")
Back to Journals » Clinical Ophthalmology » Volume 16
Ophthalmological Manifestations of Oculocutaneous and Ocular Albinism: Current Perspectives
Authors Neveu MM, Padhy SK, Ramamurthy S, Takkar B, Jalali S, CP D, Padhi TR, Robson AG
Received 7 February 2022
Accepted for publication 14 April 2022
Published 24 May 2022 Volume 2022:16 Pages 1569—1587
DOI https://doi.org/10.2147/OPTH.S329282
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Scott Fraser
Magella M Neveu,1,2 Srikanta Kumar Padhy,3 Srishti Ramamurthy,4 Brijesh Takkar,4 Subhadra Jalali,4 Deepika CP,4 Tapas Ranjan Padhi,3 Anthony G Robson1,2
1Department Electrophysiology, Moorfields Eye Hospital, London, EC1V 2PD, UK; 2Institute of Ophthalmology, University College London, London, UK; 3Anant Bajaj Retina Institute, LV Prasad Eye Institute, Bhubaneswar, India; 4Anant Bajaj Retina Institute, LV Prasad Eye Institute, Hyderabad, India
Correspondence: Anthony G Robson, Department of Electrophysiology, Moorfields Eye Hospital, 162 City Road, London, EC1V 2PD, UK, Tel +44 20 7566 2554, Email [email protected]
Abstract: Albinism describes a heterogeneous group of genetically determined disorders characterized by disrupted synthesis of melanin and a range of developmental ocular abnormalities. The main ocular features common to both oculocutaneous albinism (OCA), and ocular albinism (OA) include reduced visual acuity, refractive errors, foveal hypoplasia, congenital nystagmus, iris and fundus hypopigmentation and visual pathway misrouting, but clinical signs vary and there is phenotypic overlap with other pathologies. This study reviews the prevalence, genetics and ocular manifestations of OCA and OA, including abnormal development of the optic chiasm. The role of visual electrophysiology in the detection of chiasmal dysfunction and visual pathway misrouting is emphasized, highlighting how age-associated changes in visual evoked potential (VEP) test results must be considered to enable accurate diagnosis, and illustrated further by the inclusion of novel VEP data in genetically confirmed cases. Differential diagnosis is considered in the context of suspected retinal and other disorders, including rare syndromes that may masquerade as albinism.
Keywords: albinism, visual electrophysiology, misrouting, foveal hypoplasia, VEP
Introduction
Albinism describes a heterogeneous group of genetically determined disorders characterized by disrupted synthesis of melanin pigmentation or melanosome maturation during development. In the eye cup, this results in a cascade of ocular abnormalities and intracranial visual pathway misrouting, common to both oculocutaneous albinism (OCA) and ocular albinism (OA). The hallmark clinical features of OCA include congenital hypopigmentation of the skin, hair and eyes, with an autosomal recessive inheritance mode.1 OCA can also occur in several syndromic disorders with systemic complications posing significant morbidity, highlighting the importance of early diagnosis. Ocular albinism is distinguished by predominant involvement of the ocular tissues and X-linked inheritance, with the ocular hypopigmentation occurring to a lesser degree than in OCA.2 Ocular signs such as foveal hypoplasia and nystagmus are common in both OCA and OA but there is wide phenotypic variation and overlap. Visual electrophysiology can play an important role in the detection of visual pathway misrouting, helping to determine the diagnosis, particularly if the phenotype is mild or “sub-clinical”. The increasing availability of genetic testing provides the possibility of a definitive molecular diagnosis and subtyping, informing clinical management and the further understanding of developmental eye disease. This study reviews the prevalence, genetic causes and main ophthalmological manifestations of OCA and OA, including novel genotype-phenotype analysis based on electrophysiological data.
Genetic Classification and Prevalence
Non-Syndromic OCA
Oculocutaneous albinism can be classified into one of several genetic subtypes, all involving disruption of melanin or tyrosinase (TYR) synthesis (Figure 1). The commonest forms are TYR-related and include OCA type 1A (OMIM #203100) and OCA type 1B (OMIM #606952). TYR spans 65kb of genomic DNA and codes for the protein tyrosinase, a copper-containing enzyme that facilitates the first two steps in melanin biosynthesis, involving conversion of tyrosine to DOPAquinone. In OCA type 1A (tyrosine negative; complete OCA) an inactive protein is encoded and there is an absence of tyrosinase catalytic function. In OCA type 1B (tyrosinase positive, partial OCA) there is residual enzyme activity and a milder clinical phenotype.3 The overall prevalence of OCA is estimated to be around 1/17,000 but incidence varies according to ethnic background. OCA1 is the commonest subtype in the non-Hispanic Caucasian population with a prevalence of approximately 1 in 40,000.4
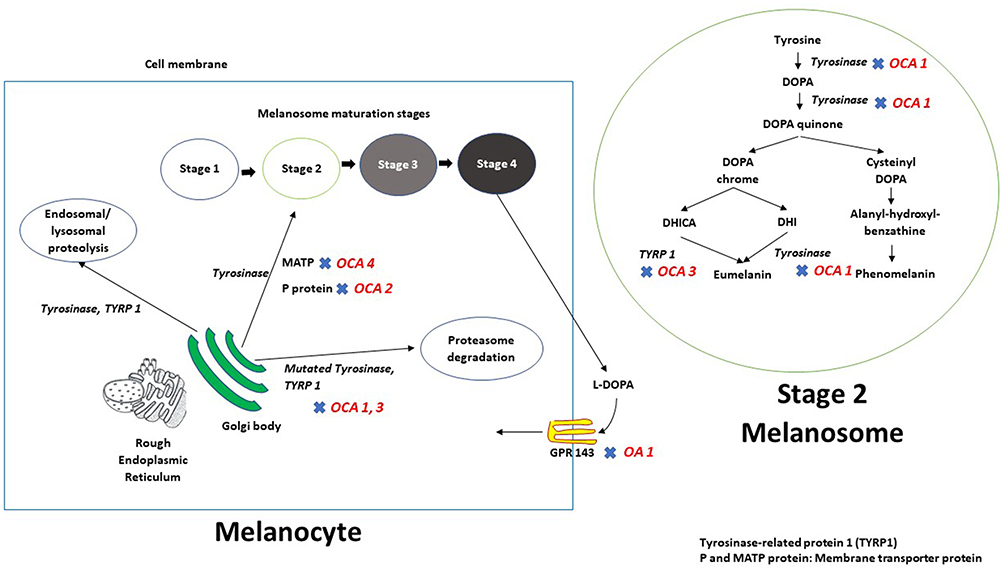 |
Figure 1 Schematic representation of melanin biosynthesis in the melanocyte. After synthesis and subsequent processing in the Golgi body - rough endoplasmic reticulum (ER) complex, tyrosinase and tyrosinase-related protein 1 (TYRP1) are trafficked to the developing melanosome via membrane transporter proteins P and MATP. Mutations in tyrosinase or in TYRP1 result in the retention of these mutant proteins in the ER, and these are subsequently degraded using proteasome mediated pathways. The rest of the tyrosinase and TYRP 1 proteins undergo endosomal/lysosomal proteolysis. Retinal pigment epithelium makes and releases L-DOPA during the procerss of melanin biosynthesis; L-DOPA is an endo agonist for GPR 143 (expressed at the membrane of the melanosomes) thus create an autocrine loop. GPR143 is the protein product of OA1 and mutations at this locus result in ocular albinism type 1. |
OCA2 (OMIM #203200) is caused by mutations in OCA which spans 345kb of genomic DNA and encodes P protein. The P protein stabilizes tyrosinase and enables transport of melanosomal tyrosine. Mutations involving OCA2 thereby disturb pigmentation by affecting normal functioning of melanosomal tyrosine/tyrosinase. OCA2 is the commonest globally with a prevalence of almost 1 in 3900 in southern parts of Africa, 1 in 10,000 in African Americans and 1 in 30,000 in Caucasians.5–8
Mutations in tyrosinase-related protein 1 (TYRP1) result in OCA3 (“Rufous oculocutaneous albinism”). TYRP1 spans 17KB genomic DNA and catalyzes the oxidation of DHICA (5,6-dihydroxyindole-2-carboxylic acid) monomers to melanin. Mutations cause early degradation of tyrosinase, negatively influencing the maturation of melanosomes and melanin biosynthesis.9–13 OCA3 occurs in about 1 in 8500 patients of African ethnicity and is associated with a relatively mild OCA phenotype.5,14
OCA4 (OMIM #606574) is caused by mutations in SLC45A2, which spans 40kb of genomic DNA and encodes a membrane-associated transporter protein (MATP). This subtype is largely confined to Japanese patients, accounting for approximately 25–27% of cases.15–17
Other forms of OCA are rare but include OCA5 (OMIM #615312; gene unknown); OCA6 (OMIM #113750; SLC24A5-related); OCA7 (OMIM #615179), reported in a Faroese cohort and associated with LRMDA or c10orf11, and OCA8 (OMIM #619165), caused by mutation in DCT.
Syndromic OCA and Syndromic Hypopigmentation Disorders
The rarer syndromic forms of OCA are associated with bi-allelic variants in one or more genes associated with lysosomal protein trafficking (Table 1). Hermansky–Pudlak syndrome (HPS), an autosomal recessive multisystem disorder, is associated with 11 genetic subtypes that impair lysosomal related organelles (LROs) synthesis, causing interstitial pulmonary fibrosis, granulomatous colitis and platelet alterations. The subtypes include HPS1 (HPS1), HPS2 (AP3B1), HPS3 (HPS3), HPS4 (HPS4), HPS5 (HPS5), HPS6 (HPS6), HPS7 (DTNBP1 or BLOC1S8), HPS8 (BLOC1S3), HPS9 (BLOC1S6), HPS10 (AP3D1), and HPS11 (BLOC1S5). Chédiak–Higashi syndrome is caused by mutations in LYST (OMIM #21450), resulting in hematologic causes of bleeding and infection. There are three forms of Griscelli syndrome, an autosomal recessive disorder caused by variants of MYO5A (GS1; OMIM #214450), Rab27A (GS2; OMIM #607624) or MLPH or MYO5A (GS3; OMIM #609227). The syndrome is associated with immunodeficiency, pancytopenia and demyelination of the cerebral white matter.
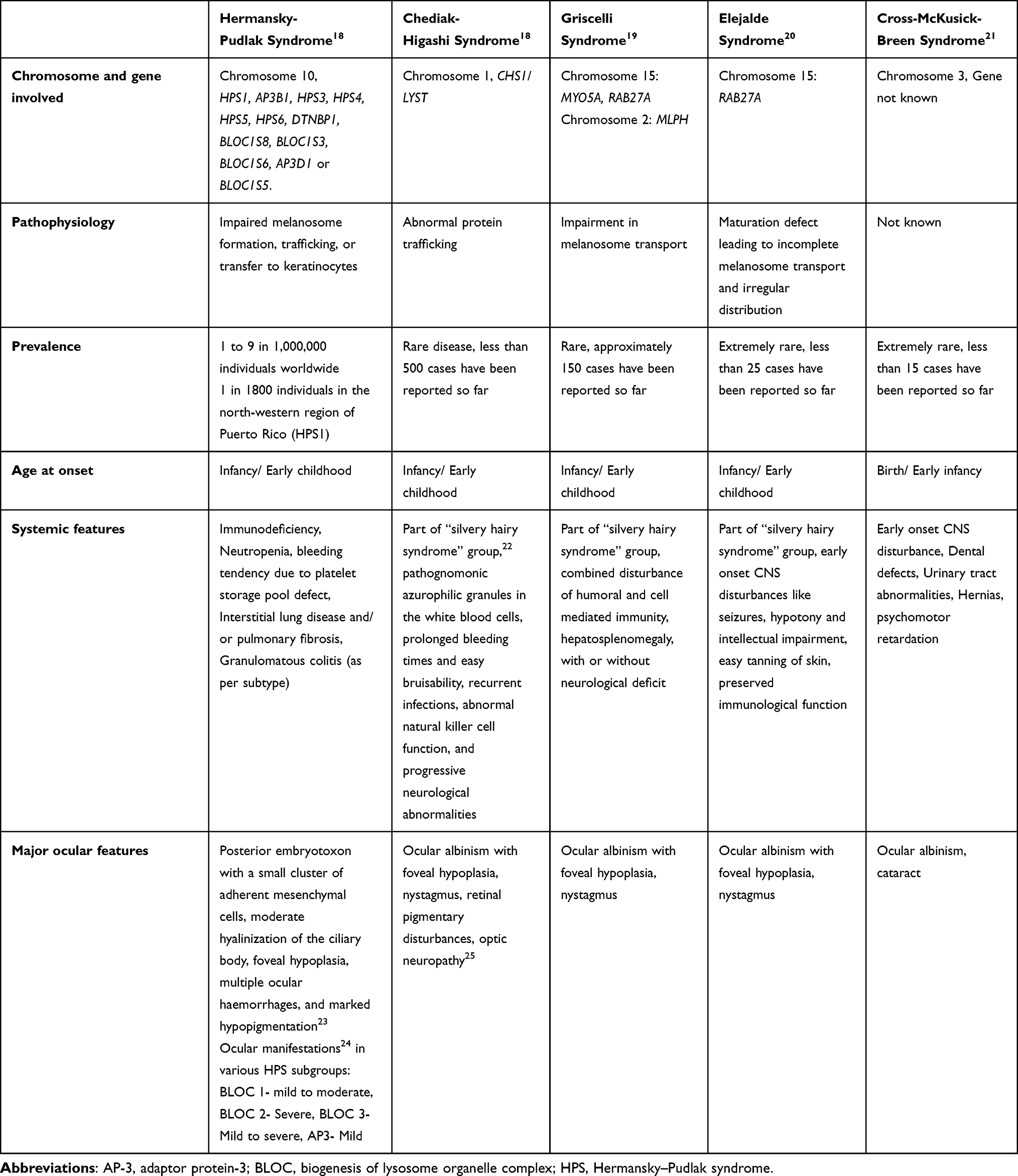 |
Table 1 Summary of Genetic, Pathophysiologic and Clinical Features Associated with Syndromic OCA |
Mutation or deletion of chromosome 15 (the locus for OCA2) can cause Prader Willi Syndrome (paternal deletion of 15q11-q13 or maternal disomy for chromosome 15) or Angelman Syndrome (UBE3A).26–29 Hypopigmentation of the skin, hair and eyes are common features from birth that may lessen during adolescence. There are contradictory reports of visual pathway misrouting in Prada Willi syndrome, seen in some28 but not all cases.30,31 The latter is consistent with the authors’ experience (MMN; AGR, personal observations), and the proposal that visual pathway misrouting in this syndrome may be explained by two separate disorders.29 Angelman syndrome varies in severity and electrophysiological evidence of optic nerve misrouting has been described in cases without the typical clinical signs of albinism.32
The extremely rare and life limiting Vici Syndrome is an autosomal recessive multisystem disorder resulting from a mutation in EPG5, a key autophagy regulator in higher organisms. The disorder is characterized by agenesis of the corpus callosum, hypopigmentation of the eyes and hair, cardiomyopathy, immunodeficiency and often delayed motor development.33 A report on a child with Vici Syndrome34 showed that the ophthalmological features of the syndrome included ocular hypopigmentation, foveal hypoplasia and intracranial misrouting of the visual pathways.
Waardenburg Syndrome (WS)35–37 is a rare autosomal dominant disorder (prevalence 1/40,000) due to a mutation in PAX338 or MITF (microphthalmia-associated transcription factor).39 There are 4 types (WS1-4),39–41 mainly characterized by congenital sensorineural hearing loss, and the presence (WS1 and WS3) or absence of dystopia canthorum (WS4). A striking feature of WS is heterochromia iridium, where one iris is heavily pigmented (brown iris) and the other relatively hypopigmented (blue or green iris). The iris hypopigmentation is typically associated with hypopigmentation of the fundus and “albino-like” features, whereas the other eye has relatively normal pigmentation. Although WS has some features common to albinism, the reported cases of reduced visual acuity, nystagmus, and foveal hypoplasia result from a digenic interaction between MITF (WS2) and TYR (AROA/OCA1). There are no reports of Waardenburg Syndrome being associated with misrouting of the visual pathways at the optic chiasm, and visual electrophysiology may enable the distinction from OCA in clinically equivocal cases (see Visual Electrophysiology in Albinism).
Ocular Albinism
Ocular albinism (OA, OMIM #300500) is an X-linked disorder with a prevalence of 1 in 60,000 males42 and is caused by mutations in GPR143 (OA1), expressed in melanocytes. The disorder primarily affects the eyes and genetic diagnosis is of particular value given the milder phenotype compared with OCA, including normal or near-normal skin and hair pigmentation and cases of “sub-clinical” disease. Mutations in GPR143 result in the formation of enlarged melanosomes with reduced motility, with reduced numbers in the melanocytes and RPE.43,44 In milder phenotypes of OA, ocular development fails in the presence of normal or near normal pigmentation, suggesting a spatio-temporal deficit in retinal development independent of RPE pigmentation.45,46
Clinical Features of Albinism
Cutaneous Involvement
Patients with OCA1A typically present with white hair and skin at birth, and the ability to tan is lacking. Residual tyrosinase function in OCA1B can result in the development of pigmentation with time, with skin appearing cream in color. Nevi are commonly noted. Variants, which are temperature sensitive, have pigmented hair on their extremities. OCA2 is milder in presentation with creamy skin color and blonde to red hair. OCA3, also termed ‘Rufous’ or red OCA, is associated with reddish hair and skin. OCA4 is often indistinguishable from OCA2 based on clinical findings.6,47 OCA predisposes to squamous cell carcinoma of sun-exposed areas such as head and neck, with high recurrence rates noted in sub-Saharan Africa.48 In darkly pigmented races, as in some parts of South India, it may require due diligence to distinguish the “fair baby” in the family from the “dark-pigmented” parents.
Ocular Involvement
The main ocular abnormalities associated with OCA subtypes and OA are similar and are detailed below. Symptoms include reduced visual acuity (range 20/60 to 20/400) and color vision impairment. It is important that visual acuity is recorded on non-illuminated charts, as glare can result in a lower visual acuity than when non-illuminated charts are used. Visual acuity has been correlated with the degree of melanin pigmentation at the macula, worse in those with the least or no pigment. The commonest signs are detailed below, including foveal hypoplasia and varying severity of congenital nystagmus, iris hypopigmentation and iris translucency and hypopigmentation of the retinal pigment epithelium (RPE). There may be positive angle kappa and reduced or absent stereopsis and strabismus. Fundus examination may also reveal abnormal retinal blood vessel patterns such as wide exit angles from the optic nerve head and vessels that encroach upon the central macula area.49
The discriminant feature of albinism is misrouting of optic nerve fibres with an excess of decussating fibres at the chiasm,6,50,51 typically revealed using visual electrophysiology (see below) and of particular value if the clinical features are equivocal or absent, or if ophthalmic examination is difficult, eg, in young children.
Foveal Hypoplasia
Albinism is commonly associated with foveal hypoplasia (Figure 2), where there is complete or partial failure of foveal pit formation and specialization. The severity of foveal hypoplasia varies, and detection may be enhanced with OCT imaging, including hand-held devices in the paediatric population.52 Rudimentary foveal development with thinning noted on OCT has been documented in a few OCA cases, associated with better visual acuity (≥20/50).53
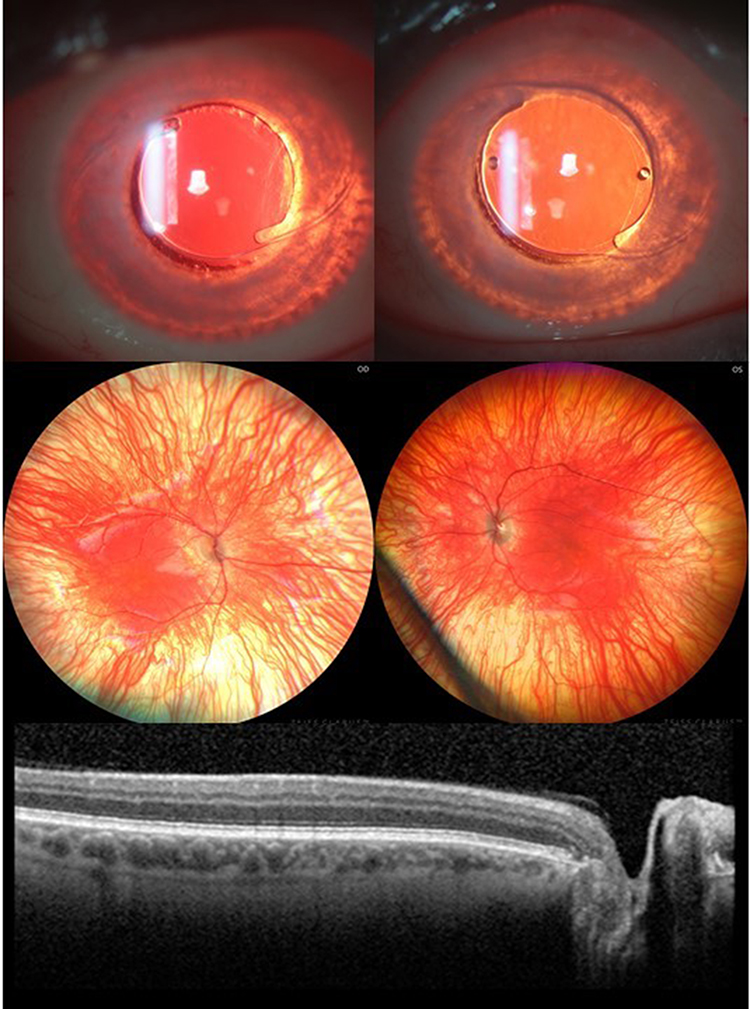 |
Figure 2 Iris transillumination defect, albinotic fundus and foveal hypoplasia in pseudophakic eyes of a 41-year-old patient. Note that major retinal blood vessels show a relatively wide exit angle as they emerge from the optic disc. |
Lack of tyrosinase activity and DOPA is hypothesized to result in increased mitosis and cell death during retinal development. Directional OCT studies have revealed both a reduction in the outer nuclear layer thickness and increase in the Henle fibre layer, the latter attributed to an increase in foveal cone packing.54
Congenital Nystagmus
Nystagmus in albinism is conjugate pendular or jerk type and develops within the first few weeks of life. Presentation in infancy is usually with large amplitude nystagmus that diminishes with age. Compensatory head posture, reduced stereovision and strabismus are commonly associated features. Mild albinism phenotypes may be mistaken as infantile nystagmus syndrome55 or retinal dystrophy (see later). The presence of nystagmus does not limit the reading speed in albinism if words are presented with sufficient magnification. Rather, it is primarily the sensory visual impairments, such as reduced visual acuity, that limits reading ability.56
Refractive Errors
High hyperopia and astigmatism are commonly reported in albinism. Refractive errors have been found to correlate with axial length, which may be causative.57 Patients with OCA have been reported to have impaired emmetropization due to foveal hypoplasia-related nystagmus leading to the perception of smeared image motion by the retina.58 Hyperopic eyes in albinism show meridional emmetropization with a normal rate of emmetropization in the less hyperopic meridian, in contrast to an abnormal emmetropization in the more hyperopic meridian.59 When compared with other types, subjects with OCA1A-related albinism show highest increase in astigmatism with age, worst visual acuity and photophobia.58,59 Ocular associations of keratoconus or pellucid marginal corneal degeneration in albinism can also contribute to the variable astigmatism in certain cohorts.60
Iris and RPE Hypopigmentation
Iris hypopigmentation (Figure 2) occurs secondary to reduced or nil production of melanin in the pigmented epithelium and stromal melanocytes of the iris. In OCA, melanocytes are present within the posterior iris and RPE, but the formation of melanin within the melanosome is defective. This leads to a blue or light brown iris and imparts the characteristic features of iris transillumination on slit lamp examination or on globe transillumination.61 The iris in OCA1A is typically pink and translucent. OCA1B may be associated with minimal pigmentation, which increases with age. Iris color in the OCA2 type may vary, and a pink iris need not be present to diagnose OCA2.7 Decreased melanin formation in RPE results in the classical albinotic fundus with enhanced visualization of deeper choroidal vessels. Eyes with reduced pigmentation reflect light to a greater extent resulting in severe photophobia in patients with OCA.62
Clinical Features in Carriers of X-Linked OA
Obligate female carriers of ocular albinism can manifest an abnormal fundus appearance due to random X chromosome inactivation (lyonization), with characteristic “mud-splatter” pigmentation on fundus examination and dark radial streaks against a bright background on fundus autofluorescence imaging. There may be mild iris transillumination but a normal-appearing retinal structure on OCT without foveal hypoplasia. There is no evidence of visual pathway misrouting, as observed in affected males.
Misrouting of Retinocortical Fibres at the Optic Chiasm
Normal Development of the Chiasm and Optic Nerve Projection
The mammalian retina and RPE are formed from the neural ectoderm. The inner surface of the optic cup gives rise to the retina and the outer surface to the RPE.63–65 Early in development, the fovea and the spatio-temporal characteristics of retinal cell proliferation centred on this region, determine the retinotopic order of the visual field, which is then projected via the optic nerve to the chiasm.66–69
The optic chiasm is the stage in the mammalian visual system, where retinal ganglion cell axons from each eye either cross the chiasmal midline and project to the contralateral cortical hemisphere or remain uncrossed and project to the ipsilateral cortical hemisphere, with approximately 45% of retinal ganglion axons projecting ipsilaterally.70–72 The segregation of these projections is critical to binocular vision and representation of the temporal and nasal visual field in each hemisphere of the visual cortex.
There is a distinct segregation of the crossed and uncrossed projection across the naso-temporal division. The first ganglion cells to be generated span the presumptive foveal region72,73 and the retinotopic order is largely preserved along the optic nerve and into the chiasm. The first axons to enter the chiasm give rise to both the crossed and uncrossed projection and as ganglion cell generation expands from central retina towards the periphery,72 elements of both the crossed and uncrossed projections are added to the chiasm. The control of this process is not fully understood although it has been noted that numerous genes are likely to be implicated.67,68
The Development of the Chiasm and Optic Nerve Projection in Albinism
A lack of pigment in the RPE of albino mammals disrupts the development of neural retina,74–84 resulting in spatio-temporal defects in patterns of cell production.85 The naso-temporal division is shifted toward temporal retina, there is misrouting of the visual pathway (Figure 3) 86 and a reduction of the uncrossed in favour of the crossed projection.87–93
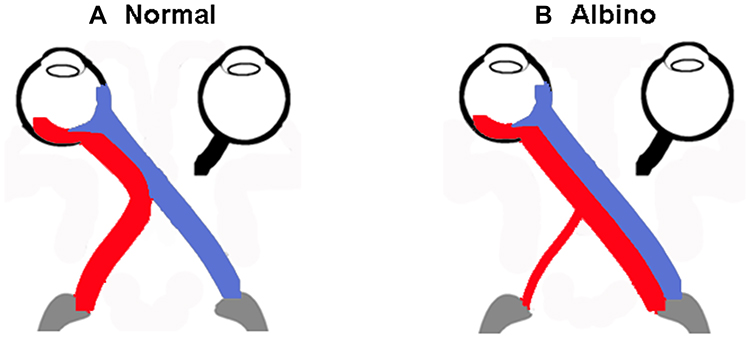 |
Figure 3 Schematic of normal and albino visual pathways. (A) Normal visual pathways. Nerve fibres originating from temporal retina project to the ipsilateral hemisphere (red line). Nerve fibres originating from predominantly nasal retina, cross at the chiasm and project to the contralateral hemisphere (blue line). (B) Albino misrouting. The majority of optic nerve fibres decussate to the contralateral hemisphere (red and blue lines). Adapted from Neveu MM, Jeffery G. Chiasm formation in man is fundamentally different from that in the mouse. Eye. 2007;21(10):1264–1270.86 |
The developmental process linking melanin synthesis with formation of the naso-temporal division is not fully understood and misrouting of the visual pathways can occur in the presence of normal pigmentation.94–97 Studies on rodents and ferrets suggest that the uncrossed visual pathway, which is located temporally and peripherally in the retina, is relatively small and fragile, making it more susceptible to changes in retinal hypopigmentation69,79,98–100 and there is evidence that the degree to which the naso-temporal division is shifted is proportional to the degree of retinal hypopigmentation.101,102 Localized differences in the concentration of melanin in the RPE affect retinal development to varying degrees103 and the regulatory influence of the RPE over the developing retina depends on localized interactions between the RPE and neural retina.
Visual Electrophysiology in Albinism
The abnormal chiasmal decussation of nerve fibres in albinism can be detected by comparing multi-channel visual evoked potentials (VEP) from each eye, recorded over both hemispheres using scalp electrodes.104–112
Multi-channel VEPs normally show a symmetrical distribution across the occiput (Figures 4-6A). In albinism monocular VEPs show bilateral contralateral predominance (Figure 6B and C). The dominant response is elicited in the cortical hemisphere opposite to the eye being stimulated and when the other eye is stimulated, the dominant response is elicited in the other hemisphere (a “crossed” asymmetry). It is highlighted that achiasma is also associated with congenital nystagmus and chiasmal dysfunction, but in contrast to albinism, monocular VEPs show evidence of bilateral ipsilateral predominance, caused by reduced or absent optic nerve decussation.113,114 Pattern reversal VEPs,115 used routinely in many laboratories to assess optic nerve function, are significantly affected by nystagmus, and have been shown to be equivocal or unreliable in detecting misrouting.116 Evidence of albino visual pathway misrouting on VEP testing requires age-appropriate use of both flash and pattern onset-offset (“appearance-disappearance”) stimulation.110,117 The most consistent VEP abnormality in young albino children is bilateral contralateral predominance of flash VEP in terms of amplitude (Figure 7). The inter-hemispheric amplitude asymmetry in the flash VEP lessens with increasing age and can resolve by the teenage years. This is shown in Figure 8, including novel data in genetically confirmed cases of OCA (N = 17), OA (N = 3), HP syndrome (N=5), and Chediak-Higashi syndrome (N = 1). Monocular pattern onset-offset VEPs in children are of shorter peak time over the contralateral compared with ipsilateral hemisphere. In contrast to the flash VEP, the pattern onset-offset VEP peak time difference between hemispheres increases with age, resulting from increasing peak time of the ipsilateral response. This is shown in Figure 9, including novel data in genetically confirmed cases of OCA (N = 8), OA (N = 3), HP syndrome (N = 2) and Chediak-Higashi syndrome (N = 1). The magnitude of the pattern onset-offset VEP peak time difference between hemispheres correlates with the clinical features of albinism including foveal hypoplasia, iris transillumination, nystagmus, reduced visual acuity, and reduced stereoacuity. Inter-hemispheric flash VEP amplitude asymmetries also show correlation with clinical features but only in children under the age of 7 years.109 The sensitivity of VEPs in the detection of albino misrouting has been estimated to be about 80%,104,109,116–119 although typical VEP abnormalities can occur in the absence of classical clinical features109 and the diagnosis of albinism is usually also informed by medical and family history, and detailed ophthalmic evaluation.
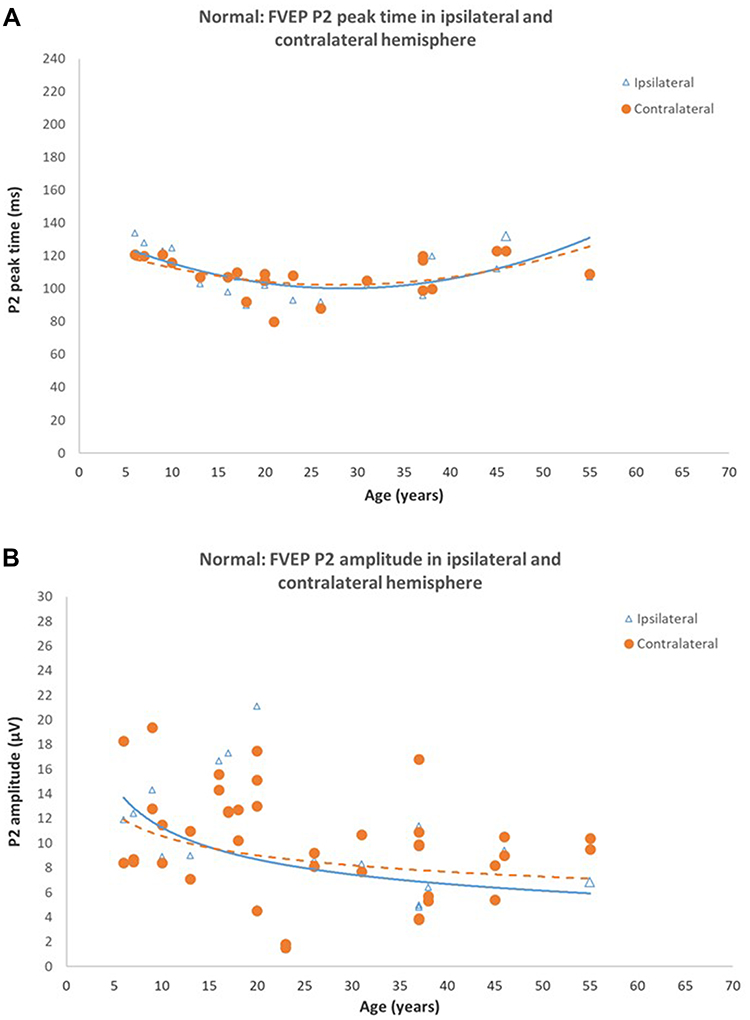 |
Figure 4 Normal Flash VEPs (A) P2 peak time in ipsilateral and contralateral hemispheres of control subjects. Peak times from both hemispheres are symmetrical and shorten until approximately 18–20 years of age, do not change between 20–40 years of age, then increases beyond 40 years of age (B) P2 amplitude in ipsilateral and contralateral hemispheres of control subjects. P2 amplitudes from both hemispheres are symmetrical and reduce significantly with age (P < 0.05). Reprinted from Neveu MM, Jeffery G, Burton LC, Sloper JJ, Holder GE. Age-related changes in the dynamics of human albino visual pathways. Eur J Neurosci. 2003;18(7):1939–1949.110 |
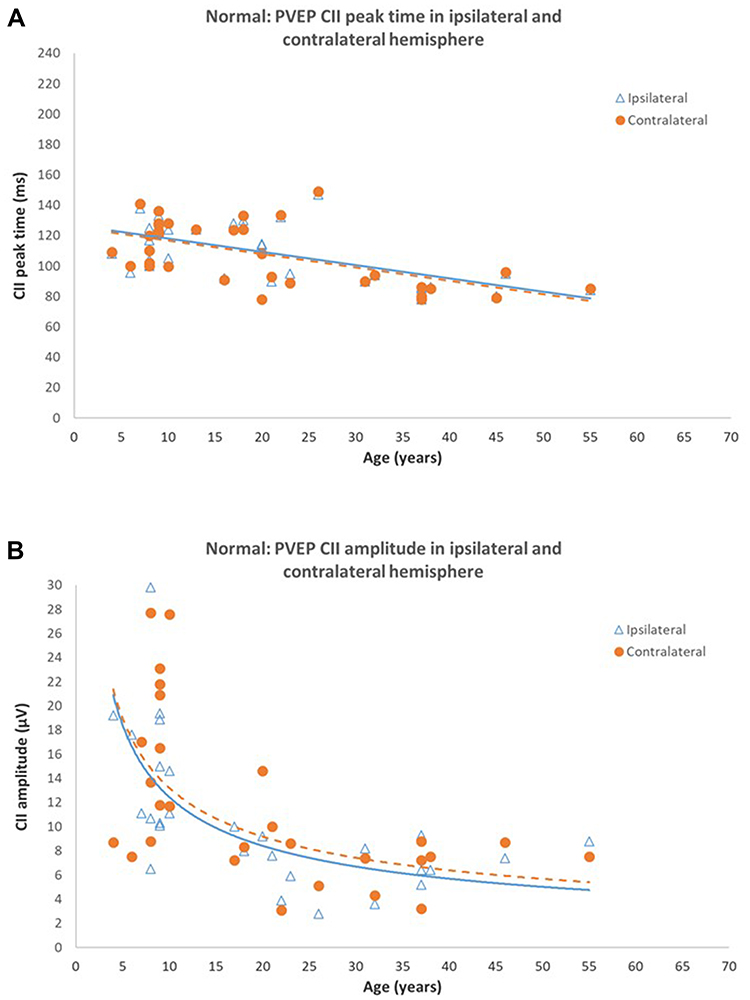 |
Figure 5 Normal Pattern onset-offset VEPs (A) CII peak times in ipsilateral and contralateral hemispheres of control subjects. CII peak times from both hemispheres are symmetrical and significantly shorten with increasing age (P < 0.0001). (B) CII amplitude in ipsilateral and contralateral hemispheres of control subjects. CII amplitudes from both hemispheres are symmetrical. P2 amplitude reduces significantly with age (P < 0.005). Reprinted from Neveu MM, Jeffery G, Burton LC, Sloper JJ, Holder GE. Age-related changes in the dynamics of human albino visual pathways. Eur J Neurosci. 2003;18(7):1939–1949.110 |
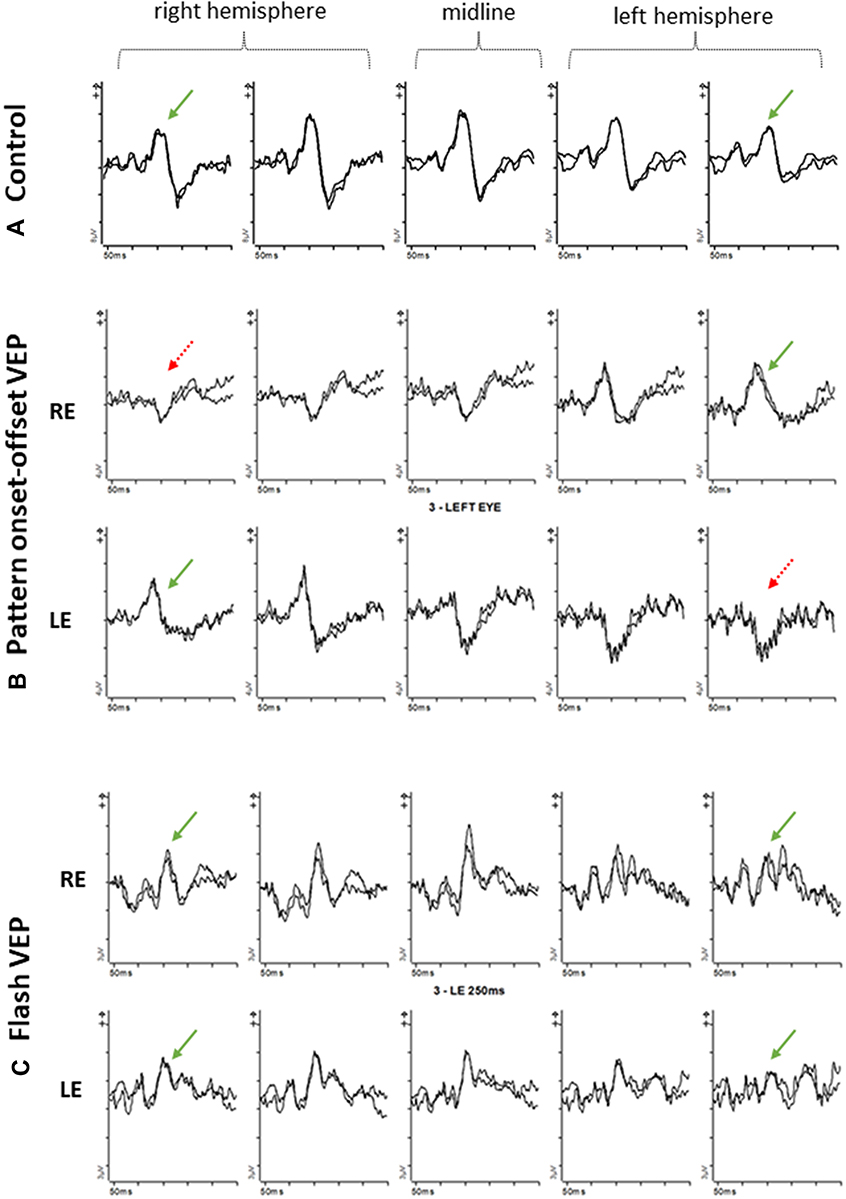 |
Figure 6 Pattern onset-offset VEPs and Flash VEPs from an age-matched control (A) and a 60 year old patient with albinism (B, C) using five scalp electrodes positioned over the occiput; 2 electrodes over the right hemisphere; two electrodes over the left hemisphere; 1 electrode over the midline. (A) VEP responses from control subject are symmetrical and of similar amplitude and peak time from the left and right hemisphere. (B). Pattern onset-offset VEP responses from a 60-year-old patient with albinism. Right eye (rows 2) VEP responses are of shorter peak and/or larger amplitude from the left hemisphere compared with the right hemisphere. Left eye (rows 3) VEP responses are of shorter peak and/or larger amplitude from the right hemisphere compared with the left hemisphere. (C) Flash VEP responses from a 60-year-old patient with albinism. VEP responses from both eyes are symmetrical and of similar amplitude and peak time from the left and right hemisphere. |
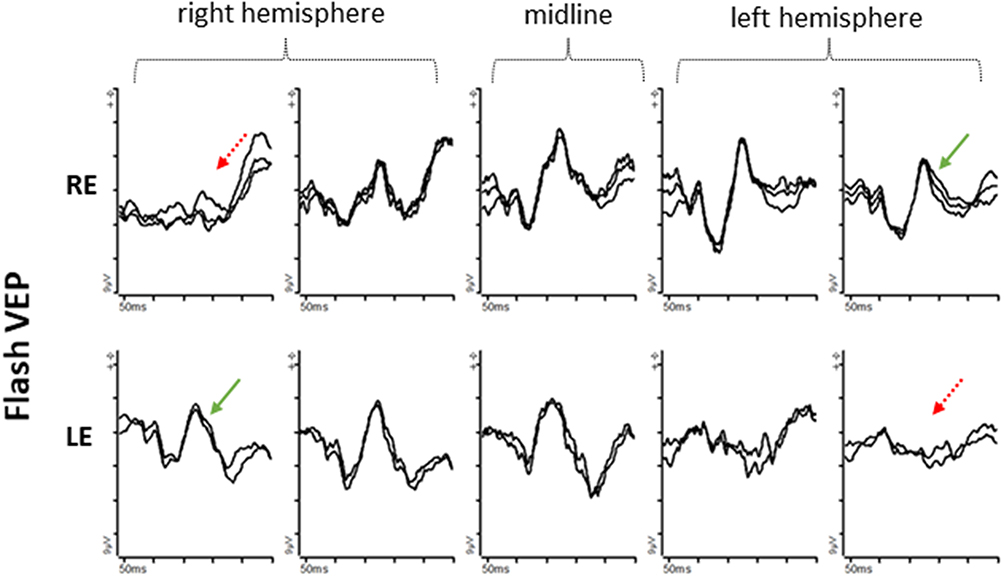 |
Figure 7 Flash VEPs from an 8-month-old patient with albinism using five occipital scalp electrodes; 2 electrodes over the right hemisphere; 2 electrodes over the left hemisphere; 1 electrode over the midline. Right eye (row 1) VEP responses are of higher amplitude from the left hemisphere compared with the right hemisphere. Left eye (row 2) VEP responses are of higher amplitude from the right hemisphere compared with the left hemisphere. |
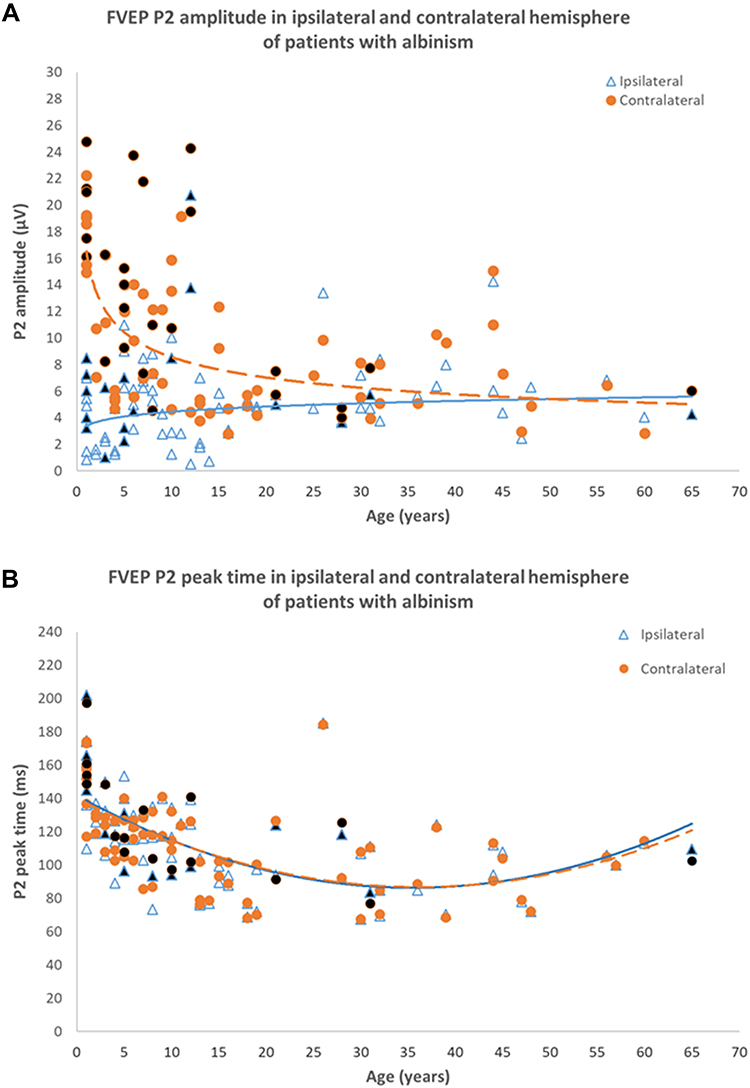 |
Figure 8 Flash VEP characteristics in 83 patients with albinism including 17 genetically confirmed cases of OCA, 3 cases of OA, 5 cases of HP syndrome and 1 case of CH Syndrome. (A) P2 amplitude in ipsilateral (bold line) and contralateral (dashed line) hemispheres. There is a significant inter-hemispheric amplitude difference up until ~18 years of age. There is no significant inter-hemispheric difference beyond 18 years. (B) Flash VEP P2 peak time in ipsilateral (bold line) and contralateral (dashed line) hemispheres of patients with albinism. P2 peak times in both hemispheres are symmetrical and similar to those in control subjects. Black filled data points are from genetically confirmed patients. Data from Neveu MM, Jeffery G, Burton LC, Sloper JJ, Holder GE. Age-related changes in the dynamics of human albino visual pathways. Eur J Neurosci. 2003;18(7):1939-1949110, with additional patients and highlighting of genetically confirmed cases. |
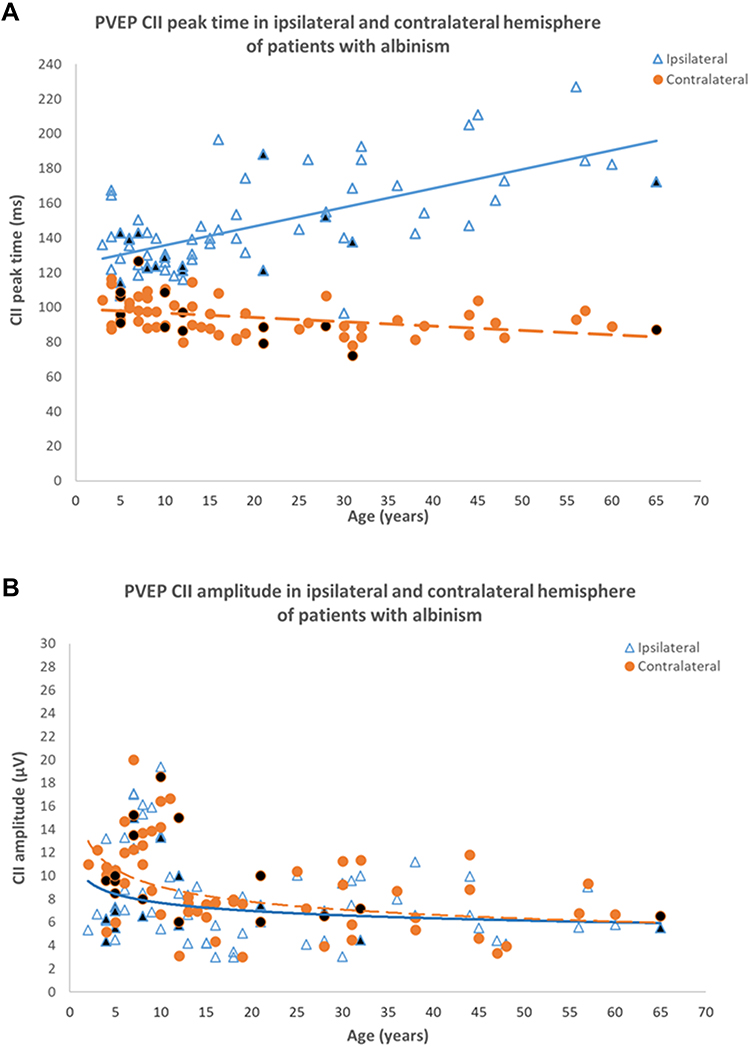 |
Figure 9 Pattern onset-offset VEP characteristics in 66 patients with albinism including 8 genetically confirmed cases of OCA, 3 cases of OA, 2 cases of HP syndrome and 1 case of CH Syndrome. (A) CII peak time in ipsilateral (bold line) and contralateral hemispheres (dashed line). CII peak time in the contralateral hemisphere is shorter than peak time in the ipsilateral hemisphere for all ages. The inter-hemispheric peak time difference increases with age. (B) CII amplitude in ipsilateral (bold line) and contralateral (dashed line) hemispheres of patient with albinism. CII amplitudes in both hemispheres are symmetrical, similar to that in control subjects. Black filled data points are from genetically confirmed patients. Data from Neveu MM, Jeffery G, Burton LC, Sloper JJ, Holder GE. Age-related changes in the dynamics of human albino visual pathways. Eur J Neurosci. 2003 Oct;18(7):1939-1949110, with additional patients and highlighting of genetically confirmed cases. |
In a small number of cases, particularly in individuals with a mild phenotype, contralateral predominance of the VEP is confined to one eye. The reason for this is unknown although it is highlighted that albino misrouting of the visual pathways correlates with ocular signs, including pigmentation120,121 and typical VEP abnormalities are greatest in patients with the most ocular features of albinism.109 Localized differences in the concentration of melanin in the RPE affect retinal development to varying degrees,103 and comparisons between hypopigmented mice mutants have shown that the size of the uncrossed projection decreases with decreasing amounts of pigment in the eye.101,102 This may have relevance to unilateral cases, consistent with some of these individuals having inter-ocular asymmetries in hypopigmentation and foveal architecture.121
Maturational and degenerative processes in the normal population do not fully account for the opposing age-related changes in the pattern onset-offset VEP and flash VEP in albinism. The horizontal and interlaminar connections and synaptogenesis of the retinocortical hemispheric projections are fully established in infants and the anatomical pattern of chiasmal misrouting in albinos should not change with age.63,122–126 However, structural imaging studies have shown that intracortical myelin in the visual cortex continues to increase well into adulthood, peaking between 30 and 40 years of age127 and declines beyond 50 years of age. Development of V1 in infants may favour flash over pattern onset-offset activation, whereas developmental changes in contrast sensitivity (up to 8 years of age) and contour integration (up to 15 years of age) may favour pattern onset-offset over flash activation.128–130 The presence of misrouted fibres and temporal deficits during the critical periods of visual development may contribute to differences in synaptogenesis between normally pigmented and albino individuals, accounting for some of the age-related changes in the VEP. This does not explain why the ipsilateral cortical projection shows a greater degree of abnormality compared to the contralateral, irrespective of the stimulus modality, and developmental irregularities of the hypopigmented retina may be relevant. Normal retinal development in mammals follows a centre-periphery gradient originating at the presumptive foveal region131–136 and at birth the foveal pit is formed. The specialization of the fovea, however, occurs postnatally, reaching maturity at 15–45 months.137–139 Thereafter, plasticity and maturation of the visual system is highly dependent upon visual experience.140 Similar developmental profiles are observed in hypopigmented animals but the gradient is shallower82,141 and there is an increased number of mitotic profiles along the horizontal meridian due to delay in retinal mitosis.142 This suggests that ipsilateral RGCs originating in temporal retina are less developed than contralateral RGCs originating in nasal retina. Therefore, abnormal retinocortical projections in the albino may be further compounded by underdevelopment of the ipsilateral pathway.
Visual Fields
In individuals with albinism, visual field representation has been studied using static perimetry, MRI and fMRI143–149 but there is conflicting evidence of visual field abnormalities. Most studies show that the abnormal visual pathway projection in albinism results in an altered retinotopic map, where a mirror-image map from temporal retina is superimposed on the normal retinotopic map from nasal retina.143,144 This mirroring of the abnormal projection in human albinos is made available for visual perception and is consistent with a detailed study of an albino monkey.78
Differential Diagnosis
The clinical diagnosis of albinism can be clear in patients where most of the clinical signs are present or misrouting is confirmed on VEP testing. However, OCA is a heterogeneous disorder and the presence and severity of ocular and cutaneous features can show inter- and intra-familial variation.9,150,151 On clinical examination, the typical characteristics can be absent or equivocal and it may be difficult to detect foveal hypoplasia or iris transillumination, particularly in patients with nystagmus or in young children. Symptoms and signs such as poor visual acuity and nystagmus are non-specific, and milder cases of albinism may be mistaken for relatively common disorders such as infantile onset or congenital motor nystagmus.152,153 It is essential to exclude progressive retinal, neurological and syndromic causes154–159 and visual electrophysiology plays an important role.104–110 Syndromes that include “albino-like” signs such as iris or skin hypopigmentation may lack VEP evidence of optic nerve misrouting (see Syndromic OCA and Syndromic Hypopigmentation Disorders). Inherited retinal causes of nystagmus include Leber congenital amaurosis, cone and cone rod dystrophy and stationary disorders such as achromatopsia, S-cone monochromacy and complete and incomplete congenital stationary night blindness, distinguished by abnormal functional phenotypes evident on full-field flash electroretinography (ERG).158–160 In albinism, the ERGs fall within the “normal” reference range, which excludes a retinal cause for the nystagmus. This is often reassuring as progressive photoreceptor dystrophies or severe cone dysfunction disorders may be associated with a worse visual prognosis.
Summary
Albinism is a heterogeneous disorder with ocular features common to OA and OCA. Accurate diagnosis informs clinical management and is essential to enable counselling and in patients that require medical intervention or monitoring, such as those with some syndromic forms of OCA. Recent advances in genetics offer the possibility of precise diagnosis if genetic testing is indicated, but the availability of genotyping may be limited, and early diagnosis confounded if symptoms and signs are mild or non-specific. Visual electrophysiology can play a pivotal role in distinguishing albinism from other disorders including those associated with ocular hypopigmentation. An important characteristic of albinism is the presence of intra-cranial visual pathway misrouting, as revealed by multi-channel VEPs. The sensitivity of VEPs to misrouting has been shown to depend on age-appropriate choice of flash or pattern onset-offset stimulation, as corroborated in this study by novel VEP data in a range of genetically confirmed albino cases.
Acknowledgments
AGR is supported by the National Institute for Health Research (NIHR) Biomedical Research Centre based at Moorfields Eye Hospital NHS Foundation Trust and UCL Institute of Ophthalmology. The views expressed are those of the authors and not necessarily those of the NHS, the NIHR or the Department of Health.
Funding
There is no funding to report.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Biswas S, Lloyd IC. Oculocutaneous albinism. Arch Dis Child. 1999;80(6):565–569.
2. Marti A, Lasseaux E, Ezzedine K, et al. Lessons of a day hospital: comprehensive assessment of patients with albinism in a European setting. Pigment Cell Melanoma Res. 2018;31(2):318–329.
3. Kamaraj B, Purohit R. Mutational analysis of oculocutaneous albinism: a compact review. BioMed Res Int. 2014;2014:905472.
4. Hutton SM, Spritz RA. Comprehensive analysis of oculocutaneous albinism among non-hispanic caucasians shows that OCA1 is the most prevalent OCA type. J Invest Dermatol. 2008;128:2442–2450.
5. Manga P, Kerr R, Ramsay M, Kromberg JGR. Biology and genetics of oculocutaneous albinism and vitiligo-common pigmentation disorders in Southern Africa. S Afr Med J. 2013;103:984-988.
6. Grønskov K, Brondum-Nielsen K. Oculocutaneous albinism. Orphanet J Rare Dis. 2007;2:43.
7. Simeonov DR, Wang X, Wang C, et al. DNA variations in oculocutaneous albinism: an updated mutation list and current outstanding issues in molecular diagnostics. Hum Mutat. 2013;34:827-835.
8. Chan HW, Schiff ER, Tailor VK, et al. Prospective study of the phenotypic and mutational spectrum of ocular albinism and oculocutaneous albinism. Genes (Basel). 2021;12(4):508.
9. Camand O, Marchant D, Boutboul S, et al. Mutation analysis of the tyrosinase gene in oculocutaneous albinism. Hum Mutat. 2001;17(4):352.
10. Olivares C, Jiménez-Cervantes C, Lozano JA, Solano F, García-Borrón JC. The 5,6-dihydroxyindole-2-carboxylic acid (DHICA) oxidase activity of human tyrosinase. Biochem J. 2001;354(1):131–139.
11. Urabe K, Aroca P, Hearing VJ. From gene to protein: determination of melanin synthesis. Pigment Cell Res. 1993;6(4):186–192.
12. Schiaffino MV. Signaling pathways in melanosome biogenesis and pathology. Int J Biochem Cell Biol. 2010;42:1094–1104.
13. David CV. Oculocutaneous albinism. Cutis. 2013;3:91.
14. Kromberg JGR, Bothwell J, Kidson SH, Manga P, Kerr R, Jenkins T. Types of Albinism in the Black Southern Africa Population. East Afr Med J. 2012;89:20–27.
15. Suzuki T, and Tomita Y. Recent advances in genetic analyses of oculocutaneous albinism types 2 and 4. J Dermatol Sci. 2008 Jul;51(1):1-9.
16. Inagaki K, Suzuki T, Shimizu H, et al. Oculocutaneous albinism type 4 is one of the most common types of albinism in Japan. Am J Hum Genet. 2004;74(3):466–471.
17. Okamura K, Suzuki T. Current landscape of Oculocutaneous Albinism in Japan. Pigment Cell Melanoma Res. 2021;34:190–203.
18. Scheinfeld NS. Syndromic albinism: a review of genetics and phenotypes. Dermatol Online J. 2003;9(5):5.
19. Mansouri Nejad SE, Yazdan Panah MJ, Tayyebi Meibodi N, et al. Griscelli Syndrome: a Case Report. Iran J Child Neurol. 2014;8(4):72–75.
20. Najmuddin F, Rai R, Lahiri K, Cholera PP. Elejalde Syndrome: the Silvery Hair Syndrome. Int J Genet Sci. 2015;2(1):734.
21. Tezcan I, Demir E, Aşan E, Kale G, Müftüoğlu SF, Kotiloğlu E. A new case of oculocerebral hypopigmentation syndrome (Cross syndrome) with additional findings. Clin Genet. 1997;51(2):118–121.
22. Reddy RR, Babu BM, Venkateshwaramma B, Hymavathi C. Silvery hair syndrome in two cousins: Chediak-Higashi syndrome vs Griscelli syndrome, with rare associations. Int J Trichology. 2011;3(2):107–111.
23. Zhou M, Gradstein L, Gonzales JA, Tsilou ET, Gahl WA, Chan -C-C. Ocular Pathologic Features of Hermansky-Pudlak Syndrome Type 1 in an Adult. Arch Ophthalmol. 2006;124(7):1048–1051.
24. Power B, Ferreira CR, Chen D, et al. Hermansky-Pudlak syndrome and oculocutaneous albinism in Chinese children with pigmentation defects and easy bruising. Orphanet J Rare Dis. 2019;14(1):52.
25. Desai N, Weisfeld-Adams JD, Brodie SE, et al. Optic neuropathy in late-onset neurodegenerative Chédiak-Higashi Syndrome. Br J Ophthalmol. 2016;100(5):704–707.
26. Spritz RA, Bailin T, Nicholls RD, et al. Hypopigmentation in the Prader-Willi syndrome correlates with P gene deletion but not with haplotype of the hemizygous P allele. Am J Med Genet. 1997;71(1):57–62.
27. Saitoh S, Oiso N, Wada T, et al. Oculocutaneous albinism type 2 with a P gene missense mutation in a patient with Angelman syndrome. JMed Genet. 2000;37:392–394.
28. Creel DJ, Bendel CM, Wiesner GL, Wirtschafter JD, Arthur DC, King RA. Abnormalities of the central visual pathways in Prader-Willi syndrome associated with hypopigmentation. N Engl J Med. 1986;314(25):1606–1609.
29. Ramsay M, Colman MA, Stevens G, et al. The tyrosinase-positive oculocutaneous albinism locus maps to chromosone 15q11.2-q12. Am J Hum Genet. 1992;51(4):879-84.
30. Apkarian P, Spekreijse H, Van Swaay E, Van schooneveld M. Visual evoked potentials in Prader-Willi syndrome. Doc Ophthalmol. 1989;71:355-367.
31. Fitzgerald K, Cibic GW. The vlaue of flash visual evoked potentials in albinism. J Pediatr Ophthalmol Strabismus. 1994;31(1):18-25.
32. Thompson DA, Kriss A, Cottrell S, Taylor D. Visual evoked potential evidence of albino-like chiasmal misrouting in a patient with Angelman syndrome with no ocular features of albinism. Dev Med Child Neurol. 1999;41(9):633-638.
33. Byrne S, Dionisi-Vici C, Smith L, et al. Vici Syndrome: a review. Orphanet J Rare Dis. 2016;11:21.
34. Filloux FM, Hoffman RO, Viskochil DH, Jungbluth H, Creel DJ. Ophthalmologic features of Vici syndrome. J Pediatr Ophthalmol Strabismus. 2014;51(4):214-220.
35. Goldberg MF. Waardenburg's syndrome with fundus and other anomalies. Arch Ophthalmol. 1996;76(6):797-810.
36. Ahmed Jan N, Mui RK, Masood S. Waardenburg Syndrome. Treasure Island (FL) StatPearls Publishing; 2021.
37. Newton VE. Clinical features of the Waardenburg syndromes. Adv Otorhinolaryngol. 2002;61:201-208.
38. Wollnik B, Tukel T, Uyguner O, et al. Homozygous and heterozygous inheritance of PAX3 mutations causes different types of Waardenburg syndrome. Am J Med Genet A.2003;122(1):42-45.
39. Tassabehji M, Newton VE, Read AP. Waardenburg syndrome type 2 caused by mutations in the human microphthalmia (MITF) gene. Nat Genet. 1994;8(3):251-255.
40. Mullner-Eidenbock A, Moser E, Frisch H, Read AP. Waardenburg syndrome type 2 in a Turkish family: implications for the importance of the pattern of fundus pigmentation. Br J Ophthalmol. 2001;85(11):1384-1386.
41. Ohno N, Kiyosawa M, Mori H, Wang WF, Takase H, Mochizuki M. Clinical findings in Japanese patients with Waardenburg syndrome type 2. Jpn J Ophthalmol. 2003;47(1):77-84.
42. Tsang SH, Sharma T. X-linked Ocular Albinism. Tissue Eng. 2018;1085:49–52.
43. Garner A, Jay BS. Macromelanosomes in X-linked ocular albinism. Histopathology. 1980;4:243–254.
44. Palmisano I, Bagnato P, Palmigiano A, et al. The ocular albinism type 1 protein, an intracellular G protein-coupled receptor, regulates melanosome transport in pigment cells. Hum Mol Genet. 2008;17:3487–3501.
45. Schiaffino MV, D’Addio M, Alloni A, et al. Ocular albinism: evidence for a defect in an intracellular signal transduction system. Nat Genet. 1999;23(1):108–112.
46. McKay BS. Pigmentation and vision: is GPR143 in control? J Neurosci Res. 2019;97(1):77–87.
47. Summers CG. Albinism: classification, clinical characteristics, and recent findings. Optom Vis Sci off Publ Am Acad Optom. 2009;86(6):659–662.
48. Lekalakala PT. Oculocutaneous Albinism and Squamous Cell Carcinoma of the Skin of the Head and Neck in Sub-Saharan Africa. J Skin Cancer. 2015;2015:167847.
49. Neveu MM, Holder GE, Sloper JJ, Jeffery G. Optic chiasm formation in humans is independent of foveal development. Eur J Neurosci. 2005;22(7):1825–1829.
50. Oetting WS, Summers CG, King RA. Albinism and the associated ocular defects. Metab Pediatr Syst Ophthalmol. 1994;17(1–4):5–9.
51. King RA, Summers CG. Albinism. Dermatol Clin. 1988;6(2):217–228.
52. Mallipatna A, Vinekar A, Jayadev C, et al. The use of handheld spectral domain optical coherence tomography in pediatric ophthalmology practice: our experience of 975 infants and children. Indian J Ophthalmol. 2015;63(7):586–593.
53. Lee DJ, Woertz EN, Visotcky A, et al. The Henle Fiber Layer in Albinism: comparison to Normal and Relationship to Outer Nuclear Layer Thickness and Foveal Cone Density. Invest Ophthalmol Vis Sci. 2018;59(13):5336–5348. doi:10.1167/iovs.18-24145
54. Wilk MA, McAllister JT, Cooper RF, et al. Relationship between foveal cone specialization and pit morphology in albinism. Invest Ophthalmol Vis Sci. 2014;55(7):4186–4198.
55. Hertle RW. Albinism: particular Attention to the Ocular Motor System. Middle East Afr J Ophthalmol. 2013;20(3):248–255.
56. Dysli M, Abegg M. Nystagmus Does Not Limit Reading Ability in Albinism. PLoS One. 2016;11(7):e0158815.
57. Yahalom C, Tzur V, Blumenfeld A, et al. Refractive profile in oculocutaneous albinism and its correlation with final visual outcome. Br J Ophthalmol. 2012;96(4):537–539.
58. Wildsoet CF, Oswald PJ, Clark S. Albinism: its Implications for Refractive Development. Invest Ophthalmol Vis Sci. 2000;41(1):1–7.
59. Schweigert A, Lunos S, Connett J, Summers CG. Changes in refractive errors in albinism: a longitudinal study over the first decade of life. J AAPOS off Publ Am Assoc Pediatr Ophthalmol Strabismus. 2018;22(6):462–466.
60. Kaur A, Akhila K, Sahu SK, Padhy SK. Bilateral pellucid marginal degeneration with oculocutaneous albinism. BMJ Case Rep. 2021;14(6):e243640.
61. King RA, Summers CG. Albinism: Ocular And Oculocutaneous Albinism and Hermansky–Pudlak Syndrome. Manag Genet Syndr 2010:53-68.
62. Okulicz JF, Shah RS, Schwartz RA, Janniger CK. Oculocutaneous albinism. J Eur Acad Dermatol Venereol. 2003;17(3):251–256.
63. Mann I. The Development of the Human Eye. London: British Medical Association; 1964.
64. Casey MA, Lusk S, Kwan KM. Build me up optic cup: intrinsic and extrinsic mechanisms of vertebrate eye morphogenesis. Dev Biol. 2021;476:128–136.
65. Heavner W, Pevny L. Eye development and retinogenesis. Cold Spring Harb Perspect Biol. 2012;4(12):76.
66. Guillery RW, Mason CA, Taylor JS. Developmental determinants at the mammalian optic chiasm. J Neurosci. 1995;15(7 Pt 1):4727–4737.
67. Murcia-Belmonte V, Erskine L. Wiring the Binocular Visual Pathways. Int J Mol Sci. 2019;20(13):3282.
68. Williams SE, Mason CA, Herrera E. The optic chiasm as a midline choice point. Curr Opin Neurobiol. 2004;14(1):51–60.
69. Colello RJ, Guillery RW. The early development of retinal ganglion cells with uncrossed axons in the mouse: retinal position and axonal course. Development. 1990;108(3):515–523.
70. Fukuda Y, Sawai H, Watanabe M, Wakakuwa K, Morigiwa K. Nasotemporal overlap of crossed and uncrossed retinal ganglion cell projections in the Japanese monkey (Macaca fuscata). J Neurosci. 1989;9(7):2353–2373.
71. Chalupa LM, Lia B. The nasotemporal division of retinal ganglion cells with crossed and uncrossed projections in the fetal rhesus monkey. J Neurosci. 1991;11(1):191–202.
72. Rapaport DH, Fletcher JT, LaVail MM, Rakic P. Genesis of neurons in the retinal ganglion cell layer of the monkey. J Comp Neurol. 1992;322(4):577–588.
73. Provis JM, van Driel D, Billson FA, Russell P. Development of the human retina: patterns of cell distribution and redistribution in the ganglion cell layer. J Comp Neurol. 1985;233(4):429–451.
74. Kinnear PE, Jay B, Witkop CJ. Albinism. Surv Ophthalmol. 1985;30(2):75–101.
75. Jeffery G. The albino retina: an abnormality that provides insight into normal retinal development. Trends Neurosci. 1997;20(4):165–169.
76. Stone J, Campion JE, Leicester J. The nasotemporal division of retina in the Siamese cat. J Comp Neurol. 1978;180(4):783–798.
77. Ilia M, Jeffery G. Delayed neurogenesis in the albino retina: evidence of a role for melanin in regulating the pace of cell generation. Brain Res Dev Brain Res. 1996;95(2):176–183.
78. Guillery RW, Hickey TL, Kaas JH, Felleman DJ, Debruyn EJ, Sparks DL. Abnormal central visual pathways in the brain of an albino green monkey (Cercopithecus aethiops). J Comp Neurol. 1984;226(2):165–183.
79. Drager UC, Olsen JF. Origins of crossed and uncrossed retinal projections in pigmented and albino mice. J Comp Neurol. 1980;191(3):383–412.
80. Shatz CJ, Kliot M. Prenatal misrouting of the retinogeniculate pathway in Siamese cats. Nature. 1982;300(5892):525–529.
81. Thompson ID, Morgan JE. The development of retinal ganglion cell decussation patterns in postnatal pigmented and albino ferrets. Eur J Neurosci. 1993;5(4):341–356.
82. Webster MJ, Rowe MH. Disruption of developmental timing in the albino rat retina. J Comp Neurol. 1991;307(3):460–474.
83. Baker GE, Jeffery G. Distribution of uncrossed axons along the course of the optic nerve and chiasm of rodents. J Comp Neurol. 1989;289(3):455–461.
84. Iwai-Takekoshi L, Ramos A, Schaler A, Weinreb S, Blazeski R, Mason C. Retinal pigment epithelial integrity is compromised in the developing albino mouse retina. J Comp Neurol. 2016;524(18):3696–3716.
85. Rachel RA, Dolen G, Hayes NL, et al. Spatiotemporal features of early neuronogenesis differ in wild-type and albino mouse retina. J Neurosci. 2002;22(11):4249–4263.
86. Neveu MM, Jeffery G. Chiasm formation in man is fundamentally different from that in the mouse. Eye. 2007;21(10):1264–1270.
87. Lund R. Uncrossed visual pathways of hooded and albino rats. Science. 1965;149(3691):1506–1507.
88. Hubel DH, Wiesel TN. Aberrant visual projections in the Siamese cat. J Physiol. 1971;218(1):33–62.
89. Leventhal AG, Creel DJ. Retinal projections and functional architecture of cortical areas 17 and 18 in the tyrosinase-negative albino cat. J Neurosci. 1985;5(3):795–807.
90. Guillery RW. Visual pathways in albinos. Scientific American. 1974;230(5):44–54.
91. Guillery RW, Okoro AN, Witkop CJ. Abnormal visual pathways in the brain of a human albino. Brain Res. 1975;96(2):373–377.
92. Kirk DL, Levick WR, Cleland BG, Wassle H. Crossed and uncrossed representation of the visual field by brisk-sustained and brisk-transient cat retinal ganglion cells. Vision Res. 1976;16(3):225–231.
93. Leventhal AG. Morphology and distribution of retinal ganglion cells projecting to different layers of the dorsal lateral geniculate nucleus in normal and Siamese cats. J Neurosci. 1982;2(8):1024–1042.
94. Kruijt CC, Gradstein L, Bergen AA, Florijn RJ, Arveiler B, Lasseaux E, et al. The Phenotypic and Mutational Spectrum of the FHONDA Syndrome and Oculocutaneous Albinism: Similarities and Differences. Invest Ophthalmol Vis Sci. 2022;63(1):19.
95. van Genderen MM, Riemslag FC, Schuil J, Hoeben FP, Stilma JS, Meire FM. Chiasmal misrouting and foveal hypoplasia without albinism. Br J Ophthalmol. 2006;90(9):1098–1102.
96. Poulter JA, Al-Araimi M, Conte I, et al. Recessive mutations in SLC38A8 cause foveal hypoplasia and optic nerve misrouting without albinism. Am J Hum Genet. 2013;93:1143–1150.
97. Schiff ER, Tailor VK, Chan HW, Theodorou M, Webster AR, Moosajee M. Novel Biallelic Variants and Phenotypic Features in Patients with SLC38A8-Related Foveal Hypoplasia. Int J Mol Sci. 2021;22(3):1130.
98. Jeffery G, Perry VH. Evidence for ganglion cell death during development of the ipsilateral retinal projection in the rat. Brain Res. 1981;254(1):176–180.
99. Perry VH, Henderson Z, Linden R. Postnatal changes in retinal ganglion cell and optic axon populations in the pigmented rat. J Comp Neurol. 1983;219(3):356–368.
100. Henderson Z. Distribution of ganglion cells in the retina of adult pigmented ferret. Brain Res. 1985;358(1–2):221–228.
101. LaVail JH, Nixon RA, Sidman RL. Genetic control of retinal ganglion cell projections. J Comp Neurol. 1978;182(3):399–421.
102. Balkema GW, Drager UC. Origins of uncrossed retinofugal projections in normal and hypopigmented mice. Vis Neurosci. 1990;4(6):595–604.
103. Gimenez E, Lavado A, Jeffery G, Montoliu L. Regional abnormalities in retinal development are associated with local ocular hypopigmentation. J Comp Neurol. 2005;485(4):338–347.
104. Apkarian P. A practical approach to albino diagnosis. VEP misrouting across the age span. Ophthalmic Paediatr Genet. 1992;13(2):77–88.
105. Kriss A, Russell-Eggitt I, Harris CM, Lloyd IC, Taylor D. Aspects of albinism. Ophthalmic Paediatr Genet. 1992;13(2):89–100.
106. Creel D, Witkop CJ, King RA. Asymmetric visually evoked potentials in human albinos: evidence for visual system anomalies. Invest Ophthalmol. Vis. Sci. 1974;13(6):430–440.
107. Pott JW, Jansonius NM, Kooijman AC. Chiasmal coefficient of flash and pattern visual evoked potentials for detection of chiasmal misrouting in albinism. Doc Ophthalmol. 2003;106(2):137–143.
108. Liasis A, Handley SE, Nischal KK. Occipital Petalia and Albinism: A Study of Interhemispheric VEP Asymmetries in Albinism with No Nystagmus. J Clin Med. 2019;8(6):802.
109. Dorey SE, Neveu MM, Burton LC, Sloper JJ, Holder GE. The clinical features of albinism and their correlation with visual evoked potentials. Br J Ophthalmol. 2003;87(6):767–772.
110. Neveu MM, Jeffery G, Burton LC, Sloper JJ, Holder GE. Age-related changes in the dynamics of human albino visual pathways. Eur J Neurosci. 2003;18(7):1939–1949.
111. Puzniak RJ, Ahmadi K, Kaufmann J, et al. Quantifying nerve decussation abnormalities in the optic chiasm. Neuroimage Clin. 2019;3:24.
112. Liasis A, Handley SE, Nischal KK. Occipital Petalia and Albinism: a Study of Interhemispheric VEP Asymmetries in Albinism with No Nystagmus. J Clin Med. 2019;8(6):802.
113. Sami DA, Saunders D, Thompson DA, et al. The achiasmia spectrum: congenitally reduced chiasmal decussation. Br J Ophthalmol. 2005;89(10):1311–1317.
114. Brecelj J, Sustar M, Pečarič-Meglič N, Skrbec M, Stirn-Kranjc B. VEP characteristics in children with achiasmia, in comparison to albino and healthy children. Doc Ophthalmol. 2012;124(2):109–123.
115. Odom JV, Bach M, Brigell M, et al. International Society for Clinical Electrophysiology of Vision. ISCEV standard for clinical visual evoked potentials: (2016 update). Doc Ophthalmol. 2016;133(1):1–9.
116. Creel D, Spekreijse H, Reits D. Evoked potentials in albinos: efficacy of pattern stimuli in detecting misrouted optic fibers. Electroencephalogr Clin Neurophysiol. 1981;52(6):595–603.
117. Kruijt CC, de Wit GC, Talsma HE, Schalij-Delfos NE, van Genderen MM. The Detection Of Misrouting In Albinism: evaluation of Different VEP Procedures in a Heterogeneous Cohort. Invest Ophthalmol Vis Sci. 2019;60(12):3963–3969.
118. Pott J, Jansonius N, Kooijman A. Chiasmal coefficient of flash and pattern visual evoked potentials for detection of chiasmal misrouting in albinism. Doc Ophthalmol. 2003;106:137–143.
119. Russell-Eggitt I, Kriss A, Taylor DS. Albinism in childhood: a flash VEP and ERG study. Br J Ophthalmol. 1990;74(3):136–140.
120. von Dem Hagen EA, Houston GC, Hoffmann MB, Morland AB. Pigmentation predicts the shift in the line of decussation in humans with albinism. Eur J Neurosci. 2007;25(2):503–511.
121. NeveuMM, Sloper JJ, Moore AT, Jeffery G, Holder GE. Asymmetric Hypopigmentation in Albinism. Invest Ophthalmol Vis Sci. 2011;52(14): 6086.
122. Polyak S. The Vertebrate Visual System. Chicago: University of Chicago Press; 1957a.
123. Bourgeois JP, Rakic P. Changes of synaptic density in the primary visual cortex of the macaque monkey from fetal to adult stage. J Neurosci. 1993;13(7):2801–2820.
124. Missler M, Wolff A, Merker HJ, Wolff JR. Pre- and postnatal development of the primary visual cortex of the common marmoset. II. Formation, remodelling, and elimination of synapses as overlapping processes. J Comp Neurol. 1993;333(1):53–67.
125. Gilmore JH, Knickmeyer RC, Gao W. Imaging structural and functional brain development in early childhood. Nat Rev Neurosci. 2018;19(3):123–137.
126. Gibaldi A, Benson NC, Banks MS. Crossed–uncrossed projections from primate retina are adapted to disparities of natural scenes. Proc Natl Acad Sci. 2021;4:118.
127. Rowley CD, Sehmbi M, Bazin PL, et al. Age-related mapping of intracortical myelin from late adolescence to middle adulthood using T1 -weighted MRI. Hum Brain Mapp. 2017;38(7):3691–3703.
128. Owsley C. Aging and vision. Vision Res. 2011;51(13):1610–1622.
129. Almoqbel FM, Irving EL, Leat SJ. Visual acuity and contrast sensitivity development in children: sweep visually evoked potential and psychophysics. Optom Vis Sci. 2017;94(8):830–837.
130. Allard R, Renaud J, Molinatti S, Faubert J. Contrast sensitivity, healthy aging and noise. Vision Res. 2013;92:47–52.
131. Hoon M, Okawa H, Della Santina L, Wong RO. Functional architecture of the retina: development and disease. Prog Retin Eye Res. 2014;42:44–84.
132. Bringmann A, Syrbe S, Görner K, et al. The primate fovea: structure, function and development. Prog Retin Eye Res. 2018;66:49–84.
133. Provis JM, Diaz CM, Dreher B. Ontogeny of the primate fovea: a central issue in retinal development. Prog Neurobiol. 1998;54(5):549–580.
134. Reese BE, Johnson PT, Baker GE. Maturational gradients in the retina of the ferret. J Comp Neurol. 1996;375(2):252–273.
135. Rapaport DH, Stone J. The site of commencement of maturation in mammalian retina: observations in the cat. Brain Res. 1982;281(3):273–279.
136. Stone J, Egan M, Rapaport DH. The site of commencement of retinal maturation in the rabbit. Vision Res. 1985;25(3):309–317.
137. Springer AD, Hendrickson AE. Development of the primate area of high acuity, 3: temporal relationships between pit formation, retinal elongation and cone packing. Vis Neurosci. 2005;22(2):171–185.
138. Provis JM. Development of the primate retinal vasculature. Prog Retin Eye Res. 2001;20(6):799–821.
139. O’Brien KMB. Development of the Foveal Specialization. In: Tombran-Tink J, Barnstable CJ, editors. Visual Transduction and Non-Visual Light Perception. Humana Press; 2008:17-33.
140. Sengpiel F, Kind PC. The role of activity in development of the visual system. Curr Biol. 2002;12(23):R818–826.
141. Braekevelt CR, Hollenberg MJ. The development of the retina of the albino rat. Am J Anat. 1970;127(3):281–301.
142. Ilia M, Jeffery G. Retinal mitosis is regulated by dopa, a melanin precursor that may influence the time at which cells exit the cell cycle: analysis of patterns of cell production in pigmented and albino retinae. J Comp Neurol. 1999;405(3):394–405.
143. Guillery RW. Normal and abnormal visual field maps in albinos. Central Effects Non Matching Maps. Ophthalmic Paediatr Genet. 1990;11(3):177–183.
144. Hoffmann MB, Tolhurst DJ, Moore AT, Morland AB. Organization of the visual cortex in human albinism. J Neurosci. 2003;23(26):8921–8930.
145. Hoffmann MB, Seufert PS, Schmidtborn LC. Perceptual relevance of abnormal visual field representations: static visual field perimetry in human albinism. Br J Ophthalmol. 2007;91(4):509–513.
146. Ather S, Proudlock FA, Welton T, et al. Aberrant visual pathway development in albinism: from retina to cortex. Hum Brain Mapp. 2019;40(3):777–788.
147. Sheth V, Gottlob I, Mohammad S, McLean RJ, Proudlock FA. Visual field deficits in albinism. Invest Ophthalmol Vis Sci. 2014;55(13):2659.
148. Alvarez I, Smittenaar R, Handley SE, et al. Altered visual population receptive fields in human albinism. Cortex. 2020;128:107–123.
149. Duwell EJ, Woertz EN, Mathis J, Carroll J, DeYoe EA. Aberrant visual population receptive fields in human albinism. J Vis. 2021;21(5):19.
150. Preising MN, Forster H, Gonser M, Lorenz B. Screening of TYR, OCA2, GPR143, and MC1R in patients with congenital nystagmus, macular hypoplasia, and fundus hypopigmentation indicating albinism. Mol Vis. 2011;17:939–948.
151. Trebušak Podkrajšek K, Stirn Kranjc B, Hovnik T, Kovač J, Battelino T. GPR143 gene mutation analysis in pediatric patients with albinism. Ophthalmic Genet. 2012;33(3):167–170.
152. Simon JW, Kandel GL, Krohel GB, Nelsen PT. Albinotic characteristics in congenital nystagmus. Am J Ophthalmol. 1984;97:320–327.
153. Abadi RV, Pascal E. Periodic alternating nystagmus in humans with albinism. Invest Ophthalmol Vis Sci. 1994;35(12):4080–4086.
154. Gregory-Evans K, Kelsell RE, Gregory-Evans CY, et al. Autosomal dominant cone-rod retinal dystrophy (CORD6) from heterozygous mutation of GUCY2D, which encodes retinal guanylate cyclase. Ophthalmology. 2000;107(1):55–61.
155. Lorenz B, Gyürüs P, Preising M, et al. Early-onset severe rod-cone dystrophy in young children with RPE65 mutations. Invest Ophthalmol Vis Sci. 2000;41(9):2735–2742.
156. Michaelides M, Aligianis IA, Ainsworth JR, et al. Progressive cone dystrophy associated with mutation in CNGB3. Invest Ophthalmol Vis Sci. 2004;45(6):1975–1978.
157. Papageorgiou E, McLean RJ, Gottlob I. Nystagmus in childhood. Pediatr Neonatol. 2014;55:341–351.
158. Robson AG, Nilsson J, Li S, et al. ISCEV guide to visual electrodiagnostic procedures. Doc Ophthalmol. 2018;136(1):1–26.
159. Kurent A, Stirn-Kranjc B, Brecelj J. Electroretinographic characteristics in children with infantile nystagmus syndrome and early-onset retinal dystrophies. Eur J Ophthalmol. 2015 Jan-Feb;25(1):33-42.
160. Aboshiha J, Dubis AM, Carroll J, Hardcastle AJ, Michaelides M. The cone dysfunction syndromes. Br J Ophthalmol. 2016;100(1):115-121.
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.