hypertension. J Am Coll Cardiol. 2011;58(8): 863–867. | ||
Humbert M, Sitbon O, Chaouat A, et al. Survival in patients with idiopathic, familial, and anorexigen-associated pulmonary arterial hypertension in the modern management era. Circulation. 2010;122(2): 156–163. | ||
Sitbon O, Humbert M, Nunes H, et al. Long-term intravenous epoprostenol infusion in primary pulmonary hypertension: prognostic factors and survival. J Am Coll Cardiol. 2002;40(4):780–788. | ||
McLaughlin VV, Shillington A, Rich S. Survival in primary pulmonary hypertension: the impact of epoprostenol therapy. Circulation. 2002;106(12):1477–1482. | ||
Dimopoulos K, Inuzuka R, Goletto S, et al. Improved survival among patients with Eisenmenger syndrome receiving advanced therapy for pulmonary arterial hypertension. Circulation. 2010;121(1):20–25. | ||
Launay D, Sitbon O, Le Pavec J, et al. Long-term outcome of systemic sclerosis-associated pulmonary arterial hypertension treated with bosentan as first-line monotherapy followed or not by the addition of prostanoids or sildenafil. Rheumatology (Oxford). 2010;49(3):490–500. | ||
ATS Committee on Proficiency Standards for Clinical Pulmonary Function Laboratories. ATS statement: guidelines for the six-minute walk test. Am J Respir Crit Care Med. 2002;166(1):111–117. | ||
Provencher S, Sitbon O, Humbert M, Cabrol S, Jaïs X, Simonneau G. Long-term outcome with first-line bosentan therapy in idiopathic pulmonary arterial hypertension. Eur Heart J. 2006;27(5):589–595. | ||
Wensel R, Opitz CF, Anker SD, et al. Assessment of survival in patients with primary pulmonary hypertension: importance of cardiopulmonary exercise testing. Circulation. 2002;106(3):319–324. | ||
McLaughlin VV, Gaine SP, Howard LS, et al. Treatment goals of pulmonary hypertension. J Am Coll Cardiol. 2013;62(25 Suppl):D73–D81. | ||
McLaughlin VV, Archer SL, Badesch DB, et al; ACCF/AHA. ACCF/AHA 2009 expert consensus document on pulmonary hypertension: a report of the American College of Cardiology Foundation Task Force on Expert Consensus Documents and the American Heart Association: developed in collaboration with the American College of Chest Physicians, American Thoracic Society, Inc., and the Pulmonary Hypertension Association. Circulation. 2009;119(16):2250–2294. | ||
Nickel N, Golpon H, Greer M, et al. The prognostic impact of follow-up assessments in patients with idiopathic pulmonary arterial hypertension. Eur Respir J. 2012;39(3):589–596. | ||
Miura Y, Fukumoto Y, Sugimura K, et al. Identification of new prognostic factors of pulmonary hypertension. Circ J. 2010;74(9):1965–1971. | ||
Hoeper MM, Pletz MW, Golpon H, Welte T. Prognostic value of blood gas analyses in patients with idiopathic pulmonary arterial hypertension. Eur Respir J. 2007;29(5):944–950. | ||
Badano LP, Ginghina C, Easaw J, et al. Right ventricle in pulmonary arterial hypertension: haemodynamics, structural changes, imaging, and proposal of a study protocol aimed to assess remodelling and treatment effects. Eur J Echocardiogr. 2010;11(1):27–37. | ||
Miller D, Farah MG, Liner A, Fox K, Schluchter M, Hoit BD. The relation between quantitative right ventricular ejection fraction and indices of tricuspid annular motion and myocardial performance. J Am Soc Echocardiogr. 2004;17(5):443–447. | ||
Forfia PR, Fisher MR, Mathai SC, et al. Tricuspid annular displacement predicts survival in pulmonary hypertension. Am J Respir Crit Care Med. 2006;174(9):1034–1041. | ||
Tei C, Dujardin KS, Hodge DO, et al. Doppler echocardiographic index for assessment of global right ventricular function. J Am Soc Echocardiogr. 1996;9(6):838–847. | ||
Yeo TC, Dujardin KS, Tei C, Mahoney DW, McGoon MD, Seward JB. Value of a Doppler-derived index combining systolic and diastolic time intervals in predicting outcome in primary pulmonary hypertension. Am J Cardiol. 1998;81(9):1157–1161. | ||
Grapsa I, Pavlopoulos H, Dawson D, Gibbs JS, Nihoyannopoulos P. Retrospective study of pulmonary hypertensive patients: is right ventricular myocardial performance index a vital prognostic factor? Hellenic J Cardiol. 2007;48(3):152–160. | ||
López-Candales A, Rajagopalan N, Saxena N, Gulyasy B, Edelman K, Bazaz R. Right ventricular systolic function is not the sole determinant of tricuspid annular motion. Am J Cardiol. 2006;98(7):973–977. | ||
Ghio S, Klersy C, Magrini G, et al. Prognostic relevance of the echocardiographic assessment of right ventricular function in patients with idiopathic pulmonary arterial hypertension. Int J Cardiol. 2010;140(3): 272–278. | ||
Torbicki A. Cardiac magnetic resonance in pulmonary arterial hypertension: a step in the right direction. Eur Heart J. 2007;28(10): 1187–1189. | ||
van Wolferen SA, Marcus JT, Boonstra A, et al. Prognostic value of right ventricular mass, volume, and function in idiopathic pulmonary arterial hypertension. Eur Heart J. 2007;28(10):1250–1257. | ||
Yamada Y, Okuda S, Kataoka M, et al. Prognostic value of cardiac magnetic resonance imaging for idiopathic pulmonary arterial hypertension before initiating intravenous prostacyclin therapy. Circ J. 2012;76(7): 1737–1743. | ||
Torbicki A, Kurzyna M, Kuca P, et al. Detectable serum cardiac troponin T as a marker of poor prognosis among patients with chronic precapillary pulmonary hypertension. Circulation. 2003;108(7):844–848. | ||
Nagaya N, Nishikimi T, Uematsu M, et al. Plasma brain natriuretic peptide as a prognostic indicator in patients with primary pulmonary hypertension. Circulation. 2000;102(8):865–870. | ||
Mauritz GJ, Rizopoulos D, Groepenhoff H, et al. Usefulness of serial N-terminal pro-B-type natriuretic peptide measurements for determining prognosis in patients with pulmonary arterial hypertension. Am J Cardiol. 2011;108(11):1645–1650. | ||
Williams MH, Handler CE, Akram R, et al. Role of N-terminal brain natriuretic peptide (N-TproBNP) in scleroderma-associated pulmonary arterial hypertension. Eur Heart J. 2006;27(12):1485–1494. | ||
Nagaya N, Uematsu M, Satoh T, et al. Serum uric acid levels correlate with the severity and the mortality of primary pulmonary hypertension. Am J Respir Crit Care Med. 1999;160(2):487–492. | ||
Taguchi H, Kataoka M, Yanagisawa R, et al. Platelet level as a new prognostic factor for idiopathic pulmonary arterial hypertension in the era of combination therapy. Circ J. 2012;76(6):1494–1500. | ||
Price LC, Wort SJ, Perros F, et al. Inflammation in pulmonary arterial hypertension. Chest. 2012;141(1):210–221. | ||
Cracowski JL, Chabot F, Labarère J, et al. Proinflammatory cytokine levels are linked to death in pulmonary arterial hypertension. Eur Respir J. 2014;43(3):915–917. | ||
Heresi GA, Aytekin M, Hammel JP, Wang S, Chatterjee S, Dweik RA. Plasma interleukin-6 adds prognostic information in pulmonary arterial hypertension. Eur Respir J. 2014;43(3):912–914. | ||
Benza RL, Miller DP, Barst RJ, Badesch DB, Frost AE, McGoon MD. An evaluation of long-term survival from time of diagnosis in pulmonary arterial hypertension from the REVEAL Registry. Chest. 2012;142(2):448–456. | ||
Benza RL, Gomberg-Maitland M, Miller DP, et al. The REVEAL Registry risk score calculator in patients newly diagnosed with pulmonary arterial hypertension. Chest. 2012;141(2):354–362. | ||
Humbert M, Sitbon O, Yaïci A, et al; French Pulmonary Arterial Hypertension Network. Survival in incident and prevalent cohorts of patients with pulmonary arterial hypertension. Eur Respir J. 2010;36(3): 549–555. | ||
Thenappan T, Shah SJ, Rich S, Tian L, Archer SL, Gomberg-Maitland M. Survival in pulmonary arterial hypertension: a reappraisal of the NIH risk stratification equation. Eur Respir J. 2010;35(5):1079–1087. | ||
Barst RJ, Gibbs JS, Ghofrani HA, et al. Updated evidence-based treatment algorithm in pulmonary arterial hypertension. J Am Coll Cardiol. 2009;54(1 Suppl):S78–S84. | ||
Galiè N, Torbicki A, Barst R, et al; Task Force. Guidelines on diagnosis and treatment of pulmonary arterial hypertension. The Task Force on Diagnosis and Treatment of Pulmonary Arterial Hypertension of the European Society of Cardiology. Eur Heart J. 2004;25(24): 2243–2278. | ||
Badesch DB, Abman SH, Simonneau G, Rubin LJ, McLaughlin VV. Medical therapy for pulmonary arterial hypertension: updated ACCP evidence-based clinical practice guidelines. Chest. 2007;131(6): 1917–1928. | ||
Kataoka M, Nagaya N, Satoh T, et al. A long-acting prostacyclin agonist with thromboxane inhibitory activity for pulmonary hypertension. Am J Respir Crit Care Med. 2005;172(12):1575–1580. | ||
Giaid A, Yanagisawa M, Langleben D, et al. Expression of endothelin-1 in the lungs of patients with pulmonary hypertension. N Engl J Med. 1993;328(24):1732–1739. | ||
Mehta S. Sildenafil for pulmonary arterial hypertension: exciting, but protection required. Chest. 2003;123(4):989–992. | ||
Barst RJ, Rubin LJ, Long WA, et al; Primary Pulmonary Hypertension Study Group. A comparison of continuous intravenous epoprostenol (prostacyclin) with conventional therapy for primary pulmonary hypertension. N Engl J Med. 1996;334(5):296–301. | ||
Sitbon O, McLaughlin VV, Badesch DB, et al. Survival in patients with class III idiopathic pulmonary arterial hypertension treated with first line oral bosentan compared with an historical cohort of patients started on intravenous epoprostenol. Thorax. 2005;60(12):1025–1030. | ||
Channick RN, Simonneau G, Sitbon O, et al. Effects of the dual endothelin-receptor antagonist bosentan in patients with pulmonary hypertension: a randomised placebo-controlled study. Lancet. 2001;358(9288): 1119–1123. | ||
Rubin LJ, Badesch DB, Barst RJ, et al. Bosentan therapy for pulmonary arterial hypertension. N Engl J Med. 2002;346(12):896–903. | ||
Galiè N, Brundage BH, Ghofrani HA, et al; Pulmonary Arterial Hypertension and Response to Tadalafil (PHIRST) Study Group. Tadalafil therapy for pulmonary arterial hypertension. Circulation. 2009;119(22):2894–2903. | ||
Barst RJ, Langleben D, Badesch D, et al; STRIDE-2 Study Group. Treatment of pulmonary arterial hypertension with the selective endothelin-A receptor antagonist sitaxsentan. J Am Coll Cardiol. 2006;47(10): 2049–2056. | ||
Galiè N, Rubin Lj, Hoeper M, et al. Treatment of patients with mildly symptomatic pulmonary arterial hypertension with bosentan (EARLY study): a double-blind, randomised controlled trial. Lancet. 2008;371(9630):2093–2100. | ||
Humbert M, Barst RJ, Robbins IM, et al. Combination of bosentan with epoprostenol in pulmonary arterial hypertension: BREATHE-2. Eur Respir J. 2004;24(3):353–359. | ||
Hoeper MM, Leuchte H, Halank M, et al. Combining inhaled iloprost with bosentan in patients with idiopathic pulmonary arterial hypertension. Eur Respir J. 2006;28(4):691–694. | ||
Simonneau G, Rubin LJ, Galiè N, et al; PACES Study Group. Addition of sildenafil to long-term intravenous epoprostenol therapy in patients with pulmonary arterial hypertension: a randomized trial. Ann Intern Med. 2008;149(8):521–530. | ||
Sitbon O, Jaïs X, Savale L, et al. Upfront triple combination therapy in pulmonary arterial hypertension: a pilot study. Eur Respir J. 2014;43(6): 1691–1697. | ||
Sitbon O, Simonneau G. Optimal management of severe pulmonary arterial hypertension. Eur Respir Rev. 2011;20(122):254–261. | ||
Kurzyna M, Dabrowski M, Bielecki D, et al. Atrial septostomy in treatment of end-stage right heart failure in patients with pulmonary hypertension. Chest. 2007;131(4):977–983. | ||
Reichenberger F, Pepke-Zaba J, McNeil K, Parameshwar J, Shapiro LM. Atrial septostomy in the treatment of severe pulmonary arterial hypertension. Thorax. 2003;58(9):797–800. | ||
Doyle RL, McCrory D, Channick RN, Simonneau G, Conte J; American College of Chest Physicians. Surgical treatments/interventions for pulmonary arterial hypertension: ACCP evidence-based clinical practice guidelines. Chest. 2004;126(1 Suppl):63S–71S. | ||
Keogh AM, Mayer E, Benza RL, et al. Interventional and surgical modalities of treatment in pulmonary hypertension. J Am Coll Cardiol. 2009;54(1 Suppl):S67–S77. | ||
Sandoval J, Rothman A, Pulido T. Atrial septostomy for pulmonary hypertension. Clin Chest Med. 2001;22(3):547–560. | ||
McLaughlin VV, Shillington A, Rich S. Survival in primary pulmonary hypertension: the impact of epoprostenol therapy. Circulation. 2002;106(12):1477–1482. | ||
Sitbon O, Humbert M, Simonneau G. Primary pulmonary hypertension: current therapy. Prog Cardiovasc Dis. 2002;45(2):115–128. | ||
Trulock EP, Christie JD, Edwards LB, et al. Registry of the International Society for Heart and Lung Transplantation: twenty-fourth official adult lung and heart-lung transplantation report-2007. J Heart Lung Transplant. 2007;26(8):782–795. | ||
Orens JB, Estenne M, Arcasoy S, et al; Pulmonary Scientific Council of the International Society for Heart and Lung Transplantation. International guidelines for the selection of lung transplant candidates: 2006 update – a consensus report from the Pulmonary Scientific Council of the International Society for Heart and Lung Transplantation. J Heart Lung Transplant. 2006;25(7):745–755. | ||
Davis SQ, Garrity ER. Organ allocation in lung transplant. Chest. 2007;132(5):1646–1651. | ||
Iribarne A, Russo MJ, Davies RR, et al. Despite decreased wait-list times for lung transplantation, lung allocation scores continue to increase. Chest. 2009;135(4):923–928. | ||
Sitbon O, Galiè N. Treat-to-target strategies in pulmonary arterial hypertension: the importance of using multiple goals. Eur Respir Rev. 2010;19(118):272–278. | ||
Hoeper MM, Markevych I, Spiekerkoetter E, Welte T, Niedermeyer J. Goal-oriented treatment and combination therapy for pulmonary arterial hypertension. Eur Respir J. 2005;26(5):858–863. | ||
Mubarak KK. A review of prostaglandin analogs in the management of patients with pulmonary arterial hypertension. Respir Med. 2010; 104(1):9–21. | ||
Christman BW, McPherson CD, Newman JH, et al. An imbalance between the excretion of thromboxane and prostacyclin metabolites in pulmonary hypertension. N Engl J Med. 1992;327(2):70–75. | ||
Tuder RM, Cool CD, Geraci MW, et al. Prostacyclin synthase expression is decreased in lungs from patients with severe pulmonary hypertension. Am J Respir Crit Care Med. 1999;159(6):1925–1932. | ||
Abramovitz M, Adam M, Boie Y, et al. The utilization of recombinant prostanoid receptors to determine the affinities and selectivities of prostaglandins and related analogs. Biochim Biophys Acta. 2000;1483(2): 285–293. | ||
Kuwano K, Hashino A, Asaki T, et al. 2-[4-[(5,6-diphenylpyrazin-2-yl)(isopropyl)amino] butoxy]-N-(methylsulfonyl)acetamide (NS-304), an orally available and long-acting prostacyclin receptor agonist prodrug. J Pharmacol Exp Ther. 2007;322(3):1181–1188. | ||
Tapson VF, Torres F, Kermeen F, et al. Oral treprostinil for the treatment of pulmonary arterial hypertension in patients on background endothelin receptor antagonist and/or phosphodiesterase type 5 inhibitor therapy (the FREEDOM-C study): a randomized controlled trial. Chest. 2012;142(6):1383–1390. | ||
Tapson VF, Jing ZC, Xu KF, et al; FREEDOM-C2 Study Team. Oral treprostinil for the treatment of pulmonary arterial hypertension in patients receiving background endothelin receptor antagonist and phosphodiesterase type 5 inhibitor therapy (the FREEDOM-C2 study): a randomized controlled trial. Chest. 2013;144(3):952–958. | ||
Jing ZC, Parikh K, Pulido T, et al. Efficacy and safety of oral treprostinil monotherapy for the treatment of pulmonary arterial hypertension: a randomized, controlled trial. Circulation. 2013;127(5):624–633. | ||
Rich J, Hoeper MM. The search for an oral prostanoid to treat pulmonary arterial hypertension continues. Are we getting any closer? Int J Clin Pract Suppl. 2009;(161):17–18. | ||
Ikeda D, Tsujino I, Sakaue S, et al. Pilot study of short-term effects of a novel long-acting oral beraprost in patients with pulmonary arterial hypertension. Circ J. 2007;71(11):1829–1831. | ||
Armstrong DJ, Benza RL, Delcroix M, et al. Clinical pharmacology and safety of beraprost sodium modified release (BPS-MR), an oral twice daily prostacyclin analogue – a phase II study [abstract]. Am J Respir Crit Care Med. 2010;181:A3360. | ||
Morrison K, Ernst R, Hess P, Studer R, Clozel M. Selexipag: a selective prostacyclin receptor agonist that does not affect rat gastric function. J Pharmacol Exp Ther. 2010;335(1):249–255. | ||
Simonneau G, Torbicki A, Hoeper MM, et al. Selexipag: an oral, selective prostacyclin receptor agonist for the treatment of pulmonary arterial hypertension. Eur Respir J. 2012;40(4):874–880. | ||
Dupuis J. Endothelin: setting the scene in PAH. Eur Respir Rev. 2007;16(102):3–7. | ||
Galié N, Manes A, Branzi A. The endothelin system in pulmonary arterial hypertension. Cardiovasc Res. 2004;61(2):227–237. | ||
Iglarz M, Clozel M. At the heart of tissue: endothelin system and end-organ damage. Clin Sci (Lond). 2010;119(11):453–463. | ||
Pulido T, Adzerikho I, Channick RN, et al; SERAPHIN Investigators. Macitentan and morbidity and mortality in pulmonary arterial hypertension. N Engl J Med. 2013;369(9):809–818. | ||
Bruderer S, Aänismaa P, Homery MC, et al. Effect of cyclosporine and rifampin on the pharmacokinetics of macitentan, a tissue-targeting dual endothelin receptor antagonist. AAPS J. 2012;14(1):68–78. | ||
Sidharta PN, Atsmon J, Dingemanse J. Investigation of the effect of ketoconazole on the pharmacokinetics of macitentan in healthy male subjects. Br J Clin Pharmacol. 2010;70:930–931. | ||
Ignarro LJ, Buga GM, Wood KS, Byrns RE, Chaudhuri G. Endothelium-derived relaxing factor produced and released from artery and vein is nitric oxide. Proc Natl Acad Sci U S A. 1987;84(24):9265–9269. | ||
Arnold WP, Mittal CK, Katsuki S, Murad F. Nitric oxide activates guanylate cyclase and increases guanosine 3′:5′-cyclic monophosphate levels in various tissue preparations. Proc Natl Acad Sci U S A. 1977;74(8):3203–3207. | ||
Ghofrani HA, Osterloh IH, Grimminger F. Sildenafil: from angina to erectile dysfunction to pulmonary hypertension and beyond. Nat Rev Drug Discov. 2006;5(8):689–702. | ||
Grimminger F, Weimann G, Frey R, et al. First acute haemodynamic study of soluble guanylate cyclase stimulator riociguat in pulmonary hypertension. Eur Respir J. 2009;33(4):785–792. | ||
Ghofrani HA, Galiè N, Grimminger F, et al; PATENT-1 Study Group. Riociguat for the treatment of pulmonary arterial hypertension. N Engl J Med. 2013;369(4):330–340. | ||
Ghofrani HA, D’Armini AM, Grimminger F, et al; CHEST-1 Study Group. Riociguat for the treatment of chronic thromboembolic pulmonary hypertension. N Engl J Med. 2013;369(4):319–329. | ||
Barst RJ. PDGF signaling in pulmonary arterial hypertension. J Clin Invest. 2005;115(10):2691–2694. | ||
Panos RJ, Baker SK. Mediators, cytokines, and growth factors in liver-lung interactions. Clin Chest Med. 1996;17(1):151–169. | ||
Schermuly RT, Dony E, Ghofrani HA, et al. Reversal of experimental pulmonary hypertension by PDGF inhibition. J Clin Invest. 2005; 115(10):2811–2821. | ||
ten Freyhaus H, Dumitrescu D, Berghausen E, Vantler M, Caglayan E, Rosenkranz S. Imatinib mesylate for the treatment of pulmonary arterial hypertension. Expert Opin Investig Drugs. 2012;21(1):119–134. | ||
Hoeper MM, Barst RJ, Bourge RC, et al. Imatinib mesylate as add-on therapy for pulmonary arterial hypertension: results of the randomized IMPRES study. Circulation. 2013;127(10):1128–1138. | ||
Song KW, Rifkind J, Al-Beirouti B, et al. Subdural hematomas during CML therapy with imatinib mesylate. Leuk Lymphoma. 2004;45(8): 1633–1636. | ||
Kerkelä R, Grazette L, Yacobi R, et al. Cardiotoxicity of the cancer therapeutic agent imatinib mesylate. Nat Med. 2006;12(8):908–916. | ||
Galiè N, Corris PA, Frost A, et al. Updated treatment algorithm of pulmonary arterial hypertension. J Am Coll Cardiol. 2013; 62(25 Suppl):D60–D72. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.