Back to Journals » Cancer Management and Research » Volume 18

Oncolytic Vaccinia Virus-Engineered EVs Convert Tumor-Promoting Macrophages into Anti-Tumor Effectors
Authors Chen L, Ye L, Bao L, Wang J, Tan M ![]()
Received 17 December 2025
Accepted for publication 10 April 2026
Published 20 April 2026 Volume 2026:18 589766
DOI https://doi.org/10.2147/CMAR.S589766
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Chien-Feng Li
Liqiong Chen,1 Luyi Ye,2 Liuqian Bao,1 Junwei Wang,1 Mingming Tan1
1Emergency Medicine, Tiantai People’s Hospital of Zhejiang Province (Tiantai Branch of Zhejiang Provincial People’s Hospital), Hangzhou Medical College, Taizhou, Zhejiang, 317200, People’s Republic of China; 2Joint Centre of Translational Medicine, Wenzhou Institute, University of Chinese Academy of Sciences, Wenzhou, Zhejiang, 325000, People’s Republic of China
Correspondence: Mingming Tan, Email [email protected]
Introduction: Extracellular vesicles (EVs) play a critical role in shaping the tumor immune microenvironment (TME) by mediating intercellular communication and transferring oncogenic proteins, microRNAs, and immunosuppressive molecules. Tumor-derived EVs (TDEVs) typically promote immune evasion by inducing M2 macrophage polarization, suppressing cytotoxic T-cell activity, and enhancing angiogenesis. Recent evidence suggests that oncolytic viruses (OVs) can reprogram EV secretion, generating OV-derived EVs (OV-EVs) with immunostimulatory properties capable of activating antitumor immune responses.
Methods: In this study, we investigated how infection with oncolytic vaccinia virus (OVV) alters the EV secretion profile of colon cancer cells. EVs derived from OVV-infected tumor cells were isolated and characterized, and their effects on macrophage polarization were evaluated using in vitro assays.
Results: Our results demonstrate that OVV infection significantly alters the composition and immunological properties of tumor-derived EVs. OV-EVs were enriched with pathogen-associated and damage-associated molecular patterns (PAMPs/DAMPs), which promoted macrophage reprogramming toward a pro-inflammatory M1 phenotype and enhanced immune activation associated with antitumor responses.
Discussion: These findings indicate that OVV-induced EVs can reshape the tumor immune microenvironment by modulating macrophage polarization. This study provides new insights into the EV-mediated mechanisms of oncolytic virotherapy and highlights the potential of OV-derived EVs as a novel strategy for EV-based cancer immunotherapy.
Keywords: extracellular vesicles, tumor immune microenvironment, oncolytic viruses, macrophage polarization
Introduction
In the dynamic regulatory network of the tumor immune microenvironment (TME), extracellular vesicles (EVs), as nanoscale membrane-bound communication vehicles, play a pivotal role in tumor initiation, progression, and metastasis by transporting proteins, nucleic acids, lipids, and other bioactive molecules.1–3 Numerous studies have demonstrated that tumor-derived EVs (TDEVs) often act as facilitators of tumor progression by delivering oncogenic proteins, microRNAs (miRNAs), and immunosuppressive molecules such as PD-L1 and TGF-β, as well as M2-polarization-promoting factors like miR-21 and HIF-1α. These cargos can drive macrophage polarization toward the pro-tumorigenic M2 phenotype, suppress CD8⁺ T cell activity, promote angiogenesis and tumor metastasis, and ultimately dampen anti-tumor immune responses, thereby fostering an environment conducive to tumor growth and immune evasion.2,4–9 For example, breast cancer cell-derived EVs deliver miR-1246, which downregulates BATF3 expression in macrophages, thereby inhibiting their polarization toward the anti-tumor M1 phenotype.10,11 Additionally, tumor-secreted EVs can transfer PD-L1 to immune cells, suppressing T and NK cell cytotoxic activity.12 TDEVs carrying oncogenic miRNAs such as miR-21 further enhance tumor cell proliferation, migration, and invasion, and angiogenesis.7 Recent studies have revealed that viral infection can reshape the EV secretion profile of tumor cells, potentially converting EVs from immunosuppressive carriers into immunostimulatory messengers.13 Oncolytic viruses (OVs) can influence the composition and biological functions of tumor-derived EVs through multiple mechanisms, including virus-induced cellular stress responses and immune activation.14,15 Virus-induced endoplasmic reticulum stress and calcium dysregulation can enhance EVs release efficiency.16,17 oncolytic vaccinia virus (OVV) can generate “oncolytic virus-derived EVs (OV-EVs)”18,19 carrying viral pathogen-associated molecular patterns (PAMPs, such as dsRNA, CpG DNA) and host damage-associated molecular patterns (DAMPs, such as HSP70, HMGB1), thereby exerting anti-tumor effects.20–22 Engineered OVs can deliver misfolded proteins to tumor cells, activating dendritic cells and CD8⁺ T cell-mediated immunity.23,24 Furthermore, biomaterials with cytocompatibility and bioactivity have been explored as delivery platforms for OVs, improving targeting and enhancing immune activation.25 Despite the encouraging progress of oncolytic virotherapy, several challenges remain for its clinical translation, including limited systemic delivery efficiency, antiviral immune clearance, tumor heterogeneity, and the fact that currently approved therapies such as T-VEC are mainly administered through intratumoral injection, which restricts their application to accessible lesions.
Macrophages are the most abundant immune cells infiltrating solid tumors (often exceeding 50%). M2-type macrophages promote tumor progression and treatment resistance by secreting IL-10, TGF-β, and arginase-1,26,27 whereas M1-type macrophages can recruit CD8⁺ T cells and improve the adhesion ability of tumor vascular endothelial cells to immune cells by secreting chemokines such as CXCL9/10,28,29 thereby transforming the tumor microenvironment from an “immune desert” to an “immune inflammatory” state. Their remarkable plasticity is guided by metabolic reprogramming, which serves as a central determinant of functional polarization.30 Although existing studies have preliminarily confirmed the potential of OV-EVs to induce M1 polarization,31–33 different oncolytic viruses exhibit distinct EV secretion characteristics.
Although previous studies have demonstrated that oncolytic viruses can reshape the extracellular vesicle landscape of tumor cells, most investigations have focused on a limited number of viral platforms, and the virus-specific characteristics of EV modulation remain insufficiently understood. In particular, the mechanisms by which OVV influences the composition and immunological function of tumor-derived EVs have not been fully elucidated. The specific molecular components of OVV-derived EVs responsible for macrophage reprogramming remain largely unknown. Understanding these mechanisms could provide a basis for developing OVV-EV-based immunotherapies.
As an oncolytic adenovirus (Ads) obtained by genetically engineering the wild-type human adenovirus type 5 to knock out the E1B-55KD gene fragment and part of the E3 region, H101 is currently the only approved oncolytic virus-based anti-tumor drug in China, used for the treatment of advanced nasopharyngeal carcinoma.34 OVV is the first OV proven to form infections in tumor foci through intravenous injection. OVV selectively replicates and lyses tumor cells within tumors, triggers anti-tumor immunity in the body, and interacts with the immune system in various forms and pathways, improving the tumor immune microenvironment, enhancing immune cell infiltration, and activating tumor-specific immune responses.23 However, whether infection with OVV alters the immunological properties of tumor-derived EVs and thereby regulates macrophage polarization remains poorly understood. In this study, we aim to explore how OVV infection affects the characteristics of EVs secreted by colon cancer cells, isolate EVs via ultracentrifugation, and assess their potential impact on macrophage polarization, providing theoretical support for novel EV-based tumor immunotherapy strategies (Scheme 1).
 |
Scheme 1 Schematic diagram of the improvement of the tumor immunosuppressive microenvironment by extracellular vesicles secreted by tumor cells infected with oncolytic virus. After being infected with oncolytic viruses, tumor cells release exosomes/vesicles carrying the virus and related immunomodulatory factors through vesicle secretion. After separating these vesicles by ultracentrifugation, they were applied to tumor-associated macrophages (TAMs) to promote their transformation from pro-tumor M2 type to anti-tumor M1 type, thereby reshaping the tumor immune microenvironment and enhancing the body’s immune response to tumors. |
Materials and Methods
Cell Culture and Reagents
CT26 (mouse colon cancer cells) were cultured in DMEM (with 100 IU/mL penicillin-streptomycin added and 10% fetal bovine serum) at 37°C and 5% CO2. RAW 264.7 (mouse mononuclear macrophage leukemia cells) were cultured in DMEM with 10% fetal bovine serum added at a temperature of 37°C and 5% CO2. The cell lines used in this study were obtained from American Type Culture Collection (ATCC). F4/80, CD86 and CD206 antibodies used for flow cytometry analysis were derived from BD Pharmingen (USA) and Thermo Fisher (USA).
EV Production and Isolation
For the infection experiment, confluence tumor cells were infected with MOI = 0.1 and cultured in serum-free medium for 24 hours. Collect the culture supernatant, centrifuge the conditioned medium to remove cells (500 xg, 5 min), cell debris (2000 xg, 10 min), and large vesicles (10,000 xg, 30 min), and filter under a 0.22 μm filter membrane. Then perform ultracentrifugation until the precipitate EV (100,000 xg, 2 h). Resuspend EV in 100 μL of 0.22 μm filtered PBS and use immediately or store at -80°C. The extracellular vesicles (EVs) secreted by tumor cells and the exosomes (OV-EVs) released by tumor cells after oncolytic virus infection were collected and extracted, and the total amount of exosome proteins was quantified by the BCA method. Polarized RAW264.7 macrophages were treated with 10 μg/mL EV or OV-EV for 24 h. The PBS treatment group was used as the negative control.
Tem
Use tweezers to pick up the copper mesh. Place the Formvar/carbon membrane copper mesh (200 mesh) with the front side up on the filter paper and let it stand at room temperature for 5 minutes. Drop 10 μL of 0.1% polylysine solution onto the surface of the copper mesh, absorb the excess liquid with filter paper, and dry at room temperature for 10 minutes for later use. Draw 10 μL of exosome suspension and drop it onto a copper mesh, RT, let it stand for 5–10 minutes, and then dry the liquid with filter paper. Add 5 μL of uranium acetate negative staining solution drop by drop, at room temperature for 30s-1 min, absorb dry with filter paper, and bake in an oven at 37°C overnight.
Cytotoxicity Determination
RAW264.7 cells were respectively induced or not induced into M1 and M2 phenotypes. Then, different treatment groups of EVs and OV-EVs were added. After incubation for 24 h, the supernatant was discarded. CCK8 reagent was added and incubated at 37°C for 1–2 h, avoiding light. Absorbance was measured at a wavelength of 450 nm.
Flow Cytometry Detection
The mouse macrophage cell line RAW264.7 was cultured in high glucose DMEM medium containing 10% fetal bovine serum and 1% streptomycin and placed in a constant temperature incubator at 37 °C with 5% CO2. Cells were inoculated in 24-well plates and subjected to polarization treatment when the fusion degree reached approximately 70–80%. To induce the M1 phenotype, 100 ng/mL lipopolysaccharide (LPS) and 20 ng/mL interferon-γ (IFN-γ) were added for stimulation for 24 h. To induce the M2 type phenotype, 10 ng/mL interleukin 4 (IL-4) and 10 ng/mL interleukin 13 (IL-13) were added for 24 h, while EVs and OV-EVs were added for treatment. After the treatment was completed, the cells were collected and washed twice with PBS, and then stained with fluorescence-labeled antibodies, including F4/80 (PE), CD86 (FITC), and CD206 (APC), all of which were purchased from BioLegend. Staining was carried out in the dark at 4 °C for 30 min. CD206 was stained after membrane rupture. After staining, the cells were washed again and resuspended in PBS containing 2% FBS. Data collection was performed using a Beckman flow cytometer.
qPCR
RAW264.7 was stimulated with 100 ng/mL LPS and 20 ng/mL IFN-γ, while different treatment groups of EVs and OV-EVs were added respectively. After incubation for 24 h, the supernatant was discarded and RNA of each group was extracted using the RNA Extraction Kit (Vazyme). The concentration and purity of RNA (the ratio of 260/280 was between 1.8 and 2.0) were detected by the NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific). 1 μg of total RNA was taken for cDNA synthesis using the reverse transcription kit (Takara), and the reaction system and conditions were carried out according to the instructions. Real-time quantitative PCR reactions were performed on the qPCR instrument using SYBR Green qPCR Master Mix (Takara). The total amount of each reaction system was 20 μL, including 10 μL of 2×SYBR Premix, 0.4 μL of upstream primers, 0.4 μL of downstream primers, 2 μL of template cDNA, and 7.2 μL of RNase free water. The amplification conditions are as follows: Pre-denaturation at 95 °C for 30s; Then 40 cycles: 95 °C for 5s, 60 °C for 30s. After the reaction was completed, the melting curve analysis was conducted to verify the specificity of the amplification products. All samples were repeated three times using the technique, and the expression levels were calculated using the 2^−ΔΔCt method. The internal reference gene selected is β-actin.
Statistical Analysis
All the data were statistically analyzed using GraphPad Prism8.0 software. Mean ± standard deviation (SD) is presented for all values. All data were obtained from at least three independent biological replicates. The statistical differences between the two groups were analyzed using the unpaired t-test, and one-way analysis of variance (Graphpad Prism 8) was used among multiple groups with differences. The statistical significance was *P < 0.05, **P < 0.01, and ***P < 0.001 respectively.
Results
Characteristics of Tumor-Derived Extracellular Vesicles
To explore the effect of oncolytic virus infection on the secretion profile of tumor-derived extracellular vesicles (EVs), we infected the murine colorectal cancer cell line CT26 with an OVV. Virus-modulated EVs (OV-EVs) were isolated from the culture supernatant at 24 hours post-infection, a time point selected prior to the onset of notable cytopathic effects to reduce the potential contribution of vesicles released during extensive cell lysis. We first performed a physicochemical characterization of both EVs and OV-EVs. Compared to EVs from uninfected cells, OV-EVs exhibited an approximately 15% increase in particle size (Figure 1A), along with a marked shift in zeta potential (Figure 1B), indicating that viral infection may remodel surface charge properties. Transmission electron microscopy (TEM) confirmed the typical vesicular morphology in both groups (Figure 1C). Classical exosomal markers such as CD63, CD81, and TSG101 were not evaluated in this study, and EV identification was based on TEM morphology, particle size distribution, and zeta potential analysis. Subsequently, we assessed the cytotoxicity of EVs and OV-EVs toward macrophages with distinct phenotypes. Both EV preparations showed minimal toxicity, with no significant adverse effects on macrophage viability (Figure 1D). Interestingly, morphological analysis of RAW264.7 macrophages 24 hours after treatment revealed divergent polarization responses: conventional tumor-derived EVs induced an elongated spindle-like morphology characteristic of M2-like macrophages, while OV-EVs promoted a stellate, pro-inflammatory morphology indicative of M1 polarization (Figure 1E). These findings support our hypothesis that oncolytic virus infection reprograms the molecular content and functional properties of tumor-derived EVs, potentially enabling them to deliver immunostimulatory signals that shift the tumor microenvironment from immunosuppressive to immunoreactive.
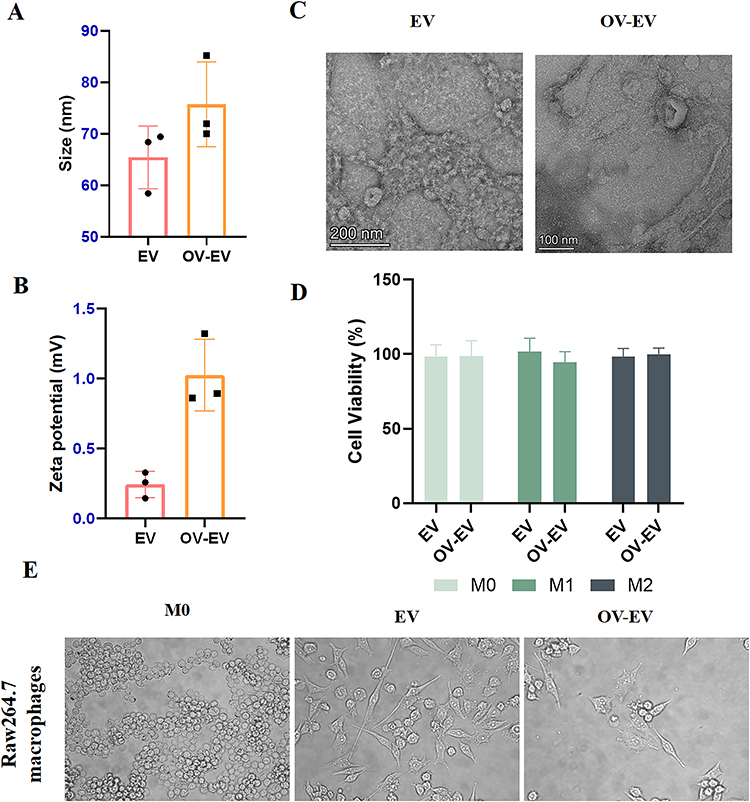 |
Figure 1 Characterization of extracellular vesicles derived from tumors. (A) Particle size diagrams of EV and OV-EV. n=3. (B) Zeta potentials of EV and OV-EV. n=3. (C) Transmission electron microscope image of EV. (D) Cytotoxicity of EV administration under the three states of M0, M1 and M2. (E) Bright-field cell morphology of RAW264.7 cells after treatment with different administration groups. Data were expressed as mean ± SD. |
Tumor-Derived EVs Promote M2-Like Macrophage Polarization
To investigate the role of tumor-derived extracellular vesicles (EVs) in regulating macrophage polarization, we collected OV-EVs secreted by CT26 cells 24 hours post-infection with OVV. M0-type macrophages were subsequently treated with EVs derived from both infected and uninfected cells. Flow cytometry analysis (Figure 2A) revealed that tumor-derived EVs significantly promoted macrophage polarization toward the immunosuppressive M2 phenotype, as evidenced by a marked increase in the proportion of CD206⁺ F4/80⁺ cells. This finding suggests that EVs possess the potential to facilitate the establishment of a tumor-associated immunosuppressive microenvironment. Conversely, treatment with OV-EVs markedly upregulated the proportion of CD86⁺ F4/80⁺ cells, indicating their capacity to drive macrophages toward the pro-inflammatory M1 phenotype (Figure 2B and C). Quantitative analysis further corroborated these findings, demonstrating that OV-EV treatment increased the proportion of CD86⁺ macrophages to approximately 12%, which was significantly higher than that observed in the EV-treated group (Figure 2B). Additionally, the proportion of CD206⁺ cells was notably reduced compared to the EV treatment group (Figure 2C). Collectively, these results suggest that EVs derived from uninfected tumors strongly induce macrophages to polarize toward the M2-type immunosuppressive phenotype, whereas OV infection can reprogram EV function, reverse their inducing effect, promote the formation of the M1-type pro-inflammatory phenotype, and potentially enhance the tumor immune microenvironment.
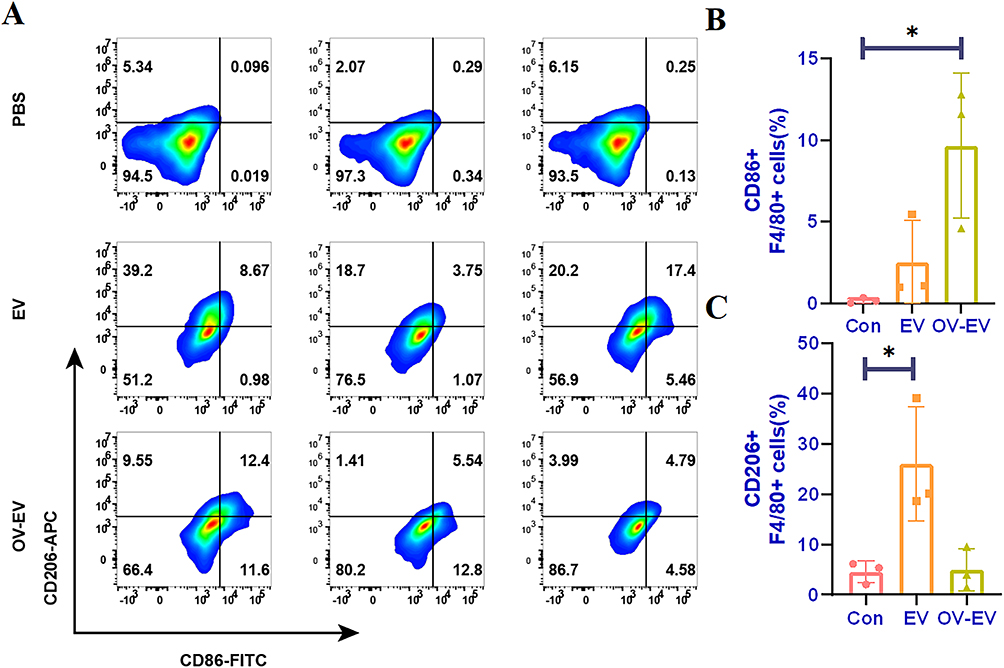 |
Figure 2 Tumor-derived EVs can induce M0 type RAW264.7 cells to polarize towards pro-inflammatory macrophages. (A) The expression of CD86 and CD206 after treatment in different groups of PBS, EV and OV-EV, and (B) quantitative analysis of CD86 and (C) CD206. n=3. Data were expressed as mean ± SD. *p < 0.05. |
OV Infection Reprograms Immunosuppressive EV Signaling
To determine whether tumor-derived EVs influence macrophage polarization, we employed a classical induction model in which M0 macrophages were polarized toward the M1 phenotype under LPS and IFN-γ stimulation, while simultaneously treated with EVs from different experimental groups. It is well established that M0 macrophages can differentiate into immunosuppressive M2 macrophages under IL-4 and IL-13 stimulation, characterized by elevated expression of CD206, Arg-1, and IL-10. In contrast, exposure to LPS and IFN-γ drives polarization toward the pro-inflammatory M1 phenotype, typically marked by upregulation of CD86, TNF-α, and IL-6 (Figure 3A). Using this model, we observed that co-incubation with tumor-derived EVs during M1 induction led to a significant increase in the proportion of CD206⁺ macrophages, suggesting that these EVs disrupted M1 polarization and instead promoted a shift toward the M2 phenotype. This indicates that tumor-derived EVs may skew macrophages toward an anti-inflammatory or tissue-repair phenotype (Figure 3B), further supporting their role in establishing an immunosuppressive tumor microenvironment. In contrast, EVs derived from OV-EVs did not exhibit this M2-inducing effect. Instead, they enhanced M1-associated markers and inflammatory responses (Figure 3C–E). These findings suggest that OV infection reprograms the immunomodulatory properties of tumor-derived EVs, effectively disrupting their capacity to promote M2 polarization, and thereby playing a pivotal role in remodeling the tumor immune microenvironment.
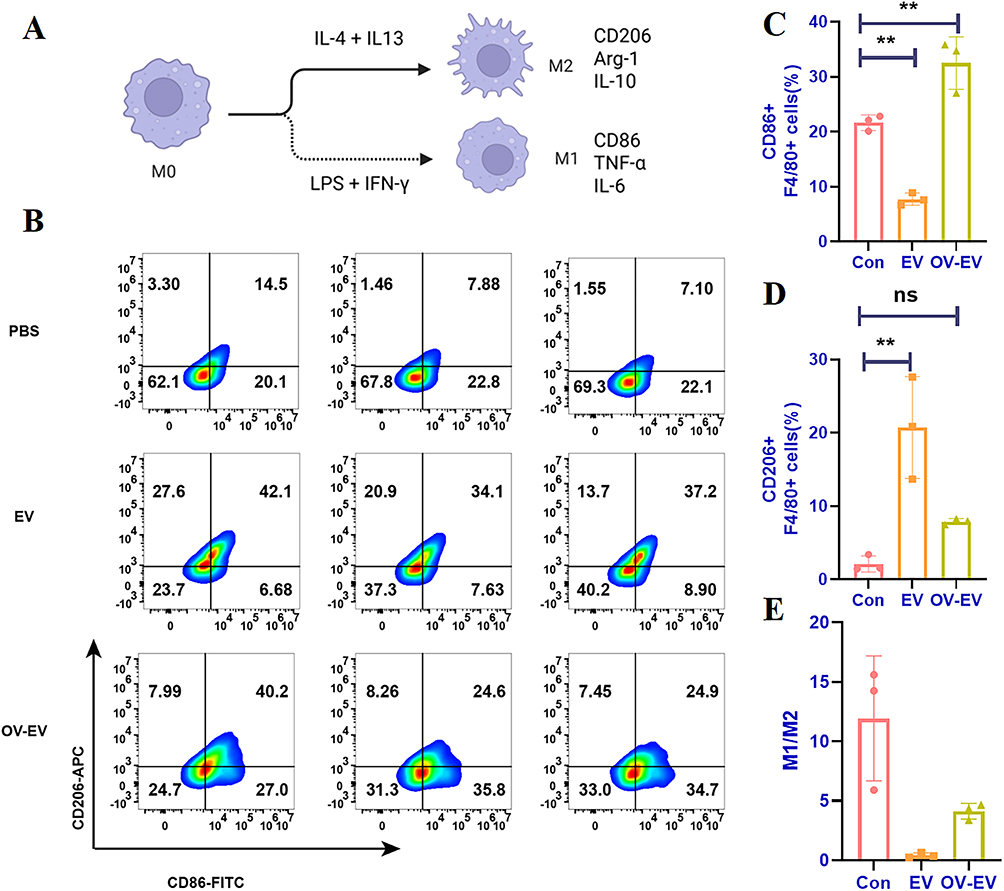 |
Figure 3 The EV secreted by tumor cells infected with OV partially enhances the polarization of macrophages towards type M1. (A) Schematic diagram of the classical model of macrophage polarization. (B) Flow cytometry detection of CD86 and CD206 induced by different administration groups of RAW264.7 induced M1-type macrophages and (C) quantitative analysis of CD86 and (D) CD206. (E) Proportional analysis of M1/M2. n=3. Data were expressed as mean ± SD. **p < 0.01, and ns indicated no statistical significance. |
OV-EVs Disrupt Tumor EV-Induced M2 Polarization of Macrophages
To mimic the macrophage phenotype typically found in the tumor microenvironment, we first induced M0 macrophages to polarize toward the M2 phenotype using IL-4 and IL-13. During the polarization process, EVs or OV-EVs from different treatment groups were added to assess their effects on macrophage phenotype. Flow cytometry analysis revealed that co-treatment with IL-4/IL-13 and tumor-derived EVs led to a two-fold increase in the proportion of CD206⁺ macrophages compared to the control group (Figure 4A). In contrast, treatment with OV-EVs significantly reduced the proportion of CD206⁺ cells while simultaneously increasing CD86 expression (Figure 4B–D). These findings suggest that OV infection may alter the cargo composition of EVs, such as by incorporating pro-inflammatory miRNAs or viral protein fragments—which in turn disrupts the immunosuppressive M2 polarization program and promotes a shift toward a pro-inflammatory M1-like phenotype.
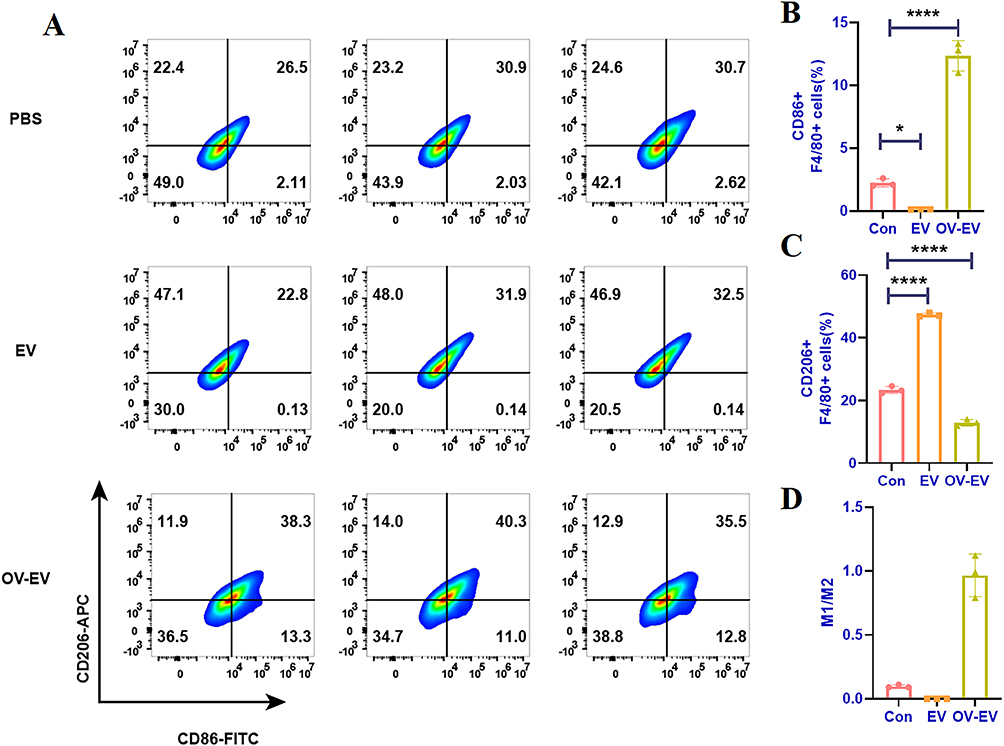 |
Figure 4 The EV secreted by tumor cells infected with OV reverses the immunosuppression of macrophages. (A) Flow cytometry detection of CD86 and CD206 induced by different administration groups of RAW264.7-induced M2-type macrophages and (B) quantitative analysis of CD86 and (C) CD206. (D) Proportional analysis of M1/M2. n=3. Data were expressed as mean ± SD. *p < 0.05, ****p < 0.0001. |
EV-Mediated Cytokine Modulation Following OV Infection
To further elucidate the mechanisms by which EVs and OV-EVs modulate macrophage inflammatory states, we assessed the expression of key genes associated with M1 (TNF-α, IL-6, CD86) and M2 (IL-10, Arg-1, CD206) polarization. OV-EV treatment markedly increased the mRNA levels of CD86, TNF-α, and IL-6 compared to both control and EV-treated groups (Figure 5A–C), indicating enhanced M1-like activation. In contrast, the expression of M2-associated markers CD206, Arg-1, and IL-10 was significantly reduced in the OV-EV group relative to the EV group (Figure 5D–F). These expression patterns were further confirmed by heatmap analysis (Figure 5G), reinforcing the notion that OV-EVs reprogram macrophage polarization toward a pro-inflammatory M1 phenotype. Collectively, these findings suggest that OV-EVs not only promote M1-type inflammatory responses but also actively suppress M2-type immunosuppressive signaling, thereby shifting macrophages toward an immunostimulatory state. This highlights the therapeutic potential of OV-EVs as immune-modulating agents in the context of cancer immunotherapy.
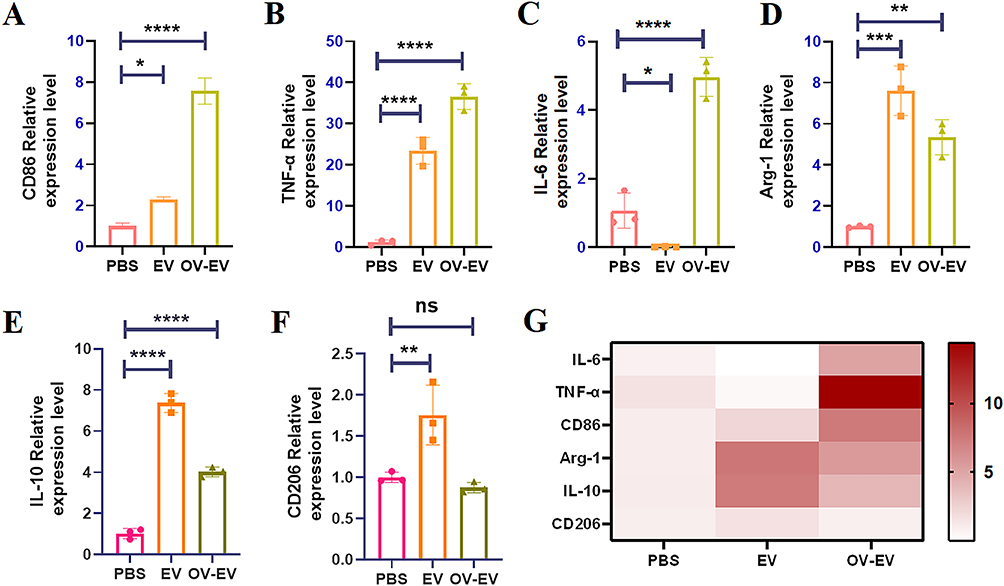 |
Figure 5 Extracellular vesicles of tumor cells induce M2 polarization, and extracellular vesicles infected with viruses can reverse the macrophage phenotype. The secretion levels of CD86 (A), TNF-α (B), IL-6 (C), Arg-1 (D), IL-10 (E) and CD206 (F) in Raw264.7 cells after different treatments were measured by qPCR. (G) The mRNA expression levels of CD86, TNF-α, IL-6, Arg-1, IL-10 and CD206 are shown as heat maps. n=3. Data were expressed as mean ± SD. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 and ns indicated no statistical significance. |
Conclusion
In this study, we observed that although tumor-derived EVs carry tumor antigens capable of initiating anti-tumorimmune responses, growing evidence suggests that tumor cells can also evade immune surveillance by releasing EVs enriched with immunosuppressive molecules.13 EVs have been widely recognized as pivotal mediators of communication between tumors and the host immune system.15 For instance, Wieckowski et al14 demonstrated that tumor-derived microvesicles promote the expansion of regulatory T cells (Tregs) and induce apoptosis of tumor-reactive CD8⁺ T cells, thereby enhancing tumor immune evasion. Interestingly, EVs released by tumor cells following OV infection may carry viral nucleic acids and immune-regulatory factors that reshape the immunosuppressive tumor microenvironment. By encapsulating OVs along with immune-stimulating components—such as tumor antigens, interferon-β,and DAMPs—within small EVs (sEVs), these vesicles may exert synergistic effects on both tumor and immune cells, ultimately boosting anti-tumor immune responses.16 Moreover, the anti-tumor efficacy of certain OVs may involve modulation of the protein cargo within tumor-derived EVs, affecting both anti-tumor and antiviral immunity. Notably, OV-infected tumor cells can release EVs enriched with viral components and immune-stimulatory molecules, which play a critical role in reprogramming the tumor immune microenvironment. Previous studies have shown that OV-EVs are enriched in pro-inflammatory microRNAs such as miR-155 and miR-21, which can promote macrophage polarization toward the pro-inflammatory M1 phenotype, thereby contributing to the restoration of anti-tumor immunity.
Recent studies have also demonstrated that extracellular vesicles derived from tumor cells infected with other OVs exhibit similar immunomodulatory properties. For example, EVs released from tumor cells infected with oncolytic adenoviruses have been reported to carry viral components and immune-stimulatory molecules capable of activating dendritic cells and promoting anti-tumor immune responses. Likewise, extracellular vesicles associated with herpes simplex virus (HSV) infection contain viral nucleic acids, proteins, and host-derived immune regulators that can modulate innate immune signaling pathways. These observations suggest that virus-modulated EVs may represent a common mechanism through which different OVs communicate with and reshape the tumor immune microenvironment. In this context, our findings further support that OVV infection can similarly reprogram tumor-derived EVs to enhance pro-inflammatory macrophage polarization.
Our study further confirms that tumor-derived EVs, in the absence of viral infection, significantly increase the proportion of CD206⁺ cells across different macrophage phenotypes, indicating a strong tendency to drive polarization toward the immunosuppressive M2 phenotype. This process not only inhibits the activation of anti-tumor immune responses but also potentially facilitates immune evasion, sustained tumor growth, and metastasis. In contrast, macrophages treated with OV-EVs exhibited a distinct shift in phenotype, characterized by a significantly increased M1/M2 ratio and a higher proportion of pro-inflammatory M1-like macrophages, suggesting a heightened potential to elicit anti-tumor immune responses.
Collectively, our findings demonstrate that OV infection alters the immunological properties of tumor-derivedEVs, transforming them from immunosuppressive vesicles that promote M2 polarization into immune-activating carriers capable of driving M1 polarization. This reprogramming effect significantly influences macrophage polarization and highlights the critical role of EVs in modulating tumor immunity. The high sensitivity of macrophage phenotypes to EV-mediated signals underscores the potential of targeting EV function to optimize OV-based immunotherapy. However, several limitations of the present study should be acknowledged. First, the experiments were primarily conducted using in vitro macrophage models and a single colon cancer cell line, which may not fully recapitulate the complexity and heterogeneity of the tumor immune microenvironment in vivo. Second, although OV-EVs were shown to modulate macrophage polarization, the specific molecular components responsible for this effect were not comprehensively identified in the current study. Building on these results, future investigations should focus on the identification and functional characterization of key immunoregulatory components-such as miRNAs, proteins, and lipids-within OV-EVs that contribute to macrophage polarization. Additionally, mechanistic studies comparing OVV-EVs with other OV-EVs, including adenovirus and HSV-derived EVs, will be critical to elucidate virus-specific immunomodulatory effects. Elucidating the molecular mechanisms underlying this process will provide a solid theoretical foundation and technical basis for the development of OV-EV-based combination immunotherapies.
Abbreviations
EVs, Extracellular vesicles; OVV, Oncolytic vaccinia virus; OV, Oncolytic virus; TME, Tumor microenvironment; M1, Classically activated macrophages; M2, Alternatively activated macrophages; TDEVs, Tumor-derived EVs; MiRNAs, MicroRNAs; ICD, Immunogenic cell death; PAMPs, Pathogen-associated molecular patterns; DAMPs, Damage-associated molecular patterns; Ads, Adenovirus; Tregs, Regulatory T cells; SEVs, Small EVs.
Declaration of Generative AI and AI-Assisted Technologies in the Writing Process
During the preparation of this work the authors used Deepseek in order to proof the grammar and language. After using this tool/service, the authors reviewed and edited the content as needed and take full responsibility for the content of the publication.
Data Sharing Statement
The datasets generated and/or analyzed during the current study are not publicly available but are available from the corresponding author upon reasonable request.
Consent for Publication
All authors agreed with the content and all gave explicit consent to submit the manuscript.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
There is no funding to report.
Disclosure
The authors declare that they have no known competing financial interests or personal relationships that might influence the work reported in this paper.
References
1. Paskeh MDA, Entezari M, Mirzaei S, et al. Emerging role of exosomes in cancer progression and tumor microenvironment remodeling. J Hematol Oncol. 2022;15(1):83. doi:10.1186/s13045-022-01305-4
2. Marar C, Starich B, Wirtz D. Extracellular vesicles in immunomodulation and tumor progression. Nat Immunol. 2021;22(5):560. doi:10.1038/s41590-021-00899-0
3. van Niel G, D’Angelo G, Raposo G. Shedding light on the cell biology of extracellular vesicles. Nat Rev Mol Cell Biol. 2018;19(4):213. doi:10.1038/nrm.2017.125
4. Capik O, Karatas OF. Pathways and outputs orchestrated in tumor microenvironment cells by hypoxia-induced tumor-derived exosomes in pan-cancer. Cell Oncol. 2025;48(3):539. doi:10.1007/s13402-025-01042-z
5. Wang B, Cheng D, Ma D, et al. Mutual regulation of PD-L1 immunosuppression between tumor-associated macrophages and tumor cells: a critical role for exosomes. Cell Commun Signal. 2024;22(1):21. doi:10.1186/s12964-024-01473-5
6. Li Q, He G, Yu Y, Li X, Peng X, Yang L. Exosome crosstalk between cancer stem cells and tumor microenvironment: cancer progression and therapeutic strategies. Stem Cell Res Ther. 2024;15(1):449. doi:10.1186/s13287-024-04061-z
7. Ye B, Duan Y, Zhou M, et al. Hypoxic tumor-derived exosomal miR-21 induces cancer-associated fibroblast activation to promote head and neck squamous cell carcinoma metastasis. Cell Signal. 2023;108:110725. doi:10.1016/j.cellsig.2023.110725
8. Buzas EI. The roles of extracellular vesicles in the immune system. Nat Rev Immunol. 2023;23(4):236. doi:10.1038/s41577-022-00763-8
9. Garofalo M, Villa A, Crescenti D, et al. Heterologous and cross-species tropism of cancer-derived extracellular vesicles. Theranostics. 2019;9(19):5681. doi:10.7150/thno.34824
10. Parashar D, Mukherjee T, Gupta S, Kumar U, Das K. MicroRNAs in extracellular vesicles: a potential role in cancer progression. Cell Signal. 2024;121:111263. doi:10.1016/j.cellsig.2024.111263
11. Ghafouri-Fard S, Khoshbakht T, Hussen BM, Taheri M, Samadian M. A review on the role of miR-1246 in the pathoetiology of different cancers. Front Mol Biosci. 2021;8:771835. doi:10.3389/fmolb.2021.771835
12. Dou X, Hua Y, Chen Z, Chao F, Li M. Extracellular vesicles containing PD-L1 contribute to CD8+ T-cell immune suppression and predict poor outcomes in small cell lung cancer. Clin Exp Immunol. 2022;207(3):307. doi:10.1093/cei/uxac006
13. Hirigoyen U, Guilbaud C, Krejbich M, et al. Oncolytic viruses alter the biogenesis of tumor extracellular vesicles and influence their immunogenicity. Mol Ther Oncol. 2024;32(4):200887. doi:10.1016/j.omton.2024.200887
14. Bommareddy PK, Shettigar M, Kaufman HL. Integrating oncolytic viruses in combination cancer immunotherapy. Nat Rev Immunol. 2018;18(8):498. doi:10.1038/s41577-018-0014-6
15. Kaufman HL, Kohlhapp FJ, Zloza A. Erratum: oncolytic viruses: a new class of immunotherapy drugs. Nat Rev Drug Discovery. 2016;15(9):660. doi:10.1038/nrd.2016.178
16. Moulin C, Crupi MJF, Ilkow CS, Bell JC, Boulton S. Extracellular vesicles and viruses: two intertwined entities. Int J Mol Sci. 2023;24(2):1036. doi:10.3390/ijms24021036
17. Raab-Traub N, Dittmer DP. Viral effects on the content and function of extracellular vesicles. Nat Rev Microbiol. 2017;15(9):559. doi:10.1038/nrmicro.2017.60
18. Kakiuchi Y, Kuroda S, Kanaya N, et al. Local oncolytic adenovirotherapy produces an abscopal effect via tumor-derived extracellular vesicles. Mol Ther. 2021;29(10):2920. doi:10.1016/j.ymthe.2021.05.015
19. Saari H, Turunen T, Lõhmus A, et al. Extracellular vesicles provide a capsid-free vector for oncolytic adenoviral DNA delivery. J Extracell Vesicles. 2020;9(1):1747206. doi:10.1080/20013078.2020.1747206
20. Cordelier P. Unveiling the nexus: oncolytic viruses, extracellular vesicles, and immune modulation. Mol Ther Oncol. 2025;33(1):200940. doi:10.1016/j.omton.2025.200940
21. Zhang J, Chen J, Lin K. Immunogenic cell death-based oncolytic virus therapy: A sharp sword of tumor immunotherapy. Eur J Pharmacol. 2024;981:176913. doi:10.1016/j.ejphar.2024.176913
22. Feng X, Liu W, Jia X, et al. Antitumor effect and immunomodulatory mechanism of “oncolytic extracellular vesicles”. Nano Lett. 2024;24(31):9598. doi:10.1021/acs.nanolett.4c02279
23. DePeaux K, Gunn WG, Rivadeneira DB, Delgoffe GM. Treatment with oncolytic vaccinia virus infects tumor-infiltrating regulatory and exhausted T cells. J Immunother Cancer. 2024;12(8):e009062. doi:10.1136/jitc-2024-009062
24. Kim GB, Kim S, Hwang YH, et al. Harnessing oncolytic extracellular vesicles for tumor cell-preferential cytoplasmic delivery of misfolded proteins for cancer immunotherapy. Small. 2023;19(37):e2300527. doi:10.1002/smll.202300527
25. Bernardi S, Marchetti E, Torge D, Simeone D, Macchiarelli G, Bianchi S. Ultrastructural assessment of human periodontal ligament fibroblast interaction with bovine pericardium membranes: An in vitro study. Histol Histopathol. 2025;40(8):1185–12. doi:10.14670/HH-18-860
26. Jumaniyazova E, Lokhonina A, Dzhalilova D, Miroshnichenko E, Kosyreva A, Fatkhudinov T. The role of macrophages in various types of tumors and the possibility of their use as targets for antitumor therapy. Cancers. 2025;17(3):342. doi:10.3390/cancers17030342
27. Kazakova A, Sudarskikh T, Kovalev O, Kzhyshkowska J, Larionova I. Interaction of tumor‑associated macrophages with stromal and immune components in solid tumors: research progress (Review). Int J Oncol. 2023;62(2). doi:10.3892/ijo.2023.5480
28. Zhang W, Wang M, Ji C, Liu X, Gu B, Dong T. Macrophage polarization in the tumor microenvironment: emerging roles and therapeutic potentials. Biomed Pharmacother. 2024;177:116930. doi:10.1016/j.biopha.2024.116930
29. Chu X, Tian Y, Lv C. Decoding the spatiotemporal heterogeneity of tumor-associated macrophages. Mol Cancer. 2024;23(1):150. doi:10.1186/s12943-024-02064-1
30. Li Y, Liu Y, Wei X, Wang C, Zahoor Khan M, Ma Q. Probiotic extracellular vesicles reprogram macrophage immunometabolism: From gut crosstalk to host health. Gut Microbes. 2026;18(1). doi:10.1080/19490976.2026.2614115
31. Wang S, Kong L, Wang L, et al. Viral expression of NE/PPE enhances anti-colorectal cancer efficacy of oncolytic adenovirus by promoting TAM M1 polarization to reverse insufficient effector memory/effector CD8+ T cell infiltration. J Exp Clin Cancer Res. 2025;44(1):97. doi:10.1186/s13046-025-03358-y
32. Lecoultre M, Walker PR, Helali AE. Oncolytic virus and tumor-associated macrophage interactions in cancer immunotherapy. Clin Exp Med. 2024;24(1):202. doi:10.1007/s10238-024-01443-8
33. Chatelain C, Berland L, Grard M, et al. Interplay between oncolytic measles virus, macrophages and cancer cells induces a proinflammatory tumor microenvironment. Oncoimmunology. 2024;13(1):2377830. doi:10.1080/2162402x.2024.2377830
34. Zhang Y, Qian L, Chen K, et al. Oncolytic adenovirus in treating malignant ascites: a Phase II trial and longitudinal single-cell study. Mol Ther. 2024;32(6):2000. doi:10.1016/j.ymthe.2024.04.029
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.