")
Back to Archived Journals » Oncolytic Virotherapy » Volume 7
Oncolytic myxoma virus synergizes with standard of care for treatment of glioblastoma multiforme
Authors Burton C, Das A , McDonald D, Vandergrift III WA, Patel SJ, Cachia D , Bartee E
Received 6 July 2018
Accepted for publication 19 September 2018
Published 19 November 2018 Volume 2018:7 Pages 107—116
DOI https://doi.org/10.2147/OV.S179335
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Faris Farassati
Chase Burton,1,* Arabinda Das,2,* Daniel McDonald,3 William A Vandergrift III,2 Sunil J Patel,2 David Cachia,2 Eric Bartee1
1Department of Microbiology and Immunology, Medical University of South Carolina, SC, USA; 2Department of Neurosurgery, Medical University of South Carolina, SC, USA; 3Department of Radiation Oncology, Medical University of South Carolina, SC, USA
*These authors contributed equally to this work
Background: Glioblastoma multiforme (GBM) is an aggressive form of brain cancer which is associated with poor prognosis. A variety of oncolytic viruses have previously shown positive efficacy against GBM, potentially offering new treatment options for patients. One such oncolytic virus is Myxoma virus (MYXV), a rabbit-specific poxvirus that has been shown to be efficacious against a variety of tumor models including GBM.
Purpose: The purpose of this study was to test the efficacy of MYXV combined with current treatment regimens for GBM in both established cell lines as well as patient biopsy samples.
Materials and methods: U118 gliobastoma cell lines were treated under various standard of care combinations (untreated, radiation and chemotherapeutic) prior to infection with MYXV. Infection was then monitored for differences in rate of infection, titer and rate of spread. Cellular death was measured by MTT assay and Caspase-3 colorimetric assay. Patient biopsies were harvested and treated under similar treatment conditions.
Results: The addition of GBM standard of care to MYXV infection resulted in an increased rate of spread compared to single treatment with either radiation or chemotherapeutic alone. SOC did not alter viral replication or infection rates. Similar effects were seen in ex vivo patient biopsies. Cellular viability was significantly decreased with the combination therapy of SOC and MYXV infection compared to any other treatment outcome. Caspase-3 activity was also significantly increased in samples treated with combination therapy when compared to any other treatment combination.
Conclusion: Our results show that the combination of MYXV with current SOC results in both increased killing of GBM cells compared to either treatment regime alone as well as increased spread of MYXV infection. These findings lay the foundation for future in vivo studies on combining MYXV with GBM SOC.
Keywords: myxoma virus, glioblastoma multiforme, standard of care, oncolytic virotherapy
Introduction
Glioblastoma multiforme (GBM) is a form of brain cancer which arises from the glial cells of the brain and is characterized by aggressive spread.1 As with most cancers, the onset of GBM occurs later in life with most cases presenting between the ages of 45 and 75. The disease is often difficult to diagnose due to the generality of its symptoms, which can include headaches, nausea and blurred vision. The challenge of a definitive GBM diagnosis often leads to disease progression without the patient’s knowledge2 resulting in an average survival of only 12–36 months after the onset of treatment.
Currently, the accepted standard of care (SOC) for GBM focus on the use of three therapeutic options: surgery, radiation, and chemotherapeutic drugs.3–6 Surgery is often used first to remove the bulk of the tumor mass; however, this method is typically not efficient due to the invasion of malignant cells into the normal neural networks which prevents complete surgical resection.7 After surgery, patients typically undergo treatment with the chemotherapeutic drugs Temozolomide or Bevacizumab3,8 in combination with radiation treatment. The combination of these treatments serves to eliminate what remains of the non-excised tumor.9
Unfortunately, since GBM develops in the brain, it presents a unique set of therapeutic challenges not commonly associated with other types of cancer.10 One such obstacle is the cell type from which the tumors originate. Both neurons and glial cells are slow dividing cells that can evade traditional cancer treatments, such as chemotherapy and radiation, which focus on killing rapidly dividing cells. An additional challenge lies in the inherent structure of the brain and cranial cavity, which has a minimal volume making it susceptible to increased pressure and/or inflammation. These risks limit the use of directly injected chemotherapeutic drugs, since the injections can result in increased pressure unless delivered in very low dosages. In addition, the blood–brain barrier, which functions to prevent the transfer of substances either into or out of the brain, presents a challenge in delivering chemotherapeutic agents to the tumor and prevents many drugs from being used against GBM.11 Due to these therapeutic challenges, GBM prognosis has remained poor and the disease has become a focus of research to identify novel treatments which can improve the overall patient survival.12,13
One such novel treatment is oncolytic virotherapy (OV).14 OV works through injection of a non-pathogenic virus into the tumor mass.15 This injection reduces tumor burden through two primary mechanisms. The first is through the direct oncolytic effect by which infected cells are eliminated due to either lytic viral replication or the induction of programmed cell death.16,17 The second mechanism is the induction of an antitumor immune response. This response occurs due to the presence of viral pathogen associated molecular patterns that activate antigen-presenting cells. This activation primes adaptive immune responses against both viral and tumor antigens.18,19
Several oncolytic viruses have been previously tested as treatments for GBM.20 One such virus is Myxoma virus (MYXV), a normally rabbit specific member of the Leporipoxviridae. While not tested in a human model to date, MYXV is an attractive oncolytic candidate due to its inherent ability to specifically infect tumor cells as well as its non-pathogenic nature towards non-rabbit species. This results in an effective yet safe agent for use during OV in humans.21 Previous studies examining MYXV-based treatment in GBM models demonstrated a surprising effectiveness in reducing tumor burden and decreasing cell viability against established tumors in both mouse-22 and patient-derived xenograft models.23 These previous studies, however, did not examine how MYXV may interact with the current GBM SOC. The purpose of this paper is to determine how the addition of MYXV to SOC for GBM impacts the efficacy of treatment. For the purposes of this paper, SOC is defined as treatment with 1 Gray (Gy) and one of several chemotherapeutic drugs, including Temozolomide, Crizotinib, and Bevacizumab.
Materials and methods
Cell lines and reagents
BSC40 cells (Cat# CRL-2761) were purchased from American Type Culture Collection (Manassas, VA, USA). U118, GL261 and T9 cells were purchased from Mediatech (Herndon, VA, USA). All cells were cultured in DMEM (10% fetal bovine serum +1 × penicillin–streptomycin–l–glutamine Corning (Oneonta, NY, USA). Temozolomide was purchased from Sigma-Aldrich Co. (St Louis, MO, USA). Crizotinib was purchased from Selleckchem (Houston, TX, USA). Bevacizumab was purchased from Genentech, Inc. (South San Francisco, CA, USA). Western blot samples were prepared as described by Bartee et al.24 Cell viability was measured using the CellTiter-96 Non-Radioactive Cell Proliferation Assay (Promega Corporation, Fitchburg, WI, USA), referred to as the MTT assay for the purposes of this paper, according to manufacturer’s recommendations. Caspase-3 activity was measured using the commercially available Colorimetric Caspase-3 Assay Kit (CASP3C, Sigma-Aldrich Co.) according to the manufacturer’s protocol. Antibodies used for this study were purchased from Santa Cruz Biotechnology Inc., Dallas, TX, USA and include GAPDH (clone G-9, sc-365062), Caspase-3 (clone 3C119, sc-70497), p-AKT (Clone 104A282, sc-52940).
Virus preparation and infection
MYXV (strain Lausanne) expressing GFP from the intergenic region between the m135r and m136r viral reading frames (vGFP) has been previously described.25 The virus was amplified in BSC40 cells and purified using sucrose gradient centrifugation.26 For all experiments, cells were infected for one hour at room temperature according to the multiplicity of infection (MOI) stated for each experiment. Where noted, cells were treated with 1 Gy of radiation 12 hours prior to infection. After infecting for one hour, the media was replaced and therapeutic drug was added at a concentration of 10 mg/kg assuming a mass of 20 g (Figure 1A). Single step growth curves, foci forming assays, and initial infection assays were each performed as previously described.27–29
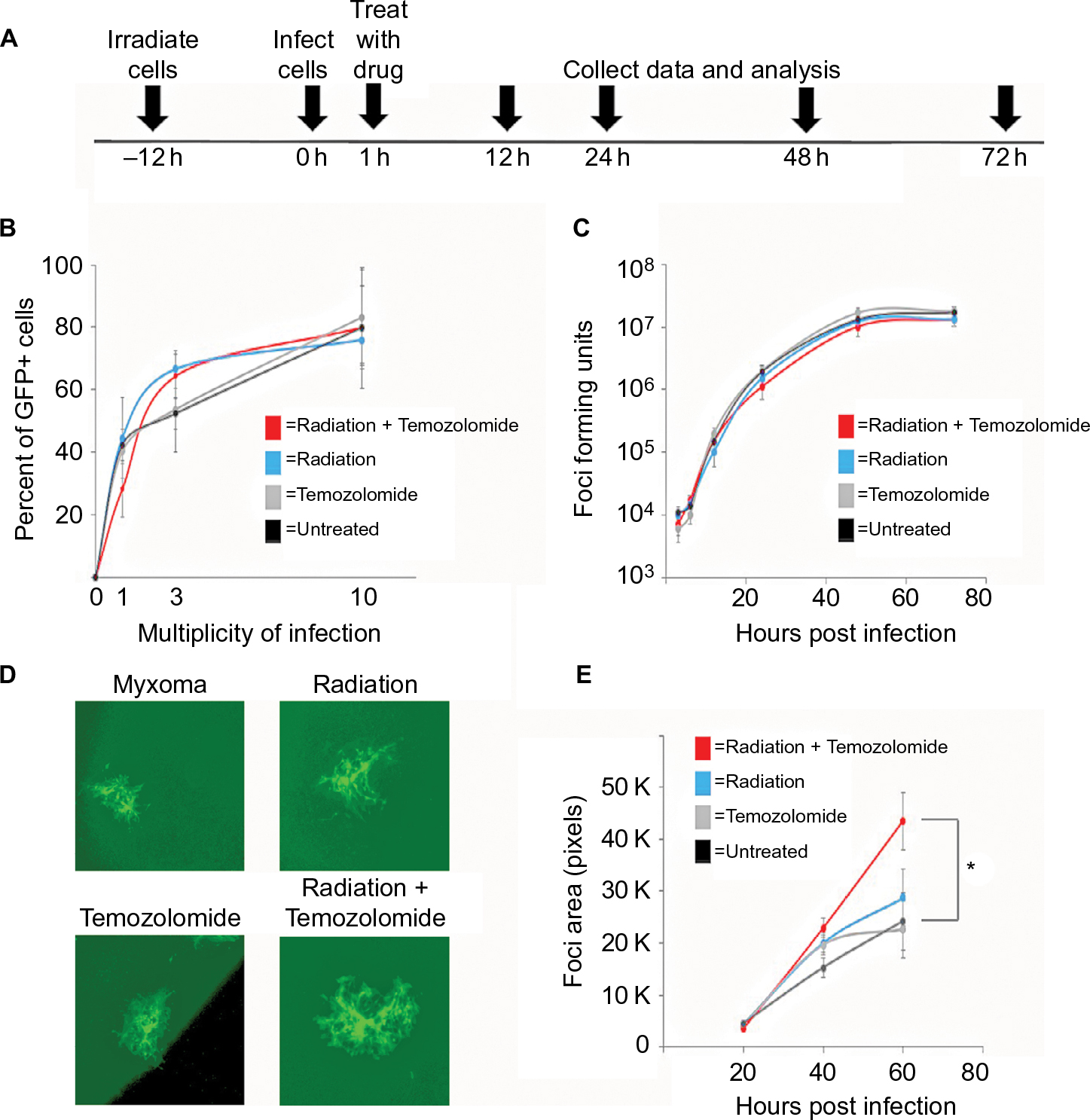 | Figure 1 Standard of care increases Myxoma virus (MYXV) spread. Notes: (A) Timeline of treatment protocol for all experiments. (B) Cells were plated in replicates onto 12-well plates. Percent of GFP+ cells were analyzed by flow cytometry 12 hours after infection with vGFP and treatment as indicated in the protocol. Data represent summation of three independent experiments (P<0.05). (C) Cells were plated into replicates on to 12-well plates. The numbers of replication competent MYXV particles were determined by titering after harvesting from cells pretreated with the indicated treatment protocols in duplicate. Data represent the summation of two independent experiments. (D) Cells were plated onto 6-well plates in replicates. Images of GFP positive foci, infected with vGFP and treated as indicated, at 4× magnification 72 hours after infection under fluorescent microscopy. (E) Mean foci area was assessed at 24-hour intervals after infection with vGFP and treated as indicated for 72 hours. There was a significant difference between cells treated with 1 Gy of radiation and untreated cells (*=P<0.011). Data are representative of three independent experiments. |
GBM patient samples
Patient samples were collected surgically from patients diagnosed with GBM according to the Response Assessment in Neuro-Oncology Criteria. Patients were evaluated based on the MRI, as well as clinical and pathological examination. Patients who received neoadjuvant chemotherapy and/or radiotherapy prior to surgery were excluded. GBM samples from three male patients (confirmed by histopathological examination by a pathologist) were analyzed. All tissue was collected and handled according to procedures approved by the MUSC Institutional Review Board.
Slice cultures were prepared from fresh human GBM specimens. Following surgical excision, the tissue was transported to the laboratory in minimal essential medium (MEM; Thermo Fisher Scientific, Waltham, MA, USA). The tissue was prepared using the standard Stoppini Slice Culture Technique at a thickness of 250 µm.30 The slices were then transferred onto membrane culture inserts (EMD Millipore, Billerica, MA, USA) in 6-well plates at a maximum of three slices per insert. Slices were cultured in MEM (Gibco, Waltham, MA, USA), supplemented with 25% Hank’s Balanced Salt Solution (with Ca and Mg; Gibco), 20% horse serum (Gibco), 2 mM l-glutamine (Braun, Bethlehem, PA, USA), 0.5% glucose, and 1% penicillin/streptomycin (Sigma-Aldrich Co.). Slices were then cultured on a liquid/air interface in a humidified incubator at 37°C and 5% CO2. The medium was changed three times a week.
Results
GBM SOC increases MYXV spread
In order to test the efficacy of combining MYXV with the SOC for GBM, we first looked at how SOC affected the ability of MYXV to establish initial infection, produce new infectious viral progeny, and spread to neighboring cells. To measure how SOC impacted initial infection, U118 cells were inoculated with varying MOIs (1, 3, 10) of vGFP in either the presence or absence of radiation (1 Gy) and/or temozolomide. Infections were allowed to progress for 12 hours after which the initial infection of cells was determined by analyzing the percent of GFP+ cells using flow cytometry. Not surprisingly, we observed an increase in GFP+ cells correlated to an increase in MOI. However, the number of GFP+ cells was not significantly impacted by addition of radiation, temozolomide, or SOC (Figure 1B). To analyze the impact of SOC on the production of new infectious progeny virus, U118 cells were either mock treated or treated with 1 Gy of radiation and then infected at an MOI of 15. Following infection, cells were treated with either saline or temozolomide. Samples were harvested at several time points postinfection (3, 6, 12, 24, 48 and 72 hours) and the number of new infectious progeny virus was analyzed by titering each sample on BSC40 cells. The results indicated that treatment with radiation, temozolomide, or SOC did not impact the production of new infectious progeny in MYXV infected U188 cells (Figure 1C). Finally, to analyze the effect of SOC on the ability of MYXV to spread from cell to cell, U188 cells were treated as above and infected with various low MOIs (0.01–0.0001). At 24-hour intervals postinfection, pictures of individual GFP+foci were taken and analyzed in Photoshop. No obvious morphological differences were observed in foci formed following any of the treatment conditions (Figure 1D). A significant increase in foci size, however, was observed 72 hours postinfection between mock and cells treated with GBM SOC (Figure 1E, P<0.05). Interestingly, this difference was not observed in groups treated with either Temozolomide or radiation alone, suggesting that it was specific to triple combination therapy. Similarly, experiments were conducted to determine whether SOC containing bevacizumab or Crizotinib also enhanced viral spread. However, these combinations were found to be too toxic to allow for accurate analysis of foci formation (Figure S1). To determine whether this increase in viral spread also occurred in primary GBM patient samples, we obtained GBM patient biopsies and tested how addition of SOC would impact MYXV infection in these samples. Each sample was either mock treated or irradiated and then infected with 1×107 FFU of vGFP. Initiation and spread of infection was then assayed by monitoring GFP expression every 24 hours after infection. While individual foci could not be identified in primary slice cultures, the results clearly indicated increased GFP expression in GBM biopsies pretreated with radiation compared to the non-irradiated samples 48 hours postinfection (Figure 2).
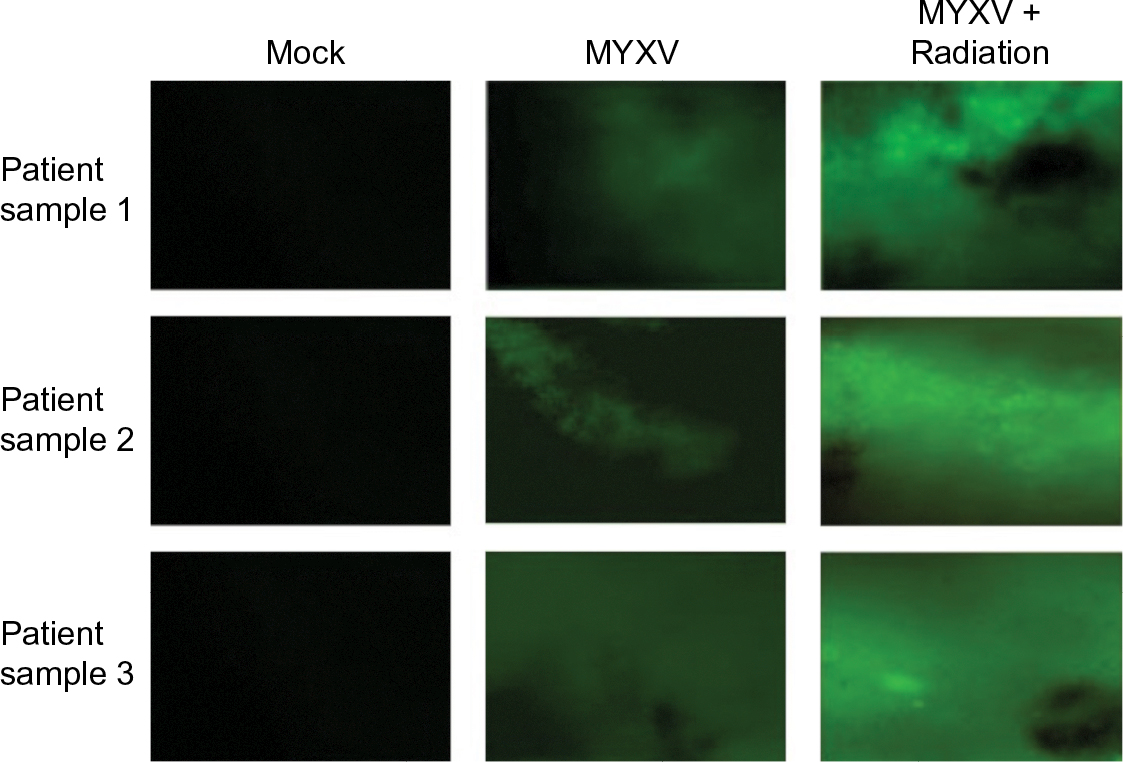 | Figure 2 Infection of primary GBM biopsies. Notes: Biopsies were collected from three patients with GBM. Samples were prepared via the standard Stoppini Slice Culture as outlined. Samples were then treated as indicated and infected with vGFP for 48 hours prior to imaging at a magnification of 4×. Images taken via fluorescent microscopy. Abbreviations: GBM, glioblastoma multiforme; MYVX, Myxoma virus. |
MYXV combined with SOC decreases GBM cell viability
We next set out to determine if the combination of viral treatment and SOC would increase killing of GBM. In order to assess this, we assayed cellular viability and Caspase-3 activity following treatment with MXYV in either the presence or absence of SOC. U118, T9 and GL261 cells were infected at an MOI of 15 in either the presence or absence of radiation, chemotherapeutic agent, or SOC. Measurements were taken at 72 hours post treatment. While all mono-therapies decreased cellular viability to some extent, we observed that the combination therapy, including MYXV, radiation and a chemotherapeutic drug resulted in significantly higher decrease in cell viability compared to any other condition (Figure 3A). These results were consistent across all three chemotherapeutic drugs tested. Similarly, when using either Bevacizumab or Crizotinib as the chemotherapeutic drug, we observed a significant increase in Caspase-3 activity when cells were treated with the triple combination of MYXV, radiation, and drug (Figure 3B). Interestingly, we observed a much lower increase in Caspase-3 activity with Temozolomide with either of the two other chemotherapeutic agents, though it was still significantly different from that with SOC combined with radiation and MYXV (Figure 3B). Consistent with these results, we observed both increased levels of cleaved Caspase-3 and decreased phosphorylation of AKT in samples treated with MYXV and SOC (Figure 3C). To again confirm the previous results in primary GBM samples, experiments similar to those described above were performed in primary GBM patient biopsies. Similar to what was seen in cell lines, the results demonstrated that the combination of MYXV and SOC significantly reduced cellular viability of primary GBM cells, more than other treatment regimens (Figure 4A). This reduction in cellular viability was again correlated with increased Caspase-3 activity and cleavage as well as decreased phosphorylation of AKT (Figure 4B and C).
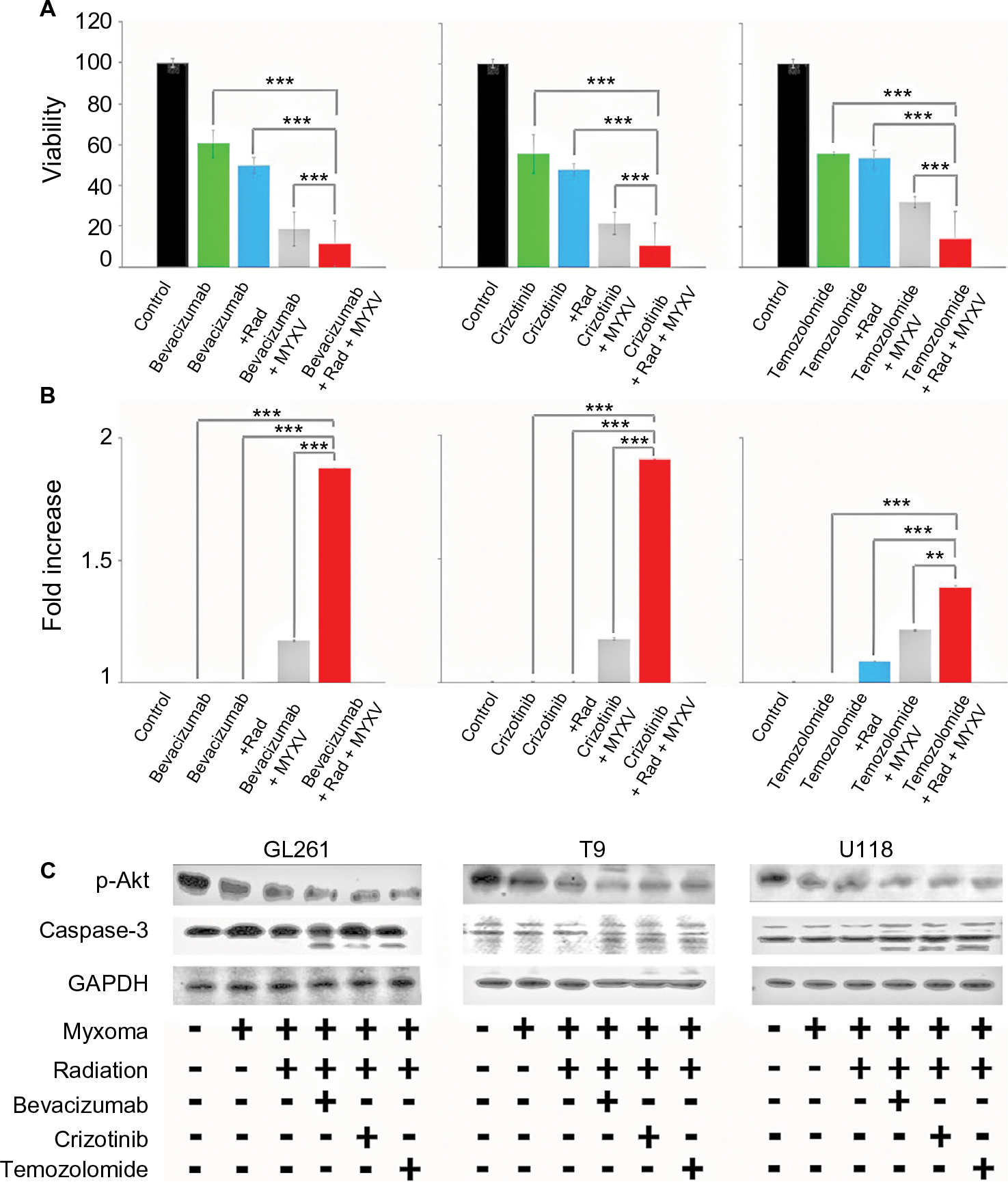 | Figure 3 In vitro analysis of cell viability of combined treatment conditions. Notes: (A) Cells were plated in replicates and treated as indicated prior to infection with vGFP. Cell viability of U118 tumor cells measured by MTT assay as percent of control at 72 hours postinfection. All treatments resulted in significant decrease in viability compared to untreated cells (P<0.001, individual values not shown). Combination therapy of chemotherapeutic drug, MXYV and radiation significantly reduced cell viability compared to all other treatment groups at 72 hours (***<0.001). Data are the summation of three independent experiments. (B) Cells were plated in replicates and treated as indicated prior to infection with vGFP. Fold increase of Caspase-3 activity compared to control was measured using a commercially available kit, as described, at 24-hour intervals postinfection. Combination treatment of chemotherapeutic drug, MXYV and radiation significantly increased Caspase-3 activity compared to any other treatment group at 72 hours (***<0.001, **<0.01). Data are the summation of three independent experiments. (C) Western blot analysis of p-Akt, Caspase-3 and actin in GL261 mouse brain tumor cells, T9 rat brain tumor cells and U118 human tumor cells. Samples were treated with each chemotherapeutic agent either with or without radiation and MYXV. Abbreviations: MYXV, Myxoma virus; Rad, radiation; . |
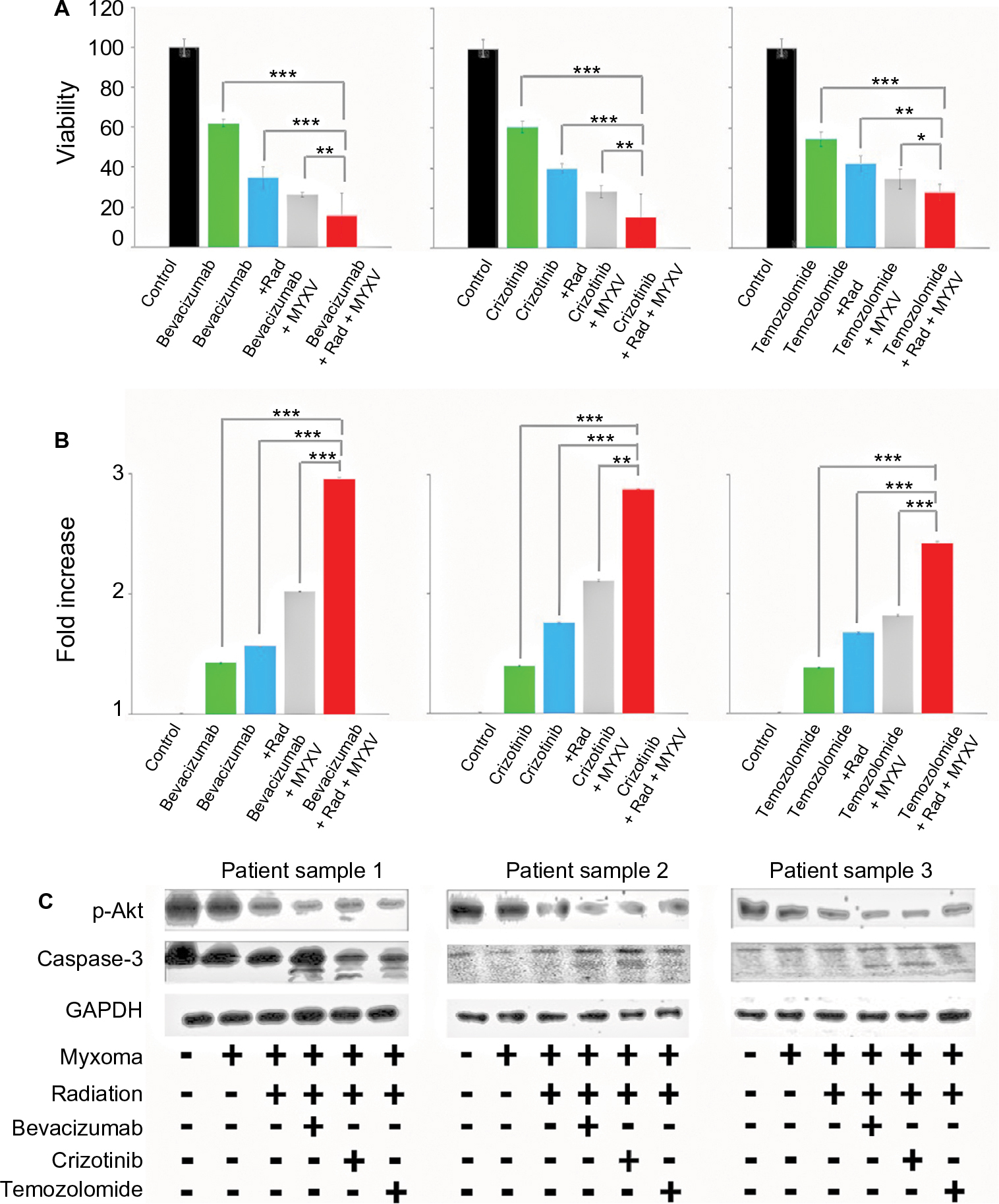 | Figure 4 Ex vivo analysis of cell viability of combined treatment conditions. Notes: (A) Patient biopsies were harvested, cultured in duplicate 96-well plates, infected with vGFP and treated as indicated. Cell viability of patient biopsies was measured by MTT assay as described at 24-hour intervals postinfection. Addition of all treatments resulted in a significant decrease in cell viability (P<0.01, individual values not shown). Combination treatment of chemotherapeutic agent, radiation and MYXV showed a significant decrease in cell viability compared to any other treatment combination (***<0.001, **<0.01, *<0.05). Data are the summation of three independent experiments. (B) Patient biopsies were harvested, cultured in duplicate 96-well plates, infected with vGFP and treated as indicated. Caspase-3 activity of patient biopsies was measured by a colorimetric kit, as described, at 24-hour intervals postinfection. Fold increase of Caspase-3 activity was calculated as compared to control. Addition of all treatments resulted in a significant increase in Caspase-3 activity (P<0.01, individual values not shown). Combination treatment of chemotherapeutic agent, radiation and MYXV showed a significant increase in Caspase-3 activity compared to any other treatment combination (***<0.001, **<0.01). Data are the summation of three independent experiments. (C) Western blot analysis of p-Akt, Caspase-3 and actin in three patient GBM biopsies. Samples were treated with each chemotherapeutic agent either with or without radiation and MYXV. Abbreviations: GBM, Glioblastoma; MYXV, Myxoma virus; Rad, radiation; MYXV, Myxoma virus; GBM, Glioblastoma. |
Discussion
The poor outcomes of a GBM diagnosis have remained static over the years. However, OV is to be considered a promising novel treatment option for GBM.20 Current oncolytic viruses, including adenovirus, newcastle disease virus, herpes simplex virus, poliovirus, measles virus, H1 parvovirus, reovirus and vaccinia virus have been tested against GBM models and have shown therapeutic efficacy.31,32 While these viruses have shown oncolytic potential, there is risk associated with their natural human pathogenicity. In light of this, MYXV a non-human pathogenic virus which has previously been shown to be an effective oncolytic virus against multiple myeloma,33 has shown efficacy as a potential GBM treatment.34 While there is some literature which suggests that the combination of MYXV and chemotherapeutics, such as rapamyacin,22 may increase the efficacy of treatment, little work has been done to elucidate the interaction between MYXV and SOC for GBM. In this study, we were able to show that MYXV is a suitable oncolytic virus for GBM in vitro and synergistically increases efficacy when combined with SOC.
In order to be effective, oncolytic viruses must initiate infection and successfully replicate within tumor cells, resulting in cell death. Our results indicate that combining MYXV with SOC does not alter initial infection or the ability of MYXV to replicate in GBM cells. Importantly, we did observe that viral spread was increased upon MYXV infection with both radiation and Temozolomide. Furthermore, a similar result was observed in patient biopsies in terms of fluorescence, both with and without radiation. Interestingly, we observe a discrepancy between the in vitro cell lines and ex vivo patient biopsies in terms of viral spread in the presence of radiation. We hypothesize that this discrepancy is due to the makeup of the patient biopsies compared to cells grown in culture. While cultured cells offer a fairly homozygous population, patient biopsies are comprised of a diverse cellular makeup. It is possible that the patient biopsies then react differently to radiation and viral treatment than how cultured cells do, resulting in the observed discrepancy of spread between samples. Additionally, differences between these two samples sets could be related to tumor treatment prior to the harvesting of the biopsy. It is possible that part of the initial treatment primed the cells in the biopsies to allow for increased spread, priming which cultured cells would not have undergone. The differences between initial infection and spread are curious, and merit further inquiry, particularly in terms of patient biopsies, as they are the most clinically relevant model. This could be due to a number of factors, including alterations in cellular morphology, such as cellular junctions, which allows MYXV to spread between the cells more effectively than under normal conditions. Additionally, treatment of the cells with SOC could result in the increased ability of the virus to egress from infected cells, either through changes in cellular permeability or by some other mechanism, and would explain the differences in the foci forming assay but not in the single step growth curve or the initial infection assay.
We next decided to check the effect that our treatment protocol had on cell viability by both, an MTT assay and Caspase-3 colorimetric assay. The results of our MTT assay show that the combination of SOC and MYXV greatly reduced cell viability compared to either treatment alone. This increase correlated with similar increases in Caspase-3 activation following SOC using all three chemotherapeutic drugs. Interestingly, while all drugs showed an increase in Caspase-3 activity when combined with MYXV, a markedly smaller increase was observed when using temozolomide compared to either Bevacizumab or Crizotinib. We hypothesize that this is the result of different mechanisms of action for each chemotherapeutic drug. Temozolomide methylates the DNA of cells, whereas Bevacizumab targets the VEGF/VEGF-R interaction to prevent cell survival. Another possibility is the half-life of Temozolomide, which is 1.8 hours in vitro. This could potentially hinder treatment outcomes, especially over the course of experiments which extend out several days. These differences could lead to cell death through pathways other than Caspase-3, and thus explain the differences we observed between p-Akt and Caspase-3 levels. Future studies are needed to find the true cause of this difference.
In addition to our work in cell lines, we also tested the combination of MYXV and SOC in primary GBM patient biopsies. We first saw that the addition of radiation to the patient samples resulted in increased GFP expression in tumor slices, even in the absence of chemotherapeutic drug. These results differ somewhat from our results with cultured cells where the addition of both radiation and drug was needed to increase viral spread. This could be due to the makeup of patient biopsies, as they contain more heterogeneous cell populations than purified cultured cell lines. SOC may therefore affect individual populations of cells differently and may explain the inconsistent results between the biopsies and cultured cells. Further investigation into in vivo models is needed in order to determine the potential cause of this difference and make the results more clinically relevant to patient care.
Replication of the MTT and Caspase-3 assays in our ex vivo model showed similar results as observed in vitro. We did, however, observe that the Caspase-3 assay showed a larger fold change in activity than the ex vivo samples. This could be due to the heterogenicity of the tumor cells in the patient biopsy vs that of the cultured cells that could be undergoing death by different mechanisms. These results could also be a product of the harvesting process of patient tumors where the stimuli of the patient-specific tumor microenvironment may affect how these cells react to different treatments when compared to the cultured U118 cell lines.
Taken together, this data suggests that MYXV combined with SOC can increase efficacy of treatment for GBM by both increasing the apoptosis of tumor cells as well as by enhancing the spread of viral infection. Whether this data suggests that such effects would also deliver significant benefits in vivo, where the immunological impact of SOC must be taken into account, remains to be determined. We hope that this data provides the basis for subsequent studies looking at the combination of MYXV and SOC in a biologically relevant model of GBM.
Compliance with ethical standards
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or National Research Committee and with the 1964 Declaration of Helsinki and its later amendments or comparable ethical standards as determined by the MUSC Institutional Review Board. Written informed consent was obtained from all individual participants included in the non-therapeutic human study.
Acknowledgments
Completion of this project was made possible by funding to Dr Eric Bartee from NIH-NIAID (1K22AI095372-01A1), NIH-NCI (1R01CA194090-01A1), and ACS (RSG-17-047-01-MPC). This work was also supported in part by grants from the Hollings Cancer Center, including a support grant (P30 CA138313), the Fund for Brain Tumor Research, and Funds from the Department of Neurosurgery.
Disclosure
The authors report no conflicts of interest in this work.
References
Carlsson SK, Brothers SP, Wahlestedt C. Emerging treatment strategies for glioblastoma multiforme. EMBO Mol Med. 2014;6(11):1359–1370. | ||
Phillips HS, Kharbanda S, Chen R, et al. Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell. 2006;9(3):157–173. | ||
Stupp R, Mason WP, van den Bent MJ, et al; European Organisation for Research and Treatment of Cancer Brain Tumor and Radiotherapy Groups; National Cancer Institute of Canada Clinical Trials Group. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352(10):987–996. | ||
Wong ET, Lok E, Swanson KD, et al. Response assessment of NovoTTF-100A versus best physician’s choice chemotherapy in recurrent glioblastoma. Cancer Med. 2014;3(3):592–602. | ||
Wong ET, Yamaguchi NH. Treatment advances for glioblastoma. Expert Rev Neurother. 2011;11(10):1343–1345. | ||
Omuro A, DeAngelis LM. Glioblastoma and other malignant gliomas: a clinical review. JAMA. 2013;310(17):1842–1850. | ||
Baumert BG, Hegi ME, van den Bent MJ, et al. Temozolomide chemotherapy versus radiotherapy in high-risk low-grade glioma (EORTC 22033-26033): a randomised, open-label, phase 3 intergroup study. Lancet Oncol. 2016;17(11):1521–1532. | ||
Hartmann C, Hentschel B, Simon M, et al; German Glioma Network. Long-term survival in primary glioblastoma with versus without isocitrate dehydrogenase mutations. Clin Cancer Res. 2013;19(18):5146–5157. | ||
Venur VA, Peereboom DM, Ahluwalia MS. Current medical treatment of glioblastoma. Cancer Treat Res. 2015;163:103–115. | ||
Fiorentino A, De Bonis P, Chiesa S, Balducci M, Fusco V. Elderly patients with glioblastoma: the treatment challenge. Expert Rev Neurother. 2013;13(10):1099–1105. | ||
Kim SS, Harford JB, Pirollo KF, Chang EH. Effective treatment of glioblastoma requires crossing the blood-brain barrier and targeting tumors including cancer stem cells: The promise of nanomedicine. Biochem Biophys Res Commun. 2015;468(3):485–489. | ||
Patil CG, Yi A, Elramsisy A, et al. Prognosis of patients with multifocal glioblastoma: a case-control study. J Neurosurg. 2012;117(4):705–711. | ||
Dolecek TA, Propp JM, Stroup NE, Kruchko C. CBTRUS statistical report: primary brain and central nervous system tumors diagnosed in the United States in 2005-2009. Neuro Oncol. 2012;14(Suppl 5):v1–v49. | ||
Smith ER, Chiocca EA. Oncolytic viruses as novel anticancer agents: turning one scourge against another. Expert Opin Investig Drugs. 2000;9(2):311–327. | ||
Russell SJ, Peng KW. Viruses as anticancer drugs. Trends Pharmacol Sci. 2007;28(7):326–333. | ||
Ogbomo H, Zemp FJ, Lun X, et al. Myxoma virus infection promotes NK lysis of malignant gliomas in vitro and in vivo. PLoS One. 2013;8(6):e66825. | ||
Sze DY, Reid TR, Rose SC. Oncolytic virotherapy. J Vasc Interv Radiol. 2013;24(8):1115–1122. | ||
Miyamoto S, Inoue H, Nakamura T, et al. Coxsackievirus B3 is an oncolytic virus with immunostimulatory properties that is active against lung adenocarcinoma. Cancer Res. 2012;72(10):2609–2621. | ||
Ahmad Z, Kratzke RA. Novel oncolytic viral therapies in patients with thoracic malignancies. Oncolytic Virother. 2016;6:1–9. | ||
Wollmann G, Ozduman K, van den Pol AN. Oncolytic virus therapy for glioblastoma multiforme: concepts and candidates. Cancer J. 2012;18(1):69–81. | ||
Stanford MM, Shaban M, Barrett JW, et al. Myxoma virus oncolysis of primary and metastatic B16F10 mouse tumors in vivo. Mol Ther. 2008;16(1):52–59. | ||
Zemp FJ, Lun X, McKenzie BA, et al. Treating brain tumor-initiating cells using a combination of myxoma virus and rapamycin. Neuro Oncol. 2013;15(7):904–920. | ||
McKenzie BA, Zemp FJ, Pisklakova A, et al. In vitro screen of a small molecule inhibitor drug library identifies multiple compounds that synergize with oncolytic myxoma virus against human brain tumor-initiating cells. Neuro Oncol. 2015;17(8):1086–1094. | ||
Bartee MY, Dunlap KM, Bartee E. Myxoma virus induces ligand independent extrinsic apoptosis in human myeloma cells. Clin Lymphoma Myeloma Leuk. 2016;16(4):203–212. | ||
Bartee MY, Dunlap KM, Bartee E. Tumor-Localized Secretion of Soluble PD1 Enhances Oncolytic Virotherapy. Cancer Res. 2017;77(11):2952–2963. | ||
Smallwood SE, Rahman MM, Smith DW, McFadden G. Myxoma virus: propagation, purification, quantification, and storage. Curr Protoc Microbiol. 2010;17(1):14A.1.1–14A.1.20. | ||
Wolfe AM, Dunlap KM, Smith AC, Bartee MY, Bartee E. Myxoma Virus M083 is a virulence factor which mediates systemic dissemination. J Virol. 2018;92(7):e02186-e02217. | ||
Chan WM, Bartee EC, Moreb JS, Dower K, Connor JH, McFadden G. Myxoma and vaccinia viruses bind differentially to human leukocytes. J Virol. 2013;87(8):4445–4460. | ||
Bartee E, Chan WM, Moreb JS, Cogle CR, McFadden G. Selective purging of human multiple myeloma cells from autologous stem cell transplantation grafts using oncolytic myxoma virus. Biol Blood Marrow Transplant. 2012;18(10):1540–1551. | ||
Seidl AH, Rubel EW. A simple method for multiday imaging of slice cultures. Microsc Res Tech. 2010;73(1):37–44. | ||
Desai R, Suryadevara CM, Batich KA, Farber SH, Sanchez-Perez L, Sampson JH. Emerging immunotherapies for glioblastoma. Expert Opin Emerg Drugs. 2016;21(2):133–145. | ||
Shchelkunov SN, Razumov IA, Kolosova IV, Romashchenko AV, Zavjalov EL. Virotherapy of the malignant U87 human glioblastoma in the orthotopic xenotransplantation mouse SCID model. Dokl Biochem Biophys. 2018;478(1):30–33. | ||
Bartee E, Bartee MY, Bogen B, Yu XZ. Systemic therapy with oncolytic myxoma virus cures established residual multiple myeloma in mice. Mol Ther Oncolytics. 2016;3:16032. | ||
Lun X, Yang W, Alain T, et al. Myxoma virus is a novel oncolytic virus with significant antitumor activity against experimental human gliomas. Cancer Res. 2005;65(21):9982–9990. |
Supplementary material
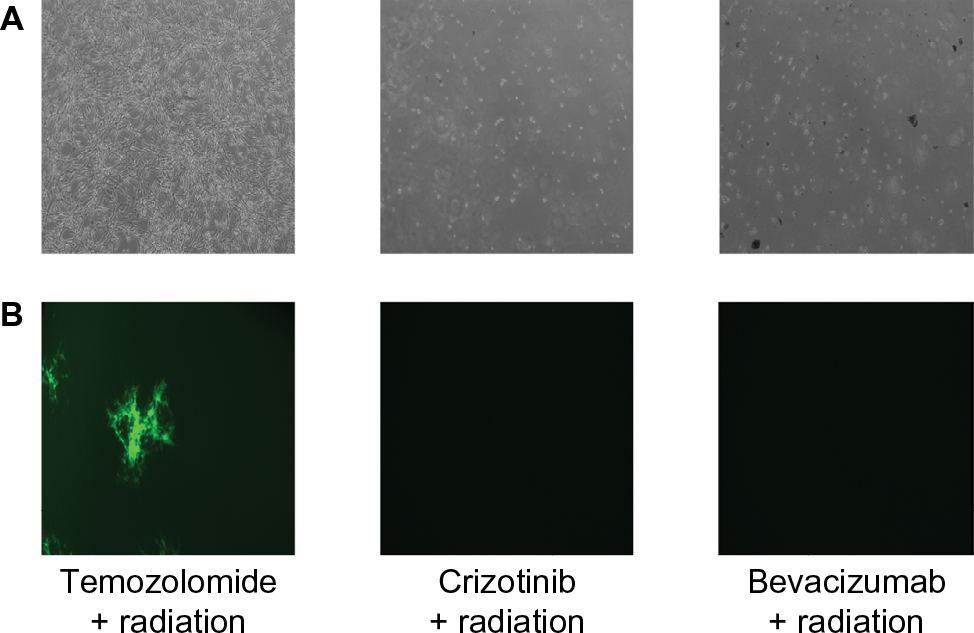 | Figure S1 (A) Images of U118 cells treated with the corresponding treatment viewed under phase at 72 hours after infection. (B) GFP positive foci of infected U118 cells treated with the corresponding treatment regimen 72 hours after infection. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.