Back to Archived Journals » Oncolytic Virotherapy » Volume 4
Oncolytic virotherapy for head and neck cancer: current research and future developments
Authors Malhotra A, Sendilnathan A, Old M, Wise-Draper T
Received 26 March 2015
Accepted for publication 12 May 2015
Published 20 July 2015 Volume 2015:4 Pages 83—93
DOI https://doi.org/10.2147/OV.S54503
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Faris Farassati
Akshiv Malhotra,1 Arun Sendilnathan,1 Matthew O Old,2 Trisha M Wise-Draper1
1Division of Hematology-Oncology, Department of Internal Medicine, University of Cincinnati, Cincinnati, 2Department of Otolaryngology-Head and Neck Surgery, Ohio State University, Columbus, OH, USA
Abstract: Head and neck cancer (HNC) is the sixth most common malignancy worldwide. Despite recent advancements in surgical, chemotherapy, and radiation treatments, HNC remains a highly morbid and fatal disease. Unlike many other cancers, local control rather than systemic control is important for HNC survival. Therefore, novel local therapy in addition to systemic therapy is urgently needed. Oncolytic virotherapy holds promise in this regard as viruses can be injected intratumorally as well as intravenously with excellent safety profiles. This review will discuss the recent advancements in oncolytic virotherapy, highlighting some of the most promising candidates and modifications to date.
Keywords: head and neck cancer, virotherapy, oncolytic viruses
Introduction
Head and neck cancer (HNC) remains a global health concern accounting for 650,000 new cases each year, resulting in 350,000 deaths worldwide. The identification of human papillomavirus (HPV) infection as a favorable prognostic factor has fueled clinical studies investigating deintensification strategies in this HPV-positive disease subset. However, the overall survival of HPV-negative tumors remains poor despite intensive therapy. Early stage localized tumors are often cured with a single modality, including surgical resection or radiation therapy; however, locally advanced HNC often requires multimodality treatment. Despite primary intensive treatment of locally advanced disease, relapse rates at 2 years remain at 30%–50%,1–3 resulting in 5-year survival of less than 10% in the relapsed and metastatic group.4 Even though the addition of targeted agents, such as the epidermal growth factor receptor (EGFR) inhibitor, cetuximab, has resulted in incremental rise in overall survival both in locally advanced and metastatic disease, the overall impact has been minimal.4,5 Novel therapeutic strategies are desperately required to further advance the success in HNC treatment. Several exciting areas of investigation are ongoing, including other molecular-targeted therapy, immunotherapy, gene therapy, as well as oncolytic virotherapy, as means of improving treatment outcomes and reducing morbidity in HNC treatment. Here, we review the background, feasibility, and potential role for oncolytic virotherapy in the treatment in HNC.
Oncolytic virotherapy
Oncolytic virotherapy is a promising approach as a cancer therapeutic, especially in its potential for local tumor control and low morbidity. Oncolytic viruses have the ability to selectively replicate in tumor cells via deletion of critical virulence genes that are required for replication in the normal host. Several oncolytic viruses have been tested both preclinically and in clinical trials (Tables 1 and 2), demonstrating safety and, in some instances, activity in human tumors. HNC allows for a unique opportunity for the application of these viruses as locoregional control is often most critical in this disease. Here, we will review the most common oncolytic viruses and their potential use as therapy in HNC.
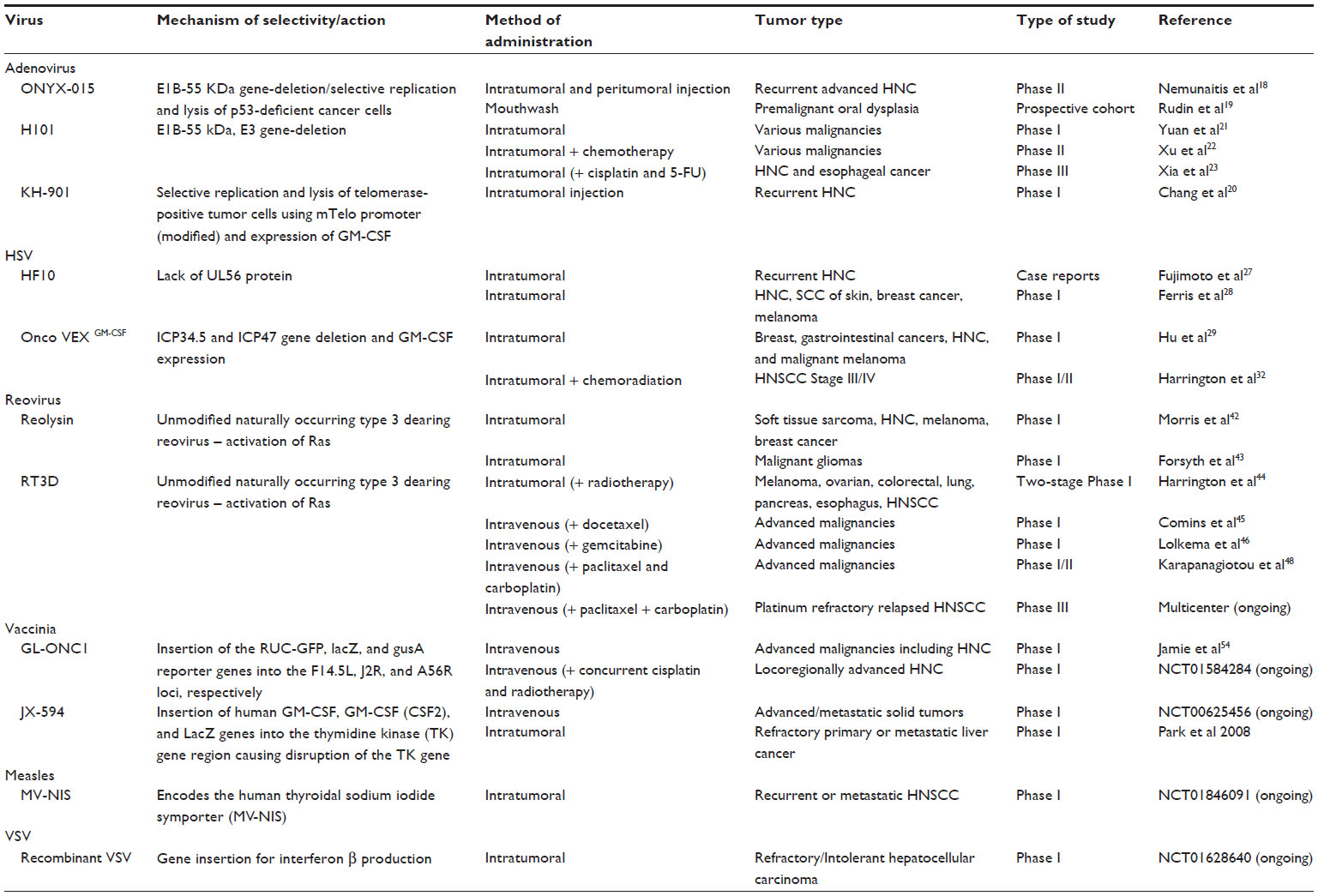 | Table 1 Summary of oncolytic viruses in clinical studies |
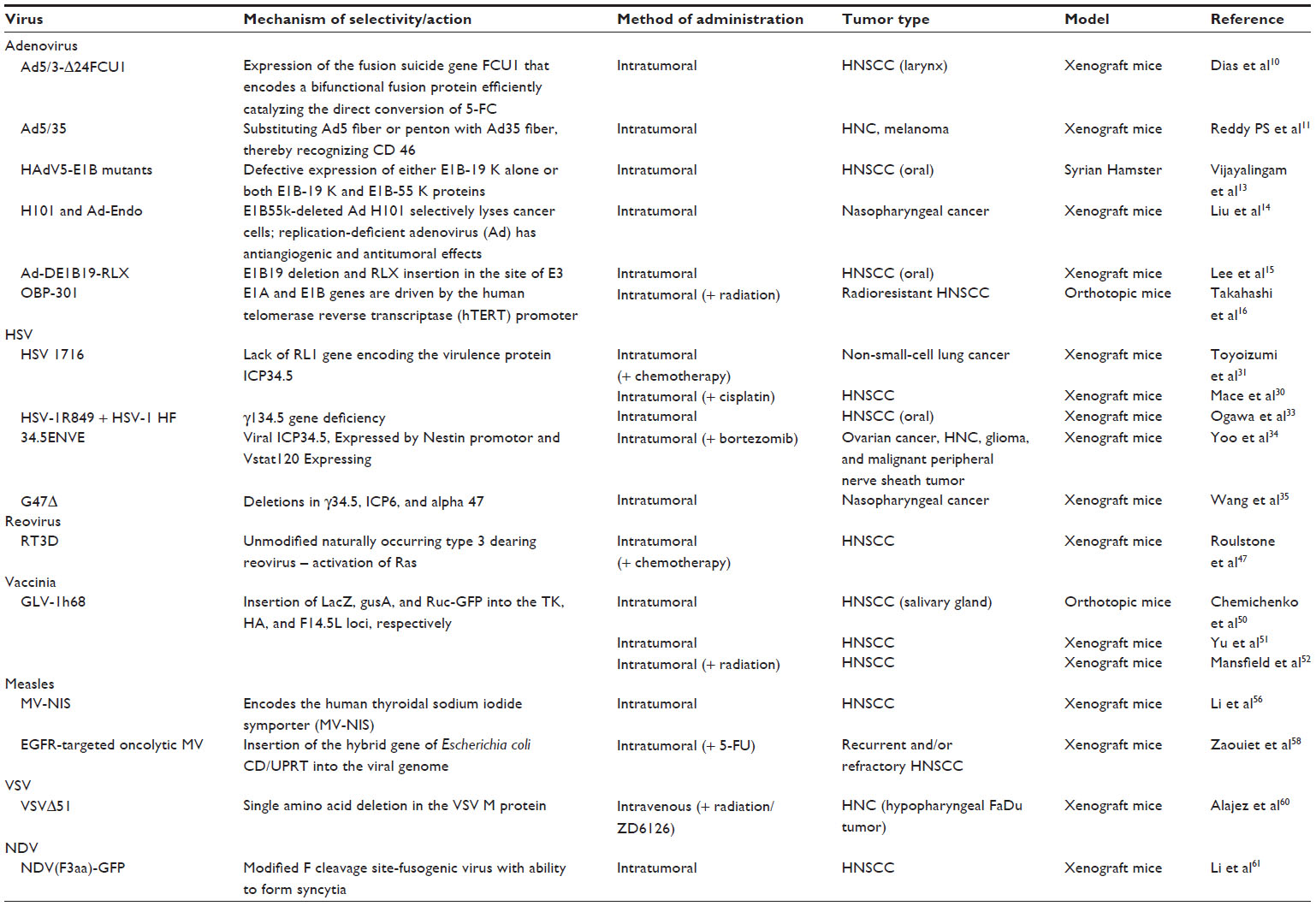 | Table 2 Summary of oncolytic viruses in preclinical studies |
Oncolytic adenovirus
One of the first engineered replication selective viruses to be tested in humans in clinical trials are the adenoviruses. The adenoviruses are naturally cytolytic, nonenveloped, double-stranded DNA viruses. Oncolytic adenoviruses contain modifications conferring tumor-selective replication. Modifications include deletion of the p300/CBP-binding or the pRB-binding region of E1A (resulting in selective killing of tumor cells with defects in the Rb pathway), or deletion/mutation of the E1B-55 K gene resulting in the loss of p53 degradation and late viral RNA export.6–8
Several oncolytic adenoviruses have been tested in preclinical models, including those with viral modifications to enhance tumor cell killing and delivery, as well as in combination with conventional treatments. Ad5/3-delta24, a capsid-modified oncolytic adenovirus, was synergistic with conventional chemotherapy, including cisplatin and 5-flurouracil (5-FU), as well as with radiation in a laryngeal squamous cell carcinoma model in vivo.9 Interestingly, the addition of cetuximab to chemotherapy and radiation resulted in complete prevention of tumor relapse in xenografts compared to cetuximab alone. In order to limit toxicity of 5-FU selectively to tumor cells, oncolytic adenoviruses were genetically engineered to express an FCU1 protein, which allows for conversion of 5-fluorocytosine to the active drug 5-fluorouridine.10 Despite decreased viral DNA replication in the tumors, there continued to be increased levels of apoptosis providing evidence for conversion to 5-FU selectively in the tumor.
Due to upregulation of CD46 receptor on many tumors, the Addl1520 (ONYX-015, Onyx Pharmaceuticals, California, USA) virus was genetically modified to create an Ad5/35 chimeric virus by substituting the Ad5 fiber or penton for the Ad35 fiber, which recognizes CD46, to enhance potency.11 This virus has shown good tumor selectivity and safety, but durable objective responses with this virus as a single agent have been limited, and this could be partly due to the loss of other essential functions of the deleted genes.12 However, the Ad5/35 chimeric oncolytic adenovirus in combination with cisplatin demonstrated delayed tumor progression compared to Ad5/35 alone or the combination of Ad5 and cisplatin.13 Similar results were also seen when Ad5/35 was combined with radiation, suggesting that combining this virus with current treatment may be an effective therapeutic strategy.
In an immunocompetent Syrian hamster animal model of HNC, Vijayalingam et al14 evaluated the effect of two apoptogenic human adenovirus (HAdV) vectors. They established two cell lines of hamster cheek pouch squamous cell carcinomas, induced by treatment with 9,10-dimethyl-1,2-benzanthracene. These cell lines, when infected with HAdV5 mutants lp11w and lp11w/D55 K (which are defective in the expression of either E1B-19 K alone or both E1B-19 K and E1B-55 K proteins), exhibited enhanced apoptotic and cytotoxic responses. Intratumoral administration of HAdV5-E1B mutants efficiently suppressed the growth of tumors, suggesting the potential of these viruses in HNC treatment. These tumors also exhibited increased caspase-3 levels, suggesting that viral-induced apoptosis may contribute to tumor growth suppression.
In a nasopharyngeal carcinoma (NPC) model, the replication-deficient adenovirus encoding human endostatin (Ad-Endo) demonstrated to have promising antiangiogenic and antitumoral effects. In addition, the combination of H101 (an E1B-deleted adenovirus) and Ad-Endo significantly inhibited CNE-2 xenografts growth through increased endostatin expression and Ad replication.15 Similar results were obtained using the E1B-55 K–attenuated adenovirus, dl1520 in neural progenitor cells (NPCs) and xenografts in nude mice. Importantly, the antitumor activity of dl1520 was augmented by the addition of cisplatin both in vitro and in vivo, showing that dl1520 and cisplatin have a synergistic anti-NPC effect through oncolysis and the induction of apoptosis.
Lee et al16 described a genetically modified adenovirus (Ad-DE1B19-RLX), in which the E1B19 region was deleted and relaxin (RLX) was inserted in the site of the E3 region to express RLX, affecting collagen metabolism and improving intratumoral spread of the virus. In animal experiments, Ad-DE1B19-RLX significantly reduced the size of oral squamous cell carcinoma (OSCC) tumors. There was no significant difference between radiosensitive and radioresistant OSCC.16 Additionally, the OBP-301, a telomerase-specific replication-selective adenovirus, was also able to overcome radioresistance of HNC in an orthotopic nude mouse model.17 Infection with OBP-301 was found to enhance the antitumor efficacy of radiation, both in vitro and in vivo, by inhibiting MRN complex expression and increasing apoptosis induction. Additionally, OBP-301 either alone or in combination with paclitaxel or cisplatin has been found to exert significant antitumor effects on head and neck squamous cell carcinoma (HNSCC) cells both in vitro and in vivo.18
Though many adenoviruses have been studied in animal models, only a few of them have entered clinical trials in humans, namely ONYX 015, H101, and KH 901. ONYX-015 is an E1B-55 kDa gene deleted adenovirus engineered to replicate in and lyse p53-deficient cancer cells. However, it should be noted that ONYX-15 was later shown to lyse tumor cells independent of p53 status, and therefore, the exact mechanism in which ONYX-15 exerts its selectivity remains to be elucidated. A Phase II clinical study including peritumoral and intratumoral ONYX-015 injection in 24 patients with recurrent HNC showed highly selective tissue destruction with significant tumor regression (>50%) in 21% of evaluable patients, with no toxicity to injected normal peritumoral tissues. Interestingly, p53 mutant tumors underwent significantly more necrosis compared to p53 wild-type tumors. High-neutralizing antibody titers did not prevent infection or replication within tumors.19 ONYX-015 was also found to be effective as a mouthwash in premalignant oral dysplasia, with specific cytotoxicity to cells carrying defects in p53 signaling pathways. Histologic resolution of dysplasia was observed in 7 (37%) of 19 patients, and the grade of dysplasia improved in one additional patient. Though the responses were transient, it was concluded to be a tolerable and feasible treatment modality with a favorable toxicity profile.20
KH901 is a conditionally replicating oncolytic adenovirus, which selectively replicates in and lyses telomerase-positive tumor cells and expresses granulocyte-macrophage colony-stimulating factor (GM-CSF). A Phase I trial involving 23 patients demonstrated that the intratumoral administration of KH901 in recurrent HNC patients was feasible, well tolerated, and associated with biological activity.21
H101, an E1B-deleted adenovirus, which is very similar to ONYX-015 but lacks all E3 proteins, was found to be safe when injected intratumorally in 15 patients in a dose escalation manner over 5 consecutive days. There was no observed dose-limiting toxicity or serious adverse events.22 Additionally, various Phase II and Phase III clinical trials, involving a combination of H101 and standard chemotherapy, demonstrated improved overall response rates in HNC.23,24 These studies eventually led to China’s State Food and Drug Administration approving Shanghai Sunway Biotech’s genetically modified adenovirus-H101 in November 2005, the first of its kind in oncolytic virotherapy for the treatment of HNC.25
Oncolytic herpes simplex viruses
Herpes simplex virus (HSV) is a double-stranded DNA virus surrounded by a capsid and enveloped in a lipid bilayer. The HSV consists of both a long unique region (UL) containing 56 genes and a short unique region (US) containing 12 genes. Entry is mediated through receptor-mediated endocytosis involving fusion of the envelope membrane and host membrane, allowing viral entry through pores.26 Once entry is achieved, HSV hijacks the host machinery for viral replication. The host antiviral defense machinery would normally inhibit production of virions through phosphorylation of protein kinase R (PKR) and downstream phosphorylation of the translation initiation factor, eIF2α, effectively shutting off protein translation. However, viral protein, ICP 34.5, antagonizes PKR by directly dephosphorylating eIF2α. ICP 34.5 is also important for downregulating the immune response by downmodulation of MHC (major histocompatibility complex) II expression. Deletion of ICP 34.5 results in the inability of HSV to replicate in normal cells. However, in cancer cells that have upregulated Ras, which inhibits the phosphorylation of PKR,27 this pathway is already bypassed, allowing translation of viral proteins and production of virions, leading to lysis of the cancer cell. HSV is also able to evade the immune system through expression of ICP 47 which inhibits MHC class 1 from binding antigen to TAP (transporter associated with antigen processing). This prevents a cytotoxic T-cell response and clearance of the virus. Oncolytic herpes simplex viruses (oHSVs) are found to be highly oncolytic to tumor cells and are either naturally occurring mutants or genetically engineered by deleting virulence-dependent proteins, such as those mentioned earlier, allowing for selective replication in tumor cells while providing safety to normal bystander cells. The first oHSV, which was engineered with a deletion in thymidine kinase, was shown to be active in cancer in 1991 in a high-grade glioma mouse model.28 Since then, oHSV has been investigated in many solid tumors including HNC.
HF10 is a spontaneously occurring mutant form of the HSV-1 virus lacking the UL56 protein, which is thought to be important for cytoplasmic transport and release of virions. An early proof of principle study showed that intratumoral injections in two patients with metastatic subcutaneous nodules were safe and demonstrated efficient spread and cell death in the tumor.29 At the 2014 American Society of Clinical Oncology (ASCO) meeting, Ferris et al30 demonstrated that multiple intratumoral injections of HF-10 in refractory and superficial cancers (HNC, squamous cell carcinoma of skin, breast cancer, and melanoma) were safe in an ongoing Phase I study, with most common adverse events of flu-like symptoms and pain at injection site. Follow-up of this study will be interesting to determine if these patients experience clinical responses.
oHSVs have been generated to express transgenes including Onco-VEXGM-CSF (JS1/34.5-/47-/GM-CSF), which in addition to deletion of ICP 34.5 and ICP 47 is an oHSV expressing GM-CSF. A Phase I study of Onco-VEXGM-CSF showed safety in solid tumors as well as local tumor activity and suggested activity in noninjected tumors as well.31 Local reaction at the injection site of the latter study was dose limiting.
However, it is unlikely that intratumoral injection of oHSV alone will be sufficient for total eradication of tumor. Preclinical studies have shown possible benefit of combining oHSV with chemotherapy. Mace et al32 showed that the addition of cisplatin in vitro to HSV1716 was not cytotoxic to the virus and that efficacy was in fact additive with the combination. In non-small-cell cancer cell lines, the addition of chemotherapy to HSV1716 was synergistic.33 Due to preclinical efficacy, clinical studies with oHSV in combination with chemotherapy and radiation were initiated. Impressive responses were seen when Onco-VEXGM-CSF was added to standard cisplatin and radiation in locally advanced HNC.34 Patients with stage III/IVA or B were enrolled and received escalating doses of virus (4 injections total) in 3 cohorts, followed by an expansion phase with a total of 17 patients treated. All patients received concurrent cisplatin 100 mg/m2 every 3 weeks and 70 Gy of radiation over 35 fractions and they also underwent neck dissection at the conclusion of treatment to assess response. The pathologic complete response rate was very high at 93% in injected tumors. Just less than half of the patients were found to be positive for HPV. At median follow-up of 29 months, 100% of patients remained free of any locoregional disease. Disease-specific survival was 82.4%, relapse-free rate was 76.5%, and overall survival was 70.5%. However, it is important to note that these high rates of survival may be skewed due to neck dissection required in all patients, which is not the usual treatment in patients who had received a radiologic complete response and had not relapsed. Despite clinical efficacy seen in Phase I/II, the Phase III study, in which Onco-VEXGM-CSF was being tested in combination with definitive cisplatin and radiation vs chemotherapy and radiation alone (clinicaltrials.gov NCT01161498), was terminated early likely due to new prognostic implications of HPV status and changing landscape of HNC treatments. Amgen (Thousand Oaks, CA, USA), who now own the virus under the new name T-VEC, are working on new strategies for the use of the virus as treatment for HNC.
Other strategies to enhance oHSV oncolysis antitumor efficacy and safety have also been studied. Dual infection with HSV-1 mutants, γ134.5 gene-deficient HSV-1 R849, and HSV-1 HF (which induces cell fusion) in a human oral SCC cell xenograft model led to enhancement of the oncolytic ability of the γ134.5 gene-deficient HSV-1 R849, suggesting that fusogenic viruses may be beneficial in HNC.35 Combining bortezomib treatment with oHSV (34.5ENVE) in vivo significantly increased viral replication, at least partially, through Hsp90 as well as enhanced antitumor efficacy.36 G47Δ, a third-generation oHSV with deletions in γ34.5, ICP6, and α47, proved to be efficacious in a nasopharyngeal cancer model in vivo.37 Deletion in the latter virus makes the virus more sensitive to acyclovir and ganciclovir as an extra safety measure.
Another strategy that is being explored is copper chelation. Physiologic levels of copper are known to inhibit HSV and have been found to abolish replication and effectiveness of oHSV in preclinical models in HNC, glioma, ovarian cancer, and various other models. Plans are being made to move this strategy into Phase I trials.38
Reovirus
Reovirus (respiratory enteric orphan virus) is a member of the Reoviridae family. Although it is not associated with a named human disease, it can infrequently cause flu-like upper respiratory tract symptoms or a mild diarrheal illness. Exposure is very common with up to 100% of healthy human adults showing seropositivity.39 There are three serotypes of reovirus, which are designated type 1 (Lang), type 2 (Jones), and type 3 (Dearing). The type 3 serotype has been extensively tested as a proprietary formulation developed by Oncolytics Biotech (Calgary, Canada). Reovirus virions consist of a nonenveloped, icosahedral capsid with a double shell of proteins. It binds to JAM-1 or sialic acid receptors in the infected cell, and subsequent endocytosis initiates cleavage of the outer shell proteins to release the core into the cytoplasm. Viral RNA is then copied within the core, and mRNA is released and translated into viral proteins using the host cell machinery. Subsequently, viral particles assemble and are released for reinfection by cell lysis.
Reovirus first sparked interest as a cancer-killing therapeutic agent in the 1970s, when WT reovirus was shown to replicate in transformed, but not normal, cells.40 Reovirus-resistant NIH3T3 cells became sensitive upon transformation with activated Sos or Ras, suggesting that reovirus may exploit elements of the Ras pathway to enhance infection.41 Cells overexpressing the EGFR, upstream of Ras, were also found to be sensitive to reovirus. The Ras pathway is frequently activated in cancers, either through mutation of the Ras gene or overexpression/mutational activation of the upstream EGFR or other receptor tyrosine kinases.42 In HNC, up to 90% of tumors overexpress EGFR.43 The mechanism by which reovirus is selective for Ras-mutated/activated cancer cells has been shown to involve PKR similar to oHSV.
The first Phase I clinical trial involving reovirus therapy studied the safety of an intratumoral injection of reovirus to accessible subcutaneous tumors in patients who had exhausted all other standard cancer treatments. Doses up to 1×1010 tissue culture infectious dose (TCID50) were administered without encountering dose-limiting toxicity and, as a secondary endpoint of the study, 61% of patients responded to the treatment (REO001). These data demonstrated that single-agent intratumoral treatment with reovirus is well tolerated with predictable side effects of headache and flu-like symptoms.44,45 Subsequently, a Phase I trial of intratumoral reovirus combined with radiotherapy was conducted. A total of 23 patients received treatment, and three had HNC. The combination treatment was well tolerated, and 14 patients had either partial response or disease stabilization as their best treatment response.46 Phase I studies of intravenous reovirus in patients with advanced solid tumors reported similar side effects, an increase of antireoviral antibodies with treatment, and few positive viral shedding results. A Phase I study of intravenous reovirus and docetaxel in 25 patients with various advanced cancers showed that treatment with the combination was well tolerated and a disease-control rate (combined complete response, partial response, and stable disease) of 88% was reported.47 A Phase I study of reovirus in combination with gemcitabine showed that of ten patients who were evaluable for response, one had a partial response, one a minor response, and six patients had stable disease.48
Triple therapy with reovirus, cisplatin, and paclitaxel demonstrated synergistic cytotoxicity in vitro in both malignant and nonmalignant cell lines. In HNC cell lines, this was associated with enhanced caspase-3 and -7 cleavage, but no increase in viral replication. Triple therapy was significantly more effective than reovirus or cisplatin–paclitaxel in athymic nude mice. These data suggest that the combination of reovirus plus platinum–taxane doublet chemotherapy has significant activity in HNC.49 In a recently completed Phase I/II study, a platinum–taxane doublet regimen in combination with reovirus was tested. Treatment was well tolerated, and a dose was recommended for subsequent Phase II evaluation (full dose chemotherapy plus 3×1010 TCID50 on days 1–5). In the Phase I part of the study, treatment responses were particularly impressive in patients with incurable or relapsed/metastatic HNC. Of 19 heavily pretreated patients, there was 1 complete response, 6 partial responses, and 2 clinically meaningful responses (resolution of cutaneous disease). The median overall survival was an impressive 8.9 months.50 These data have led to a randomized, multicenter Phase III study of paclitaxel and carboplatin with or without intravenous reovirus in patients with platinum-refractory relapsed HNSCC.
Poxviruses with a focus on vaccinia virus
The poxviruses are a group of viruses that can infect both vertebrates and invertebrates. Several poxviruses have been tested in HNC cell lines including myxoma, vaccinia, raccoonpox, and tanapox viruses. Preclinically, using cell-based assays, vaccinia virus was shown to have the greatest oncolytic activity.51 Utilizing the vaccinia virus as an oncolytic viral therapy is attractive due to its extensive safety profile, its use for eradication of small pox, its ability to infect a wide variety of hosts, and its potent oncolytic ability selective to cancer cells. Vaccinia virus has been modified for safety including GLV-1h68, which is an attenuated, replication-competent vaccinia virus. This virus has been shown to be oncolytic to salivary gland and HNC cell lines and resulted in a rapid tumor regression in mouse xenograft and orthotopic models.52,53 In a human xenograft model in nude mice, radiation in combination with vaccinia virus resulted in higher rates of tumor regression,54 suggesting that further exploration of this combination may be efficacious clinically. Vaccinia viruses have also been engineered to contain transgenes, including the vaccinia virus containing the endostatin–angiostatin fusion gene (VVhEA), which was shown to be superior to ONYX-015 and the control vaccinia virus in vivo.55
Clinically, JX-594 or Pexa-Vec (Sillajen, Busan, South Korea), which is a thymidine-kinase-deleted vaccinia virus plus GM-CSF, has been tested in solid tumors including HNC in a Phase I study (clinicaltrials.gov, NCT00625456). Results from this study have yet to be published, but at least in hepatocellular carcinoma, the decision has been made to move forward to Phase II based on responses in this subset. Interestingly, the vaccinia virus, GL-ONC1, which is a genetically modified virus for improved safety (deletion of thymidine kinase) and that contains an imaging transgene (genelux, San Diego, CA, USA) is currently being tested intravenously in combination with cisplatin and radiation in HNC based on preclinical evidence to evaluate the safety of this combination (clinicaltrials.gov, NCT01584284). Prior Phase I data as a single agent were presented at ASCO 2012 and showed safety data and specific tumor delivery in multiple tumor types including HNC.56
Paramyxoviridae
The measles virus is a single-stranded, negative-sense, enveloped RNA virus in which humans are the natural host. MV-NIS, which is a genetically modified form of the oncolytic MV-Edm virus and contains the sodium iodide symporter (NIS), has been tested preclinically both in anaplastic thyroid cancer cells and in HNC cell lines.57–59 MV-NIS was shown to have oncolytic efficacy in both anaplastic thyroid cancer cell lines and xenografts, and efficacy could be boosted with cyclophosphamide, radioiodine (131I), as well as a Chk-1 inhibitor (SAR-020106) in HNC cell lines in vitro. These preclinical data have led to a Phase I clinical study, investigating intratumoral MV-NIS in HNC (clinicaltrials.gov NCT01846091).
Another genetically modified measles virus that was targeted to EGFR, which as mentioned earlier is highly expressed in HNC, and that contains a bifunctional enzyme cytosine deaminase/uracil phosphoribosyltransferase (CD/UPRT) has also been tested in HNC in vitro and in vivo.60 CD/UPRT converts 5-fluorocytosine into the chemotherapeutic 5-FU, which is often used in HNC. Interestingly, intratumoral injection of this virus resulted in cytotoxic cell death of the tumor, but also a bystander effect on noninfected tumor cells likely through 5-FU toxicity. Per authors, a Phase I clinical trial is planned.
The Newcastle disease virus (NDV) is a paramyxovirus and has been genetically modified to enhance tumor selective killing. The naturally occurring NDV is a single-stranded RNA virus, which was shown to lyse tumor cells as early as 1964 and is advantageous in that the natural virus is pathogenic in birds but only causes mild flu-like symptoms in humans. NDV(F3aa)-GFP is a genetically modified NDV that is fusogenic allowing syncytia formation and has been tested in the treatment of HNC both in vitro and in vivo.61 NDV(F3aa)-GFP was found to be highly tumor cytotoxic in the intratumoral model without associated toxicity.
Vesicular stomatitis virus
Vesicular stomatitis virus (VSV) has been shown to have some oncolytic activity. Unfortunately, many tumors are resistant to VSV, prohibiting its use. Interestingly, one study in HNC cell lines showed that Janus kinase (JAK) 1/2 inhibitors reversed the resistance, resulting in enhanced viral activity.62 This may be one strategy in boosting VSV efficacy and transition to clinical studies. Another mutant virus, VSVΔ51 (single mutation in M protein), was tested in the human FaDu hypopharyngeal SCC model either as a single agent, combined with radiation therapy, or with the vascular disrupting agent ZD6126 which all demonstrated enhanced efficacy of the virus.63 There currently is one Phase I clinical trial in liver cancer in which the VSV is genetically engineered to express interferon β, which alone has efficacy in hepatocellular carcinoma (NCT01628640).
Other oncolytic viruses continue to emerge in the treatment of HNC, and further modifications of these viruses hold promise for enhanced efficacy. Although there have been failures and some of the Phase I studies have been withdrawn, there continues to be promising new developments.
Oncolytic virotherapy drawbacks and hurdles
Several obstacles continue to make generating effective oncolytic viruses difficult. Although deletion of certain genes are necessary for tumor selectivity, some of these mutations result in decreased antitumor efficacy either due to inefficient entry, replication, or lysis of tumor cells, some examples of which were already described. It remains difficult to both genetically modify the viruses for safety while continuing to enhance efficacy. Other hurdles include maintaining efficient viral spread and avoidance of the host immune system.
Virus delivery and spread
The tumor microenvironment is critical in promoting malignant cell growth and progression, as well as restricting virus spread especially in areas of fibrosis and necrosis. In addition, viruses are larger than other anticancer agents such as chemicals and antibodies. The matrix metalloproteinases (MMPs) are proteolytic enzymes that degrade the extracellular matrix and are essential for tumor spread and neovascularization. Oncolytic viruses with genes that encode MMP inhibitors have shown good results in delaying tumor growth and angiogenesis,64 providing one mechanism to overcome the microenvironment hurdle. In addition, coadministration of the enzyme hyaluronidase with oncolytic Ads during intratumoral injection was studied by Ganesh et al.65 Hyaluronidase degraded the major constituent of the extracellular matrix, hyaluronan, resulting in enhanced virus spread in vivo. Also, induction of cancer cell death with an apoptosis-inducing agent prior to injection of oncolytic HSV could result in effective virus spread.66 Okunaga et al67 showed that ultrasound with microbubbles promoted the entry of HSV-1 into oral SCC cells and increased the number and size of the RH2 antigen-positive foci within treated tumors. This suppressed the growth of nude mouse tumors. This method may be useful for enhancing the antitumor ability of oncolytic HSV-1 for HNC.67
Elevated interstitial hydrostatic pressure as a result of fibrosis and vessel abnormalities poses another physical barrier to successful virus delivery.68 The viruses could escape by drainage into the circulation or back through the injection site, resulting in reduced efficacy and increased systemic toxicities. Price et al69 recently demonstrated that a silk-elastin-like protein polymer (SELP) gel matrix significantly potentiates the efficacy of intraoperative oncolytic vaccinia therapy following incomplete tumor resection. The SELP gel matrix presents many advantages, including prolonged physical contact of virus against the tumor surface, effective viral infection of the tumor surface, protection of viral integrity within the SELP gel, and slow release of virus as the SELP gradually biodegrades over time.69
A virus that has successfully entered the cell needs to replicate for efficient cell lysis and virus spread. The protein p21CIP1/WAF normally inhibits cyclin-dependent kinase 2 and blocks the progression of the cell cycle from G1 to S phase. siRNA knockdown of p21CIP1/WAF was shown to increase Ad replication and oncolysis by Shiina et al.70 Jung et al71 evaluated antitumor efficacy by sustained delivery of oncolytic Ad containing shRNA, using SELP hydrogels for treatment of HNC. The combination of oncolytic Ad with shRNA targeted to C-Met expression resulted in antitumor efficacy. SELP hydrogels maximize the potential of Ad-mediated cancer gene therapy by spatiotemporal control and preservation of biological activity in the tumor tissue. The matrix-mediated delivery of oncolytic Ad containing shRNA has potential for therapy of HNC with maximized efficacy and reduced toxicity.71
Yoo et al38 demonstrated that antiangiogenic strategies increased viral replication and promoted systemic administration of oHSV. ATN-224 is a second-generation analog of tetrathiomolybdate which is used to treat Wilson’s disease. ATN-224 and tetrathiomolybdate are copper chelators that also inhibit superoxide dismutase leading to potent antiangiogenic and antitumoral effects in addition to increasing oHSV efficacy.
Host immune response against the virus
Intravenous virus delivery could be affected by neutralizing antibodies, complement activation, poor virus escape from the vascular compartment, as well as nonspecific uptake by other tissues such as the liver and spleen. For Ad, adhesion to blood cells could also lead to therapeutic hindrance.72 To modify the immune response in favor of virus replication and tumor lysis, one method is to use an immunosuppressive agent, such as cyclophosphamide, that has been shown to improve virus spread and antitumoral efficacy.73 Antibodies prevent possible toxicity,74 but they also reduce oncolytic efficacy. Preexisting antibodies decrease virus spread after intravenous delivery,75 but have a lesser effect on intratumoral injection.76 To circumvent these antibodies, plasmapheresis can be used to deplete the antibodies prior to virus injection. Another way is to use viral strains with a lower prevalence of antibodies in the human population. One example is Ad11, with a reported antibody prevalence of 10%–31% compared to 45%–90% for Ad5.77
Interestingly, despite the drawback of the activation of the immune response resulting in clearance of the virus, in some situations it is advantageous, in that an immune response directed to the virus may also result in tumor cell killing. This has led to genetic modifications of the oncolytic viruses as described earlier to produce immune growth factors. It continues to be a challenge to maintain a balance of viral potency and propagation while taking full advantage of the host immune response directed to tumor.
Conclusion
Oncolytic viruses are promising as future HNC therapy, both alone and in combination with standard treatments. With the discovery of immunotherapy, and the activation of the immune response by viral oncotherapy, it will be interesting to determine the impact of these therapeutic combinations. The results of many ongoing clinical studies are anxiously anticipated to understand the potential use of these treatments in the future.
Disclosure
Dr Wise-Draper is supported by the Clinical Scientist Training Program at the University of Cincinnati. The authors report no other conflicts of interest in this work.
References
Bourhis J, Sire C, Graff P, et al. Concomitant chemoradiotherapy versus acceleration of radiotherapy with or without concomitant chemotherapy in locally advanced head and neck carcinoma (GORTEC 99-02): an open-label phase 3 randomised trial. Lancet Oncol. 2012;13(2):145–153. | |
Bernier J, Domenge C, Ozsahin M, et al. Postoperative irradiation with or without concomitant chemotherapy for locally advanced head and neck cancer. N Engl J Med. 2004;350(19):1945–1952. | |
Cooper JS, Pajak TF, Forastiere AA, et al. Postoperative concurrent radiotherapy and chemotherapy for high-risk squamous-cell carcinoma of the head and neck. N Engl J Med. 2004;350(19):1937–1944. | |
Burtness B, Goldwasser MA, Flood W, Mattar B, Forastiere AA; Eastern Cooperative Oncology Group. Phase III randomized trial of cisplatin plus placebo compared with cisplatin plus cetuximab in metastatic/recurrent head and neck cancer: an eastern cooperative oncology group study. J Clin Oncol. 2005;23(34):8646–8654. | |
Bonner JA, Harari PM, Giralt J, et al. Radiotherapy plus cetuximab for locoregionally advanced head and neck cancer: 5-year survival data from a phase 3 randomised trial, and relation between cetuximab-induced rash and survival. Lancet Oncol. 2010;11(1):21–28. | |
Bischoff JR, Kirn DH, Williams A, et al. An adenovirus mutant that replicates selectively in p53-deficient human tumor cells. Science. 1996;274(5286):373–376. | |
O’Shea CC, Johnson L, Bagus B, et al. Late viral RNA export, rather than p53 inactivation, determines ONYX-015 tumor selectivity. Cancer Cell. 2004;6(6):611–623. | |
Fueyo J, Gomez-Manzano C, Alemany R, et al. A mutant oncolytic adenovirus targeting the Rb pathway produces anti-glioma effect in vivo. Oncogene. 2000;19(1):2–12. | |
Dias JD, Guse K, Nokisalmi P, et al. Multimodal approach using oncolytic adenovirus, cetuximab, chemotherapy and radiotherapy in HNSCC low passage tumour cell cultures. Eur J Cancer. 2010;46(3):625–635. | |
Dias JD, Liikanen I, Guse K, et al. Targeted chemotherapy for head and neck cancer with a chimeric oncolytic adenovirus coding for bifunctional suicide protein FCU1. Clin Cancer Res. 2010;16(9):2540–2549. | |
Reddy PS, Ganesh S, Yu DC. Enhanced gene transfer and oncolysis of head and neck cancer and melanoma cells by fiber chimeric oncolytic adenoviruses. Clin Cancer Res. 2006;12(9):2869–2878. | |
Nemunaitis J, Cunningham C, Buchanan A, et al. Intravenous infusion of a replication-selective adenovirus (ONYX-015) in cancer patients: safety, feasibility and biological activity. Gene Ther. 2001;8(10):746–759. | |
Ganesh S, Gonzalez-Edick M, Gibbons D, et al. Combination therapy with radiation or cisplatin enhances the potency of Ad5/35 chimeric oncolytic adenovirus in a preclinical model of head and neck cancer. Cancer Gene Ther. 2009;16(5):383–392. | |
Vijayalingam S, Kuppuswamy M, Subramanian T, et al. Evaluation of apoptogenic adenovirus type 5 oncolytic vectors in a syrian hamster head and neck cancer model. Cancer Gene Ther. 2014;21(6):228–237. | |
Liu RY, Zhou L, Zhang YL, et al. An oncolytic adenovirus enhances antiangiogenic and antitumoral effects of a replication-deficient adenovirus encoding endostatin by rescuing its selective replication in nasopharyngeal carcinoma cells. Biochem Biophys Res Commun. 2013;442(3–4):171–176. | |
Lee SY, Park HR, Rhee J, Park YM, Kim SH. Therapeutic effect of oncolytic adenovirus expressing relaxin in radioresistant oral squamous cell carcinoma. Oncol Res. 2013;20(9):419–425. | |
Takahashi H, Hyakusoku H, Horii C, et al. Telomerase-specific oncolytic adenovirus: antitumor effects on radiation-resistant head and neck squamous cell carcinoma cells. Head Neck. 2014;36(3):411–418. | |
Fujita K, Kimura M, Kondo N, et al. Anti-tumor effects of telomelysin for head and neck squamous cell carcinoma. Oncol Rep. 2008;20(6):1363–1368. | |
Nemunaitis J, Ganly I, Khuri F, et al. Selective replication and oncolysis in p53 mutant tumors with ONYX-015, an E1B-55kD gene-deleted adenovirus, in patients with advanced head and neck cancer: a phase II trial. Cancer Res. 2000;60(22):6359–6366. | |
Rudin CM, Cohen EE, Papadimitrakopoulou VA, et al. An attenuated adenovirus, ONYX-015, as mouthwash therapy for premalignant oral dysplasia. J Clin Oncol. 2003;21(24):4546–4552. | |
Chang J, Zhao X, Wu X, et al. A phase I study of KH901, a conditionally replicating granulocyte-macrophage colony-stimulating factor: armed oncolytic adenovirus for the treatment of head and neck cancers. Cancer Biol Ther. 2009;8(8):676–682. | |
Yuan ZY, Zhang L, Li S, Qian XZ, Guan ZZ. Safety of an E1B deleted adenovirus administered intratumorally to patients with cancer. Ai Zheng. 2003;22(3):310–313. Chinese. | |
Xia ZJ, Chang JH, Zhang L, et al. Phase III randomized clinical trial of intratumoral injection of E1B gene-deleted adenovirus (H101) combined with cisplatin-based chemotherapy in treating squamous cell cancer of head and neck or esophagus. Ai Zheng. 2004;23(12):1666–1670. Chinese. | |
Xu RH, Yuan ZY, Guan ZZ, et al. Phase II clinical study of intratumoral H101, an E1B deleted adenovirus, in combination with chemotherapy in patients with cancer. Ai Zheng. 2003;22(12):1307–1310. Chinese. | |
Kelly E, Russell SJ. History of oncolytic viruses: genesis to genetic engineering. Mol Ther. 2007;15(4):651–659. | |
Subramanian RP, Geraghty RJ. Herpes simplex virus type 1 mediates fusion through a hemifusion intermediate by sequential activity of glycoproteins D, H, L, and B. Proc Natl Acad Sci U S A. 2007;104(8):2903–2908. | |
Mundschau LJ, Faller DV. Oncogenic ras induces an inhibitor of double-stranded RNA-dependent eukaryotic initiation factor 2 alpha-kinase activation. J Biol Chem. 1992;267(32):23092–23098. | |
Martuza RL, Malick A, Markert JM, Ruffner KL, Coen DM. Experimental therapy of human glioma by means of a genetically engineered virus mutant. Science. 1991;252(5007):854–856. | |
Fujimoto Y, Mizuno T, Sugiura S, et al. Intratumoral injection of herpes simplex virus HF10 in recurrent head and neck squamous cell carcinoma. Acta Otolaryngol. 2006;126(10):1115–1117. | |
Ferris R, Gross ND, Nemunaitis JJ, et al. Phase I trial of intratumoral therapy using HF10, an oncolytic HSV-1, demonstrates safety in HSV+/HSV– patients with refractory and superficial cancers (Suppl; abstr 6082). J Clin Oncol. 2014;32:5s. | |
Hu JC, Coffin RS, Davis CJ, et al. A phase I study of OncoVEXGM-CSF, a second-generation oncolytic herpes simplex virus expressing granulocyte macrophage colony-stimulating factor. Clin Cancer Res. 2006;12(22):6737–6747. | |
Mace AT, Harrow SJ, Ganly I, Brown SM. Cytotoxic effects of the oncolytic herpes simplex virus HSV1716 alone and in combination with cisplatin in head and neck squamous cell carcinoma. Acta Otolaryngol. 2007;127(8):880–887. | |
Toyoizumi T, Mick R, Abbas AE, Kang EH, Kaiser LR, Molnar-Kimber KL. Combined therapy with chemotherapeutic agents and herpes simplex virus type 1 ICP34.5 mutant (HSV-1716) in human non-small cell lung cancer. Hum Gene Ther. 1999;10(18):3013–3029. | |
Harrington KJ, Hingorani M, Tanay MA, et al. Phase I/II study of oncolytic HSV GM-CSF in combination with radiotherapy and cisplatin in untreated stage III/IV squamous cell cancer of the head and neck. Clin Cancer Res. 2010;16(15):4005–4015. | |
Ogawa F, Takaoka H, Iwai S, Aota K, Yura Y. Combined oncolytic virotherapy with herpes simplex virus for oral squamous cell carcinoma. Anticancer Res. 2008;28(6A):3637–3645. | |
Yoo JY, Hurwitz BS, Bolyard C, et al. Bortezomib-induced unfolded protein response increases oncolytic HSV-1 replication resulting in synergistic antitumor effects. Clin Cancer Res. 2014;20(14):3787–3798. | |
Wang JN, Hu P, Zeng MS, Liu RB. Anti-tumor effect of oncolytic herpes simplex virus G47delta on human nasopharyngeal carcinoma. Chin J Cancer. 2011;30(12):831–841. | |
Yoo JY, Pradarelli J, Haseley A, et al. Copper chelation enhances antitumor efficacy and systemic delivery of oncolytic HSV. Clin Cancer Res. 2012;18(18):4931–4941. | |
Tai JH, Williams JV, Edwards KM, Wright PF, Crowe JE Jr, Dermody TS. Prevalence of reovirus-specific antibodies in young children in nashville, tennessee. J Infect Dis. 2005;191(8):1221–1224. | |
Hashiro G, Loh PC, Yau JT. The preferential cytotoxicity of reovirus for certain transformed cell lines. Arch Virol. 1977;54(4):307–315. | |
Duncan MR, Stanish SM, Cox DC. Differential sensitivity of normal and transformed human cells to reovirus infection. J Virol. 1978;28(2):444–449. | |
Bos JL. Ras oncogenes in human cancer: a review. Cancer Res. 1989;49(17):4682–4689. | |
Rogers SJ, Harrington KJ, Rhys-Evans P, O-Charoenrat P, Eccles SA. Biological significance of c-erbB family oncogenes in head and neck cancer. Cancer Metastasis Rev. 2005;24(1):47–69. | |
Morris DG, Feng X, DiFrancesco LM, et al. REO-001: a phase I trial of percutaneous intralesional administration of reovirus type 3 dearing (reolysin(R)) in patients with advanced solid tumors. Invest New Drugs. 2013;31(3):696–706. | |
Forsyth P, Roldan G, George D, et al. A phase I trial of intratumoral administration of reovirus in patients with histologically confirmed recurrent malignant gliomas. Mol Ther. 2008;16(3):627–632. | |
Harrington KJ, Karapanagiotou EM, Roulstone V, et al. Two-stage phase I dose-escalation study of intratumoral reovirus type 3 dearing and palliative radiotherapy in patients with advanced cancers. Clin Cancer Res. 2010;16(11):3067–3077. | |
Comins C, Spicer J, Protheroe A, et al. REO-10: a phase I study of intravenous reovirus and docetaxel in patients with advanced cancer. Clin Cancer Res. 2010;16(22):5564–5572. | |
Lolkema MP, Arkenau HT, Harrington K, et al. A phase I study of the combination of intravenous reovirus type 3 dearing and gemcitabine in patients with advanced cancer. Clin Cancer Res. 2011;17(3):581–588. | |
Roulstone V, Twigger K, Zaidi S, et al. Synergistic cytotoxicity of oncolytic reovirus in combination with cisplatin-paclitaxel doublet chemotherapy. Gene Ther. 2013;20(5):521–528. | |
Karapanagiotou EM, Roulstone V, Twigger K, et al. Phase I/II trial of carboplatin and paclitaxel chemotherapy in combination with intravenous oncolytic reovirus in patients with advanced malignancies. Clin Cancer Res. 2012;18(7):2080–2089. | |
Nichols AC, Yoo J, Um S, et al. Vaccinia virus outperforms a panel of other poxviruses as a potent oncolytic agent for the control of head and neck squamous cell carcinoma cell lines. Intervirology. 2014;57(1):17–22. | |
Chernichenko N, Linkov G, Li P, et al. Oncolytic vaccinia virus therapy of salivary gland carcinoma. JAMA Otolaryngol Head Neck Surg. 2013;139(2):173–182. | |
Yu Z, Li S, Brader P, et al. Oncolytic vaccinia therapy of squamous cell carcinoma. Mol Cancer. 2009;8:45. | |
Mansfield D, Pencavel T, Kyula JN, et al. Oncolytic vaccinia virus and radiotherapy in head and neck cancer. Oral Oncol. 2013;49(2):108–118. | |
Tysome JR, Wang P, Alusi G, et al. Lister vaccine strain of vaccinia virus armed with the endostatin-angiostatin fusion gene: an oncolytic virus superior to dl1520 (ONYX-015) for human head and neck cancer. Hum Gene Ther. 2011;22(9):1101–1108. | |
Jaime JC, Young AM, Mateo J, et al. Phase I clinical trial of a genetically modified and oncolytic vaccinia virus GL-ONC1 with green fluorescent protein imaging (Suppl; abstr 2530). J Clin Oncol. 2012;30. | |
Touchefeu Y, Khan AA, Borst G, et al. Optimising measles virus-guided radiovirotherapy with external beam radiotherapy and specific checkpoint kinase 1 inhibition. Radiother Oncol. 2013;108(1):24–31. | |
Li H, Peng KW, Russell SJ. Oncolytic measles virus encoding thyroidal sodium iodide symporter for squamous cell cancer of the head and neck radiovirotherapy. Hum Gene Ther. 2012;23(3):295–301. | |
Reddi HV, Madde P, McDonough SJ, et al. Preclinical efficacy of the oncolytic measles virus expressing the sodium iodide symporter in iodine non-avid anaplastic thyroid cancer: a novel therapeutic agent allowing noninvasive imaging and radioiodine therapy. Cancer Gene Ther. 2012;19(9):659–665. | |
Zaoui K, Bossow S, Grossardt C, et al. Chemovirotherapy for head and neck squamous cell carcinoma with EGFR-targeted and CD/UPRT-armed oncolytic measles virus. Cancer Gene Ther. 2012;19(3):181–191. | |
Li P, Chen CH, Li S, et al. Therapeutic effects of a fusogenic newcastle disease virus in treating head and neck cancer. Head Neck. 2011;33(10):1394–1399. | |
Escobar-Zarate D, Liu YP, Suksanpaisan L, Russell SJ, Peng KW. Overcoming cancer cell resistance to VSV oncolysis with JAK1/2 inhibitors. Cancer Gene Ther. 2013;20(10):582–589. | |
Alajez NM, Mocanu JD, Krushel T, Bell JC, Liu FF. Enhanced vesicular stomatitis virus (VSVDelta51) targeting of head and neck cancer in combination with radiation therapy or ZD6126 vascular disrupting agent. Cancer Cell Int. 2012;12(1):27. | |
McNally LR, Rosenthal EL, Zhang W, Buchsbaum DJ. Therapy of head and neck squamous cell carcinoma with replicative adenovirus expressing tissue inhibitor of metalloproteinase-2 and chemoradiation. Cancer Gene Ther. 2009;16(3):246–255. | |
Ganesh S, Gonzalez-Edick M, Gibbons D, Van Roey M, Jooss K. Intratumoral coadministration of hyaluronidase enzyme and oncolytic adenoviruses enhances virus potency in metastatic tumor models. Clin Cancer Res. 2008;14(12):3933–3941. | |
Nagano S, Perentes JY, Jain RK, Boucher Y. Cancer cell death enhances the penetration and efficacy of oncolytic herpes simplex virus in tumors. Cancer Res. 2008;68(10):3795–3802. | |
Okunaga S, Takasu A, Meshii N, et al. Ultrasound as a method to enhance antitumor ability of oncolytic herpes simplex virus for head and neck cancer. Cancer Gene Ther. 2015;22(3):163–168. | |
Heldin CH, Rubin K, Pietras K, Ostman A. High interstitial fluid pressure – an obstacle in cancer therapy. Nat Rev Cancer. 2004;4(10):806–813. | |
Price DL, Li P, Chen CH, et al. A silk-elastinlike protein polymer matrix for intraoperative delivery of an oncolytic vaccinia virus. Head Neck. Epub September 22, 2014. | |
Shiina M, Lacher MD, Christian C, Korn WM. RNA interference-mediated knockdown of p21(WAF1) enhances anti-tumor cell activity of oncolytic adenoviruses. Cancer Gene Ther. 2009;16(11):810–819. | |
Jung SH, Choi JW, Yun CO, et al. Sustained local delivery of oncolytic short hairpin RNA adenoviruses for treatment of head and neck cancer. J Gene Med. 2014;16(5–6):143–152. | |
Lyons M, Onion D, Green NK, et al. Adenovirus type 5 interactions with human blood cells may compromise systemic delivery. Mol Ther. 2006;14(1):118–128. | |
Li H, Zeng Z, Fu X, Zhang X. Coadministration of a herpes simplex virus-2 based oncolytic virus and cyclophosphamide produces a synergistic antitumor effect and enhances tumor-specific immune responses. Cancer Res. 2007;67(16):7850–7855. | |
Dhar D, Spencer JF, Toth K, Wold WS. Pre-existing immunity and passive immunity to adenovirus 5 prevents toxicity caused by an oncolytic adenovirus vector in the syrian hamster model. Mol Ther. 2009;17(10):1724–1732. | |
Tsai V, Johnson DE, Rahman A, et al. Impact of human neutralizing antibodies on antitumor efficacy of an oncolytic adenovirus in a murine model. Clin Cancer Res. 2004;10(21):7199–7206. | |
Herrlinger U, Kramm CM, Aboody-Guterman KS, et al. Pre-existing herpes simplex virus 1 (HSV-1) immunity decreases, but does not abolish, gene transfer to experimental brain tumors by a HSV-1 vector. Gene Ther. 1998;5(6):809–819. | |
Shashkova EV, May SM, Barry MA. Characterization of human adenovirus serotypes 5, 6, 11, and 35 as anticancer agents. Virology. 2009; 394(2):311–320. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.