Back to Archived Journals » Oncolytic Virotherapy » Volume 5
Oncolytic adenovirus-mediated therapy for prostate cancer
Received 10 March 2016
Accepted for publication 22 April 2016
Published 14 July 2016 Volume 2016:5 Pages 45—57
DOI https://doi.org/10.2147/OV.S63047
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Faris Farassati
Katrina Sweeney, Gunnel Halldén
Centre for Molecular Oncology, Barts Cancer Institute, Queen Mary University of London, London, UK
Abstract: Prostate cancer is a leading cause of cancer-related death and morbidity in men in the Western world. Tumor progression is dependent on functioning androgen receptor signaling, and initial administration of antiandrogens and hormone therapy (androgen-deprivation therapy) prevent growth and spread. Tumors frequently develop escape mechanisms to androgen-deprivation therapy and progress to castration-resistant late-stage metastatic disease that, in turn, inevitably leads to resistance to all current therapeutics, including chemotherapy. In spite of the recent development of more effective inhibitors of androgen–androgen receptor signaling such as enzalutamide and abiraterone, patient survival benefits are still limited. Oncolytic adenoviruses have proven efficacy in prostate cancer cells and cause regression of tumors in preclinical models of numerous drug-resistant cancers. Data from clinical trials demonstrate that adenoviral mutants have limited toxicity to normal tissues and are safe when administered to patients with various solid cancers, including prostate cancer. While efficacy in response to adenovirus administration alone is marginal, findings from early-phase trials targeting localized and metastatic prostate cancer suggest improved efficacy in combination with cytotoxic drugs and radiation therapy. Here, we review recent progress in the development of multimodal oncolytic adenoviruses as biological therapeutics to improve on tumor elimination in prostate cancer patients. These optimized mutants target cancer cells by several mechanisms including viral lysis and by expression of cytotoxic transgenes and immune-stimulatory factors that activate the host immune system to destroy both infected and noninfected prostate cancer cells. Additional modifications of the viral capsid proteins may support future systemic delivery of oncolytic adenoviruses.
Keywords: replication selective, virotherapy, combination therapy, clinical trials, gene deletion, transgene
Introduction
Prostate cancer is the second most frequently diagnosed cancer in men in the Western world, accounting for >13% of all new cancer cases, and is the second cause of male cancer mortality (World Health Organization, European Cancer Observatory, France) (National Cancer Institute, Surveillance, Epidemiology, and End Results Program [NIH SEER], USA).1,2 Development of prostate cancer is strongly associated with aging, and both incidence and mortality rates have been predicted to increase globally during the next decades.3 While the incidence rate is highest in Northern European countries and the US and lowest in Asia, the mortality rate is highest in less developed countries.4,5 Susceptibility to develop prostate cancer has been associated with environmental factors, dietary and hormonal agents, abdominal adiposity, diabetes, smoking, prostatitis, and genetic predisposition.6–9 An established risk factor is advanced age with >60% of all diagnosed cases in men older than 70 years.3 Men of African genetic background have two to three times higher risk of developing aggressive disease with earlier onset, compared with Caucasian men (National Cancer Institute, Surveillance, Epidemiology, and End Results Program [NIH SEER], USA).10 Almost all prostate cancers appear to be sporadic, with only 15% of cases demonstrated to have clear hereditary genetic causes.9
The majority of prostate adenocarcinomas are indolent and present as localized cancers and prostatic intraepithelial neoplasias (PINs) that are slow growing and limited to the prostate gland.11,12 Less than 30% of cases develop into locally invasive and aggressive tumors. Localized PINs and early invasive prostate cancers are dependent on androgens and potent androgen receptor (AR) activity for growth. In the normal prostate gland, AR functions as an essential differentiation factor and is expressed at low levels. In contrast, in prostate cancer, AR is frequently overexpressed and its function is deregulated to support cell proliferation and tumor growth. Therefore, the focus on emerging therapeutics has been to target the androgen–AR pathway, thereby depriving the tumors of essential growth stimuli.13 Androgen-dependent disease initially responds well to androgen-deprivation therapies (ADTs); however, tumors frequently escape and become castration resistant, developing into late-stage aggressive and metastatic disease with poor prognosis.14–16 Aggressive disease is characterized by invasion through the prostate capsule and spread to surrounding lymph nodes and distal organs, including bone, liver, and lung.17 Although many elderly men may die from other causes associated with advanced age, the metastases in vertebrae and pelvic bones cause severe morbidity, including pain and decreased quality of life.18,19 Treatment of late-stage metastatic disease includes chemotherapy and radiation therapy but have mostly palliative effects, and no curative therapies are currently available. Consequently, there is a need to develop more efficacious interventions.
In this review, we describe recent progress in the development of replication-selective (oncolytic) viruses as promising biological therapeutics targeting prostate cancer through their unique mechanisms of action that counteract drug resistance, including resistance to drugs targeting androgen signaling. Oncolytic viruses provide cancer cells with new modes of death through viral lysis and/or immune-mediated killing. The focus is on adenovirus-based vectors because of extensive and favorable clinical data, the efficient infection and replication in prostate adenocarcinomas, and the synergistic interaction with cytotoxic drugs and radiation therapy that significantly improves on antitumor efficacy.
Molecular alterations in prostate cancer and current therapies
The current treatment of choice for progressing prostate cancer is ADT, including antiandrogens (eg, flutamide and bicalutamide) and/or pituitary downregulators (eg, gonadotropin-releasing hormone agonists) or pharmaceutical estrogens.14,20 Androgen deprivation results in cell death and cell cycle arrest followed by tumor regression; however, a castration-resistant phenotype inevitably emerges, although its origin is not entirely understood.20 One possibility is that multiple independent foci arise through adaptation and expansion of transformed progenitor/stem cell clones developing into cancer stem cells that serve as a source of both androgen-dependent and -independent cells.21–23 Interestingly, a monoclonal origin of prostate tumors has also been indicated.24
In spite of castration resistance, AR continues to play a major role as a driver in prostate cancer growth and progression. Molecular alterations render the AR less dependent on androgens for activation and include AR gene amplification and development of androgen hypersensitivity, AR mutations resulting in promiscuous ligand binding or androgen-independent activation, overactive AR variants, intratumoral androgen production, and mutations and deregulation of coactivators and corepressors.20,25,26 The molecular alterations of AR regulation and signaling in prostate cancer have been thoroughly reviewed in two recent excellent reviews.15,27 Typical cancer-related genomic alterations are also present in prostate cancer, including upregulation of cell survival and growth pathways, deregulation of cell cycle control, and inhibition of apoptosis.11,28 Through extensive genomic profiling of prostate tumors, numerous alterations in copy number and chromosomal rearrangements have been identified. Amplifications in chromosome 8q and losses in 3p, 8p, 10q, 13q, and 17p are some examples. Key regulatory genes have been mapped within these chromosome regions, including the homeobox protein Nkx3.1, the phosphatase and tensin homolog deleted on chromosome 10 (PTEN), c-Myc, and cytochrome P450 family 17 (CYP17).9,15,28,29 Mutations in the breast cancer antigens 1 and 2 (BRCA-1 and -2) are associated with a higher risk of hereditary prostate cancer with earlier onset.9,30 A major chromosomal rearrangement is the generation of the fusion gene transmembrane protease, serine 2:v-ets erythroblastosis virus E26 oncogene homolog (TMPRSS2-ERG), detected in 15% of high-grade PIN lesions and 50% of localized prostate cases.31,32 The TMPRSS2-ERG fusion results in overexpression of the oncogenic transcription factor ERG from the androgen-regulated TMPRSS2-ERG promoter and demonstrates that AR is frequently overexpressed and signaling is intact in these tumors.33 There is strong evidence that the fusion is caused by AR activation of AR response elements (AREs) in the TMPRSS2-ERG promoter during the early stages of transformation.32
The first-line treatment for castration-resistant prostate cancer is, in the majority of cases, docetaxel or more recently cabazitaxel, with and without radiation therapy; however, unfortunately, resistance rapidly develops.34 Interestingly, it was recently demonstrated that simultaneous administration of docetaxel with ADT prolonged survival with 13.6 months compared with the current regimen of ADT first.35 Newer agents that overcome some of the resistance mechanisms are currently on the market or in clinical trials, for example, enzalutamide (Xtandi or MDV-3100), ARN-509, ODM-201, and the CYP17A1 inhibitor abiraterone acetate (Zytiga).36–38 Ultimately, resistance also develops to these agents since the mechanisms of action are similar to earlier ADTs.
Based on the essential role of androgen–AR signaling in prostate cancer progression, it is not surprising that gene therapy vectors and oncolytic adenoviruses targeting prostate cancer have been constructed with various combinations of AREs to either drive cytotoxic transgene expression or viral replication. The most frequently used AREs were derived from the AR-binding enhancer/promoter regulatory domains in the prostate-specific antigen (PSA), prostate-specific membrane antigen (PSMA), and the human glandular kallikrein 2 (hKLK2) genes. More recently, replication-selective AR-independent oncolytic viruses have been demonstrated to be safe and potent and to efficiently attenuate the growth of prostate cancer cells by utilizing additional cancer-specific genetic alterations.
Virotherapy
Human adenoviruses have evolved to propagate and spread in human tissues of epithelial origin and can be modified to target only cancer cells and to reinforce drug-induced apoptosis and immune-mediated tumor cell killing. Numerous viral mutants have been evaluated in clinical trials targeting solid cancers including nonreplicating vectors for expression of cytotoxic transgenes and replication-selective oncolytic viral mutants unable to propagate in normal cells. The majority of trials employed mutants from the Adenoviridae family, mainly species C serotype 5 and chimeras of serotype 5 with species B1 and B2. Oncolytic viruses derived from other DNA and RNA families have also been evaluated clinically, for example, Poxviridae (eg, Pexa-Vec;JX-594, GL-ONC1;GLV-1h68),39 Herpesviridae (eg, T-VEC),40 Paramyxoviridae (eg, Measles virus), Reoviridae (eg, Reolysin, Reosyn),41 Rhabdoviridae (eg, Newcastle disease virus), and Picornaviridae (eg, Coxsackie virus).42–45 However, the historical predominance of adenovirus as a virotherapy agent is due to its excellent clinical safety record with self-limiting, mostly flu-like side effects, no integration into the host genome, ease of manipulation of the small 36 kb DNA genome, its natural tropism to epithelial cells and adenocarcinomas, and the feasibility of high yield production under good manufacturing procedures.46 Replicating adenoviral mutants can infect, replicate in, and lyse both proliferating and nonproliferating cells, an advantage when targeting the frequently slow-growing or dormant prostate cancers, and the expression of transgenes can be engineered to occur both prior to and during viral replication. The early viral gene E1A promotes apoptosis induction in combination with other cytotoxic agents and is independent of viral replication (Figure 1A), an important feature since frequently viral propagation is attenuated when combined with chemotherapeutic drugs.47–49 Deletion of the antiapoptotic E1B19K gene further enhances drug-induced apoptosis and resensitizes drug-resistant cancer cells.47–51 Deletion of the viral immune modulatory E3gp19K gene can promote tumor antigen presentation and stimulate an immune response that targets both infected and noninfected cancer cells, a significant advantage in light of the recent discoveries of tumor-mediated immune checkpoint inhibition.45,52,53 Antibodies that target and prevent the inhibition of immune checkpoints have also been inserted as transgenes in the viral genome to further stimulate host immune cell activation to attack the cancer, for example, anti-PD1, anti-PDL1, and anti-CTLA4.52,54
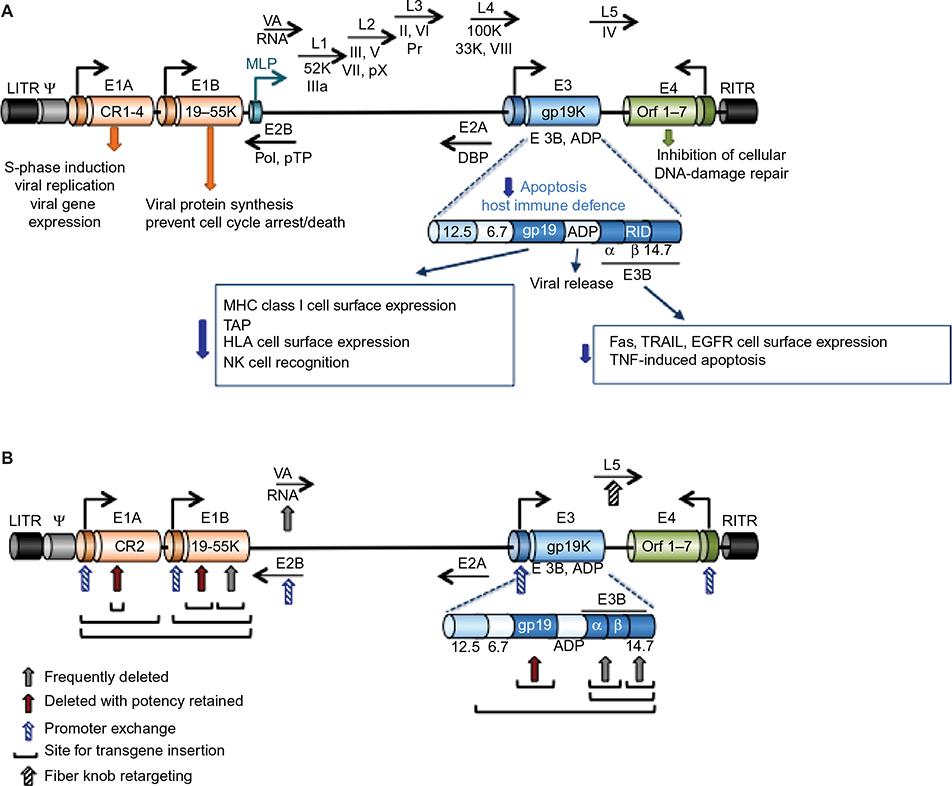 | Figure 1 Illustration of the organization and structure of the Ad5 genome. Notes: (A) Graphic representation of the Ad5 linear 36 kb genome with selected genes indicated at the approximate locations. For simplicity, only viral genes essential for viral propagation and genes relevant to the generation of viral vectors for cancer therapy are included. The E1A gene is constitutively expressed and is the first gene to be expressed after viral infection. The E1A products are essential for viral genome amplification, protein synthesis, and viral replication and drive the expression of other early viral genes (E1B–E4) that are essential for viral propagation prior to late gene expression and virion assembly. Late gene expression is initiated from the MLP with the late genes (L1–5) mostly coding for structural proteins essential for virion assembly, including hexon (L3), penton (L2), fiber (L5), and the viral protease (L3; Pr). The genes of the E3 immunomodulatory domain (E3 12.5K, E3 6.7K, E3gp19K, and the E3B proteins RIDα and β, and 14.7K) are shown in the expanded view with the most important functions indicated. The E2B genes code for the viral DNA polymerase (Pol) and the pTP, and the E2A genes code for the viral DBP. Both E2A and E2B are essential for viral DNA synthesis. VA-RNAs inhibit the cellular PKR that is activated in response to viral infection. The LITR and RITR indicate the left and right inverted terminal repeats, respectively, and Ψ is the packaging sequence. (B) Simplified view of the viral genome to illustrate sites frequently used to construct both replication-defective and replication-selective adenoviral mutants. Common sites for transgene insertion, exchange of promoters and generation of complementation-deletion mutants are indicated with arrows and lines. See text for examples of transgenes and promoters. Abbreviations: Ad5, adenovirus serotype 5; MLP, major late promoter; pTP, precursor terminal protein; DBP, DNA-binding protein; PKR, protein kinase R; ADP, adenovirus death protein; MHC, major histocompatibility complex; HLA, human leukocyte antigen; NK, natural killer; EGFR, epidermal growth factor receptor; TNF, tumor necrosis factor. |
The current lack of curative interventions for late-stage prostate cancer and the development of treatment resistance necessitate therapeutic combination strategies that may be more efficiently administered together with optimized multimodal viruses. The prostate gland is an accessible target for intratumoral administration of virotherapy through perineal or transrectal delivery. Response to therapy can be monitored through biopsy sampling through the same routes, in addition to monitoring serum levels of PSA and/or urinary prostate cancer antigen 3 (PCA3).55 Several adenoviral mutants have proven efficacy in preclinical prostate cancer models, and many of these mutants have later been pursued in early Phase I and Phase II clinical studies by direct intraprostatic injections.56–58 Although evidence of biological activity was reported in combination with radiation and chemotherapy, further improvements in efficacy are needed for patients to be cured.57–61 For example, to target metastatic disease, adenoviral binding to erythrocytes and liver tropism must also be overcome. Common approaches to generate targeted adenoviral vectors and optimize efficacy are 1) replication-selective deletion mutants complemented by altered gene expression in cancer cells, 2) replication-selective mutants with tumor/tissue specific promoters controlling early viral genes, 3) viral tropism modifications to specifically target viral uptake through tumor antigens, and 4) replication-selective mutants combined with therapeutic gene delivery, including cytotoxic transgenes, receptor ligands, and antibody fragments (Figure 1).
Adenoviral mutants and therapeutic gene delivery
A variety of transgenes, promoters, and regulatory response elements have been incorporated into the adenoviral genome including prodrug-converting enzymes, cytokines, antibody fragments, receptor ligands, short hairpin RNAs, and microRNA target sequences. The major limitation is the size of the gene to be expressed; the viral genome (∼36 kb) cannot exceed 105% to ensure proper packaging into functional virions. For large transgenes, deletion of viral genes is necessary, for example, by removing the immune-modulatory E3 genes up to 7kb-inserts can be accommodated (Figure 1). Exclusion of both E1 and E3 genes generates nonreplicating vectors that enable insertion of large expression cassettes including enhancers/promoters with relevant coding regions.62,63 In contrast, to develop efficacious replication-selective oncolytic mutants, the deletion of viral genes requires careful consideration. Importantly, the E1A gene is an absolute requirement for viral replication, except for the small 24-nucleotide sequence in conserved region 2 (E1ACR2) that is expendable in cancer cells (Figure 1B). Deletion of E1B genes renders the virus more susceptible to elimination and premature termination of its life cycle in normal cells.47,51,64 Deletion of the E3 genes may hamper efficacy in patients with intact immune system, although deletion of the E3gp19K gene alone greatly promotes tumor antigen presentation and destruction of tumor cells by activating the host immune response.65,66 Oncolytic mutants targeting prostate cancer are frequently generated wherein AREs are inserted into the genome to regulate viral replication and/or transgene expression. However, some of the most comprehensively evaluated prostate cancer-selective oncolytic mutants have utilized the cytomegalovirus (CMV) regulatory elements to control cytotoxic transgene expression, while replication selectivity was controlled by deletion of the E1B55K gene (described later).67
A common approach for adenovirus-based cancer gene therapy has been the expression of prodrug-converting enzymes. These enzymes promote the conversion of intravenously administered nontoxic prodrugs to toxic drugs in virus-transduced tumor tissue only, resulting in high local concentrations and bystander effects of the toxic drug and limited systemic toxicity. Prodrug-converting enzymes usually replace the E1A genes in nonreplicating vectors and E3 genes in replicating viruses. Some examples of commonly used enzymes are the herpes simplex virus (HSV) thymidine kinase (TK),68 bacterial or yeast-derived cytosine deaminase (CD or yCD),69 and bacterial nitroreductase (NTR).70 TK-mediated conversion of nontoxic ganciclovir or acyclovir produces toxic metabolites that compete with deoxyguanosine triphosphate for DNA incorporation, causing termination of DNA chain elongation. A nonreplicating adenoviral vector with TK expression regulated by the Rous sarcoma virus long terminal repeat promoter (ADV/RSV-TK) was demonstrated to be safe and causes some degree of prostate cancer cell killing in Phase I and Phase II trials.68 Bone metastasis was targeted in a Phase I/II trial by regulating TK expression with the osteocalcin promoter in a nonreplicating vector (Ad-OC-TK) and was reported to decrease pain in castrate-resistant prostate cancer patients.71 The CD enzyme activates the prodrug 5-fluorocytosine (5-FC) to the toxic drug 5-fluorouridine (5-FU) that is further activated by cellular enzymes to 5-FUTP and 5-FdUMP. Both 5-FUTP and 5-FdUMP block RNA synthesis and DNA replication and inhibit thymidylate synthase causing detrimental DNA damage. However, frequently the expression of thymidylate synthase is high in cancer cells resulting in decreased sensitivity to 5-FU. Therefore, the enzyme uracil phosphoribosyl transferase (UPRT) is often fused to CD (CD-UPRT), and potent conversion of 5-FU to 5-FdUMP eliminates the need for cellular enzymes.69 An adenovirus expressing the chimeric CD-UPRT was reported to be effective in 5-FU-resistant non-prostate cancer cells.72 Another chimeric enzyme, CD-TK, was evaluated in several clinical trials using a nonreplicating vector with promising results.57 The NTR enzyme derived from Escherichia coli activates the prodrug CB1954 to a toxic DNA crosslinking compound with high cell permeability that supports potent bystander effects. NTR has been inserted into nonreplicating E1- and E3-deleted adenoviruses (eg, CTL102) and in the replication-selective dl1520 mutant.73,74 In a Phase I/II trial with CTL102, definite decreases in PSA levels were demonstrated in 14 of 19 patients after intraprostatic delivery in combination with CB1954.73 The same team further improved CB1954-dependent antitumor activity in preclinical studies generating a substitution of amino acids in the active site of NTR.75 CTL102 was recently modified to express the immunostimulatory granulocyte-macrophage colony-stimulating factor (GM-CSF) and is currently being evaluated in a Phase I trial with CB1954 delivered intravenously and CTL102 perineally (Table 1).76 Outcomes are awaited.
 | Table 1 Early-phase prostate cancer clinical trials with recently developed adenoviral mutants Note: Only trials initiated or completed after 2010 are included; for earlier trials, see text with indicated references. Abbreviations: Ad5, adenovirus serotype 5; yCD, yeast-derived cytosine deaminase; hNIS, human sodium iodide transporter; SPECT, single-photon emission computed tomography; PET, positron emission tomography; FC, fluorocytosine; GCV, ganciclovir; IL, interleukin; CD, cytosine deaminase; TK, thymidine kinase; PCA, prostate cancer antigen; GM-CSF, granulocyte-macrophage colony-stimulating factor; NTR, nitroreductase; NRGM, nitroreductase and GM-CSF. |
Oncolytic viruses expressing immune-stimulatory cytokines have emerged as a superior approach to prevent cancer spread and recurrence by redirecting the host immune system to the cancer and to overcome immune tolerance. Several studies in prostate cancer models demonstrate excellent efficacy with oncolytic viruses, both promoter-driven and complementation mutants, expressing GM-CSF or interleukins IL-12, IL-24, CD40L, and FLT3L.58,77–81 Amplification of both encoded transgenes and the viral genome promotes increased viral spread, drug-induced killing of neighboring cells, and induction of tumor immunity.
Optimizing replication-selective oncolytic adenoviruses for targeting of prostate cancer
An advantage with oncolytic adenoviruses is their unique mode of cell killing, which is different from that of current anticancer therapeutics. Several mutants have also been engineered to synergize with chemotherapeutic drugs to enhance their mode of action, typically apoptotic death. Tremendous efforts have been invested in optimizing various oncolytic viruses in combination with current standard of care. The two major strategies to restrict adenovirus replication to tumor cells are inactivation of viral genes that are compensated for by the genetic alterations in cancer cells and controlling viral propagation by tissue- or tumor-specific promoters. In addition, mutants have been detargeted from the native coxsackievirus and adenovirus receptor (CAR) and retargeted to antigens expressed at high levels on the tumor cell membrane. For example, insertion of Arg-Gly-Asp (RGD) motifs in the fiber knob to bypass CAR and directly target integrins for uptake, or insertion of the epidermal growth factor receptor ligand or antibodies to bind epidermal growth factor receptor or tumor antigens, respectively, with high affinity (Figure 1B).
Promoter-regulated oncolytic adenoviral mutants
A logical approach for targeting prostate cancer is tumor-specific transcriptional control of adenoviral replication, by replacing the E1A promoter with AREs, to enable viral replication in tissues with active androgen–AR pathway. AREs have also been utilized to drive expression of the early E1B or E4 genes in addition to E1A. One of the first clinically evaluated and comprehensively investigated mutant is the CG7060 (previously CN706, CV706; Cell Genesys) with the PSA promoter/enhancer region driving E1A expression.82 Replication of CG7060 increased up to sevenfold in the presence of androgens in AR-positive cell lines. In the first Phase I trial with this mutant, safety was confirmed and evidence of prostate-specific replication and decreased PSA levels were reported in 20 patients with locally recurrent prostate cancer.59 The same team developed an improved mutant, CG7870, with the rat probasin promoter controlling E1A and the minimal PSA enhancer/promoter driving E1B expression and retention of the E3 region.83 CG7870 was evaluated in Phase I and Phase II trials targeting locally recurrent and hormone refractory metastatic disease, through brachytherapy and intravenous administration, respectively.60 Interestingly, in combination with docetaxel, PSA levels decreased in 36% of patients, and 27% of patients were free from tumor progression 6 months after systemic delivery. The CG7870 mutant was demonstrated to interact synergistically with both taxanes and radiation in preclinical models.83,84 Unfortunately, no further clinical studies have been reported for this promising mutant.
A potent mutant is Ad[I/PPT-E1A] with high selectivity for prostate cancer cells with AR activity.85,86 Viral propagation is regulated by a complex chimeric promoter/enhancer sequence comprising the T-cell receptor-gamma alternate reading frame protein promoter, the PSMA and PSA enhancers and a region of the truncated mouse H19 insulator (named I/PPT). The intact E3 genes are present to increase the viral half-life in patients.87 Despite the absence of the E1B55K gene, due to packaging limitations, the efficacy appeared to be enhanced to levels comparable to those of a CMV-driven mutant because of the potent transcription from the I/PPT promoter construct.86 Phase I studies are impending.88
Other promising transcriptionally regulated AR-dependent replication-selective mutants are Ad-IU1 and Ad-UI2 with E1A and E4 genes controlled by the prostate-specific enhancer sequence (PSES), which includes 3× AREs from the PSA enhancer and the main PSMA enhancer. Ad-UI1 also expresses the prodrug-converting enzyme HSV-TK, regulated together with E1B by the M6 enhancer including a shortened PSES with an additional AP-3 binding site.89 In Ad-UI2, the TK gene is replaced by TRAIL.90 In the oncolytic mutant Ad.DD3-E1A, E1A expression and replication are regulated by the highly prostate-specific differential display code 3 (DD3) promoter and express the secreted cytokine IL-24 or PTEN from the CMV promoter.91,92 The minimal DD3 promoter region of 214 nt was reported to have higher activity than the minimal PSA promoter/enhancer due to elimination of upstream silencer sequences. While the promoter activity is higher in AR-expressing cells, it is also active in prostate cancer cells without a functional AR but not in nonprostate cells. No clinical data have been reported for these mutants.
Prostate-selective oncolytic adenoviruses have also been generated using non-AR-dependent promoters, including regulatory sequences in genes that are typically overexpressed in both prostate and other cancers but not in normal tissue. For example, the human telomerase reverse transcriptase promoter was inserted to drive replication of OBP-301 and the osteocalcin promoter in Ad-OC-E1a (OCaP1; for bone metastasis).93,94 Both promoters control E1A expression and have been demonstrated to target prostate cancer cells in model systems but have not been evaluated clinically.
Complementation-deleted adenoviral mutants
A common strategy to generate oncolytic adenoviral mutants is by complementation deletions; viral propagation requires deregulation of the same pathways that are altered in cancer cells, including inactivation of p53 and uncoupling of pRb-p16 cell cycle control.42,95 For example, the very first oncolytic adenovirus mutant to enter clinical trials dl1520, an Ad5/2 chimeric mutant (Onyx-015), had the E1B55K gene deleted (Figure 1B). The E1B55K protein binds and inactivates p53, thereby preventing premature apoptosis and abortion of viral replication.96 The E1B55K deletion severely attenuated virus propagation in normal tissue and unexpectedly also in many tumors. It was later discovered that E1B55K was essential for viral messenger RNA nuclear export, suggesting a possible contributing cause for the poor efficacy in patients.97,98 In addition, deletion of the immune regulatory E3B genes further attenuated viral half-life and spread in patients, mainly through macrophage-mediated elimination of virus-infected cells without induction of antitumor immunity. Despite the disappointing clinical outcomes, some degree of antitumor efficacy was reported in combination with cytotoxic drugs in Phase II and III trials.99–102 H101 is a dl1520 homolog (Shanghai Sunway Biotech Co., Ltd., Shanghai, China) licensed by the Chinese Food and Drug Administration in 2005 for the treatment of head and neck cancers.100
Freytag et al57,61 have systematically modified the dl1520 virus to generate optimized prostate cancer-selective mutants expressing several cytotoxic factors for a multimodal approach to enhance the effectiveness of radiation therapy in the absence of ADT. For example, the Ad5-CD/TKrep, expressing the chimeric CD/HSV-TK enzyme from the E3 region (Figure 1B), was administered intraprostatically in patients with localized recurrent cancers in combination with systemic delivery of the prodrugs 5-FC and ganciclovir and intensity-modulated radiotherapy (IMRT) in a Phase I trial.103,104 Promising decreases in serum PSA levels were reported without major side effects. A follow-up study 5 years later revealed long-term effectiveness on decreased PSA doubling times.57 A modified, more efficacious version of the same mutant, Ad5-yCD/mutTK(SR39)rep-ADP, employed the yeast CD fused to an improved TK(SR39) mutant enzyme and expressed the adenovirus death protein (ADP).61 This mutant caused a greater degree of tumor regression in preclinical models.105 In a randomized Phase II trial with Ad5-yCD/mutTK(SR39)rep-ADP, men receiving a combination of virus plus IMRT relative to men exposed to IMRT only had a 42% reduction in biopsy positivity 2 years later.61 No additional side effects were reported than those normally seen with radiation therapy. A variant of this mutant Ad5-yCD/mutTK(SR39)rep-hNIS, coding for the human sodium iodide transporter (hNIS) under control of the CMV promoter, was evaluated in men with clinically localized prostate cancer in a Phase I trial (Table 1).67 Intraprostatic administration resulted in prostate-specific gene expression after a single-dose injection at multiple sites, followed by systemic prodrug administration with simultaneous IMRT. Decreased growth of lesions were reported, although the virus did not appear to spread throughout the prostate but remained in the areas of injection, indicating that more potent virus replication or aggressive treatment would be necessary. Advantages with this mutant are that localization and responses may be determined by noninvasive imaging using three genes, CD by magnetic resonance imaging and HSV type 1 (HSV-1) TK and hNIS by both single-photon emission computed tomography and positron emission tomography. The latest mutant generated by Freytag et al58 is the Ad5-yCD/mutTKSR39rep-hIL12, where hNIS was replaced with human IL-12, a proinflammatory cytokine, for the induction of antitumor immunity in addition to killing by prodrug activation and viral lysis (Table 1). IL-12 stimulates innate and adaptive immune responses by supporting antigen presentation and can alleviate the immunosuppressive tumor environment. Systemic delivery of IL-12 is less feasible due to its high toxicity profile, while local production by intraprostatic administration of the virus in murine models appeared to avoid systemic toxicity.58,106 Ad5-yCD/mutTKSR39rep-hIL12 is currently in a Phase I clinical trial for locally recurrent prostate cancer after definitive radiotherapy (NCT02555397). Other variants based on the E1B55K deletion in dl1520 are, for example, ZD55 (Ad5) mutants that express CD or IL-24 or more recently, short hairpin RNA targeting the special AT-rich sequence-binding protein-1 (SATB1).107 These mutants were shown to attenuate growth of prostate cancer metastasis in preclinical models.
In spite of all attempts to enhance the potency of E1B55K-deleted mutants, viral replication and spread are hampered in the majority of solid cancers, including prostate tumors. Therefore, improved deletion mutants with nonattenuating deletions are being developed. For example, deletion of the small pRb-binding E1ACR2 region does not impair viral propagation in tumor cells, while replication and spread are prevented in normal tissue (Figure 1B).108,109 The E1ACR2 deletion is complemented in cells that have deregulated cell cycle control, mainly the pRb–p16 pathway. Several mutants based on this deletion have been constructed and evaluated in clinical trials targeting a variety of tumor types. For example, dl922-947 and several Ad5ΔCR2 variants including chimeric mutants (Ad5Δ24/3) with and without an RGD sequence in the fiber knob (eg, Oncos-102 expressing GM-CSF; Oncos Therapeutics, DNX-2401; DNAtrix), with an E2F promoter regulating replication (eg, ICOVIR-7), and with expression of various transgenes (eg, Ad5/3-Cox2L-D24) have been evaluated in glioma patients.110–113 Clinical evaluation has not yet been undertaken with these mutants in prostate cancer patients, although promising efficacy in preclinical models with E1ACR2-deleted mutants has been reported.46 For example, the chimeric Ad5/3-Δ24-hNIS mutant retargeted to the Ad3 receptor and expressing hNIS from the native E3 promoter and Ad5/3Δ24hCG expressing the human beta chorionic gonadotropin as a marker were reported to attenuate prostate cancer cell growth.114,115 The genetic alterations in prostate cancer render these tumors highly susceptible to the E1ACR2-deleted mutants through the deregulation of cell cycle and cell death pathways, mainly mutations of pRb/p16, PTEN, and Bcl2 that complement the E1ACR2 deletion.11,116
We previously generated a potent mutant AdΔΔ, with deletions in both the E1ACR2 region and the antiapoptotic E1B19K gene, and retained the E3 region for improved immune protection of infected cells in vivo in immune competent hosts.51 In combination with the current standard of care for advanced prostate cancer in the UK, docetaxel and mitoxantrone, AdΔΔ has superior efficacy compared to single-drug regimens in preclinical in vivo models. The synergistic attenuation of tumor growth was caused by improved viral uptake in the presence of drugs followed by higher expression levels of the apoptosis-inducing and chemosensitizing E1A gene products that promote drug-induced apoptosis.49,51 The effectiveness and minimal toxicity to normal cells indicate that AdΔΔ is a worthy candidate for combination therapies with cytotoxic drugs to effectively kill treatment-resistant cancers. Arming AdΔΔ with therapeutic cytokines such as GM-CSF or IL-12 may further optimize effectiveness by inducing antitumor immunity in patients. In contrast, another E1ACR2-deleted mutant with the E1B55K gene deleted (AxdAdB-3) did not show further improvements of efficacy, while the addition of an RGD sequence (AxdAdB3-F/RGD) increased the uptake of virus in prostate cancer cells.117
However, to successfully eliminate metastatic prostate cancer, the viral mutants would have to be delivered systemically to reach distant and deep lesions. A major obstacle to intravenous delivery and adenovirus reaching the metastatic tumors in sufficient quantity is the high affinity binding to erythrocytes that express the native viral CAR and the complement receptor 1 (CR-1).118 The viral fiber knob binds to CAR and the capsid proteins to CR-1 in the presence of antibodies and complement. In contrast, CAR is frequently downregulated in cancer cells, including prostate cancer, contributing to low levels of viral uptake in tumor cells. Other barriers to systemic delivery are the rapid elimination of virus by hepatic Kupffer cells (KC) and hepatocyte uptake, both mediated by a number of blood factors.119–121 For example, viral KC elimination is enhanced by complement receptor 3 (CR-3) or the Fc receptor. Hepatocyte uptake is enhanced by FIX/complement-4 binding protein (C4BP) to the fiber knob or FX binding to the hexon protein that may also direct KC uptake.119,120,122–124 To overcome viral inactivation and enable systemic delivery is presently a priority when developing oncolytic adenoviral mutants, and several approaches have been reported. For example, polymer encapsidation or retargeting of viral particles by changes in fiber tropism or through generation of chimeric mutants (eg, ColoAd1 or other Ad5/species B variants).95,125 Ablation of CAR, complement binding, and hepatocyte uptake were reported for a mutant expressing an αvß6-integrin targeting peptide derived from the foot-and-mouth virus (FMDV or A20 peptide).121,126 The viral mutant Ad5-477dlTAYTA20 has the 20-amino acid RGD-binding peptide FMDV inserted in the HI-loop of the fiber knob.127 An additional base substitution in the fiber knob reduced CAR binding (Y477A), while the putative fiber–FIX/C4BP interactions were ablated through deletion of the TAYT motif.120,126,128 The altered viral tropism resulted in very promising tumor-selective uptake in preclinical studies of solid tumors, although the mutations have not yet been evaluated in prostate cancer in vivo models or in the clinic.
Future directions
A powerful approach to develop more potent and targeted tumor type-specific oncolytic adenoviral variants is the concept of “directed evolution”; by continuous passaging of pools of various adenoviral species on the cancer cells of interest, recombination events of the viral genomes are promoted. The development of the colon cancer-selective oncolytic chimeric adenovirus ColoAd1 (Enadenotucirev; PsiOxus) proved the feasibility of this strategy, and the virus is currently being evaluated clinically (NCT02028117). ColoAd1 is an Ad3/Ad11p recombinant selected on colon cancer cells.125 The viral Ad11p capsid is more resistant to elimination by human serum and blood cells than Ad5, and therefore, the viral activity is better preserved after systemic delivery. It may be possible to apply similar “directed evolutionary” recombination approaches, with genetic engineering, on prostate cancer cells to identify an optimized prostate-selective chimeric adenovirus.125,129 Ablation of the Ad5 binding to erythrocytes and hepatocyte uptake are also desirable traits that could be selected for using this strategy to enable systemic delivery.
The most promising recent development in virotherapy is the exploitation of the host immune system. Tumors frequently induce host immune tolerance, and by arming oncolytic viruses with cytokines or inhibitors that relieve the immune checkpoint inhibition, the tolerance can be reduced. Several adenoviruses have been generated to boost the oncolytic immunotherapeutic effects with promising results. Treatment of prostate cancer with similar mutants is likely to have more beneficial responses than current oncolytics expressing cytotoxic transgenes only. The effectiveness of this approach has been demonstrated by the market approval of talimogene laherparepvec (T-VEC, previously Oncovex, now Imlygic; Amgen) in 2015, first by the US Food and Drug Administration and later by the European Commission.40,130 T-VEC is a HSV-1 mutant modified to only replicate in tumor cells and to express GM-CSF for activation of antitumor immunity.131 Intratumoral administration of T-VEC caused regression of injected melanoma lesions and induced immune-mediated regression of noninjected distal lesion in late-stage melanoma patients.40,130 Although T-VEC has not yet been evaluated in prostate carcinomas, it could be expected to have similar effects by activating immune responses. More importantly, GM-CSF-expressing adenoviral mutants, such as the potent Ad5Δ24/3-RGD-GM-CSF mutant, also warrant further evaluation in prostate cancer.132 T-cells activated in response to antigen presentation after lysis of infected tumor cells would likely also target metastatic lesions, and consequently, the short adenoviral half-life in human blood after systemic injection may be less important; once an immune response is mounted to the injected tumor, growth of distant metastasis may be attenuated by improved immune recognition. GM-CSF has also been incorporated in the oncolytic poxvirus, pexastimogene devacirepvec (Pexa-Vec, previously JX-594; SillaJen).133 Pexa-Vec was reported to inhibit cancer progression in patients by both viral-induced cell lysis and antitumor immunity.39,133 So far, prostate cancer patients have not been included in the trials even though potent replication was reported in prostate cancer cell lines.134 Interestingly, severely attenuated vaccinia virus or adenovirus have been employed as prostate cancer vaccine vectors to deliver tumor antigens, including PSA, with some evidence of protection for tumor recurrence (eg, TRICOM, Prostvac, and Ad/PSA).135,136
To conclude oncolytic adenoviruses armed with immune stimulatory factors and cytotoxic transgenes are the most promising way forward to target both early- and late-stage prostate cancers, and with additional capsid modifications, these mutants may also be delivered systemically to distant metastasis. Recent findings that immune danger signals can be activated in prostate cancer resulting in exposure of tumor-associated antigens, permitting recognition by immune effector cells, render this disease an excellent target for oncolytic adenovirus-mediated immunotherapy and may partly abridge the need for systemic delivery.
Disclosure
The authors report no conflicts of interest in this work.
References
Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, 2014. CA Cancer J Clin. 2014;64(1):9–29. | ||
Ferlay J, Steliarova-Foucher E, Lortet-Tieulent J, et al. Cancer incidence and mortality patterns in Europe: estimates for 40 countries in 2012. Eur J Cancer. 2013;49(6):1374–1403. | ||
Muralidhar V, Ziehr DR, Mahal BA, et al. Association between older age and increasing Gleason score. Clin Genitourin Cancer. 2015;13(6):525–530.e521–e523. | ||
Zhou CK, Check DP, Lortet-Tieulent J, et al. Prostate cancer incidence in 43 populations worldwide: an analysis of time trends overall and by age group. Int J Cancer. 2016;138(6):1388–1400. | ||
Center MM, Jemal A, Lortet-Tieulent J, et al. International variation in prostate cancer incidence and mortality rates. Eur Urol. 2012;61(6):1079–1092. | ||
Wallstrom P, Bjartell A, Gullberg B, Olsson H, Wirfalt E. A prospective Swedish study on body size, body composition, diabetes, and prostate cancer risk. Br J Cancer. 2009;100(11):1799–1805. | ||
Kenfield SA, Batista JL, Jahn JL, et al. Development and application of a lifestyle score for prevention of lethal prostate cancer. J Natl Cancer Inst. 2016;108(3):djv329. | ||
Penney KL, Pyne S, Schumacher FR, et al. Genome-wide association study of prostate cancer mortality. Cancer Epidemiol Biomarkers Prev. 2010;19(11):2869–2876. | ||
Kral M, Rosinska V, Student V, Grepl M, Hrabec M, Bouchal J. Genetic determinants of prostate cancer: a review. Biomed Pap Med Fac Univ Palacky Olomouc Czech Repub. 2011;155(1):3–9. | ||
Ben-Shlomo Y, Evans S, Patel B, et al; PROCESS Study Group. Differences in the epidemiology and presentation of prostate cancer in Black and White men in England: lessons learnt from the process study. BJU Int. 2009;103(6):723–724. | ||
Mazaris E, Tsiotras A. Molecular pathways in prostate cancer. Nephrourol Mon. 2013;5(3):792–800. | ||
Studer UE, Whelan P, Albrecht W, et al. Immediate or deferred androgen deprivation for patients with prostate cancer not suitable for local treatment with curative intent: European Organisation for Research and Treatment of Cancer (EORTC) trial 30891. J Clin Oncol. 2006;24(12):1868–1876. | ||
Feldman BJ, Feldman D. The development of androgen-independent prostate cancer. Nat Rev Cancer. 2001;1(1):34–45. | ||
Taplin ME. Drug insight: role of the androgen receptor in the development and progression of prostate cancer. Nat Clin Pract Oncol. 2007;4(4):236–244. | ||
Chandrasekar T, Yang JC, Gao AC, Evans CP. Mechanisms of resistance in castration-resistant prostate cancer (CRPC). Transl Androl Urol. 2015;4(3):365–380. | ||
Dutt SS, Gao AC. Molecular mechanisms of castration-resistant prostate cancer progression. Future Oncol. 2009;5(9):1403–1413. | ||
Schulz WA, Burchardt M, Cronauer MV. Molecular biology of prostate cancer. Mol Hum Reprod. 2003;9(8):437–448. | ||
Body JJ, Casimiro S, Costa L. Targeting bone metastases in prostate cancer: improving clinical outcome. Nat Rev Urol. 2015;12(6):340–356. | ||
Perrault L, Fradet V, Lauzon V, LeLorier J, Mitchell D, Habib M. Burden of illness of bone metastases in prostate cancer patients in Quebec, Canada: a population-based analysis. Can Urol Assoc J. 2015;9(9–10):307–314. | ||
Chen Y, Sawyers CL, Scher HI. Targeting the androgen receptor pathway in prostate cancer. Curr Opin Pharmacol. 2008;8(4):440–448. | ||
Litvinov IV, De Marzo AM, Isaacs JT. Is the Achilles’ heel for prostate cancer therapy a gain of function in androgen receptor signaling? J Clin Endocrinol Metab. 2003;88(7):2972–2982. | ||
Zenzmaier C, Untergasser G, Berger P. Aging of the prostate epithelial stem/progenitor cell. Exp Gerontol. 2008;43(11):981–985. | ||
So A, Gleave M, Hurtado-Col A, Nelson C. Mechanisms of the development of androgen independence in prostate cancer. World J Urol. 2005;23(1):1–9. | ||
Boyd LK, Mao X, Xue L, et al. High-resolution genome-wide copy-number analysis suggests a monoclonal origin of multifocal prostate cancer. Genes Chromosomes Cancer. 2012;51(6):579–589. | ||
Gregory CW, He B, Johnson RT, et al. A mechanism for androgen receptor-mediated prostate cancer recurrence after androgen deprivation therapy. Cancer Res. 2001;61(11):4315–4319. | ||
Visakorpi T, Hyytinen E, Koivisto P, et al. In vivo amplification of the androgen receptor gene and progression of human prostate cancer. Nat Genet. 1995;9(4):401–406. | ||
Anantharaman A, Friedlander TW. Targeting the androgen receptor in metastatic castrate-resistant prostate cancer: a review. Urol Oncol. 2015. Epub 2015 Dec 16. | ||
Chen Y, Chi P, Rockowitz S, et al. ETS factors reprogram the androgen receptor cistrome and prime prostate tumorigenesis in response to PTEN loss. Nat Med. 2013;19(8):1023–1029. | ||
Eeles RA, Kote-Jarai Z, Giles GG, et al; UK Genetic Prostate Cancer Study Collaborators, British Association of Urological Surgeons’ Section of Oncology, UK ProtecT Study Collaborators. Multiple newly identified loci associated with prostate cancer susceptibility. Nat Genet. 2008;40(3):316–321. | ||
Edwards SM, Evans DG, Hope Q, et al; UK Genetic Prostate Cancer Study Collaborators and BAUS Section of Oncology. Prostate cancer in BRCA2 germline mutation carriers is associated with poorer prognosis. Br J Cancer. 2010;103(6):918–924. | ||
Tomlins SA, Rhodes DR, Perner S, et al. Recurrent fusion of TMPRSS2 and ETS transcription factor genes in prostate cancer. Science. 2005;310(5748):644–648. | ||
Bastus NC, Boyd LK, Mao X, et al. Androgen-induced TMPRSS2:ERG fusion in nonmalignant prostate epithelial cells. Cancer Res. 2010;70(23):9544–9548. | ||
Cai C, Wang H, Xu Y, Chen S, Balk SP. Reactivation of androgen receptor-regulated TMPRSS2:ERG gene expression in castration-resistant prostate cancer. Cancer Res. 2009;69(15):6027–6032. | ||
Shiota M, Yokomizo A, Eto M. Taxane chemotherapy for hormone-naive prostate cancer with its expanding role as breakthrough strategy. Front Oncol. 2015;5:304. | ||
Sweeney CJ, Chen YH, Carducci M, et al. Chemohormonal therapy in metastatic hormone-sensitive prostate cancer. N Engl J Med. 2015;373(8):737–746. | ||
Zafeiriou Z, Jayaram A, Sharp A, de Bono JS. Managing metastatic castration-resistant prostate cancer in the pre-chemotherapy setting: a changing approach in the era of new targeted agents. Drugs. 2016;76(4):421–430. | ||
Ryan CJ, Smith MR, Fizazi K, et al; COU-AA-302 Investigators. Abiraterone acetate plus prednisone versus placebo plus prednisone in chemotherapy-naive men with metastatic castration-resistant prostate cancer (COU-AA-302): final overall survival analysis of a randomised, double-blind, placebo-controlled phase 3 study. Lancet Oncol. 2015;16(2):152–160. | ||
Bellmunt J, Kheoh T, Yu MK, et al. Prior endocrine therapy impact on abiraterone acetate clinical efficacy in metastatic castration-resistant prostate cancer: post-hoc analysis of randomised phase 3 studies. Eur Urol. Epub 2015 Oct 24. | ||
Heo J, Reid T, Ruo L, et al. Randomized dose-finding clinical trial of oncolytic immunotherapeutic vaccinia JX-594 in liver cancer. Nat Med. 2013;19(3):329–336. | ||
Andtbacka RH, Kaufman HL, Collichio F, et al. Talimogene laherparepvec improves durable response rate in patients with advanced melanoma. J Clin Oncol. 2015;33(25):2780–2788. | ||
Stoeckel J, Hay JG. Drug evaluation: reolysin – wild-type reovirus as a cancer therapeutic. Curr Opin Mol Ther. 2006;8(3):249–260. | ||
Parato KA, Senger D, Forsyth PA, Bell JC. Recent progress in the battle between oncolytic viruses and tumours. Nat Rev Cancer. 2005;5(12):965–976. | ||
Fukuhara H, Homma Y, Todo T. Oncolytic virus therapy for prostate cancer. Int J Urol. 2010;17(1):20–30. | ||
Kirn DH, Thorne SH. Targeted and armed oncolytic poxviruses: a novel multi-mechanistic therapeutic class for cancer. Nat Rev Cancer. 2009;9(1):64–71. | ||
Simpson GR, Relph K, Harrington K, Melcher A, Pandha H. Cancer immunotherapy via combining oncolytic virotherapy with chemotherapy: recent advances. Oncolytic Virother. 2016;2016(5):1–13. | ||
de Vrij J, Willemsen RA, Lindholm L, et al. Adenovirus-derived vectors for prostate cancer gene therapy. Hum Gene Ther. 2010;21(7):795–805. | ||
Leitner S, Sweeney K, Oberg D, et al. Oncolytic adenoviral mutants with E1B19K gene deletions enhance gemcitabine-induced apoptosis in pancreatic carcinoma cells and anti-tumor efficacy in vivo. Clin Cancer Res. 2009;15(5):1730–1740. | ||
Cherubini G, Kallin C, Mozetic A, et al. The oncolytic adenovirus AdΔΔ enhances selective cancer cell killing in combination with DNA-damaging drugs in pancreatic cancer models. Gene Ther. 2011;18(12):1157–1165. | ||
Pantelidou C, Cherubini G, Lemoine NR, Halldén G. The E1B19K-deleted oncolytic adenovirus mutant AdΔ19K sensitizes pancreatic cancer cells to drug-induced DNA-damage by down-regulating Claspin and Mre11. Oncotarget. Epub 2016 Feb 10. | ||
Miranda E, Maya Pineda H, Oberg D, et al. Adenovirus-mediated sensitization to the cytotoxic drugs docetaxel and mitoxantrone is dependent on regulatory domains in the E1ACR1 gene-region. PLoS One. 2012;7(10):e46617. | ||
Öberg D, Yanover E, Sweeney K, et al. Improved potency and selectivity of an oncolytic E1ACR2 and E1B19K deleted adenoviral mutant (AdΔΔ) in prostate and pancreatic cancers. Clin Cancer Res. 2010;16(2):541–553. | ||
Rojas JJ, Sampath P, Hou W, Thorne SH. Defining effective combinations of immune checkpoint blockade and oncolytic virotherapy. Clin Cancer Res. 2015;21(24):5543–5551. | ||
Rajani K, Parrish C, Kottke T, et al. Combination therapy with reovirus and anti-PD-1 blockade controls tumor growth through innate and adaptive immune responses. Mol Ther. 2016;24(1):166–174. | ||
Dias JD, Hemminki O, Diaconu I, et al. Targeted cancer immunotherapy with oncolytic adenovirus coding for a fully human monoclonal antibody specific for CTLA-4. Gene Ther. 2012;19(10):988–998. | ||
Sanguedolce F, Cormio A, Brunelli M, et al. Urine TMPRSS2: ERG fusion transcript as a biomarker for prostate cancer: literature review. Clin Genitourin Cancer. 2016;14(2):117–121. | ||
Figueiredo ML, Kao C, Wu L. Advances in preclinical investigation of prostate cancer gene therapy. Mol Ther. 2007;15(6):1053–1064. | ||
Freytag SO, Stricker H, Peabody J, et al. Five-year follow-up of trial of replication-competent adenovirus-mediated suicide gene therapy for treatment of prostate cancer. Mol Ther. 2007;15(3):636–642. | ||
Freytag SO, Zhang Y, Siddiqui F. Preclinical toxicology of oncolytic adenovirus-mediated cytotoxic and interleukin-12 gene therapy for prostate cancer. Mol Ther Oncolytics. Epub 2015 Apr 29. | ||
DeWeese TL, van der Poel H, Li S, et al. A phase I trial of CV706, a replication-competent, PSA selective oncolytic adenovirus, for the treatment of locally recurrent prostate cancer following radiation therapy. Cancer Res. 2001;61(20):7464–7472. | ||
Small EJ, Carducci MA, Burke JM, et al. A phase I trial of intravenous CG7870, a replication-selective, prostate-specific antigen-targeted oncolytic adenovirus, for the treatment of hormone-refractory, metastatic prostate cancer. Mol Ther. 2006;14(1):107–117. | ||
Freytag SO, Stricker H, Lu M, et al. Prospective randomized phase 2 trial of intensity modulated radiation therapy with or without oncolytic adenovirus-mediated cytotoxic gene therapy in intermediate-risk prostate cancer. Int J Radiat Oncol Biol Phys. 2014;89(2):268–276. | ||
Russell WC. Adenoviruses: update on structure and function. J Gen Virol. 2009;90(pt 1):1–20. | ||
Nettelbeck DM. Cellular genetic tools to control oncolytic adenoviruses for virotherapy of cancer. J Mol Med (Berl). 2008;86(4):363–377. | ||
White E. Mechanisms of apoptosis regulation by viral oncogenes in infection and tumorigenesis. Cell Death Differ. 2006;13(8):1371–1377. | ||
Wang Y, Hallden G, Hill R, et al. E3 gene manipulations affect oncolytic adenovirus activity in immunocompetent tumor models. Nat Biotechnol. 2003;21(11):1328–1335. | ||
Hallden G, Hill R, Wang Y, et al. Novel immunocompetent murine tumor models for the assessment of replication-competent oncolytic adenovirus efficacy. Mol Ther. 2003;8(3):412–424. | ||
Barton KN, Stricker H, Elshaikh MA, et al. Feasibility of adenovirus-mediated hNIS gene transfer and 131I radioiodine therapy as a definitive treatment for localized prostate cancer. Mol Ther. 2011;19(7):1353–1359. | ||
Ayala G, Satoh T, Li R, et al. Biological response determinants in HSV-tk + ganciclovir gene therapy for prostate cancer. Mol Ther. 2006;13(4):716–728. | ||
Erbs P, Regulier E, Kintz J, et al. In vivo cancer gene therapy by adenovirus-mediated transfer of a bifunctional yeast cytosine deaminase/uracil phosphoribosyltransferase fusion gene. Cancer Res. 2000;60(14):3813–3822. | ||
Weedon SJ, Green NK, McNeish IA, et al. Sensitisation of human carcinoma cells to the prodrug CB1954 by adenovirus vector-mediated expression of E. coli nitroreductase. Int J Cancer. 2000;86(6):848–854. | ||
Terao S, Shirakawa T, Acharya B, et al. A pilot study of quality of life of patients with hormone-refractory prostate cancer after gene therapy. Anticancer Res. 2009;29(5):1533–1537. | ||
Richard C, Duivenvoorden W, Bourbeau D, et al. Sensitivity of 5-fluorouracil-resistant cancer cells to adenovirus suicide gene therapy. Cancer Gene Ther. 2007;14(1):57–65. | ||
Patel P, Young JG, Mautner V, et al. A phase I/II clinical trial in localized prostate cancer of an adenovirus expressing nitroreductase with CB1954 [correction of CB1984]. Mol Ther. 2009;17(7):1292–1299. | ||
Searle PF, Chen MJ, Hu L, et al. Nitroreductase: a prodrug-activating enzyme for cancer gene therapy. Clin Exp Pharmacol Physiol. 2004;31(11):811–816. | ||
Jaberipour M, Vass SO, Guise CP, et al. Testing double mutants of the enzyme nitroreductase for enhanced cell sensitisation to prodrugs: effects of combining beneficial single mutations. Biochem Pharmacol. 2010;79(2):102–111. | ||
Searle PF, Mautner V, Porfiri E, et al. The AdUP trial: combining prodrug activation with immune stimulation in locally recurrent prostate cancer. Hum Gene Ther. 2015;26:A32–A33. | ||
Pesonen S, Diaconu I, Kangasniemi L, et al. Oncolytic immunotherapy of advanced solid tumors with a CD40L-expressing replicating adenovirus: assessment of safety and immunologic responses in patients. Cancer Res. 2012;72(7):1621–1631. | ||
Mukhopadhaya A, Mendecki J, Dong X, et al. Localized hyperthermia combined with intratumoral dendritic cells induces systemic antitumor immunity. Cancer Res. 2007;67(16):7798–7806. | ||
Sarkar S, Quinn BA, Shen XN, et al. Therapy of prostate cancer using a novel cancer terminator virus and a small molecule BH-3 mimetic. Oncotarget. 2015;6(13):10712–10727. | ||
Kim JS, Lee SD, Lee SJ, Chung MK. Development of an immunotherapeutic adenovirus targeting hormone-independent prostate cancer. Onco Targets Ther. 2013;6:1635–1642. | ||
Yang YF, Xue SY, Lu ZZ, et al. Antitumor effects of oncolytic adenovirus armed with PSA-IZ-CD40L fusion gene against prostate cancer. Gene Ther. 2014;21(8):723–731. | ||
Rodriguez R, Schuur ER, Lim HY, Henderson GA, Simons JW, Henderson DR. Prostate attenuated replication competent adenovirus (ARCA) CN706: a selective cytotoxic for prostate-specific antigen-positive prostate cancer cells. Cancer Res. 1997;57(13):2559–2563. | ||
Yu DC, Chen Y, Dilley J, et al. Antitumor synergy of CV787, a prostate cancer-specific adenovirus, and paclitaxel and docetaxel. Cancer Res. 2001;61(2):517–525. | ||
Dilley J, Reddy S, Ko D, et al. Oncolytic adenovirus CG7870 in combination with radiation demonstrates synergistic enhancements of antitumor efficacy without loss of specificity. Cancer Gene Ther. 2005;12(8):715–722. | ||
Cheng WS, Dzojic H, Nilsson B, Tötterman TH, Essand M. An oncolytic conditionally replicating adenovirus for hormone-dependent and hormone-independent prostate cancer. Cancer Gene Ther. 2006;13(1):13–20. | ||
Cheng WS, Kraaij R, Nilsson B, et al. A novel TARP-promoter-based adenovirus against hormone-dependent and hormone-refractory prostate cancer. Mol Ther. 2004;10(2):355–364. | ||
Danielsson A, Dzojic H, Nilsson B, Essand M. Increased therapeutic efficacy of the prostate-specific oncolytic adenovirus Ad[I/PPT-E1A] by reduction of the insulator size and introduction of the full-length E3 region. Cancer Gene Ther. 2008;15(4):203–213. | ||
Schenk E, Essand M, Kraaij R, Adamson R, Maitland N, Bangma CH. Pre-clinical safety assessment of Ad[I/PPT-E1A], a novel oncolytic adenovirus for prostate cancer. Hum Gene Ther Clin Dev. Epub 2014 Feb 18. | ||
Ahn M, Lee SJ, Li X, et al. Enhanced combined tumor-specific oncolysis and suicide gene therapy for prostate cancer using M6 promoter. Cancer Gene Ther. 2009;16(1):73–82. | ||
Jimenez JA, Li X, Zhang YP, et al. Antitumor activity of Ad-IU2, a prostate-specific replication-competent adenovirus encoding the apoptosis inducer, TRAIL. Cancer Gene Ther. 2010;17(3):180–191. | ||
Fan JK, Wei N, Ding M, et al. Targeting Gene-ViroTherapy for prostate cancer by DD3-driven oncolytic virus-harboring interleukin-24 gene. Int J Cancer. 2010;127(3):707–717. | ||
Ding M, Cao X, Xu HN, et al. Prostate cancer-specific and potent antitumor effect of a DD3-controlled oncolytic virus harboring the PTEN gene. PLoS One. 2012;7(4):e35153. | ||
Huang P, Kaku H, Chen J, et al. Potent antitumor effects of combined therapy with a telomerase-specific, replication-competent adenovirus (OBP-301) and IL-2 in a mouse model of renal cell carcinoma. Cancer Gene Ther. 2010;17(7):484–491. | ||
Matsubara S, Wada Y, Gardner TA, et al. A conditional replication-competent adenoviral vector, Ad-OC-E1a, to cotarget prostate cancer and bone stroma in an experimental model of androgen-independent prostate cancer bone metastasis. Cancer Res. 2001;61(16):6012–6019. | ||
Alonso-Padilla J, Papp T, Kaján GL, et al. Development of novel adenoviral vectors to overcome challenges observed with HAdV-5-based constructs. Mol Ther. 2016;24(1):6–16. | ||
Bischoff JR, Kirn DH, Williams A, et al. An adenovirus mutant that replicates selectively in p53-deficient human tumor cells. Science. 1996;274(5286):373–376. | ||
O’Shea CC, Johnson L, Bagus B, et al. Late viral RNA export, rather than p53 inactivation, determines ONYX-015 tumor selectivity. Cancer Cell. 2004;6(6):611–623. | ||
Thorne SH, Brooks G, Lee YL, Au T, Eng LF, Reid T. Effects of febrile temperature on adenoviral infection and replication: implications for viral therapy of cancer. J Virol. 2005;79(1):581–591. | ||
Aghi M, Martuza RL. Oncolytic viral therapies - the clinical experience. Oncogene. 2005;24(52):7802–7816. | ||
Garber K. China approves world’s first oncolytic virus therapy for cancer treatment. J Natl Cancer Inst. 2006;98(5):298–300. | ||
Liu TC, Galanis E, Kirn D. Clinical trial results with oncolytic virotherapy: a century of promise, a decade of progress. Nat Clin Pract Oncol. 2007;4(2):101–117. | ||
Hecht JR, Bedford R, Abbruzzese JL, et al. A phase I/II trial of intratumoral endoscopic ultrasound injection of ONYX-015 with intravenous gemcitabine in unresectable pancreatic carcinoma. Clin Cancer Res. 2003;9(2):555–561. | ||
Freytag SO, Stricker H, Movsas B, Kim JH. Prostate cancer gene therapy clinical trials. Mol Ther. 2007;15(6):1042–1052. | ||
Freytag SO, Movsas B, Aref I, et al. Phase I trial of replication-competent adenovirus-mediated suicide gene therapy combined with IMRT for prostate cancer. Mol Ther. 2007;15(5):1016–1023. | ||
Barton KN, Paielli D, Zhang Y, et al. Second-generation replication-competent oncolytic adenovirus armed with improved suicide genes and ADP gene demonstrates greater efficacy without increased toxicity. Mol Ther. 2006;13(2):347–356. | ||
Leonard JP, Sherman ML, Fisher GL, et al. Effects of single-dose interleukin-12 exposure on interleukin-12-associated toxicity and interferon-gamma production. Blood. 1997;90(7):2541–2548. | ||
Mao LJ, Zhang J, Liu N, et al. Oncolytic virus carrying shRNA targeting SATB1 inhibits prostate cancer growth and metastasis. Tumour Biol. 2015;36(11):9073–9081. | ||
Page JG, Tian B, Schweikart K, et al. Identifying the safety profile of a novel infectivity-enhanced conditionally replicative adenovirus, Ad5-delta24-RGD, in anticipation of a phase I trial for recurrent ovarian cancer. Am J Obstet Gynecol. 2007;196(4):389.e381–e389. discussion 389.e389–310. | ||
Heise C, Hermiston T, Johnson L, et al. An adenovirus E1A mutant that demonstrates potent and selective systemic anti-tumoral efficacy. Nat Med. 2000;6(10):1134–1139. | ||
Jiang H, Gomez-Manzano C, Lang FF, Alemany R, Fueyo J. Oncolytic adenovirus: preclinical and clinical studies in patients with human malignant gliomas. Curr Gene Ther. 2009;9(5):422–427. | ||
Nokisalmi P, Pesonen S, Escutenaire S, et al. Oncolytic adenovirus ICOVIR-7 in patients with advanced and refractory solid tumors. Clin Cancer Res. 2010;16(11):3035–3043. | ||
Kim KH, Ryan MJ, Estep JE, et al. A new generation of serotype chimeric infectivity-enhanced conditionally replicative adenovirals: the safety profile of ad5/3-Δ24 in advance of a phase I clinical trial in ovarian cancer patients. Hum Gene Ther. 2011;22(7):821–828. | ||
Pesonen S, Helin H, Nokisalmi P, et al. Oncolytic adenovirus treatment of a patient with refractory neuroblastoma. Acta Oncol. 2010;49(1):117–119. | ||
Hakkarainen T, Rajecki M, Sarparanta M, et al. Targeted radiotherapy for prostate cancer with an oncolytic adenovirus coding for human sodium iodide symporter. Clin Cancer Res. 2009;15(17):5396–5403. | ||
Rajecki M, Kanerva A, Stenman UH, et al. Treatment of prostate cancer with Ad5/3Delta24hCG allows non-invasive detection of the magnitude and persistence of virus replication in vivo. Mol Cancer Ther. 2007;6(2):742–751. | ||
Skjøth IH, Issinger OG. Profiling of signaling molecules in four different human prostate carcinoma cell lines before and after induction of apoptosis. Int J Oncol. 2006;28(1):217–229. | ||
Shen YH, Yang F, Wang H, et al. Arg-Gly-Asp (RGD)-modified E1A/E1B double mutant adenovirus enhances antitumor activity in prostate cancer cells in vitro and in mice. PLoS One. 2016;11(1):e0147173. | ||
Carlisle RC, Di Y, Cerny AM, et al. Human erythrocytes bind and inactivate type 5 adenovirus by presenting Coxsackie virus-adenovirus receptor and complement receptor 1. Blood. 2009;113(9):1909–1918. | ||
Kalyuzhniy O, Di Paolo NC, Silvestry M, et al. Adenovirus serotype 5 hexon is critical for virus infection of hepatocytes in vivo. Proc Natl Acad Sci U S A. 2008;105(14):5483–5488. | ||
Shayakhmetov DM, Gaggar A, Ni S, Li ZY, Lieber A. Adenovirus binding to blood factors results in liver cell infection and hepatotoxicity. J Virol. 2005;79(12):7478–7491. | ||
Coughlan L, Alba R, Parker AL, et al. Tropism-modification strategies for targeted gene delivery using adenoviral vectors. Viruses. 2010;2(10):2290–2355. | ||
Xu Z, Qiu Q, Tian J, et al. Coagulation factor X shields adenovirus type 5 from attack by natural antibodies and complement. Nat Med. 2013;19(4):452–457. | ||
Waddington SN, McVey JH, Bhella D, et al. Adenovirus serotype 5 hexon mediates liver gene transfer. Cell. 2008;132(3):397–409. | ||
Parker AL, Waddington SN, Nicol CG, et al. Multiple vitamin K-dependent coagulation zymogens promote adenovirus-mediated gene delivery to hepatocytes. Blood. 2006;108(8):2554–2561. | ||
Kuhn I, Harden P, Bauzon M, et al. Directed evolution generates a novel oncolytic virus for the treatment of colon cancer. PLoS One. 2008;3(6):e2409. | ||
Coughlan L, Vallath S, Gros A, et al. Combined fiber modifications both to target α(v)β(6) and detarget the coxsackievirus-adenovirus receptor improve virus toxicity profiles in vivo but fail to improve antitumoral efficacy relative to adenovirus serotype 5. Hum Gene Ther. 2012;23(9):960–979. | ||
Dicara D, Burman A, Clark S, et al. Foot-and-mouth disease virus forms a highly stable, EDTA-resistant complex with its principal receptor, integrin alphavbeta6: implications for infectiousness. J Virol. 2008;82(3):1537–1546. | ||
Kirby I, Davison E, Beavil AJ, et al. Mutations in the DG loop of adenovirus type 5 fiber knob protein abolish high-affinity binding to its cellular receptor CAR. J Virol. 1999;73(11):9508–9514. | ||
Maheshri N, Koerber JT, Kaspar BK, Schaffer DV. Directed evolution of adeno-associated virus yields enhanced gene delivery vectors. Nat Biotechnol. 2006;24(2):198–204. | ||
Harrington KJ, Puzanov I, Hecht JR, et al. Clinical development of talimogene laherparepvec (T-VEC): a modified herpes simplex virus type-1-derived oncolytic immunotherapy. Expert Rev Anticancer Ther. 2015;15(12):1389–1403. | ||
Liu BL, Robinson M, Han ZQ, et al. ICP34.5 deleted herpes simplex virus with enhanced oncolytic, immune stimulating, and anti-tumour properties. Gene Ther. 2003;10(4):292–303. | ||
Kanerva A, Nokisalmi P, Diaconu I, et al. Antiviral and antitumor T-cell immunity in patients treated with GM-CSF-coding oncolytic adenovirus. Clin Cancer Res. 2013;19(10):2734–2744. | ||
Breitbach CJ, Burke J, Jonker D, et al. Intravenous delivery of a multi-mechanistic cancer-targeted oncolytic poxvirus in humans. Nature. 2011;477(7362):99–102. | ||
Lee HJ, Rho J, Gui SR, et al. Effect of aldosterone on the amplification of oncolytic vaccinia virus in human cancer lines. Korean J Hepatol. 2011;17(3):213–219. | ||
McNeel DG, Chen YH, Gulley JL, et al. Randomized phase II trial of docetaxel with or without PSA-TRICOM vaccine in patients with castrate-resistant metastatic prostate cancer: a trial of the ECOG-ACRIN cancer research group (E1809). Hum Vaccin Immunother. 2015;11(10):2469–2474. | ||
Lubaroff DM, Vaena D, Brown JA, et al. Vaccine immunotherapy for prostate cancer: from mice to men. Immunol Res. 2014;59(1–3):229–235. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.