Back to Journals » Journal of Multidisciplinary Healthcare » Volume 11
Optimal management of Cogan’s syndrome: a multidisciplinary approach
Authors D'Aguanno V, Ralli M ![]() , de Vincentiis M, Greco A
, de Vincentiis M, Greco A
Received 22 October 2017
Accepted for publication 25 November 2017
Published 22 December 2017 Volume 2018:11 Pages 1—11
DOI https://doi.org/10.2147/JMDH.S150940
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Scott Fraser
Vittorio D’Aguanno,1 Massimo Ralli,2 Marco de Vincentiis,2 Antonio Greco1
1Department of Sense Organs, Sapienza University of Rome, Rome, Italy; 2Department of Oral and Maxillofacial Sciences, Sapienza University of Rome, Rome, Italy
Abstract: Cogan’s syndrome (CS) is a rare disorder characterized by nonsyphilitic interstitial keratitis (IK) and audio-vestibular symptoms. CS affects mainly young Caucasian adults, mostly during their first three decades of age, and may develop into typical and atypical variants. Typical CS manifests primarily with IK and hearing loss, whereas atypical CS usually presents with inflammatory ocular manifestations in association with audio-vestibular symptoms but mostly different Ménière-like symptoms and, more frequently, with systemic inflammation (70%), of which vasculitis is the pathogenic mechanism. CS is considered as an autoimmune- or immune-mediated disease supported mainly by the beneficial response to corticosteroids. Using well-developed assays, antibodies to inner ear antigens, anti-Hsp70, and antineutrophil cytoplasmic antibodies were found to be associated with CS. Corticosteroids represent the first line of treatment, and multiple immunosuppressive drugs have been tried with variable degrees of success. Tumor necrosis factor-alpha blockers and other biological agents are a recent novel therapeutic option in CS. Cochlear implantation is a valuable rescue surgical strategy in cases with severe sensorineural hearing loss unresponsive to intensive and/or innovative immunosuppressive regimens.
Keywords: Cogan’s syndrome, hearing loss, multidisciplinary approach, autoimmunity
Introduction
Cogan’s syndrome (CS) is a rare multisystem disease of unknown origin characterized by ocular and audio-vestibular symptoms including nonsyphilitic interstitial keratitis (IK), conjunctivitis, scleritis, uveitis, retinal vasculitis, hearing loss, vertigo, and tinnitus. The association of these symptoms was first described in 1934,1 but CS was named after the American ophthalmologist David G Cogan who described a series of four cases in 1945.2 The etiology of CS is still debated. An infectious origin seems to be possible through a viral involvement that triggers a molecular mimicry mechanism.3,4 In the recent years, the role of autoimmunity has raised interest in the pathogenesis for CS; the demonstration of autoantibodies against corneal, inner ear, and endothelial antigens in some patients with CS can further support a significant role for the autoimmune nature of this disease.3,4 Other studies suggested a cell-mediated reaction.3 CS develops mainly in young Caucasian adults, mostly during their first three decades of age, but can also rarely present in childhood or later in life, with no gender-specific prevalence.5,6 However, population-based analysis of incidence and prevalence are unfeasible due to the rarity of the disease. In this review, the authors initially focus on the clinical features of CS, with a brief description of the large spectrum of clinical manifestations found in this rare condition; second, the authors discuss the pathogenetic basis of the disease, discussing the more relevant theories available in the literature. This review continues with a description of the diagnostic and, especially, therapeutic approaches to CS, with special attention to the multidisciplinary approach required for these patients. At last, a discussion of future options for diagnosis and, mainly, treatment of CS is presented.
Clinical features
CS includes a large spectrum of clinical manifestations. In 1980, Haynes et al7 defined two types of CS, a typical variant and an atypical variant. Typical CS is defined by 1) ocular symptoms, classically presenting as nonsyphilitic IK; 2) audio-vestibular symptoms similar to those of Ménière’s disease (recurrent episodes of hearing loss, tinnitus, and vertigo often associated with nausea and vomiting); and 3) an interval between the onset of ocular and audio-vestibular manifestations of <2 years. Atypical CS is characterized by 1) different inflammatory ocular manifestations, with or without IK; 2) audio-vestibular symptoms (usually progressive hearing loss); and, most important, 3) a delay of >2 years between the onset of ocular and audio-vestibular manifestations. In many cases, it is difficult to differentiate between the two types of CS because some patients do not present IK at the onset of the disease or, alternatively, develop this condition during the following years. Timing and association between symptoms of CS may be extremely variable. Systemic manifestations are much more frequent in atypical CS and can be used in the differential diagnosis between the two types.
Systemic manifestations are present in ~30%–50% of patients in addition to the typical ocular and audio-vestibular symptoms.8 Gluth et al9 reported the following systemic symptoms in a survey of 60 patients with CS: headache (40%), arthralgia (35%), fever (27%), arthritis (23%), and myalgia (22%). Vasculitis of small, medium, and large vessels is considered as the underlying pathogenic mechanism for systemic presentation of CS,8,10 which may involve the cardiovascular, neurological, and gastrointestinal systems.6 The most typical cardiovascular manifestation of CS is aortitis,9,11 leading to kidney and aortic insufficiency, reported in ~10% of patients, and congestive heart failure (Figure 1).3,12 Neurological manifestations may include hemiparesis or hemiplegia following cerebral vascular accidents, and aphasia, due to transient ischemic events.13,14 Gastrointestinal manifestations, such as diarrhea, melena, and abdominal pain, have also been reported following a possible involvement of the mesenteric arteritis.15 Inflammatory bowel disease such as ulcerative colitis and Crohn’s disease are also associated with CS.16,17
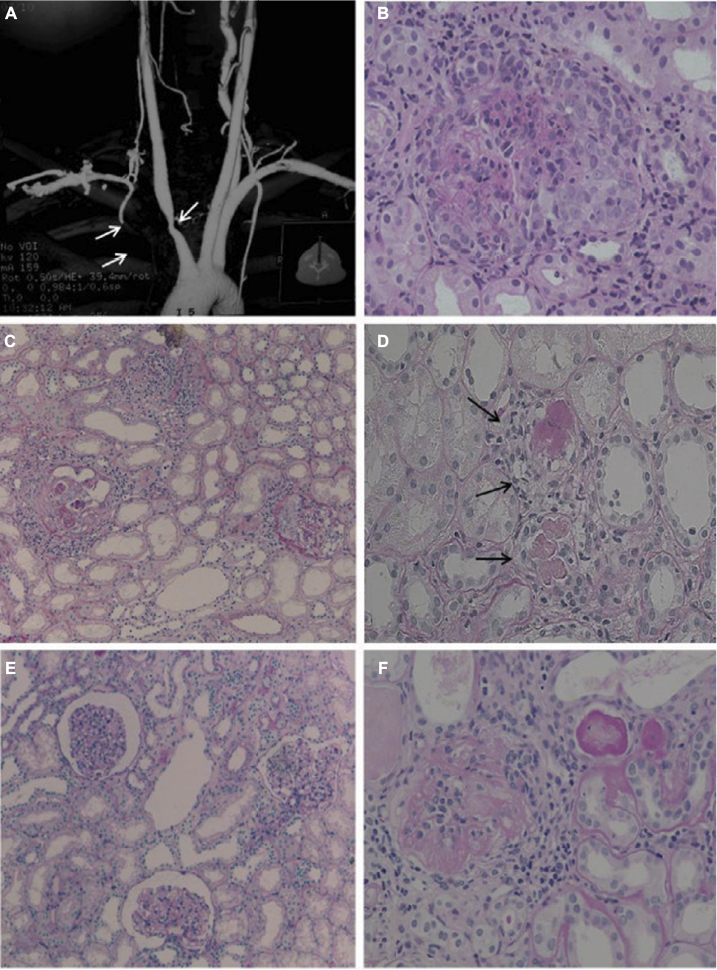 | Figure 1 The evaluation of the patient’s aortitis and renal disorder. Notes: MRA (A) and renal histologic (B–D, Periodic acid-Schiff stain) findings in the patient. MRA showed stenosis of the right brachiocephalic artery and proximal obstruction in the right subclavian artery (indicated by arrows). Renal histologic findings while proteinuria was worsening included mesangial proliferation with circumferential crescent formation (B, ×200) and periglomerular mononuclear cell infiltration (C, ×100). Small renal arteries showed medial thickening, endothelial cell proliferation, and thrombosis (D, ×100, indicated by arrows). After treatment with PSL and an immunosuppressant, glomerular and tubular interstitial lesions decreased (E, ×100). However, repeated relapses occurred, and a recent specimen showed sclerotic glomeruli (F, ×200). Reproduced from Sugimoto K, Miyazawa T, Nishi H, Izu A, Enya T, Okada M, Takemura T. Childhood Cogan syndrome with aortitis and anti-neutrophil cytoplasmic antibody-associated glomerulonephritis. Pediatr Rheumatol Online J. 2014;12:15. Copyright © 2014 Sugimoto et al.; licensee BioMed Central Ltd. Creative Commons License available at: https://creativecommons.org/licenses/by/2.0/.65 Abbreviation: MRA, magnetic resonance angiography. |
The most common audio-vestibular manifestations in CS are hearing loss, vertigo, tinnitus, ataxia, and oscillopsia. These symptoms can appear at any time during the course of the disease.9 Hearing loss may be both unilateral and bilateral and often presents as sudden sensorineural hearing loss with fluctuations or progressive worsening over time. Tinnitus, the perception of sound without an accompanying external auditory stimulus,18 is often found in patients with CS.14 Tinnitus is associated with hearing loss, ototoxic drug use, infections, psychological stress, and a range of medical conditions that can affect hearing function.18–26 In patients with CS, tinnitus appears to be a consequence of peripheral auditory deafferentation rather than a direct consequence of the pathogenic mechanisms of CS.5 Progression to complete bilateral hearing loss has been reported in ~50% of patients during the follow-up period, whereas permanent hearing loss in one ear was observed in 20% of patients. Abnormal vestibular function is found in 90% of patients with CS; at least 20% of the patients present spontaneous or gaze-induced nystagmus. Rarely, patients present with clinical symptoms of vestibulopathy that lasts for days or weeks from the time of onset without spontaneous resolution that frequently results to hospitalization.9
IK is the most frequent ophthalmologic symptom of typical CS, which appears in 80% of cases and is mostly bilateral, clinically consisting in an irregular, granular corneal infiltration, which can progress to nummular lesions.9 A deep stromal keratitis can occur following vascularization of the cornea particularly affecting the posterior part of the cornea, near the limbus. Patient often reported eye redness, pain, photophobia, and blurred vision.9 In some cases, IK develops later during the course of disease and is missed at the initial examination. Other ocular manifestations, mainly in patients with atypical CS, are acute closure angle glaucoma, retinal vasculitis, conjunctivitis, scleritis, papillitis, central vein occlusion, vasculitic optic neuropathy, and papilledema. Most of the patients maintain normal or near-normal vision, and only few develop some degree of visual loss directly attributable to inflammatory eye disease.9
Pathogenesis
The pathogenetic basis of CS is still unclear. Infections of the upper respiratory tract precede the onset of disease in 50% of cases, suggesting an infectious origin.6 Chlamydial infection in particular has received much attention for its ability to elude host defense mechanisms and cause chronic infection. However, a direct link between different Chlamydia species and CS has not been proven.27 Moreover, the antibodies against the Cogan peptide have been shown to cross-react with a structural protein of the Reovirus type III, suggesting a viral involvement in the pathogenesis of the disease possibly through a molecular mimicry mechanism, through which sequence similarities between foreign and self-peptides result in the cross-activation of autoreactive cells by pathogen-derived peptides.29 This occurs when antigenic determinants are shared between exogenous agents (viruses, bacteria, food antigens, and others) and normal human host–cell components. Molecular mimicry is one of the mechanisms whereby autoimmunity can arise.29 In fact, Reovirus type III infection causes mild rhinitis and pharyngitis and upper respiratory tract infections are frequently observed before the onset of CS.6
Immunological mechanisms play a role in CS pathogenesis; Hughes et al28 reported that serum antibodies to undefined corneal antigens and to a mixture of inner ear extracts could initiate or perpetuate the immune response in patients with the disease. Antibodies against a peptide antigen (Cogan peptide) have been found in sera from patients with CS. This peptide antigen shares sequence homology with CD148 and Connexin 26, which are expressed on endothelial cells and in the inner ear.29 Antibodies directed against the Cogan peptide showing similarities with auto-antigens, including CD148, were identified. The same antibodies also are bound to Connexin 26, which has been implicated in congenital deafness; furthermore, the loss of this protein results in potassium-mediated damage to the organ of Corti, leading to hearing loss. Connexin 26 shows similarity to Connexin 43 and Connexin 50, gap junction proteins present in corneal fibroblasts and epithelium.30 Since nonsyphilitic IK is another key feature of CS, this homology might have implications in explaining the eye involvement in the disease. The antigen is also expressed on nerve and glial cells and may explain the wide clinical spectrum of CS, including the neurological abnormalities.31
The induction of clinical features of CS in animals after either passive transfer of peptide-specific autoantibodies or active immunization with autoantigen peptides suggests that CS may be have an autoimmune basis. After passive transfer of antibodies directed against the Cogan peptide into Balb/c mice, antibodies were found within the cochlea of the tested animals, whereas antibodies against an irrelevant peptide did not bind to cochlear cells,29 six of six Balb/c mice injected with the purified antibodies developed hearing loss found via the recording of auditory brainstem responses in mice, showing that an higher stimulus intensity was necessary to obtain smaller and more delayed responses after immunization compared to pretreatment. Also, Lunardi et al29 showed that a rabbit immunized with different peptides derived from CD148 developed hearing loss and IK.
Diagnosis
The diagnosis of CS is mainly clinical and is based on audio-vestibular symptoms, ocular inflammation, and nonreactive serological tests for syphilis.32 Diagnosis requires a multidisciplinary approach, as also reported in other similar autoimmune conditions involving the auditory system.33–35 The variable onset of symptoms and the lack of specific laboratory tests contribute to challenges in making CS diagnosis that is often based on the good response to corticosteroid treatment.9,36 Clinical diagnostic criteria of CS include mandatory, prevalent, and possible additional criteria. Mandatory criteria include sensorineural hearing loss, inflammatory ocular disease, and the exclusion of alternative causes of inflammation or infection. Prevalent additional criteria include vertigo, tinnitus, ataxia, dizziness, fever, weight loss, fatigue, lymphadenopathy, and headache. Possible additional criteria include large or – rarely – medium and small vessel vasculitis and positivity to laboratory tests for systemic inflammatory markers (Table 1). Globally diagnostic criteria for CS are still missing.
 | Table 1 Criteria for clinical diagnosis of Cogan’s syndrome Note: Mandatory and on prevalent and possible additional criteria for clinical diagnosis of Cogan’s syndrome. |
Laboratory tests commonly used to detect autoimmune diseases are rarely positive in CS; a negative result does not rule out CS. No specific laboratory tests are diagnostic for CS, although the erythrocyte sedimentation rate is often elevate during active disease.9 Multiple studies indicated various antibodies directed against inner ear, corneal, and endothelial antigens as possible serological markers of the disease.15,16 In particular, antibodies against Cogan peptide, an antigen that shares sequence homology with CD148 and Connexin 26, were investigated.29 Moreover, identification of antibodies anti-heat shock protein 70 (Hsp70) was identified in patients with sensorineural hearing loss; assaying these antibodies in serum could represent a serological marker for CS.37 Some studies evaluated the rate of CS patients positive for anti-Hsp70 ranging from 45% to 50%.38,39 A prevalence of positive anti-Hsp70 was observed in patients affected by typical CS compared to those with atypical CS (66.7% vs 37.5%, respectively).39 Thus, prevalence of anti-Hsp70 in typical CS was also significant compared to patient affected from autoimmune sensorineural hearing loss and controls. In the pediatric age, typical CS may develop with negative anti-Hsp70. This clinical behavior may be explained with the uncompleted development of “immunity competence”, as proposed by some authors.40 In these cases, test repetition is recommended after the age of complete immune competence.38
Other authors suggested the association of CS and antineutrophil cytoplasmic antibodies (ANCA).41 ANCA have recently been identified in patients with systemic vasculitis, such as Wegener’s granulomatosis, Churg-Strauss syndrome, and polyarteritis nodosa. After the discovery of ANCA, myeloperoxidase (MPO) and proteinase 3 (PR3) were identified as the two major antigens.42 Normally, MPO and PR3 are localized intracellularly; however, when neutrophils are preactivated by proinflammatory cytokines, these enzymes become expressed on the cell surface and are accessible to circulating ANCA. These antibodies may be involved in the immune pathogenesis of vasculitis by the activation of primed neutrophils, leading to the release of lytic enzymes.43,44 Tervaert et al reported one case of CS that was also positive for MPO ANCA and anti-human leukocyte elastase ANCA.45 Yamanishi et al described a case of atypical CS associated with ANCA.41 To date, five cases of CS associated with ANCA have been reported41 and two of them also showed ANCA-related glomerulonephritis. Previously, Cheson et al8 reviewed 53 cases of CS; 10/18 vessel or muscle biopsy specimens showed inflammatory vascular changes, of which four were considered to be diagnostic of polyarteritis in large- and medium-sized arteries. A common pathological feature of ANCA-associated vasculitis is necrotizing vasculitis of small vessels;46 therefore, arteries of all sizes may be affected in CS.
Radiographic studies, such as cranial computed tomography (CT) and magnetic resonance imaging (MRI), are often normal in CS patients.9 Some authors have reported the presence of labyrinthine aspecific radiological abnormalities in MRI scans with gadolinium contrast like calcification or narrowing and soft tissue obliteration of the vestibular labyrinth and the cochlea.36 Positron emission tomography (PET) with 2-deoxy-2-[18F]fluoro-d-glucose (FDG-PET/CT) scanning has been reported as an additional diagnostic examination to assess involvement in large vessel vasculitis common in CS. In a case report, Balink et al supported the hypothesis that with PET, the inflammatory activity of large-vessel vasculitis could be more accurately assessed compared to laboratory acute-phase parameters (Figure 2).47,48
 | Figure 2 Aortitis in Cogan’s syndrome. Notes: (A) Transverse hybrid PET/CT slice; pathological uptake in the wall of the aortic arch, more intense in the lateral wall and perivascular space adjacent to the truncus pulmonalis. (B) Follow-up PET/CT showed clearly decreased uptake in the aortic arch after 3 weeks treatment with methyl-prednisolon intravenous and prednisolon orally. (C) Second follow-up PET/CT 6 months later (patient was in a stable condition with methotrexate and low-dose prednisone) with again high uptake in the wall of the aortic arch, with higher intensity in the lateral wall and perivascular space adjacent to the truncus pulmonalis. Methotrexate and prednisone were increased to 20 mg/day. Reproduced from Balink H, Bennink RJ, van Eck-Smit BL, Verberne HJ. The role of 18F-FDG PET/CT in large-vessel vasculitis: appropriateness of current classification criteria? Biomed Res Int. 2014;2014:687608). Copyright © 2014 H. Balink et al. Creative Commons License available at: https://creativecommons.org/licenses/by/3.0/.48 Abbreviations: CT, computed tomography; PET, positron emission tomography. |
Differential diagnoses to consider are Takayasu’s arteritis,49,50 polyarteritis nodosa, Wegener’s granulomatosis, giant cell arteritis, and rheumatic arthritis.6 It is particularly difficult to distinguish between Takayasu’s arteritis and the vasculitis of CS because they both involve medium and – mainly – large vessels. However, unlike CS, Takayasu’s arteritis does not involve the eyes.49,51 Rheumatoid factor, anti-nuclear, and anti-endothelial antibodies have also been reported in some patients with CS, further supporting the autoimmune origin theory.3
Multidisciplinary approach to treatment of CS
CS is a systemic disease that can lead to severe functional impairments; therefore, clinical management and treatment must involve by different specialists whose competencies should be coordinated.52 Preclinical therapeutic management in patients with CS is not considered in clinical practice since CS is often diagnosed following clinical manifestations.
Clinical diagnosis of CS is based on the following three mandatory criteria: sensorineural hearing loss, inflammatory ocular disease, and alternative causes of inflammation or infection ruled out. Prevalent additional criteria include vertigo, tinnitus, ataxia, dizziness, fever, weight loss, fatigue, lymphadenopathy, and headache. Among possible additional criteria, it is important to consider large or – rarely – medium and small vessel vasculitis and positivity to laboratory tests for systemic inflammatory markers (Table 1). Treatment of the CS depends on its severity and organ involvement.3,53 In cases with mild eye involvement, the treatment of choice is the application of topical glucocorticoids and cycloplegic agents, such as atropine eye drops. In a case report, topical cyclosporine A (CyA) was used successfully in the treatment of severe anterior segment inflammation.54 Patients with posterior involvement (retinal vasculitis and posterior uveitis) often require systemic treatment.
The involvement of the inner ear, a severe inflammation of the eye, and the development of systemic vasculitis require systemic immunosuppressive therapy. Corticosteroids at high dosages remain the mainstay at the beginning and during the acute phases of the disease.52
Pharmacological treatment
Corticosteroids
Though reported to be the standard of care and of promising outcome, there are no double blind controlled studies to show the beneficial effects of corticosteroids in CS management and treatment.6,7,55 A clinical response to steroids may also help a challenging diagnosis as an “ex juvantibus” criterion. In these cases, systemic corticosteroids are considered as the primary standard of care for CS. In response to corticosteroid therapy, the ocular symptoms have been reported to be more responsive than the audio-vestibulary symptoms and a poor clinical response to this therapy may indicate a possible misdiagnosis rather than a resistant form.52
In early stages of the disease, a limited vasculitis results in labyrinthine ischemia, with a beneficial response to steroid treatment. Over the long term, the organ of Corti can degenerate, with the development of fibrosis and osteoneogenesis within the perilymphatic space; in such cases, significant improvement should not be expected.10
At the onset of audio-vestibular dysfunction, rapid initiation of high dose corticosteroids (1–1.5 mg/kg of prednisone daily) is recommended, expecting a beneficial response to be noticed within 2–3 weeks.7,56 Cabezas-Rodriguez et al57 reported significant hearing improvements after treatment with intravenous methylprednisolone. This single-drug regimen, however, should last no longer than 2–3 weeks due to either the development of significant glucocorticoid-induced toxicity or the inability to taper the dosage maintaining the control of the disease.52 When hearing improves, based on the responses of pure tone audiometry, steroids should be tapered slowly and usually continued for 2–6 months.52
The importance of corticosteroid therapy was confirmed in a 5-year follow-up study conducted by Haynes in which 95% of untreated patients with CS developed permanent hearing loss compared to only 55% of patients treated with systemic steroids within 2 weeks of initial hearing loss.7 Nevertheless, some authors have noted that although hearing loss progression could be slowed with corticosteroid treatment, bilateral audio-vestibular dysfunction in CS could not be prevented.58,59
The vasculitis and other prominent systemic complications of CS usually respond well to systemic corticosteroids, which have proven to be of short-term benefit; however, they can also be associated with side effects. Short-term corticosteroid use is associated with generally mild side effects, including cutaneous effects, electrolyte abnormalities, hypertension, hyperglycemia, pancreatitis, hematological, immunological, and neuropsychological effects. Long-term corticosteroid use may be associated with more serious sequela, including osteoporosis, aseptic joint necrosis, adrenal insufficiency, gastrointestinal, hepatic, and ophthalmological effects, hyperlipidemia, and growth suppression.
Since high doses of corticosteroids or prolonged treatment courses were required in many CS patients, other immunosuppressive agents such as cyclophosphamide (Cyc),60 azathioprine (AZA), methotrexate (MTX),61 CyA and tumor necrosis factor alpha (TNFα) blockers were often combined to steroids.43,44 Specifically, infliximab (IFX) appears to be effective when initiated at an early stage of inner ear disease and afterward in maintaining remission in patients with therapy-resistant CS.62
Cyclophosphamide
Cyc is an alkylating agent usually used for the treatment of severe uveitis/scleritis and systemic vasculitis. Cyc is an inactive drug that is converted into phosphoramide mustard and acrolein by the cytochrome P-450 oxidase system in the liver; the active form is able to introduce alkyl radicals into DNA strands. One retrospective multicentric study reported the use in CS as well as in some case series.60,63 The majority of the described patients receiving pulse intravenous Cyc combined with oral glucocorticoids with a clinical stabilization of CS. Nevertheless, some articles described only a partial or short-term response,46,59,60,64,65 or a lack of response;12,15,60,66,67 in some cases, discontinuation was warranted due to systemic side effects.60,63
Methotrexate
MTX is an inhibitor of the dihydrofolate reductase, essential for DNA replication, that interferes with the synthesis of purine nucleotides and thymidylate. MTX can be considered a second-line, steroid-sparing therapy for CS. Two small-sized prospective studies evaluate the use of MTX for refractory autoimmune inner ear disease. In both studies, the cohort included three CS patients with a clinical improvement of the disease.68,69 Thus, the efficacy and safety of MTX was assessed in several case series, case reports and one retrospective study, mainly during the maintenance phase.12,32,44,51,61,64,70,71 The use of MTX was discussed as an option also for the treatment of CS in pediatric age.72,73 Intolerance or poor effectiveness of this immunosuppressant in CD has been reported.12,15,43,70,74
Azathioprine
AZA is a prodrug that is metabolized by the liver to its active form. It interferes with DNA, RNA, and protein syntheses. It inhibits maturation of B and T lymphocytes, interfering with immunoglobulin M production and interleukin (IL)-2 synthesis. The use of AZA is reported only in several reports in CS, and efficacy and tolerability were variable.59,60,62,67,75,76 The efficacy of the sole AZA in the maintenance phase was reported only in three cases.77–79
Cyclosporine A
CyA is a cyclic peptide, which binds to cyclophilin of T lymphocytes; thus, CyA is able to reduce the release of several cytokines. The use of CyA combined with corticosteroid and other immunosuppressive agents in CS was documented in some case reports,59,63,65,76,80 two case series,60,64 and one prospective nonrandomized clinical study including four patients with typical CS.81 The efficacy was variable with good response for ocular manifestations, quite effective for possible systemic vascular involvement but less effective for the audio-vestibular symptoms.
Biological therapy
Biological therapy for the treatment of autoimmune diseases consists of genetically engineered proteins derived from human genes. These proteins are designed to inhibit specific components of the immune system that play essential roles in driving the inflammatory pathway involved in the different diseases.52 The use of biological therapy in CS patients included for anti-TNFα agents, rituximab (RTX) and tocilizumab (TCZ).
Anti-TNFα
Anti-TNFα has been introduced in early 2000s. TNFα is a cytokine released during infection and inflammation. TNFα can induce pleiotropic effects, as pyrogenic activity when affecting the hypothalamus, production of C-reactive protein, and other acute-phase reactants by the liver. It is a potent chemoattractant for innate immune cells and promotes the expression of adhesion molecules by endothelial cells.82 TNFα inhibition causes a decrease in interferon-γ and an increase in IL-4 produced by T cells, inducing a shift of the immune response, with a reduction of clinical disease activity. TNFα is also a balancing factor required for the reestablishment of physiological homeostasis and immune regulation.52
Currently available anti-TNFα agents for clinical use are one soluble TNFα receptor (etanercept [ET]) and four anti-TNFα monoclonal antibodies, such as IFX, adalimumab (ADA), golimumab, and certolizumab pegol. In some cases, the lack of efficacy of anti-TNFα monoclonal antibodies is due to the production of serum antidrug antibodies. Concomitant use of anti-TNFα with traditional immunosuppressive drugs such as MTX and AZA may prevent the production of serum antidrug antibodies. Thus, TNFα antibodies themselves may trigger neurological symptoms, which may include CS-like symptoms, as shown in cases treated with anti-TNFα for inflammatory diseases.83,84
Infliximab
IFX is a chimeric monoclonal IgG1 antibody that specifically binds to both soluble and membrane-bound TNFα with high affinity, forming stable nondisassociating immune complexes. This activity prevents the binding of TNFα to its receptors and blocks the initiation of the intracellular signaling that leads to gene transcription and subsequent biological inflammatory activity.52 Regretfully, there are limited data in the literature that does not allow drawing reliable conclusions. Only 12 cases of CS are reported in the literature, treated with intravenous infusion of IFX, previously treated with corticosteroid and other immunosuppressive drugs with poor benefits.43,62,70,82,85 The used dosage ranged between 3 mg/kg and 400 mg total. In a pediatric patient with tubulointerstitial nephritis and uveitis (TINU) syndrome, transtympanic dexamethasone injections and IFX infusions were initiated at the onset of dizziness and severe bilateral sensorineural hearing loss, with a diagnosis of atypical CS in addition to the TINU syndrome. The IFX treatment was successful except in one case, allowing corticosteroid tapering and an improvement of hearing and/or ocular disease. In six cases, IFX treatment was successful after the failure of Cyc. In one unsuccessful case, IFX was administered 30 months after the diagnosis of CS.
Etanercept
ET is a fusion protein consisting of two recombinant p75 TNFα receptors linked to the Fc portion of human IgG186 that is a powerful antagonist of TNF, binding to and inactivating this cytokine. ET is approved for the treatment of several autoimmune diseases, such as rheumatoid arthritis, ankylosing spondylitis, juvenile rheumatoid arthritis, psoriasis, and psoriatic arthritis. However, in contrast to anti-TNFα antibodies, it is less effective in inflammatory bowel disease and in autoimmunity of the eye (eg, uveitis).82 The efficacy of the ET was examined in a prospective study including patients with bilateral immune-mediated audio-vestibular disorders; of these, three patients with CS were present.87 In two patients, an improvement was assessed, although ET did not succeed in preventing hearing loss.
Adalimumab
ADA is a human recombinant monoclonal IgG1 antibody with high specificity for human TNFα, thus inhibiting the TNFα binding to surface cellular receptors p55 and p75. Subcutaneous injections of ADA were documented in two cases of multidrug-resistant CS. In one case of typical CS, a dose of 40 mg/week for 6 months associated with prednisone and MTX failed in preventing a worsening of hearing loss.88 In another case, a patient with atypical CS experienced a clinical worsening during treatment.76 However, efficacy of this drug was assessed in cases of systemic autoimmune diseases associated with sensorineural hearing loss.89–91
Tocilizumab
TCZ is a recombinant humanized monoclonal anti-IL-6 receptor antibody that inhibits both membrane-bound and soluble IL-6 receptors. TCZ action inhibits T-cell activation and antibody secretion and decreases circulating cells from myeloid lineage. Only two case reports show the use of TCZ in CS.76,92 In one case of long-standing resistant atypical CS, TCZ (8 mg/kg intravenously/month) successfully replaced ADA stabilizing the course of the disease.76 In the second case, the use of TCZ was associated with pulmonary toxicity.92
Rituximab
RTX is a chimeric murine-human monoclonal IgG1 antibody directed against lymphocyte CD20 surface antigen, inducing a depletion of B lymphocytes by various mechanisms. This activity is associated with antibody-dependent cell-mediated cytotoxicity and complement-dependent cytotoxicity, although direct growth inhibition and/or induction of apoptosis may also take place. In a case report from Orsoni et al,88 RTX was used in a patient with a multidrug-resistant form of typical CS. In this case, RTX was administered, associated with oral prednisone, at a dosage of 500 mg intravenous/week. A noticeable improvement in hearing was reported. A poor response was reported in a patient with atypical CS and a remitting non-Hodgkin lymphoma of the mastoid bone.85 The recommended dose of the drug is the 4-week division of the overall cycle dose as it appears to be particularly safe, even though its use as a first-line therapy is not recommended.88
Surgical treatment
As for other autoimmune syndromes, surgery should be postponed until a long-term medical control of the inflammatory manifestations is obtained. In cases with severe sensorineural hearing loss unresponsive to intensive and/or innovative immunosuppressive regimens, cochlear implantation is still a valuable rescue surgical strategy for functional maintenance.
Generally, postoperative hearing outcome has been reported to be good to excellent; stable postoperative hearing has been reported at follow-up after 1,93 2,94 and 5 years95 with good-to-excellent word and sentence discrimination scores. These results suggest that cochlear implantation may provide excellent and stable hearing rehabilitation in long-term follow-up in most patients with CS.
Partial obliteration or neo-ossification of the cochlea was encountered during cochlear implantation, which required an alteration of the standard surgical technique.96 A preoperative MRI scan is necessary to identify patients with obliteration or neo-ossification of the cochlea and predict the difficulty encountered during insertion of the implant electrode.
Fibrous obliteration of the scala tympani was found as early as 8 weeks after onset of deafness.96 Generally, new bone formation is seen in the cochlea after cochlear implantation for the deafness of various etiologies as well as in the cochleae deafened by the CS without cochlear implantation. Postoperative word discrimination scores have been reported to be negatively correlated with the percent volume of new bone formation within the cochlea after implantation.97
Deterioration of auditory performance after cochlear implantation has been described in two cases,98 presumably secondary to an ongoing inflammatory response due to CS pathogenic basis. In one case, deterioration in speech perception was attributed to progressive cochlear ossification. In the other case, an abrupt deterioration of loudness perception required an increase in electrical stimulation.
Conclusion
CS is a rare disorder characterized by nonsyphilitic IK and audio-vestibular symptoms. Typical CS manifests primarily with IK and hearing loss, whereas atypical CS usually presents with inflammatory ocular manifestations and, more frequently, with systemic inflammation (70%), of which vasculitis represents the main pathogenic mechanism.
CS is considered as an autoimmune- or immune-mediated disease supported mainly by the beneficial response to corticosteroids. Antibodies to inner ear antigens, anti-Hsp70, and ANCA were associated with CS. Treatment of CS is sometimes difficult and frustrating. Failure to treat hearing loss may lead to deafness, and repeated flares may lead to profound and permanent hearing loss despite initial control of hearing damage.
Corticosteroids are the first line of treatment; multiple immunosuppressive drugs have been tried with variable degrees of success. TNFα blockers and other biological drugs have added promising options to the management of severe manifestations and/or recalcitrant forms, although refractory cases are still reported, most concerning the hearing function. The use of these molecules also in the early stages of the disease should be considered in order to exploit their potentialities even in cases that are not definitely compromised. At present, the concomitant use of anti-TNFα and traditional immunosuppressive drugs, such as MTX and AZA, is considered as the preferred option. Future directions of research for CS should consider prospective randomized trials that compare the efficacy and the safety profile of different therapeutic regimens in order to optimize the approach to such a rare and kaleidoscopic disease.
Cochlear implantation is a valuable rescue surgical strategy in cases with severe sensorineural hearing loss unresponsive to intensive and innovative immunosuppressive regimens.
Disclosure
The authors report no conflicts of interest in this work.
References
Morgan RF, Baumgartner CJ. Menier’s disease complicated by recurrent interstitial keratitis. West J Surg. 1934;42:628. | ||
Cogan DG. Syndrome of nonsyphilitic interstitial keratitis and vestibuloauditory symptoms. Arch Ophthalmol. 1945;33:144–149. | ||
Kessel A, Vadasz Z, Toubi E. Cogan syndrome – pathogenesis, clinical variants and treatment approaches. Autoimmun Rev. 2014;13(4–5):351–354. | ||
Jung DH, Nadol JB Jr, Folkerth RD, Merola JF. Histopathology of the inner ear in a case with recent onset of Cogan’s syndrome: evidence for vasculitis. Ann Otol Rhinol Laryngol. 2016;125(1):20–24. | ||
Cundiff J, Kansal S, Kumar A, Goldstein DA, Tessler HH. Cogan’s syndrome: a cause of progressive hearing deafness. Am J Otolaryngol. 2006;27(1):68–70. | ||
Vollertsen RS, McDonald TJ, Younge BR, Banks PM, Stanson AW, Ilstrup DM. Cogan’s syndrome: 18 cases and a review of the literature. Mayo Clin Proc. 1986;61(5):344–361. | ||
Haynes BF, Kaiser-Kupfer MI, Mason P, Fauci AS. Cogan syndrome: studies in thirteen patients, long-term follow-up, and a review of the literature. Medicine (Baltimore). 1980;59(6):426–441. | ||
Cheson BD, Bluming AZ, Alroy J. Cogan’s syndrome: a systemic vasculitis. Am J Med. 1976;60(4):549–555. | ||
Gluth MB, Baratz KH, Matteson EL, Driscoll CL. Cogan syndrome: a retrospective review of 60 patients throughout a half century. Mayo Clin Proc. 2006;81(4):483–488. | ||
Greco A, Gallo A, Fusconi M, et al. Cogan’s syndrome: an autoimmune inner ear disease. Autoimmun Rev. 2013;12(3):396–400. | ||
Kaya M, Erkanli K, Kilinc F, Sar M, Bakir I. Surgical treatment in a case of Cogan’s syndrome complicated with proximal aortic vasculitis. Ann Thorac Surg. 2015;100(4):1467–1469. | ||
Grasland A, Pouchot J, Hachulla E, et al. Typical and atypical Cogan’s syndrome: 32 cases and review of the literature. Rheumatology (Oxford). 2004;43(8):1007–1015. | ||
Bicknell JM, Holland JV. Neurologic manifestations of Cogan syndrome. Neurology. 1978;28(3):278–281. | ||
Chynn EW, Jakobiec FA. Cogan’s syndrome: ophthalmic, audiovestibular, and systemic manifestations and therapy. Int Ophthalmol Clin. 1996;36(1):61–72. | ||
Ho AC, Roat MI, Venbrux A, Hellmann DB. Cogan’s syndrome with refractory abdominal aortitis and mesenteric vasculitis. J Rheumatol. 1999;26(6):1404–1407. | ||
Scharl M, Frei P, Fried M, Rogler G, Vavricka SR. Association between Cogan’s syndrome and inflammatory bowel disease: a case series. J Crohns Colitis. 2011;5(1):64–68. | ||
Vavricka SR, Greuter T, Scharl M, et al. Cogan’s syndrome in patients with inflammatory bowel disease – a case series. J Crohns Colitis. 2015;9(10):886–890. | ||
Baguley D, McFerran D, Hall D. Tinnitus. Lancet. 2013;382(9904):1600–1607. | ||
Ralli M, Troiani D, Podda MV, et al. The effect of the NMDA channel blocker memantine on salicylate-induced tinnitus in rats. Acta Otorhinolaryngol Ital. 2014;34(3):198–204. | ||
Fetoni AR, Ralli M, Sergi B, Parrilla C, Troiani D, Paludetti G. Protective effects of N-acetylcysteine on noise-induced hearing loss in guinea pigs. Acta Otorhinolaryngol Ital. 2009;29(2):70–75. | ||
Sheppard A, Hayes SH, Chen GD, Ralli M, Salvi R. Review of salicylate-induced hearing loss, neurotoxicity, tinnitus and neuropathophysiology. Acta Otorhinolaryngol Ital. 2014;34(2):79–93. | ||
Ralli M, Greco A, Turchetta R, Altissimi G, de Vincentiis M, Cianfrone G. Somatosensory tinnitus: current evidence and future perspectives. J Int Med Res. 2017;45(3):933–947. | ||
Ralli M, Balla MP, Greco A, et al. Work-related noise exposure in a cohort of patients with chronic tinnitus: analysis of demographic and audiological characteristics. Int J Environ Res Public Health. 2017;14(9):E1035. | ||
Ralli M, Altissimi G, Turchetta R, et al. Somatosensory tinnitus: correlation between cranio-cervico-mandibular disorder history and somatic modulation. Audiol Neurootol. 2016;21(6):372–382. | ||
Auerbach BD, Rodrigues PV, Salvi RJ. Central gain control in tinnitus and hyperacusis. Front Neurol. 2014;5:206. | ||
Heller AJ. Classification and epidemiology of tinnitus. Otolaryngol Clin North Am. 2003;36(2):239–248. | ||
St Clair EW, McCallum RM. Cogan’s syndrome. Curr Opin Rheumatol. 1999;11(1):47–52. | ||
Hughes GB, Kinney SE, Barna BP, Tomsak RL, Calabrese LH. Autoimmune reactivity in Cogan’s syndrome: a preliminary report. Otolaryngol Head Neck Surg. 1983;91(1):24–32. | ||
Lunardi C, Bason C, Leandri M, et al. Autoantibodies to inner ear and endothelial antigens in Cogan’s syndrome. Lancet. 2002;360(9337):915–921. | ||
Spanakis SG, Petridou S, Masur SK. Functional gap junctions in corneal fibroblasts and myofibroblasts. Invest Ophthalmol Vis Sci. 1998;39(8):1320–1328. | ||
Autschbach F, Palou E, Mechtersheimer G, et al. Expression of the membrane protein tyrosine phosphatase CD148 in human tissues. Tissue Antigens. 1999;54(5):485–498. | ||
Van Doornum S, McColl G, Walter M, Jennens I, Bhathal P, Wicks IP. Prolonged prodrome, systemic vasculitis, and deafness in Cogan’s syndrome. Ann Rheum Dis. 2001;60(1):69–71. | ||
Ralli M, Altissimi G, Di Stadio A, Mazzei F, Turchetta R, Cianfrone G. Relationship between hearing function and myasthenia gravis: a contemporary review. J Int Med Res. 2017;45(5):1459–1465. | ||
Di Stadio A, Ralli M. Systemic lupus erythematosus and hearing disorders: literature review and meta-analysis of clinical and temporal bone findings. J Int Med Res. 2017;45(5):1470–1480. | ||
Ralli M, Di Stadio A, Greco A, et al. Development of progressive hearing loss and tinnitus in a patient with myasthenia gravis: an overlooked comorbidity? Hear Balance Commun. In press 2017. | ||
Casselman JW, Majoor MH, Albers FW. MR of the inner ear in patients with Cogan syndrome. AJNR Am J Neuroradiol. 1994;15(1):131–138. | ||
Bloch DB, San Martin JE, Rauch SD, Moscicki RA, Bloch KJ. Serum antibodies to heat shock protein 70 in sensorineural hearing loss. Arch Otolaryngol Head Neck Surg. 1995;121(10):1167–1171. | ||
Bonaguri C, Orsoni J, Russo A, et al. Cogan’s syndrome: anti-Hsp70 antibodies are a serological marker in the typical form. Isr Med Assoc J. 2014;16(5):285–288. | ||
Bonaguri C, Orsoni JG, Zavota L, et al. Anti-68 kDa antibodies in autoimmune sensorineural hearing loss: are these autoantibodies really a diagnostic tool? Autoimmunity. 2007;40(1):73–78. | ||
Moscicki RA, San Martin JE, Quintero CH, Rauch SD, Nadol JB Jr, Bloch KJ. Serum antibody to inner ear proteins in patients with progressive hearing loss. Correlation with disease activity and response to corticosteroid treatment. JAMA. 1994;272(8):611–616. | ||
Yamanishi Y, Ishioka S, Takeda M, Maeda H, Yamakido M. Atypical Cogan’s syndrome associated with antineutrophil cytoplasmic autoantibodies. Br J Rheumatol. 1996;35(6):601–603. | ||
Lochman I, Kral V, Lochmanova A, Lupac J, Cebecauer L. ANCA in the diagnosis of neutrophil-mediated inflammation. Autoimmun Rev. 2011;10(6):295–298. | ||
Touma Z, Nawwar R, Hadi U, Hourani M, Arayssi T. The use of TNF-alpha blockers in Cogan’s syndrome. Rheumatol Int. 2007;27(10):995–996. | ||
Weyn T, Haine S, Conraads V. Cogan’s syndrome with left main coronary artery occlusion. Cardiol J. 2009;16(6):573–576. | ||
Tervaert JW, Mulder L, Stegeman C, Elema J, Huitema M, The H. Occurrence of autoantibodies to human leucocyte elastase in Wegener’s granulomatosis and other inflammatory disorders. Ann Rheum Dis. 1993;52(2):115–120. | ||
Azami A, Maleki N, Kalantar Hormozi M, Tavosi Z. Interstitial keratitis, vertigo, and vasculitis: typical Cogan’s syndrome. Case Rep Med. 2014;2014:830831. | ||
Balink H, Bruyn GA. The role of PET/CT in Cogan’s syndrome. Clin Rheumatol. 2007;26(12):2177–2179. | ||
Balink H, Bennink RJ, van Eck-Smit BL, Verberne HJ. The role of 18F-FDG PET/CT in large-vessel vasculitis: appropriateness of current classification criteria? Biomed Res Int. 2014;2014:687608. | ||
Ralli M, Greco A, Falasca V, et al. Recovery from repeated sudden hearing loss in a patient with Takayasu’s arteritis treated with hyperbaric oxygen therapy: the first report in the literature. Case Rep Otolaryngol. 2017;2017:3281984. | ||
Maruyoshi H, Toyama K, Kojima S, et al. Sensorineural hearing loss combined with Takayasu’s arteritis. Intern Med. 2005;44(2):124–128. | ||
Raza K, Karokis D, Kitas GD. Cogan’s syndrome with Takayasu’s arteritis. Br J Rheumatol. 1998;37(4):369–372. | ||
Mora P, Calzetti G, Ghirardini S, Rubino P, Gandolfi S, Orsoni J. Cogan’s syndrome: state of the art of systemic immunosuppressive treatment in adult and pediatric patients. Autoimmun Rev. 2017;16(4):385–390. | ||
Espinoza GM, Prost A. Cogan’s syndrome and other ocular vasculitides. Curr Rheumatol Rep. 2015;17(4):24. | ||
Shimura M, Yasuda K, Fuse N, Nakazawa M, Tamai M. Effective treatment with topical cyclosporin A of a patient with Cogan syndrome. Ophthalmologica. 2000;214(6):429–432. | ||
Haynes BF, Pikus A, Kaiser-Kupfer M, Fauci AS. Successful treatment of sudden hearing loss in Cogan’s syndrome with corticosteroids. Arthritis Rheum. 1981;24(3):501–503. | ||
Vollertsen RS. Vasculitis and Cogan’s syndrome. Rheum Dis Clin North Am. 1990;16(2):433–439. | ||
Cabezas-Rodriguez I, Brandy-Garcia A, Rodriguez-Balsera C, Rozas-Reyes P, Fernandez-Llana B, Arboleya-Rodriguez L. Late-onset Cogan’s syndrome associated with large-vessel vasculitis. Reumatol Clin. Epub 2017 Jun 2. | ||
Pleyer U, Baykal HE, Rohrbach JM, et al. Cogan-I-Syndrom: Zu oft zu spät erkannt? - Ein Beitrag zur Frühdiagnose des Cogan-I-Syndroms [Cogan I syndrome: too often detected too late? A contribution to early diagnosis of Cogan I syndrome]. Klin Monbl Augenheilkd. 1995;207(1):3–10. German. | ||
Migliori G, Battisti E, Pari M, Vitelli N, Cingolani C. A shifty diagnosis: Cogan’s syndrome. A case report and review of the literature. Acta Otorhinolaryngol Ital. 2009;29(2):108–113. | ||
Allen NB, Cox CC, Cobo M, et al. Use of immunosuppressive agents in the treatment of severe ocular and vascular manifestations of Cogan’s syndrome. Am J Med. 1990;88(3):296–301. | ||
Riente L, Taglione E, Berrettini S. Efficacy of methotrexate in Cogan’s syndrome. J Rheumatol. 1996;23(10):1830–1831. | ||
Ghadban R, Couret M, Zenone T. Efficacy of infliximab in Cogan’s syndrome. J Rheumatol. 2008;35(12):2456–2458. | ||
Di Leo E, Dambra PP, Capuzzimati L, et al. A case of Cogan’s syndrome resolved with mycophenolate sodium. Eur J Clin Invest. 2008;38(8):605. | ||
Orsoni JG, Zavota L, Vincenti V, Pellistri I, Rama P. Cogan syndrome in children: early diagnosis and treatment is critical to prognosis. Am J Ophthalmol. 2004;137(4):757–758. | ||
Sugimoto K, Miyazawa T, Nishi H, et al. Childhood Cogan syndrome with aortitis and anti-neutrophil cytoplasmic antibody-associated glomerulonephritis. Pediatr Rheumatol Online J. 2014;12:15. | ||
Vaiopoulos G, Sfikakis PP, Skoumas B, Kavouklis E, Doukas E, Kaklamanis P. Lack of response to corticosteroids and pulse cyclophosphamide therapy in Cogan’s syndrome. Clin Rheumatol. 1994;13(1):110–112. | ||
Murphy G, Sullivan MO, Shanahan F, Harney S, Molloy M. Cogan’s syndrome: present and future directions. Rheumatol Int. 2009;29(10):1117–1121. | ||
Matteson EL, Fabry DA, Facer GW, et al. Open trial of methotrexate as treatment for autoimmune hearing loss. Arthritis Rheum. 2001;45(2):146–150. | ||
Matteson EL, Tirzaman O, Facer GW, et al. Use of methotrexate for autoimmune hearing loss. Ann Otol Rhinol Laryngol. 2000;109(8 pt 1):710–714. | ||
Beccastrini E, Emmi G, Squatrito D, Vannucchi P, Emmi L. Infliximab and Cogan’s syndrome. Clin Otolaryngol. 2010;35(5):441–442. | ||
Pouchot J, Vinceneux P, Bouccara D, Sterkers O, Bodelet B. Methotrexate as a steroid-sparing agent in Cogan’s syndrome: comment on the concise communication by Richardson. Arthritis Rheum. 1995;38(9):1348–1349. | ||
Inoue Y, Tomemori T, Suzuki S, et al. Low-dose oral methotrexate for the management of childhood Cogan’s syndrome: a case report. Clin Rheumatol. 2007;26(12):2201–2203. | ||
Juneja M, Jain R, Chakarbarty B. Atypical Cogan syndrome mimicking acute rheumatic fever. Indian Pediatr. 2011;48(7):561–563. | ||
Baumann A, Helbling A, Oertle S, Hausler R, Vibert D. Cogan’s syndrome: clinical evolution of deafness and vertigo in three patients. Eur Arch Otorhinolaryngol. 2005;262(1):45–49. | ||
Gran JT, Nordvag BY, Storesund B. An overlap syndrome with features of atypical Cogan syndrome and Wegener’s granulomatosis. Scand J Rheumatol. 1999;28(1):62–64. | ||
Shibuya M, Fujio K, Morita K, Harada H, Kanda H, Yamamoto K. Successful treatment with tocilizumab in a case of Cogan’s syndrome complicated with aortitis. Mod Rheumatol. 2013;23(3):577–581. | ||
Lydon EJ, Barisoni L, Belmont HM. Cogan’s syndrome and development of ANCA-associated renal vasculitis after lengthy disease remission. Clin Exp Rheumatol. 2009;27(1 Suppl 52):S144. | ||
Hirvonen TP, Aalto H. Recovery of bilateral vestibular loss in Cogan’s syndrome – a case report. Otol Neurotol. 2013;34(9):1736–1738. | ||
Tirelli G, Tomietto P, Quatela E, et al. Sudden hearing loss and Crohn disease: when Cogan syndrome must be suspected. Am J Otolaryngol. 2015;36(4):590–597. | ||
Ying YL, Hirsch BE. Atypical Cogan’s syndrome: a case report. Am J Otolaryngol. 2010;31(4):279–282. | ||
Orsoni JG, Zavota L, Mora P, Rubino P, Manzotti F, Pellistri I. Discontinuous drug combination therapy in autoimmune ocular disorders. Acta Ophthalmol. 2009;87(3):340–345. | ||
Tayer-Shifman OE, Ilan O, Tovi H, Tal Y. Cogan’s syndrome – clinical guidelines and novel therapeutic approaches. Clin Rev Allergy Immunol. 2014;47(1):65–72. | ||
Mohan N, Edwards ET, Cupps TR, et al. Demyelination occurring during anti-tumor necrosis factor alpha therapy for inflammatory arthritides. Arthritis Rheum. 2001;44(12):2862–2869. | ||
Conway R, Khan S, Foley-Nolan D. Use of adalimumab in treatment of autoimmune sensorineural hearing loss: a word of caution. J Rheumatol. 2011;38(1):176. author reply 176. | ||
Kalogeropoulos C, Karachalios D, Pentheroudakis G, et al. Development of a low grade lymphoma in the mastoid bone in a patient with atypical Cogan’s syndrome: a case report. J Adv Res. 2015;6(3):523–527. | ||
Weinblatt ME, Kremer JM, Bankhurst AD, et al. A trial of etanercept, a recombinant tumor necrosis factor receptor:Fc fusion protein, in patients with rheumatoid arthritis receiving methotrexate. N Engl J Med. 1999;340(4):253–259. | ||
Matteson EL, Choi HK, Poe DS, et al. Etanercept therapy for immune-mediated cochleovestibular disorders: a multi-center, open-label, pilot study. Arthritis Rheum. 2005;53(3):337–342. | ||
Orsoni JG, Lagana B, Rubino P, Zavota L, Bacciu S, Mora P. Rituximab ameliorated severe hearing loss in Cogan’s syndrome: a case report. Orphanet J Rare Dis. 2010;5:18. | ||
Morovic Vergles J, Radic M, Kovacic J, Salamon L. Successful use of adalimumab for treating rheumatoid arthritis with autoimmune sensorineural hearing loss: two birds with one stone. J Rheumatol. 2010;37(5):1080–1081. | ||
Jachiet M, Lependu C, Fragny D, Mariette X, Lepajolec C, Seror R. Severe deafness associated with Crohn’s disease and spondylarthropathy: successful treatment with anti-TNF. Rheumatology (Oxford). 2013;52(6):1145–1147. | ||
Marsili M, Marzetti V, Lucantoni M, et al. Autoimmune sensorineural hearing loss as presenting manifestation of paediatric Behçet disease responding to adalimumab: a case report. Ital J Pediatr. 2016;42(1):81. | ||
Utz JP, Jinnur PK, Yi ES, Ryu JH, Midthun DE, Davis JM. Acute bilateral pulmonary opacities associated with use of tocilizumab. J Clin Rheumatol. 2015;21(7):382–385. | ||
Pasanisi E, Vincenti V, Bacciu A, et al. Cochlear implantation and Cogan syndrome. Otol Neurotol. 2003;24(4):601–604. | ||
Wang JR, Yuen HW, Shipp DB, et al. Cochlear implantation in patients with autoimmune inner ear disease including cogan syndrome: a comparison with age- and sex-matched controls. Laryngoscope. 2010;120(12):2478–2483. | ||
Bacciu A, Pasanisi E, Di Lella F, Guida M, Bacciu S, Vincenti V. Cochlear implantation in patients with Cogan syndrome: long-term results. Eur Arch Otorhinolaryngol. 2015;272(11):3201–3207. | ||
Aschendorff A, Lohnstein P, Schipper J, Klenzner T. [Obliterated cochlea in Cogan’s syndrome – implications for cochlear implant surgery]. Laryngorhinootologie. 2004;83(12):836–839. | ||
Kamakura T, Nadol JB Jr. Correlation between word recognition score and intracochlear new bone and fibrous tissue after cochlear implantation in the human. Hear Res. 2016;339:132–141. | ||
Bovo R, Ciorba A, Trevisi P, et al. Cochlear implant in Cogan syndrome. Acta Otolaryngol. 2011;131(5):494–497. | ||
Hur JH, Chun EJ, Kwag HJ, et al. CT features of vasculitides based on the 2012 International Chapel Hill Consensus Conference Revised Classification. Korean J Radiol. 2017;18(5):786–798. | ||
Graff JM, Freed N, Oetting TA. Cogan’s Syndrome: 42-year-old female with interstitial keratitis and vertigo. EyeRounds.org; University of Iowa. May 24, 2007. Available at: http://webeye.ophth.uiowa.edu/eyeforum/cases/69-cogansinterstitialkeratitisvestibuloauditory.htm. Accessed December 15, 2017. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.