Back to Archived Journals » Oncolytic Virotherapy » Volume 5
Oncolytic virotherapy for pediatric malignancies: future prospects
Authors Waters AM, Friedman GK, Ring EK, Beierle E ![]()
Received 10 May 2016
Accepted for publication 24 June 2016
Published 11 August 2016 Volume 2016:5 Pages 73—80
DOI https://doi.org/10.2147/OV.S96932
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Faris Farassati
Alicia M Waters,1 Gregory K Friedman,2 Eric K Ring,2 Elizabeth A Beierle1
1Department of Surgery, School of Medicine, University of Alabama at Birmingham, Birmingham, AL, USA; 2Department of Pediatrics, Division of Hematology-Oncology, School of Medicine, University of Alabama at Birmingham, Birmingham, AL, USA
Abstract: Pediatric solid tumors remain a major health concern, with nearly 16,000 children diagnosed each year. Of those, ~2,000 succumb to their disease, and survivors often suffer from lifelong disability secondary to toxic effects of current treatments. Countless multimodality treatment regimens are being explored to make advances against this deadly disease. One targeted treatment approach is oncolytic virotherapy. Conditionally replicating viruses can infect tumor cells while leaving normal cells unharmed. Four viruses have been advanced to pediatric clinical trials, including herpes simplex virus-1, Seneca Valley virus, reovirus, and vaccinia virus. In this review, we discuss the mechanism of action of each virus, pediatric preclinical studies conducted to date, past and ongoing pediatric clinical trials, and potential future direction for these novel viral therapeutics.
Keywords: oncolytic virus, herpes simplex virus, Seneca Valley virus, reovirus, vaccinia
Introduction
Cancer is rare in the pediatric population but remains the second leading cause of death in the 0- to 18-year age range, behind trauma. Cancer occurrence rates vary by age, but the overall most common cancers are acute lymphoblastic leukemia, brain and central nervous system tumors, neuroblastoma, and non-Hodgkin’s lymphoma.1 Prognosis of most cancer subtypes has improved significantly; however, long-term survival for some remains poor. Consequently, >2,000 children in the US die each year from this disease.2 Additionally, many patients who survive often suffer from life-altering morbidities secondary to treatment.
Innovative therapies and strategies are being explored for children diagnosed with solid tumors. One such targeted solution is the development of human recombinant viruses designed to attack rapidly dividing tumor cells while leaving normal healthy cells unharmed. Concerns exist as to the safety of viral therapy, especially in children, and most of the viruses currently in use have been genetically altered to be nonpathogenic or the parent virus is not pathogenic in humans. To date, four viral constructs have been shown to be safe with some evidence of efficacy in the treatment of a number of pediatric cancers in the laboratory and in early Phase I trials. In this review, we focus on these four viruses because of their proven efficacy in childhood cancer, but obviously there are other viruses in development that will likely be advanced to the pediatric population in the future.
Herpes simplex virus
Herpes simplex virus-1 (HSV-1) is a large, double-stranded DNA virus that is 152 kb in length and encodes 84 different genes.3 The genome was successfully mapped in the 1980s, and researchers began investigating HSV-1 as a possible vector for gene therapy.4 The double-stranded genome is composed of a long (UL) and short (US) genomic segment with accessory genes that can be deleted without interfering with viral replication.3 Due to the ability to easily manipulate the genome, HSV-1 has been genetically engineered to function as an oncolytic virus.
Multiple viral constructs are being investigated in preclinical studies but have yet been advanced to pediatric clinical trials. G207 is a genetically engineered HSV-1 strain F that lacks the γ134.5 gene and contains an inactivation insertion of lacZ within the UL39 gene that stops lytic activity in nondividing cells (summary of viruses discussed in the text that have been or are being advanced to clinical trial is given in Table 1). NV1020 is a similar construct but has only one of the γ134.5 loci deleted. The gene product of γ134.5 plays a role in inhibiting viral replication. To avoid viral infection, RNA-dependent protein kinase (PKR) is activated. PKR phosphorylates alpha subunit of translation initiation factor 2 (eIF-2α) and subsequently blocks translation of viral messenger RNA. ICP 34.5 counteracts this pathway by dephosphorylating eIF-2α, which prevents protein synthesis shutoff.5 Bharatan et al6 studied the sensitivity of multiple sarcoma cell lines to both G207 and NV1020. All cell lines tested demonstrated efficient viral entry and replication but were not all equally sensitive to virus killing. The rhabdomyosarcoma (RMS) and malignant fibrous histiocytoma cell lines were most sensitive followed by osteosarcoma cells. Least sensitive to both viruses were the Ewing sarcoma cells. Additionally, Parikh et al compared the efficacy of wild-type adenovirus Type 5 to the HSV-1 mutant, NV1066, on multiple neuroblastoma cell lines. The recombinant virus NV1066 lacks the γ134.5 gene but carries a transgene for enhanced green fluorescent protein, which is a visual marker of infectivity. NV1066 was able to successfully replicate and induce apoptosis in all cell lines tested. The oncolytic potency of NV1066 was much higher than adenovirus in neuroblastoma.7 Wang et al recently demonstrated that the viral construct HSV1716 was able to infect and had an antitumor response in pediatric neuroblastoma cell lines and that this response could not be predicted by the presence or absence of oncolytic engineered HSV (oHSV) entry receptors. Further, they demonstrated complete responses in six of ten animals bearing CHP-134 neuroblastoma tumors.8
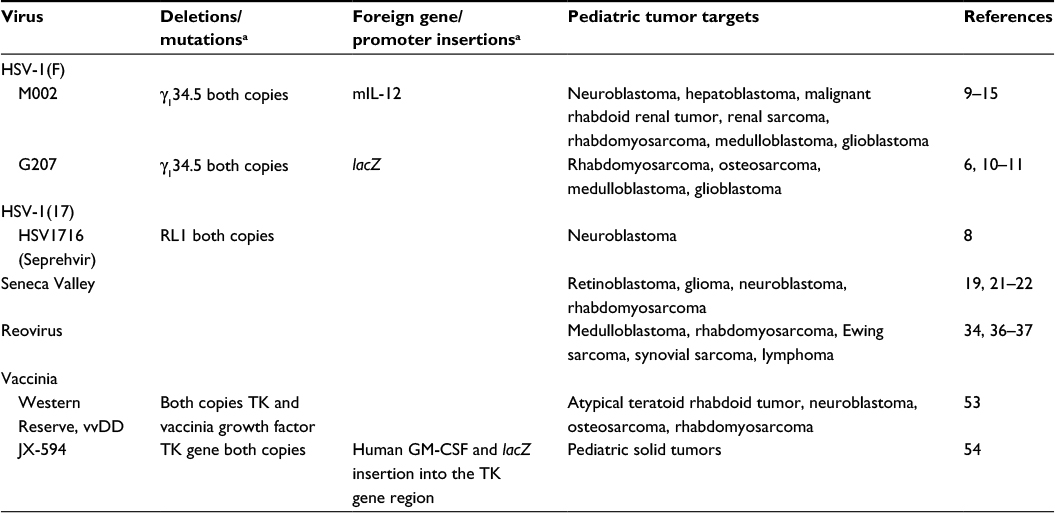 | Table 1 Oncolytic viruses that have had applications to pediatric tumors Note: aBlank spaces in the table are because they do not have deletions or foreign gene insertions. Abbreviations: HSV, herpes simplex virus; mIL-12, murine interleukin-12; vvDD, double-deleted vaccinia virus; TK, thymidine kinase; GM-CSF, granulocyte macrophage colony-stimulating factor. |
Pediatric brain tumor models have been shown to be sensitive to several oHSVs. Both G207 and M002 have been studied in these models. G207 was described earlier. M002 lacks the γ134.5 gene found within the inverted repeats of the UL segment of the parent recombinant virus HSV-1(F), rendering it unable to replicate in postmitotic cells, while the virus can replicate and rapidly lyse tumor cells. Additionally, M002 expresses both subunits of murine interleukin-12 (mIL-12) under the direct transcriptional control of the murine early-growth response-1 promoter.9 As the virus replicates, mIL-12 is produced and augments the oncolytic effect of the virus through the stimulation of cytotoxic T-cells and natural killer cells and the antiangiogenic properties of mIL-12. Friedman et al10 examined the effects of M002 and G207 on medulloblastoma (MB), including group 3 tumors that are highly resistant to conventional therapies and MB cancer stem cells with the surface proteins CD15 and CD133. All four human pediatric MB xenografts tested were highly sensitive to G207 and M002. Additionally, mice bearing these tumors intracranially lived significantly longer after treatment with the viruses.10 Friedman et al11,12 also demonstrated that the CD133+ cancer stem cells in the pediatric glioblastoma xenograft D456 were equally sensitive to virus when compared to CD133 tumor cells.
M002 has also been studied in pediatric extracranial solid tumors with encouraging results. Gillory et al demonstrated that M002 inhibited neuroblastoma growth in vitro and in vivo.13 Using two neuroblastoma cell lines, one with and another without MCYN amplification, they showed that M002 could successfully infect, replicate, and kill tumor cells. Immunocompromised mice had an increased lifespan when treated with low-dose radiation (3 Gy) in addition to M002. In vitro experiments with hepatoblastoma, malignant rhabdoid renal tumor, and renal sarcoma cell lines were likewise sensitive to treatment with M002. Viral titer assays demonstrated that the virus was able to enter and use the cell’s machinery to make progeny virus. In vivo studies demonstrated decreased tumor growth and prolonged survival in an immunocompromised xenograft mouse model.14 Furthermore, M002 was able to infect and diminish survival of both alveolar (SJCRH30) and embryonal (RD) RMS cell lines including the CD133+ cancer stem cells, and other investigators have shown encouraging results in vivo.15 The humanized version of the virus that produces human IL-12, M032, is in clinical trial in adults with recurrent glioblastoma (https://clinicaltrials.gov/ct2/show/NCT02062827). Future pediatric trials with this virus are planned.
Based on the promising preclinical data, several pediatric clinical trials in children using oHSVs are currently being conducted including two studies utilizing HSV1716 and one using G207 (summary of pediatric clinical trials based on virus type is given in Table 2). Cripe at Nationwide Children’s Hospital is currently enrolling children and young adults (7–30 years old) with relapsed solid, noncentral nervous system tumors in a Phase I dose escalation study with HSV1716 (https://clinicaltrials.gov/ct2/show/NCT00931931). This study is designed to determine the safety of intratumoral or intravenous delivery of the virus and to measure any antiviral immune response. HSV1716 is also being tested at Cincinnati Children’s Hospital in children 12–21 years old with refractory or recurrent high-grade glioma that can be removed by surgery (https://clinicaltrials.gov/ct2/show/NCT02031965). In this study, patients undergo maximal surgical resection of the tumor followed by injection of HSV1716 into the resection cavity. Friedman at the University of Alabama at Birmingham has a Phase I study open to enrollment evaluating G207 alone or combined with a single low dose of irradiation for children with recurrent or progressive supratentorial brain tumors (any malignant histological subtype) (www.clinicaltrials.gov/ct2/show/NCT02457845). The primary goal of this study is to determine safety, and the secondary aims are to obtain preliminary information on the effectiveness of and immune response to G207. In this study, patients will receive a 6-hour infusion of G207 through one to four catheters placed into the enhancing regions of the tumor. If this is proven safe, subsequent patients will receive a single 5 Gy dose of radiation to the tumor bed within 24 hours of G207 infusion. Preclinical studies indicate that low-dose radiation enhances HSV replication, and this approach was safely used in adult studies with radiographic evidence of responses.16,17 The increasing abundance of favorable preclinical trials using oHSV endorses the need for more clinical trials in this area. HSV virotherapy has the potential to target and treat rare and aggressive pediatric solid tumors.
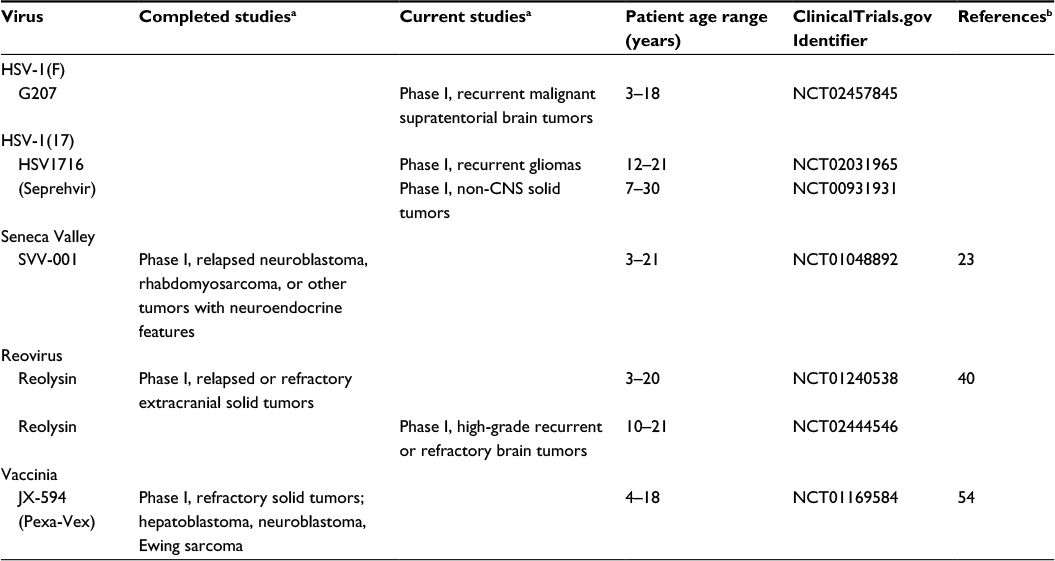 | Table 2 Clinical trials involving oncolytic viruses and pediatric tumors Notes: aBlank areas indicate there are no completed studies, no current studies, and no clinical trials. bBlank areas indicate these are ongoing phase I studies that are not completed and the data are not yet published. Abbreviations: HSV, herpes simplex virus; CNS, central nervous system; SVV, Seneca Valley virus. |
Seneca Valley virus
Nonhuman viruses have antitumor potential due to their lack of immunogenicity and capacity to cause disease in humans.18 Seneca Valley virus (SVV, SVV-001) is a small, replication-competent, single-stranded, nonenveloped RNA picornavirus, with a natural host in pigs and cows. SVV is the first oncolytic picornavirus to be tested in humans and can penetrate solid tumors through the vascular system unlike many other oncolytic viruses. Interestingly, SVV was first discovered incidentally in 2002 as a contaminant introduced in cell cultures via fetal bovine serum or porcine trypsin.19
Preclinical studies by Reddy et al19 demonstrated that SVV could be successfully delivered systemically to mice bearing retinoblastoma xenografts resulting in a remarkable decrease in tumor growth compared to control animals. This important study proved that SVV was effective via a systemic approach without deleterious effects on normal replicating cells. Viral replication assays proved that SVV was able to invade and replicate over time in tumor cells but not in normal cells such as hepatocytes. Additionally, viral levels were almost completely cleared from the blood, while high viral titers were found selectively in the tumors.
Additional preclinical studies demonstrated that SVV successfully passed the blood–brain barrier and infected and replicated in MB after a single intravenous injection, leading to further studies with SVV and pediatric gliomas.20 Liu et al21 examined the sensitivity of patient glioma tumor cells and a panel of orthotopic xenograft mouse models of pediatric malignant gliomas to SVV using Rag2/severe combined immunodeficient (SCID) mice. SVV was able to directly lyse tumor cells cultured from patient tumors and was able to infect glioblastoma cells grown as neurospheres. In the murine studies, SVV was able to eliminate 80% of xenograft tumors after 4 weeks of treatment. Animal survival was also significantly prolonged in three permissive cell types identified by in vitro studies, but no survival benefit was noted in the in vitro resistant cell lines.
Morton et al22 evaluated SVV against 23 pediatric cancer cell lines in the Pediatric Preclinical Testing Program using in vitro and in vivo models. They found that neuroblastoma and alveolar RMS were the most sensitive tumor types. These studies led to a Children’s Oncology Group study of SVV in children with solid tumors. Twenty-two children with either relapsed neuroblastoma, RMS, or other tumors with neuroendocrine features were enrolled to evaluate SVV without or with cyclophosphamide therapy in a Phase I clinical trial (https://clinicaltrial.gov/ct2/show/NCT01048892).23 SVV was successfully administered intravenously and well tolerated with only a single patient developing a dose-limiting toxicity (grade 3 pain at dose level 1). All patients cleared the virus from their blood and stool within 3 weeks of infusion. Despite the administration of low-dose cyclophosphamide, intravenous infusion of SVV did elicit the formation of neutralizing antibodies against the virus within 3 weeks.23 Further studies are needed to investigate techniques to reduce these neutralizing antibodies as well as anti-inflammatory cells such as T regulatory cells that deter a promising antitumor response. Despite promising preclinical data, no pediatric central nervous system tumor trials with SVV have been conducted to date.
Reovirus
Respiratory enteric orphan virus, or reovirus, is a double-stranded, nonenveloped RNA virus that is a member of the Reoviridiae family. The cell surface receptor for reovirus is sialic acid, which is universally expressed on mammalian cells, allowing the virus to bind to most cells; however, binding and internalization do not lead to a productive infection in normal human cells.24 Cellular activation of PKR and subsequent phosphorylation of eIF-2α lead to translational arrest and inhibition of viral protein production, resulting in an unproductive infection.25 Therefore, asymptomatic infection in humans is common with an estimated 60%–100% of adults containing anti-reovirus antibodies indicating a prior infection. When reovirus causes disease, it typically results in mild respiratory or gastrointestinal symptoms, thus making the wild-type virus attractive as an oncolytic agent.26 In children located in the US, an age-specific increase in reovirus-specific antibodies was seen with a seroprevalence of 8.2%, 32.8%, and 50% seen in children 1–2 years old, 4–5 years old, and 5–6 years old, respectively.27
Strong et al25 demonstrated that reovirus-resistant mouse fibroblast cells became susceptible when transformed with activated Ras, which prevented PKR activation and allowed for viral protein synthesis to ensue. Activated Ras signaling is common in many tumor types and has been shown to mediate tumorigenesis, thereby providing reovirus with tumor specificity.28 Coffey et al29 initially demonstrated the potential of reovirus (serotype 3, Dearing strain) as an oncolytic therapeutic agent in preclinical studies against human malignant glioma, and soon thereafter, other preclinical studies demonstrated that reovirus could target a variety of adult malignancies including lung, ovarian, colon, breast, lymphoid, and pancreatic cancers.30–35 These studies revealed that reovirus could be safely injected both intratumorally with evidence of systemic antitumor effects against distant disease and systemically to target local and metastatic disease in murine models. While intravenous reovirus effectively targeted metastatic cancer in immunocompetent mice, preexisting immunity to reovirus did limit responses. Immunosuppressive agents such as cyclosporine A or anti-CD-4/anti-CD8 antibodies could be combined with the virus to try to overcome this obstacle.31
Several preclinical studies demonstrated the effectiveness of reovirus at targeting pediatric malignancies. In MB, the most common pediatric malignant brain tumor, five of seven human cell lines, two mouse cell lines, and three primary cultures derived from surgical specimens were sensitive in vitro to reovirus infection.36 Susceptibility of the MB cells to infection was associated with levels of activated Ras. Furthermore, a single intrathecal injection of reovirus dramatically prolonged survival in an in vivo Daoy orthotopic animal model, and multiple injections reduced spinal and leptomeningeal metastases. The in vivo study was limited by the model chosen since Daoy is desmoplastic, a favorable, low-risk feature in MB. Similarly, reovirus inhibited proliferation and viability of several pediatric human sarcomas lines, including RMS, osteosarcoma, Ewing sarcoma, and synovial sarcoma.37 When injected in the tail vein every other day for three doses every 21 days, the virus inhibited xenograft tumor growth and prolonged survival in mice. The therapeutic benefit was enhanced in some of the sarcoma models when reovirus was combined with radiation and/or cisplatin. While many primary lymphoid neoplasms were sensitive to reovirus in preclinical studies, reovirus efficiently infected and lysed only two of five Burkitt lymphoma cell lines, suggesting that some tumor types may be less susceptible to killing by reovirus.34
Based on the promising preclinical pediatric data and the safety of reovirus alone and combined with chemotherapy in several adult human extracranial solid tumor clinical trials, the Children’s Oncology Group studied reovirus (Reolysin) in children with relapsed or refractory extracranial solid tumors in a Phase I study (https://clinicaltrial.gov/ct2/show/NCT01240538).38–40 A standard 3+3 design was used, and 24 patients were treated with intravenous reovirus for 5 consecutive days of a 28-day cycle alone or combined with oral cyclophosphamide (50 mg/m2/d × 21 days). Both approaches were safe, and reovirus was well tolerated up to 5×108 tissue culture inhibitor dose 50% per kg. A grade 5 respiratory failure and a grade 5 thromboembolic event were reported in the setting of disease progression, but no dose-limiting toxicities related to reovirus occurred. Correlative studies demonstrated an average time of reovirus viremia clearance of 6.5 days with no viremia seen in any patient after 17 days. Additionally, no virus was detected in the saliva or stool. While the Phase I study was not designed to demonstrate efficacy, no objective responses were seen. Three patients had stable disease and received a second cycle. Based on the lack of responses, the authors concluded that combination approaches with reovirus will be needed for efficacy.
At this time, there are no additional studies of reovirus in extracranial pediatric solid tumors, but there is an active Phase I study of reovirus in children 10 years and older with high-grade recurrent or refractory brain tumors. The study combines reovirus with sargramostim, recombinant granulocyte macrophage colony-stimulating factor, designed to enhance immune cell tumor killing induced by reovirus (https://clinicaltrial.gov/ct2/show/NCT02444546). Patients will receive sargramostim subcutaneously on days 1 and 2 and reovirus intravenously on days 3–5 of a 28-day cycle. Based on the safety profile of reovirus, opportunities exist for future studies combining reovirus with upfront therapy, other chemotherapeutic agents and/or radiation, and even other oncolytic viruses or new immune modulating agents.
Vaccinia virus
Vaccinia virus (VV), a double-stranded, nonintegrating, enveloped DNA virus in the Poxviridae family, was initially used as a vaccination to eradicate smallpox worldwide in 1979. More recently, the virus gained interest as an oncolytic agent because scientists identified nonessential genes that may be deleted to provide protection for normal cells while maintaining the oncolytic ability of the virus. Similar to HSV, these nonessential genes can be replaced with foreign DNA to augment tumor cell cytotoxicity. Several engineering approaches to attenuate VV have been used. Deletion of both copies of the thymidine kinase (TK) gene results in decreased pathogenicity for normal cells and preferential replication in tumor cells because thymidine triphosphate, which is required by a TK-deleted virus for DNA synthesis, is provided by replicating tumor cells but not normal cells.41,42 Similarly, deletion of vaccinia growth factor or hemagglutinin genes reduces VV virulence in normal cells and has been used in combination with TK deletions for additional protection of normal cells.42–45 Another mechanism to protect normal cells is through deletions in the B18R gene, the product of which neutralizes secreted type-I interferons.46 Without the gene product, the mutant virus is inactivated by interferon production in normal cells, but infection occurs in tumor cells that are often resistant to the antiviral effects of type-I interferons. Recombinant virus JX-795, which contains double deletions of B18R and TK and expresses β-interferon, demonstrated tumor-specific replication and efficacy following systemic delivery in preclinical cancer models.47 Viruses with protective gene deletions can also be delivered directly to tumors by pre-infecting cytokine-induced killer cells that are attracted to tumors and exert their cytotoxic effects following intravenous delivery.48
In addition to the β-interferon gene, several other foreign genes have been added to VV to enhance tumor cell killing. JX-594, which has been used both intratumorally and intravenously in multiple adult human solid tumor clinical trials, provides immunostimulation with increased intratumoral cytotoxic lymphocytes through the production of granulocyte macrophage colony-stimulating factor. Two viruses have been designed to target the tumor microenvironment through insertion of an anti-vascular endothelial growth factor (VEGF) foreign gene. VV GLV-1h68, a mutant virus with deletions in TK and hemagglutinin genes, was engineered to encode the anti-VEGF protein GLAF-1. The GLAF-1-producing virus demonstrated enhanced therapeutic efficacy in targeting adult human pancreatic, lung, and prostate cancer xenografts compared to the GLV-1h68 parent virus, and the treatment effect was comparable to that seen with the parent virus combined with bevacizumab.49 Similarly, vvDD, a Western Reserve VV with deletions in TK and VV growth factor, inhibited enhanced antitumor efficacy with an antiangiogenic effect in renal cell cancer models when armed with soluble VEGF receptor 1 protein.50 Other approaches for enhanced cell killing or molecular imaging have been used including the cytosine deaminase gene as suicide gene therapy with 5-fluorocytosine or the gene expressing the human somatostatin receptor type 2.51,52
Limited preclinical data exist on the efficacy of VV in pediatric tumor models. Lun et al53 demonstrated that the double-deleted VV, vvDD, effectively infected and killed nine of eleven pediatric tumor cell lines tested (three atypical teratoid rhabdoid tumors, four neuroblastomas, two osteosarcomas, and two RMSs). A neuroblastoma and an osteosarcoma cell line were more resistant to killing with ~40%–60% of the cells still alive after treatment with a high dose of the virus after 4 days. In xenograft models of intracranial atypical teratoid rhabdoid tumor, flank neuroblastoma, and metastatic osteosarcoma, intravenous vvDD decreased tumor size and prolonged survival in SCID mice supporting further evaluation of VV in pediatric brain and extracranial solid tumors.
To date, a single pediatric VV trial has been conducted (https://clinicaltrials.gov/ct2/show/NCT01169584).54 The Phase I study tested the safety of intratumoral JX-594 (Pexa-Vex) at either 106 or 107 plaque-forming units (PFU) per kg in six children (three hepatocellular carcinoma, two neuroblastoma, and a Ewing sarcoma). Five of the patients received a one-time injection in up to three tumor sites and one patient received two injections. The virus treatment was safe and tolerable; toxicities were mild (≤grade 3) with fever and sinus tachycardia being the most common symptoms. The three patients who received the 107 PFU/kg dose all developed grade 1 treatment-related skin pustules that resolved within 3–4 weeks. A strong cellular immunoreactivity to vaccinia antigens was detected in two patients who were tested. One of the six patients had radiographic evidence suggestive of antitumor activity. While a few adult cancer trials are examining VV in combination with small molecule inhibitors, there are no forthcoming pediatric trials at this time. Similar to reovirus, combination approaches with VV will likely be needed for efficacy.
Conclusion
Oncolytic recombinant viruses are promising treatments for pediatric malignancies. Several Phase I studies have been completed, and each virus was deemed safe and tolerable in the studies. Additional studies with other viruses are ongoing. Future studies of these viruses will likely include complementary combination approaches with standard chemotherapy, radiation, monoclonal antibodies or other small molecule inhibitors, or even other viruses. New viruses continue to be designed to increase tumor selectivity while enhancing virus-mediated tumor cell killing. These new viruses have likewise proven safe and efficacious in preclinical and animal studies and require testing in human pediatric clinical trials. While more clinical trials are needed to test new viruses and novel combination approaches, oncolytic virotherapy has the potential of becoming an important targeted modality of cancer treatment.
Acknowledgments
This work was supported in part by National Institutes of Health grants from the National Cancer Institute T32CQA091078 (AMW), Hyundai Hope on Wheels (EAB), and by St. Baldrick’s Foundation, Rally Foundation, The Truth 365, and Department of Defense W81XWH-15-1-CA140089 (GKF).
Disclosure
The authors report no conflicts of interest in this work.
References
Ward E, DeSantis C, Robbins A, Kohler B, Jemal A. Childhood and adolescent cancer statistics, 2014. CA Cancer J Clin. 2014;64(2):83–103. | ||
Smith MA, Seibel NL, Altekruse SF, et al. Outcomes for children and adolescents with cancer: challenges for the twenty-first century. J Clin Oncol. 2010;28(15):2625–2634. | ||
Glorioso JC. Herpes simplex viral vectors: late bloomers with big potential. Hum Gene Ther. 2014;25(2):83–91. | ||
McGeoch DJ. The genome of herpes simplex virus: structure, replication and evolution. J Cell Sci Suppl. 1987;7:67–94. | ||
Chou J, Chen JJ, Gross M, Roizman B. Association of a M(r) 90,000 phosphoprotein with protein kinase PKR in cells exhibiting enhanced phosphorylation of translation initiation factor eIF-2 alpha and premature shutoff of protein synthesis after infection with gamma 134.5- mutants of herpes simplex virus 1. Proc Natl Acad Sci U S A. 1995;92(23):10516–10520. | ||
Bharatan NS, Currier MA, Cripe TP. Differential susceptibility of pediatric sarcoma cells to oncolysis by conditionally replication-competent herpes simplex viruses. J Pediatr Hematol Oncol. 2002;24(6):447–453. | ||
Parikh NS, Currier MA, Mahller YY, et al. Oncolytic herpes simplex virus mutants are more efficacious than wild-type adenovirus Type 5 for the treatment of high-risk neuroblastomas in preclinical models. Pediatr Blood Cancer. 2005;44(5):469–478. | ||
Wang PY, Swain HM, Kunkler AL, et al. Neuroblastomas vary widely in their sensitivities to herpes simplex virotherapy unrelated to virus receptors and susceptibility. Gene Ther. 2016;23(2):135–143. | ||
Parker JN, Gillespie GY, Love CE, Randall S, Whitley RJ, Markert JM. Engineered herpes simplex virus expressing IL-12 in the treatment of experimental murine brain tumors. Proc Natl Acad Sci U S A. 2000;97(5):2208–2213. | ||
Friedman GK, Moore BP, Nan L, et al. Pediatric medulloblastoma xenografts including molecular subgroup 3 and CD133+ and CD15+ cells are sensitive to killing by oncolytic herpes simplex viruses. Neuro Oncol. 2016;18(2):227–235. | ||
Friedman GK, Gillespie GY. Cancer stem cells and pediatric solid tumors. Cancers (Basel). 2011;3(1):298–318. | ||
Friedman GK, Langford CP, Coleman JM, et al. Engineered herpes simplex viruses efficiently infect and kill CD133+human glioma xenograft cells that express CD111. J Neurooncol. 2009;95(2):199–209. | ||
Gillory LA, Megison ML, Stewart JE, et al. Preclinical evaluation of engineered oncolytic herpes simplex virus for the treatment of neuroblastoma. PLoS One. 2013;8(10):e77753. | ||
Megison ML, Gillory LA, Stewart JE, et al. Preclinical evaluation of engineered oncolytic herpes simplex virus for the treatment of pediatric solid tumors. PLoS One. 2014;9(1):e86843. | ||
Pressey JG, Haas MC, Pressey CS, et al. CD133 marks a myogenically primitive subpopulation in rhabdomyosarcoma cell lines that are relatively chemoresistant but sensitive to mutant HSV. Pediatr Blood Cancer. 2013;60(1):45–52. | ||
Bradley JD, Kataoka Y, Advani S, et al. Ionizing radiation improves survival in mice bearing intracranial high-grade gliomas injected with genetically modified herpes simplex virus. Clin Cancer Res. 1999;5(6):1517–1522. | ||
Markert JM, Razdan SN, Kuo HC, et al. A phase 1 trial of oncolytic HSV-1, G207, given in combination with radiation for recurrent GBM demonstrates safety and radiographic responses. Mol Ther. 2014;22(5):1048–1055. | ||
Koppers-Lalic D, Hoeben RC. Non-human viruses developed as therapeutic agent for use in humans. Rev Med Virol. 2011;21(4):227–239. | ||
Reddy PS, Burroughs KD, Hales LM, et al. Seneca Valley virus, a systemically deliverable oncolytic picornavirus, and the treatment of neuroendocrine cancers. J Natl Cancer Inst. 2007;99(21):1623–1633. | ||
Yu L, Baxter PA, Zhao X, et al. A single intravenous injection of oncolytic picornavirus SVV-001 eliminates medulloblastomas in primary tumor-based orthotopic xenograft mouse models. Neuro Oncol. 2011;13(1):14–27. | ||
Liu Z, Zhao X, Mao H, et al. Intravenous injection of oncolytic picornavirus SVV-001 prolongs animal survival in a panel of primary tumor-based orthotopic xenograft mouse models of pediatric glioma. Neuro Oncol. 2013;15(9):1173–1185. | ||
Morton CL, Houghton PJ, Kolb EA, et al. Initial testing of the replication competent Seneca Valley virus (NTX-010) by the pediatric preclinical testing program. Pediatr Blood Cancer. 2010;55(2):295–303. | ||
Burke MJ, Ahern C, Weigel BJ, et al. Phase I trial of Seneca Valley Virus (NTX-010) in children with relapsed/refractory solid tumors: a report of the Children’s Oncology Group. Pediatr Blood Cancer. 2015;62(5):743–750. | ||
Paul RW, Choi AH, Lee PW. The alpha-anomeric form of sialic acid is the minimal receptor determinant recognized by reovirus. Virology. 1989;172(1):382–385. | ||
Strong JE, Coffey MC, Tang D, Sabinin P, Lee PW. The molecular basis of viral oncolysis: usurpation of the Ras signaling pathway by reovirus. EMBO J. 1998;17(12):3351–3362. | ||
Selb B, Weber B. A study of human reovirus IgG and IgA antibodies by ELISA and western blot. J Virol Methods. 1994;47(1–2):15–25. | ||
Tai JH, Williams JV, Edwards KM, Wright PF, Crowe JE Jr, Dermody TS. Prevalence of reovirus-specific antibodies in young children in Nashville, Tennessee. J Infect Dis. 2005;191(8):1221–1224. | ||
Quinlan MP, Quatela SE, Philips MR, Settleman J. Activated Kras, but not Hras or Nras, may initiate tumors of endodermal origin via stem cell expansion. Mol Cell Biol. 2008;28(8):2659–2674. | ||
Coffey MC, Strong JE, Forsyth PA, Lee PW. Reovirus therapy of tumors with activated Ras pathway. Science. 1998;282(5392):1332–1334. | ||
Hirasawa K, Nishikawa SG, Norman KL, Alain T, Kossakowska A, Lee PW. Oncolytic reovirus against ovarian and colon cancer. Cancer Res. 2002;62(6):1696–1701. | ||
Hirasawa K, Nishikawa SG, Norman KL, et al. Systemic reovirus therapy of metastatic cancer in immune-competent mice. Cancer Res. 2003;63(2):348–353. | ||
Norman KL, Coffey MC, Hirasawa K, et al. Reovirus oncolysis of human breast cancer. Hum Gene Ther. 2002;13(5):641–652. | ||
Etoh T, Himeno Y, Matsumoto T, et al. Oncolytic viral therapy for human pancreatic cancer cells by reovirus. Clin Cancer Res. 2003;9(3):1218–1223. | ||
Alain T, Hirasawa K, Pon KJ, et al. Reovirus therapy of lymphoid malignancies. Blood. 2002;100(12):4146–4153. | ||
Wilcox ME, Yang W, Senger D, et al. Reovirus as an oncolytic agent against experimental human malignant gliomas. J Natl Cancer Inst. 2001;93(12):903–912. | ||
Yang WQ, Senger D, Muzik H, et al. Reovirus prolongs survival and reduces the frequency of spinal and leptomeningeal metastases from medulloblastoma. Cancer Res. 2003;63(12):3162–3172. | ||
Hingorani P, Zhang WD, Lin JA, Liu LB, Guha C, Kolb EA. Systemic administration of reovirus (Reolysin) inhibits growth of human sarcoma xenografts. Cancer. 2011;117(8):1764–1774. | ||
Comins C, Spicer J, Protheroe A, et al. REO-10: a phase I study of intravenous reovirus and docetaxel in patients with advanced cancer. Clin Cancer Res. 2010;16(22):5564–5572. | ||
Vidal L, Pandha HS, Yap TA, et al. A phase I study of intravenous oncolytic reovirus type 3 Dearing in patients with advanced cancer. Clin Cancer Res. 2008;14(21):7127–7137. | ||
Kolb EA, Sampson V, Stabley D, et al. A phase I trial and viral clearance study of reovirus (Reolysin) in children with relapsed or refractory extra-cranial solid tumors: a Children’s Oncology Group phase I consortium report. Pediatr Blood Cancer. 2014;62(5):751–758. | ||
Buller RM, Smith GL, Cremer K, Notkins AL, Moss B. Decreased virulence of recombinant vaccinia virus expression vectors is associated with a thymidine kinase-negative phenotype. Nature. 1985;317(6040):813–815. | ||
McCart JA, Ward JM, Lee J, et al. Systemic cancer therapy with a tumor-selective vaccinia virus mutant lacking thymidine kinase and vaccinia growth factor genes. Cancer Res. 2001;61(24):8751–8757. | ||
Zhang Q, Yu YA, Wang E, et al. Eradication of solid human breast tumors in nude mice with an intravenously injected light-emitting oncolytic vaccinia virus. Cancer Res. 2007;67(20):10038–10046. | ||
Buller RM, Chakrabarti S, Cooper JA, Twardzik DR, Moss B. Deletion of the vaccinia virus growth factor gene reduces virus virulence. J Virol. 1988;62(3):866–874. | ||
Shida H, Hinuma Y, Hatanaka M, et al. Effects and virulences of recombinant vaccinia viruses derived from attenuated strains that express the human T-cell leukemia virus type I envelope gene. J Virol. 1988;62(12):4474–4480. | ||
Symons JA, Alcami A, Smith GL. Vaccinia virus encodes a soluble type I interferon receptor of novel structure and broad species specificity. Cell. 1995;81(4):551–560. | ||
Kirn DH, Wang Y, Le Boeuf F, Bell J, Thorne SH. Targeting of interferon-beta to produce a specific, multi-mechanistic oncolytic vaccinia virus. PLoS Med. 2007;4(12):e353. | ||
Thorne SH, Negrin RS, Contag CH. Synergistic antitumor effects of immune cell-viral biotherapy. Science. 2006;311(5768):1780–1784. | ||
Frentzen A, Yu YA, Chen N, et al. Anti-VEGF single-chain antibody GLAF-1 encoded by oncolytic vaccinia virus significantly enhances antitumor therapy. Proc Natl Acad Sci U S A. 2009;106(31):12915–12920. | ||
Guse K, Sloniecka M, Diaconu I, et al. Antiangiogenic arming of an oncolytic vaccinia virus enhances antitumor efficacy in renal cell cancer models. J Virol. 2010;84(2):856–866. | ||
Gnant MF, Puhlmann M, Alexander HR Jr, Bartlett DL. Systemic administration of a recombinant vaccinia virus expressing the cytosine deaminase gene and subsequent treatment with 5-fluorocytosine leads to tumor-specific gene expression and prolongation of survival in mice. Cancer Res. 1999;59(14):3396–3403. | ||
McCart JA, Mehta N, Scollard D, et al. Oncolytic vaccinia virus expressing the human somatostatin receptor SSTR2: molecular imaging after systemic delivery using 111In-pentetreotide. Mol Ther. 2004;10(3):553–561. | ||
Lun X, Ruan Y, Jayanthan A, et al. Double-deleted vaccinia virus in virotherapy for refractory and metastatic pediatric solid tumors. Mol Oncol. 2013;7(5):944–954. | ||
Cripe TP, Ngo MC, Geller JI, et al. Phase 1 study of intratumoral Pexa-Vec (JX-594), an oncolytic and immunotherapeutic vaccinia virus, in pediatric cancer patients. Mol Ther. 2015;23(3):602–608. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.