Back to Journals » Therapeutics and Clinical Risk Management » Volume 11
OnabotulinumtoxinA for chronic migraine: a critical appraisal
Received 17 April 2015
Accepted for publication 10 June 2015
Published 29 June 2015 Volume 2015:11 Pages 1003—1013
DOI https://doi.org/10.2147/TCRM.S76964
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Editor who approved publication: Professor Garry Walsh
Rubesh Gooriah, Fayyaz Ahmed
Department of Neurology, Hull Royal Infirmary, Kingston Upon Hull, UK
Abstract: Chronic migraine (CM) is a severe disabling condition with a few available evidence-based management options. OnabotulinumtoxinA (onaBoNTA) is approved for use in a number of disorders. Its benefits and potential use in migraine were observed incidentally while treating patients cosmetically for wrinkles. The mechanism of action of onaBoNTA in CM is not fully understood, but there is evidence that this involves axonal transport via sensory fibers. The Phase III REsearch Evaluating Migraine Prophylaxis Therapy trials have established the efficacy as well as the long-term safety and tolerability of onaBoNTA in CM. This review will discuss the evidence behind its use in this setting.
Keywords: migraine pathophysiology, botulinum toxin, botox, migraine prophylaxis, chronic daily headache
Introduction
Migraine, the seventh disabler,1 has a lifetime prevalence of around 15% of the population.2 It can significantly limit the daily activities of sufferers as well as having considerable psychological3 and economic impacts, with costs amounting to €27 billion in the European countries.4 Some patients progress from having episodic migraine (EM) to chronic migraine (CM), the latter affecting 1%–2% of the population.5 This does not happen suddenly, but is a gradual process, initially changing from low-frequency EM to a high-frequency stage and eventually to CM.6 CM is defined as a headache on ≥15 days per month for ≥3 months, of which ≥8 days meet the criteria for migraine with or without aura or responds to migraine-specific treatment.7 Approximately 88% of CM sufferers will seek help from a health professional; however, the majority of patients are underdiagnosed.8 Only 20% of those with CM receive a diagnosis of CM, chronic daily headache, or transformed migraine.8 Patients with CM use health care resources to a larger extent than those with EM. People with CM seek medical attention more often and are also admitted to hospital, require diagnostic tests and migraine drugs more frequently.9,10 This reflects the fact that CM belongs to a more severe end of the spectrum compared to EM.
The primary objective of migraine prophylaxis is to reduce the frequency, duration, and severity of migraine attacks. There are a number of oral prophylactics that are effective against EM. However, their benefit in the treatment of CM is lacking, except for topiramate, which has a potential role. OnabotulinumtoxinA (onaBoNTA) is used in the treatment of a variety of neurological and nonneurological conditions.11 Its potential use for migraines was incidentally noted in patients who were cosmetically treated for wrinkles.12 Subsequent trials confirmed its safety and efficacy for this indication. It was approved by the Medicines and Healthcare Products Regulatory Agency (MHRA) in July 2010 followed by the US Food and Drug Administration later in the same year for the prophylaxis of CM. The National Institute for Health and Care Excellence (NICE) endorsed its use for adults with CM in May 2012.13 This article will discuss the pathophysiology of CM and the mechanism of action of onaBoNTA in CM and will focus on the evidence behind the use of onaBoNTA for this indication.
Risk factors for transformation
Approximately 2.5% of people with EM will experience transformation to CM annually.14,15 Given the considerable socioeconomic impact of CM, it is important for public health authorities to recognize the risk factors associated with migraine chronification, especially the modifiable ones, as early intervention could potentially prevent transformation. Several factors are known to influence migraine chronification (Table 1). These include some nonmodifiable ones such as age, female sex, genetic factors, low socioeconomic status, and head injury.16 Among the modifiable risk factors, psychiatric disorders such as depression, anxiety, bipolar disorder, and panic disorder were three times more common in those affected by CM.17 Obesity is a strong risk factor for CM.18 Individuals who are overweight, obese, and morbidly obese are all more likely to develop CM (odds ratios of 1.4, 1.7, and 2.2, respectively).18 Medication overuse is also a significant risk factor for CM.19,20 Frequent use of analgesics can transform some patients with EM to CM or chronic daily headache. The use of nonsteroidal anti-inflammatory drugs fewer than 10 times per month is paradoxically protective against the development of medication overuse headache (MOH), whereas more frequent use has the opposite effect.21 It is however unclear if MOH is a distinct biological entity or whether medication overuse acts as a trigger to the underlying biology of headache.20 Other risk factors for transformation include sleep-related problems such as insomnia, habitual snoring, sleep bruxism, and daytime sleepiness; caffeine overuse; high baseline frequency of headaches; and cutaneous allodynia.16
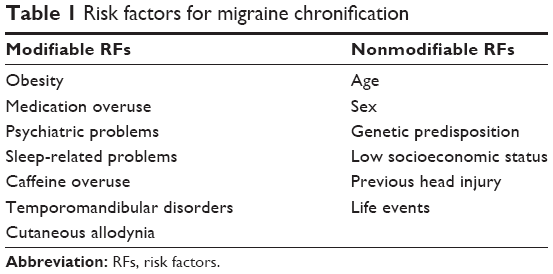 | Table 1 Risk factors for migraine chronification |
Pathophysiology of CM
Little was known about the pathophysiology of CM and the associated brain changes when it was first described. However, our understanding of this condition has improved considerably with research in the past two decades. The pathophysiology of CM is still not fully understood. It is likely that multiple factors and different parts of the brain are involved. The widely accepted theory for migraine with aura suggests that cortical spreading depression, a wave of neuronal hyperactivity followed by an area of cortical depression, accounts for the aura22,23 and that the headache depends on activation of the trigeminovascular pain pathway.24,25 In CM, atypical pain processing, central and peripheral sensitization, cortical hyperexcitability, and neurogenic inflammation all have a role to play.26 The pain of migraine suggests an important role of the nociceptive activation of the trigeminovascular pathway.27 In migraine sufferers, pain-induced functional activation of pain-inhibiting brainstem regions and the functional connectivity of brainstem pain modulatory regions are atypical.26,28 This atypical pain modulation appears to play a role in transformation of EM to CM.
Central and peripheral sensitization occur during migraine. Central sensitization refers to a condition in which nociceptive neurons in the spinal and medullary dorsal horn exhibit increased excitability, increased synaptic strength, and enlargement of their receptive fields beyond the original site of inflammation or injury.29 Peripheral sensitization defines a state in which primary afferent nociceptive neurons exhibit increased responsiveness to external mechanical or thermal stimuli at the site of inflammation or injury.29 Peripheral sensitization of the trigeminal nerve, and the blood vessels supplied by them, leads to a throbbing pain. This stage of migraine is termed first-order neuron sensitization.30 Second-order neuron sensitization occurs when sensitization spreads to the second-order trigeminovascular neurons in the spinal trigeminal nucleus, causing scalp hypersensitivity or cutaneous allodynia.30 Third-order sensitization is the result of sensitization spreading to the thalamus, which causes extracephalic hypersensitivity.30 Allodynia is therefore the clinical manifestation of second- and third-order neuron sensitization and a sign of migraine progression.31 There is evidence that allodynia symptoms occur more commonly in patients who have an extensive history of CM.31 Migraines are mostly nonallodynic initially but become allodynic after a few years with repeated attacks due to sensitization of the trigeminovascular pathway, which results in a lower threshold for activation and therefore more frequent migraine attacks.32 As mentioned earlier, this makes allodynia a marker of chronification.32
Cortical hyperexcitability is thought to be another major factor precipitating transformation of EM to CM.33 Increased cortical excitability, compared to subjects with EM and migraine-free controls, has been shown in subjects with CM.34,35 Aurora et al36 demonstrated that magnetic suppression of perceptual accuracy was significantly reduced in 25 patients with CM compared with subjects with EM and migraine-free controls, indicating increased cortical excitability. Positron emission tomography scan studies were also performed in ten of the patients with CM, and enhanced metabolism was observed in the pons and right temporal cortex compared to global cerebral metabolism.36
Neurogenic inflammation refers to an array of events, including local increase in blood flow, leakage of plasma protein from blood vessels, mast cell degranulation, and platelet aggregation.37 Several messengers that activate or sensitize pain-signaling pathways have been found in relation to cortical spreading depression in animal models.38 A dense network of dural nerve fibers that react with substance P, calcitonin gene-related peptide (CGRP), and vasoactive intestinal peptide (VIP) has been found.39,40 In a study of 103 women with CM, CGRP levels were found to be significantly increased in CM subjects compared with healthy controls, subjects with EM, and those with episodic cluster headaches,41 providing further support of the role of neurogenic inflammation in CM. Interictal VIP levels have also been found to be raised in subjects with CM, supporting a role of VIP in the sensitization of pain circuits in CM.42
Mechanisms of action of botulinum toxin
Botulinum neurotoxins (BoNTs) are produced by strains of the bacillus Clostridium botulinum.43 Seven serotypes, classified A–G based on their immunological properties, are known.44 BoNT is chemically inactive when synthesized, consists of a heavy chain and a light chain (LC). The role of the heavy chain is to facilitate uptake of the whole molecule into the cytosol.45 Irrespective of the route by which BoNTs enter the body, they are transported to the neuromuscular junction. They are then internalized by binding to different gangliosides, namely synaptic vesicle-2, synaptotagmin I, or synaptotagmin II.46–49 In the cytosol, the LC then cleaves soluble N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE) complex in motor neurons. These SNARE molecules are central to the mechanism that mediates the fusion of synaptic vesicles with the presynaptic plasma membrane, resulting in the release of neurotransmitter.50 The LC of BoNT-A and BoNT-E specifically targets synaptosome-associated protein of 25 kDa (SNAP25), while BoNT serotypes B, D, F, and G cleave synaptobrevin, a vesicle-associated membrane protein.51,52 BoNT-C cleaves both SNAP25 and syntaxin, another plasma membrane-anchored SNARE.51,52 The SNARE complex is essential for acetylcholine release at the presynaptic nerve endings.53 Its inhibition therefore results in flaccid paralysis. Recovery occurs through sprouting of nerve terminals, thus re-establishing synaptic contacts.54 This takes about 3 months, which usually coincides with the wearing off effect seen following injection of botulinum toxin in clinical practice.
While it is well understood how BoNTs cause paralysis, the exact mechanism by which onaBoNTA relieves CM is less clear. There is evidence that onaBoNTA has a direct antinociceptive effect, which is independent of its muscular-relaxing properties.55 This effect is likely dependent on several mechanisms and is believed to involve the inhibition of neurotransmitter release from motor neurons and from sensory nociceptive neurons of muscle fibers.56 Uptake of the neurotoxin in sensory neurons that innervate the skin and muscles is thought to inhibit the release of inflammatory mediators at several sites within the sensory neuron.57,58 Animal studies have shown that onaBoNTA can block the stimulated release of CGRP, glutamate, and substance P from trigeminal neurons. onaBoNTA has also recently been shown to decrease CGRP plasma levels in patients with CM.59 However, a local extracranial effect of BoNTs is unlikely to account for all the antimigrainous effects. It is likely that onaBoNTA reduces peripheral sensitization. Given that central sensitization results from ongoing input from pain fibers, the inhibition of these peripheral signals indirectly inhibits central sensitization. Matak et al60 have also demonstrated that the antinociceptive effects of onaBoNTA are likely to involve suppression of second-order nociceptive neurons by blocking the release of CGRP and glutamate from primary nociceptors that terminate in the medullary dorsal horn, thus providing evidence for axonal transport of onaBoNTA via sensory fibers.
Efficacy
Early trials
The prophylactic effects of onaBoNTA for migraines were initially observed by a plastic surgeon treating patients cosmetically for wrinkles, and an open-label study involving 106 patients with headaches was conducted.61 In those with clinically definite migraines, 51% reported a complete remission lasting an average of 4.1 months and 38% responded partially for a mean duration of 2.7 months.61 In the same year, Silberstein et al62 led a double-blind, randomized, vehicle-controlled study of 123 patients with a history of two to eight moderate-to-severe migraine attacks per month, with or without aura. Eligible patients were randomized to single administrations of vehicle or botulinum toxin type A, 25 U or 75 U, injected into multiple sites of pericranial muscles at the same visit. The primary efficacy end point was a change from baseline in the frequency of moderate-to-severe migraines. Those who received botulinum toxin A showed significantly fewer migraine attacks per month, reduced maximum severity of migraines, a reduced number of days using acute migraine medications, and reduced incidence of migraine-associated vomiting compared to the vehicle group. Botulinum toxin A treatment was well tolerated. However, the 75-U treatment group displayed a significantly higher rate of treatment-related adverse events, in the form of blepharoptosis, diplopia, and injection site weakness, compared to the vehicle group.62
Meanwhile, it became apparent that botulinum toxin A injections were not effective in the treatment of chronic tension headaches. In 2001, Schmitt et al63 conducted a randomized, placebo-controlled trial to study the efficacy of 20 U of botulinum toxin injected into frontal and temporal muscles in 59 patients (30 received botulinum toxin and 29 received placebo). At 8 weeks post-treatment, pain intensity, number of pain-free days, and consumption of analgesics were not statistically different between the groups. Rollnik et al64 used higher doses of botulinum toxin A (200 U) in a double-blind, placebo-controlled trial involving 21 patients and reassessed the patients at 4 weeks, 8 weeks, and 12 weeks and found no difference in clinical outcome between the placebo and botulinum toxin A groups. A further study involving eight patients assigned to either placebo or even higher doses of botulinum toxin A (500 U) failed to show any significant differences in outcome despite electromyographic evidence of a reduction in resting muscle activity in the botulinum toxin A group, suggesting that muscle tone plays a minor role in the pathophysiology of chronic tension-type headaches.65
Botulinum toxin A was then studied in the setting of chronic daily headaches. In 2004, Ondo et al66 enrolled 60 patients with headaches on more than 15 days per month to either 200 U of botulinum toxin A or placebo in a 1:1 ratio, after which they were assessed at 4 weeks and 12 weeks. All patients then entered an open-label phase and received botulinum toxin A. They were again assessed at 4 weeks and 12 weeks following the second set of injections. Forty-six of those patients had chronic tension-type headaches, while the rest had CM. Fewer headache days, which was the primary end point, was significant in the botulinum toxin group from week 8 to week 12 compared with placebo (P<0.05, t-test).66 Medication overuse was a predictor of better response in the placebo-controlled phase but not in the open-label phase.66
Larger studies were needed to confirm these results. In 2005, Mathew et al67 enrolled 355 patients with chronic daily headache in a randomized, double-blind, placebo-controlled trial. Following a placebo-response period to identify placebo responders and nonresponders, patients in each group were randomized to either botulinum toxin A or placebo. In all, 279 patients (79%) were classified as placebo nonresponders and 76 (21%) as placebo responders. The primary end point was a change from baseline in the frequency of headache-free days in a 30-day period for the placebo nonresponder group at day 180. At day 180, placebo nonresponders treated with botulinum toxin A experienced a mean improvement of 6.7 days from baseline compared to a mean of 5.2 days in the placebo-treated patients. This difference of 1.5 days was not statistically significant. The treatment did meet secondary efficacy outcome measures, including the percentage of patients experiencing a 50% or more decrease in the frequency of headache days, in addition to statistically significant reductions in headache frequency. Results of a further large, double-blind, place-controlled trial with a similar methodology of identifying placebo responders and nonresponders, but this time involving 702 patients with chronic daily headache, became available in the same year.68 Different doses of botulinum toxin A were given: 225 U, 150 U, and 75 U. Patients returned for treatment at days 90 and 180. The primary efficacy end point was a mean change from baseline in the frequency of headache-free days at day 180 for the placebo nonresponder group. This was however not met. At day 180, mean improvements from baseline of 6.0, 7.9, 7.9, and 8.0 headache-free days per month were observed in the placebo nonresponder group treated with botulinum toxin A at 225 U, 150 U, 75 U, or placebo, respectively (P=0.44). These two studies suggested that botulinum toxin A was not effective for chronic daily headaches. However, it is important to note that the term CM was only defined as an entity in 2004, and prior to this, patients suffering from CM were either merged under the umbrella of chronic daily headaches/mixed headache syndrome or the term transformed migraine was used. It is therefore probable that trials enrolling patients with chronic daily headache before 2004 also included patients with CM, which would account for the small, albeit statistically insignificant, improvements seen in the two trials described above.
Subsequent trials
In 2006, Silberstein et al69 published the results of a multicenter, randomized, double-blind, placebo-controlled, parallel-group study of botulinum toxin A for the prophylactic treatment of chronic tension-type headache. Three hundred patients were enrolled. Injections were given in five muscle groups, and there were six treatment arms. Three of them received botulinum toxin A but at different doses (50 U, 100 U and 150 U), two received botulinum toxin (86 U and 100 U) in three muscle groups and placebo in the other two muscle groups, and one received placebo only. The primary efficacy end point was a mean change from baseline in the number of tension type headache-free days per month. This was not met, and the study also failed to meet a few other secondary efficacy variables. It appeared that the momentum gathered from early, yet small, studies supporting the use of botulinum toxin A for headaches was rapidly waning. This even led Evers and Olesen to announce the end of the road for botulinum toxin A.70 However, some investigators turned their attention to CM. In 2008, Freitag et al71 carried out a randomized, double-blind, placebo-controlled trial studying botulinum toxin A for CM. In all, 60 patients were randomized and 100 U of botulinum toxin A was administered in a fixed dose and site paradigm. Botulinum toxin A was found to be significantly superior to placebo for the primary end point of reduction in migraine headache episodes. However, larger and more robust trials were still needed to confirm these findings.
PREEMPT trials
Results of the Phase III REsearch Evaluating Migraine Prophylaxis Therapy 1 (PREEMPT 1) became available in 2010.72 This was a phase III multicenter, double-blind, placebo-controlled study conducted to evaluate the efficacy and safety of onaBoNTA for the prophylaxis of headaches in patients with CM. PREEMPT 1 had a 28-day baseline screening period followed by a 24-week double-blind, parallel-group, placebo-controlled phase during which two injection cycles of medications (onaBoNTA or placebo) were given. After this, all patients entered a 32-week, open-label phase with three injection cycles. A total of 679 patients were enrolled. The primary efficacy end point was mean change from baseline in frequency of headache episodes for the 28-day period ending with week 24. Secondary efficacy end points were frequency of headache days, migraine days, migraine episodes, and overall acute headache pain medication use. The trial however failed to achieve its primary end point, that is, no significant difference was detected for the mean change from baseline in the frequency of headache episodes at week 24. The investigators however found a significant difference in baseline headache frequency between the two groups (P=0.023) and therefore postulated that this anomaly could explain the failure in meeting the primary end point. A post hoc analysis was performed and this showed significant reduction in headache episodes with onaBoNTA compared to placebo at weeks 4, 8, and 20.
With regard to secondary efficacy end points, there was a significant between-group difference in the mean decrease from baseline in the frequency of headache days observed at all time points (−7.8 onaBoNTA vs −6.4 placebo, P=0.006). Although large improvements for mean change from baseline in frequencies of migraine episodes and acute pain medication intakes were noted, these did not achieve statistically significant between-group differences. However, a further post hoc analysis identified a significant reduction in the frequency of triptan intake in the onaBoNTA group at week 24 (−3.3 onaBoNTA vs −2.5 placebo, P=0.023). In addition, a significant decrease in disability and improvement in functioning were observed in the onaBoNTA group compared to placebo as demonstrated by a mean change in total Headache Impact Test-6 (HIT-6) score at all time points (P<0.001).
PREEMPT 2 was another Phase III study, with a 24-week, double-blind, placebo-controlled phase, followed by a 32-week, open-label phase.73 It was methodologically quite similar to PREEMPT 1 with 705 patients randomized to onaBoNTA (n=347) or placebo (n=358). However, it appeared that the investigators had taken preemptive measures, given the failure of PREEMPT 1 to meet its primary efficacy end point. The primary efficacy end point of PREEMPT 2 was switched to a mean change from baseline in the frequency of headache days instead of headache episodes. Nonetheless, this change, from a purely methodological point of view, is unlikely to majorly impact on the treatment outcome, although it is criticized because it was made after the results of PREEMPT 1 became available. PREEMPT 2 therefore met its primary efficacy end point with a statistically significant mean change from baseline in frequency of headache days from the first post-treatment study visit (week 4) to week 24 (−9.0 days onaBoNTA vs −6.7 days placebo, P<0.001; 95% confidence interval [−3.25, −1.31]). This was anticipated given that PREEMPT 1 had already revealed these findings. Results of several other secondary efficacy end points included in PREEMPT 1 were also replicated in PREEMPT 2, as expected. Interestingly, the mean change from baseline in headache episodes in PREEMPT 2, a secondary efficacy variable, did reach statistical significance in the onaBoNTA-treated group compared to placebo (P=0.003). Both PREEMPT studies showed a similar mean change from baseline in frequency of headache episodes in the onaBoNTA group (−5.2 and −5.3, respectively). The mean change from baseline for the placebo groups in these trials, on the other hand, was different (−5.3 and −4.6, respectively). This further supports the possibility that the higher mean number of headache episodes in the placebo group than the onaBoNTA group in PREEMPT 1 confounded the post-treatment effect as this anomaly was not observed in PREEMPT 2.
A pooled analysis (n=1,384) showed a statistically significant mean reduction from baseline in frequency of headache days favoring onaBoNTA over placebo at week 24 (−8.4 vs −6.6; P<0.01) and all other time points.74 In a further pooled analysis, there were statistically significant reductions in secondary efficacy variables at 56 weeks favoring those who received onaBoNTA/onaBoNTA over placebo/onaBoNTA (Aurora et al).75 These included frequencies of migraine days (−11.2 vs −10.3, P=0.018) and moderate/severe headache days (−10.7 vs −9.9, P=0.027) (Aurora et al).75 In addition, after the open-label phase, statistically significant within-group changes from baseline were observed for efficacy variable.75 What also granted the PREEMPT results more clinical importance was the fact that relevant improvements in functioning and health-related quality of life were observed with onaBoNTA compared with placebo in both trials.72,73 The impact of headache on functioning and health-related quality of life was assessed using HIT-6 and Migraine-Specific Quality of Life questionnaire, respectively. These improvements were even more manifest in the pooled analyses. In relation to declining disability, there was both a significant reduction in the proportion of patients with a severe (≥60) HIT-6 score and a mean decrease from baseline in total HIT-6 score in the onaBoNTA group compared to the placebo group at all time points through week 24.72–75 PREEMPT was the largest clinical program to study the use of onaBoNTA as a prophylactic treatment for CM according to an established set of diagnostic criteria and specific clinically relevant outcome measures. The two PREEMPT trials were methodologically robust. They were well-designed studies with large numbers of patients and sufficiently long-blinded and open-label phases. The trials also used an electronic diary because of its superior reliability over paper diaries. This allowed for greater compliance, and data were retrieved without requiring a long recall time for patients. The PREEMPT trials have also established the treatment paradigm used in clinical practice. A total of 155 U of onaBoNTA divided equally into 31 injection sites is administered.
Efficacy end points in CM trials
Guidelines for the controlled trials of prophylactic treatment of CM patients have been published to assist in the design of well-controlled clinical trials.76,77 Selecting appropriate primary efficacy end points depending on the nature of the trial is essential. This should be done a priori and should be influenced by the objective of the study.76 It therefore seems logical that a study on CM should not use frequency of migraine episodes as the primary efficacy end point because subjects could potentially have a single episode that lasts for several days. The use of migraine or headache days would appear to be more appropriate. They are also easier to record on headache diaries. However, migraine days, unlike migraine attacks, represent a compound end point as it includes attack duration. This in turn is influenced by acute (symptomatic) treatment of the migraine and the therapy for recurrence/relapse, and neither is standardized in migraine prevention randomized controlled trials.77 Therefore, the efficacy end point migraine days is neither as sensitive nor as specific as migraine attacks when the primary study objective is the evaluation of a preventative agent. Moreover, the main objectives of a prophylactic agent should be to reestablish the patient’s ability to function as well as to improve their overall quality of life. However, there seems to be a lack of powerful tools to assess the changes in quality of life as an end point in migraine trials.
Shortcomings of the PREEMPT trials
onaBoNTA was statistically superior to placebo in numerous efficacy variables. However, statistical superiority does not necessarily correlate with clinical improvement. In fact, it is unclear when a statistically significant change equates with definite clinical benefit. The primary end point of PREEMPT 1, which was headache episodes, is probably not an adequate outcome measure in patients who have near-daily headaches, as discussed above. If one also looks at the absolute gain of headache-free days, the primary efficacy end point of PREEMPT 2, this was only 2.3 days on average over a 28-week period.
One of the major criticisms of the PREEMPT trials is the high placebo effect observed in both studies. In previous studies, placebo rates in migraine prophylaxis have ranged from 20% to 49%.78 However, in the PREEMPT pooled analyses, >50% responder rates at week 24 with placebo ranged from 35.1% to 43.4% for efficacy variables such as headache days, headache episodes, migraine days, and migraine episodes and were as high as 68% at week 56 for migraine episodes. The high placebo effect could represent spontaneous improvement. It is known that the natural course of CM is to revert to the episodic form in about 78% of patients over a 3-year period.79 Placebo analgesia is accompanied by an intrinsic expectation of pain relief, and Diener et al rightly pointed out the need to standardize this expectation across clinical trials.80 In a study comparing zolmitriptan, sumatriptan, and placebo, subjects were informed that the probability of receiving an active drug to placebo was 16:1.81 No difference for any end point was seen with zolmitriptan or sumatriptan compared to placebo. This is a classic example of how expectation can impact on placebo response. It has also been argued that, in the PREEMPT trials, effective blinding might not have been maintained because the placebo group would not observe the changes in the appearance of the forehead, which would occur with onaBoNTA injections. The level of unblinding was not mentioned in the PREEMPT trials. However, the authors called attention to the high placebo rate, which suggests that effective blinding must have been maintained, as the reverse would be true if unblinding had occurred. It is therefore unlikely that the lack of changes in forehead appearance had contributed to significant unblinding.
Another concern is that both PREEMPT trials enrolled a majority of subjects with concomitant MOH. This is probably representative of real-life patients. Observational and clinical trials have shown that 50%–80% of patients with CM overuse acute medication. It is however likely that a substantial proportion of subjects had a secondary headache and not CM alone. MOH can respond following a period of detoxification. This high proportion of MOH could partly be the reason for the improvement seen in both treatment and placebo arms, given that there was a reduction in the frequency of triptan and other acute headache pain medication intakes. The investigators have also failed to indicate if the response was any different in those with or without MOH, which would have been relevant to clinical practice. Moreover, 40% of the subjects in the PREEMPT trials had never received a preventative treatment, and this is even more surprising given the fact that the mean age of the subjects was about 40 years, having suffered from CM for a mean duration of about 20 years. This, however, is not reflective of a real-life setting, whereby most of these patients would have tried a migraine prophylaxis. In the UK, onaBoNTA for CM is recommended by NICE only for those who have previously failed three different migraine prophylaxis medications.13
onaBoNTA versus oral prophylactics
Unfortunately, in the case of CM, there is no clearly effective comparator for head-to-head trials, hence the comparison with placebo in the PREEMPT trials. onaBoNTA is the only drug approved by the US Food and Drug Administration for headache prophylaxis in CM. There have however been two randomized, double-blind trials comparing botulinum toxin A with oral prophylactics. Mathew and Jaffri82 randomized 60 patients to either onaBoNTA, maximum 200 U at baseline and 3 months, plus an oral placebo or topiramate, 4-week titration to 100 mg/day, plus placebo saline injections. The primary end point was treatment responder rate assessed using the Physical Global Assessment 9-point scale. Secondary end points included the change from baseline in the number of headache/migraine days per month (headache diary) and disability measured using HIT-6. Of the 60 patients, 36 completed the study at the end of the 9 months of active treatment. The majority of patients in both groups reported moderate-to-marked improvement at all time points. No significant between-group difference was observed, except for statistically significant improvement favoring topiramate at month 9. However, 24.1% of patients in the topiramate group reported adverse effects that required permanent discontinuation of treatment compared to 2.7% of patients in the onaBoNTA group.
Cady et al83 used a similar methodology to Mathew et al and randomized 59 patients to either onaBoNTA plus placebo tablets or topiramate plus placebo injections. Subjects not reporting a >50% reduction in headache frequency at 12 weeks were invited to participate in a 12-week open-label extension study with onaBoNTA. Twenty patients volunteered for this extension (nine from the topiramate arm and eleven from the onaBoNTA arm). Overall, the study showed statistically significant within-group differences but not between groups with onaBoNTA and topiramate demonstrating similar efficacy. When compared to amitriptyline in a trial involving 72 patients, >50% reduction in headache days was recorded in 67.8% of patients receiving onaBoNTA and 72% of patients receiving amitriptyline (P=0.78).84 There were also no statistically significant between-group differences for efficacy variables, such as reduction in pain intensity and reduction in analgesic doses. Although all three trials showed botulinum toxin A to be as effective as the oral prophylactic being studied, they were small and none were designed to detect small between-group differences.
Safety and tolerability
The PREEMPT trials have also confirmed the long-term safety and tolerability of onaBoNTA after five treatment cycles. Adverse event rates were 28.5% for onaBoNTA and 12.4% for placebo in the double-blind phase. This was 34.8% for patients treated only with onaBoNTA for the full five cycles lasting 56 weeks.85 Neck pain was the most common side effect reported, occurring in 4.3% of patients. Other frequently reported side effects include injection site pain (2.1%), eyelid ptosis (1.9%), and muscular weakness (1.6%). onaBoNTA for CM has not been studied in pregnant and lactating subjects, and therefore, its safety in this group is not established.
Cost-effectiveness
While the PREEMPT program has established the efficacy, safety, and tolerability of onaBoNTA, less is known of its effects on health resources. Rothrock et al86 recently published data from an open-label study involving 230 patients with CM refractory to two or more oral prophylactics who were treated with two cycles of onaBoNTA. Treatment costs of onaBoNTA were compared to the direct medical costs related to migraine in untreated patients (emergency department visits and hospitalizations). Treatment-refractory CM patients who started onaBoNTA experienced a significant cost-offset through reduced migraine-related emergency department visits, urgent care visits, and hospitalizations in the 6 months following treatment initiation of onaBoNTA.84 This study was performed in the US, and caution should be exercised when extrapolating these results to other health care systems. Batty et al have evaluated the cost-effectiveness of onaBoNTA for CM in the UK.87 The Markov model was used and efficacy data and utility values were taken from the pooled PREEMPT program. At 2 years, treatment with onaBoNTA resulted with an increase in costs of £1,367 and an increase in quality-adjusted life years of 0.1 compared to placebo, resulting in an incremental cost-effectiveness ratio of £15,028.87 NICE’s committee concluded that the most likely cost-effectiveness estimate was £18,900 per quality-adjusted life year gained. This is lower than the £20,000–£30,000 range, which NICE would typically deem to be a cost-effective use of National Health Service (NHS) resources.13
What still needs to be addressed?
We conquer with the general consensus that onaBoNTA is effective for the treatment of CM as evidenced by its superiority over placebo for a number of efficacy variables in the PREEMPT trials. However, some unanswered questions still remain. Do those with or without MOH respond differently, and should MOH be dealt with before trying onaBoNTA? NICE certainly recommends that MOH should be tackled before patients are offered onaBoNTA. However, preliminary data from 370 patients treated with a total of 1,266 cycles of onaBoNTA show that patients with CM respond to onaBoNTA irrespective of their consumption of analgesics.88 It is unclear which subgroup of patients is more likely to respond and how to identify them. NICE advises discontinuation of treatment with onaBoNTA if the patient fails to respond to two treatment cycles (negative stopping rule) or successfully converts to EM, ie, headaches of less than 15 days (positive stopping rule). This means that a patient with 14 headache days would have to stop treatment, whereas a patient with 16 headache days would be allowed to continue. Our early experience in treating CM also suggested that some patients experienced a reduction in migraine days (hence, reverting to EM) without any reduction in headache days, leading to early discontinuation. This led us to propose our own responder criteria (Hull criteria) whereby a responder is defined as any patient with either a 50% reduction in either headache or migraine days or an increment in crystal clear days twice that of the baseline in a 30-day period (where pretreatment crystal clear days were at least 3 days; otherwise, patient has to achieve at least six crystal clear days post-treatment to be a responder).89 We found that an extra 17% of patients, who would have previously been classified as a nonresponder under the NICE criteria and would have had to stop treatment, improved with reduction in the severity of their headaches and number of migraine days.90 Questions surrounding optimal duration of treatment also need to be addressed as these will impact on cost-effectiveness. Our 2-year follow-up data demonstrated that out of our patients who were successfully converted to EM, 21% relapsed after a mean of 7 months (range 4–10 months) and recommenced treatment.91 The only other available data are from Rothrock et al92 whereby 67% of patients continued to receive treatment after 2 years, although these patients were receiving treatment through insurance reimbursement and criteria for the continuation of treatment were unclear. We have also proposed our modified stopping rule whereby we use a reduction of headache days to less than 10 days per month for 3 months, given that those with a high frequency of headaches of 10–14/month carry a higher risk of relapse to CM. Data from a real-life setting in the UK have confirmed the results of PREEMPT;86 however, the authors agree that onaBoNTA should be retained for those who have failed three oral migraine prophylactics as recommended by NICE, and until further data on its cost-effectiveness suggest otherwise, its position in the care pathway of chronic migraineurs in the UK will remain unchanged.
Conclusion
CM is a debilitating condition with significant socioeconomic impact. onaBoNTA is of proven benefit for the management of this condition. Data from the PREEMPT trials suggest that onaBoNTA not only reduces headache days but also leads to improvement in functioning and quality of life. It is safe and well tolerated in the long term. However, there are still some unanswered questions surrounding identification of responders, duration of treatment, as well as which patient subgroup is more or less likely to respond. Further studies are required to address these questions.
Disclosure
Fayyaz Ahmed has received honorarium to deliver training workshops for Allergan paid to British Association for the Study of Headache (BASH) and received honorarium to attend Allergan Advisory Board meetings. He is on the standing committee of the Headache Guidelines (CG150 Revision) for the National Institute of Clinical Excellence, trustee of the Migraine Trust, and educational officer for the BASH. The authors report no other conflicts of interest in this work.
References
Steiner TJ, Stovner LJ, Birbeck GL. Migraine the 7th disabler. J Headache Pain. 2013;14:1. | ||
Steiner TJ, Scher AI, Stewart WF, Kolodner K, Liberman J, Lipton RB. The prevalence and disability burden of adult migraine in England and their relationship to age, gender and ethnicity. Cephalalgia. 2003;23:519–527. | ||
Clarke CE, MacMillan L, Sondhi S, Wells NE. Economic and social impact of migraine. Q J Med. 1996;89:77–84. | ||
Stovner LJ, Andree C. Impact of headache in Europe: a review for the Eurolight project. J Headache Pain. 2008;9:139–146. | ||
Natoli JL, Manack A, Dean B, et al. Global prevalence of chronic migraine: a systematic review. Cephalalgia. 2010;30:599–609. | ||
Bigal ME. The paradoxical effects of analgesics and the development of chronic migraine. Arq Neuropsiquiatr. 2011;69(3):544–551. | ||
IHS. The international classification of headache disorders: 3rd edition (beta version). Cephalalgia. 2013;33(9):629–808. | ||
Bigal ME, Serrano D, Reed DM, Lipton RB. Chronic migraine in the population: burden, diagnosis, and satisfaction with treatment. Neurology. 2008;71:559–566. | ||
Stokes M, Becker WJ, Lipton RB, et al. Cost of health care among patients with chronic and episodic migraine in Canada and the USA: results from the International Burden of Migraine Study (IBMS). Headache. 2011;51:1058–1077. | ||
Blumenfeld AM, Varon SF, Wilcox TK, et al. Disability, HRQoL and resource use among chronic and episodic migraineurs: results from the International Burden of Migraine Study (IBMS). Cephalalgia. 2011;31: 301–315. | ||
Gooriah R, Ahmed F. Therapeutics uses of botulinum toxin. J Clin Toxicol. 2015;5:225. | ||
Binder WJ, Blitzer A, Brin MF. Treatment of hyperfunctional lines of the face with botulinum toxin A. Dermatol Surg. 1998;24: 1198–1205. | ||
NICE technology appraisal guidance 260. Botulinum toxin type A for the prevention of headaches in adults with chronic migraine; 2012. Available from: http://publications.nice.org.uk/botulinum-toxin-type-a-for-the-prevention-ofheadaches-n-adults-with-chronic-migraine-ta260 | ||
Scher AI, Stewart WF, Liberman J, Lipton RB. Prevalence of frequent headache in a population sample. Headache. 1998;38(7):497–506. | ||
Scher A, Stewart W, Ricci J, Lipton R. Factors associated with the onset and remission of chronic daily headache in a population-based study. Pain. 2003;106(1):81–89. | ||
Cho SJ, Chu MK. Risk factors for chronic daily headaches or chronic migraine. Curr Pain Headache Rep. 2015;19:465. | ||
Chen YC, Tang CH, Ng K, Wang SJ. Comorbidity profiles of chronic migraine sufferers in a national database in Taiwan. J Headache Pain. 2012;13(4):311–319. | ||
Bigal ME, Lipton RB. Obesity is a risk factor for transformed migraine but not chronic tension-type headache. Neurology. 2006;67(2): 252–257. | ||
Bigal ME, Rapoport AM, Sheftell FD, Tepper SJ, Lipton RB. Transformed migraine and medication overuse in a tertiary headache centre: clinical characteristics and treatment outcomes. Cephalalgia. 2004;24: 483–490. | ||
Goadsby PJ. Is medication-overuse headache a distinct biological entity? Nat Clin Pract Neurol. 2006;2:401. | ||
Starling AJ, Hoffman-Snyder C, Halker RB, et al. Risk of development of medication overuse headache with nonsteroidal anti-inflammatory drug therapy for migraine: a critically appraised topic. Neurologist. 2011; 17(5):297–299. | ||
Lauritzen M. Pathophysiology of the migraine aura. The spreading depression theory. Brain. 1994;117(pt 1):199–210. | ||
Ayata C. Cortical spreading depression triggers migraine attack: pro. Headache. 2010;50:725–730. | ||
Olesen J, Burstein R, Ashina M, Tfelt-Hansen P. Origin of pain in migraine: evidence for peripheral sensitisation. Lancet Neurol. 2009;8: 679–690. | ||
Levy D. Migraine pain and nociceptor activation – where do we stand? Headache. 2010;50:909–916. | ||
Goadsby PJ, Charbit AR, Andreou AP, Akerman S, Holland PR. Neurobiology of migraine. Neuroscience. 2009;161:327–341. | ||
Moulton EA, Burstein R, Tully S, Hargreaves R, Becerra L, Borsook D. Interictal dysfunction of a brainstem descending modulatory center in migraine patients. PLoS One. 2008;3:e3799. | ||
Schwedt TJ, Larson-Prior L, Coalson RS, et al. Allodynia and descending pain modulation in migraine: a resting state functional connectivity analysis. Pain Med. 2014;15:154–165. | ||
Burstein R, Zhang X, Levy D, Aoki KR, Brin MF. Selective inhibition of meningeal nociceptors by botulinum neurotoxin type A: therapeutic implications for migraine and other pains. Cephalalgia. 2014;34:853–869. | ||
Mathew NT. Pathophysiology of chronic migraine and mode of action of preventive medications. Headache. 2011;51:84–92. | ||
Mathew NT, Kailasam J, Seifert T. Clinical recognition of allodynia in migraine. Neurology. 2004;63:848–852. | ||
Louter MA, Bosker JE, van Oosterhout WP, et al. Cutaneous allodynia as a predictor of migraine chronification. Brain. 2013;136:3489–3496. | ||
Aurora SK, Kulthia A, Barrodale PM. Mechanism of chronic migraine. Curr Pain Headache Rep. 2011;15:57–63. | ||
Aurora SK. Is chronic migraine one end of a spectrum of migraine or a separate entity? Cephalalgia. 2009;29:597–605. | ||
Aurora SK. Spectrum of illness: understanding biological patterns and relationships in chronic migraine. Neurology. 2009;72(suppl 5): S8–S13. | ||
Aurora SK, Borrodale PM, Tipton RL, Khodavirdi A. Brainstem dysfunction in chronic migraine as evidenced by neurophysiological and positron emission tomography studies. Headache. 2007;47:996–1003. | ||
Bernstein C, Burstein R. Sensitization of the trigeminovascular pathway: perspective and implications to migraine pathophysiology. J Clin Neurol. 2012;8(2):89–99. | ||
Charles AC, Baca SM. Cortical spreading depression and migraine. Nat Rev Neurol. 2013;9:637–644. | ||
Messlinger K, Hanesch U, Baumgartel M, Trost B, Schmidt RF. Innervation of the dura mater encephali of cat and rat: ultrastructure and calcitonin gene-related peptide-like and substance P-like immunoreactivity. Anat Embryol. 1993;188:213–219. | ||
Messlinger K, Fischer MJ, Lennerz JK. Neuropeptide effects in the trigeminal system: pathophysiology and clinical relevance in migraine. Keio J Med. 2011;60(3):82–89. | ||
Cernuda-Morollon E, Larrosa D, Ramon C, Vega J, Martinez-Camblor P, Pascual J. Interictal increase of CGRP levels in peripheral blood as a biomarker for chronic migraine. Neurology. 2013;81:1191–1196. | ||
Cernuda-Morollon E, Martinez-Camblor P, Alvarez R, Larrosa D, Ramon C, Pascual J. Increased VIP levels in peripheral blood outside migraine attacks as a potential biomarker of cranial parasympathetic activation in chronic migraine. Cephalalgia. 2015;35:310–316. | ||
Kostrzewa R, Segura-Aguilar J. Botulinum neurotoxin: evolution from poison, to research tool – onto medicinal therapeutic and future pharmaceutical panacea. Neurotox Res. 2007;12:275–290. | ||
Schantz EJ, Johnson EA. Properties and use of botulinum toxin and other microbial neurotoxins in medicine. Microbiol Rev. 1992;56(1): 80–99. | ||
Ramachandran R, Yaksh TL. Therapeutic use of botulinum toxin in migraine: mechanisms of action. Br J Pharmacol. 2014;171(18): 4177–4192. | ||
Dong M, Yeh F, Tepp WH, et al. SV2 is the protein receptor for botulinum neurotoxin A. Science. 2006;312:592–596. | ||
Fu Z, Chen C, Barbieri JT, Kim JJ, Baldwin MR. Glycosylated SV2 and gangliosides as dual receptors for botulinum neurotoxin serotype F. Biochemistry. 2009;48:5631–5641. | ||
Peng L, Berntsson RP, Tepp WH, et al. Botulinum neurotoxin D-C uses synaptotagmin I/II as receptors and human synaptotagmin II is not an effective receptor for type B, D-C, and G toxins. J Cell Sci. 2012;125: 3233–3242. | ||
Brunger AT, Rummel A. Receptor and substrate interactions of clostridial neurotoxins. Toxicon. 2009;54:550–560. | ||
Risselada HJ, Grubmuller H. How SNARE molecules mediate membrane fusion: recent insights from molecular simulations. Curr Opin Struct Biol. 2012;22:187–196. | ||
Rossetto O, Montecucco C. Presynaptic neurotoxins with enzymatic activities. Handb Exp Pharmacol. 2008;184:129–170. | ||
Rummel A, Karnath T, Henke T, Bigalke H, Binz T. Synaptotagmins I and II act as nerve cell receptors for botulinum neurotoxin G. J Biol Chem. 2004;279:30865–30870. | ||
Barnes M. Botulinum toxin – mechanisms of action and clinical use in spasticity. J Rehabil Med. 2003;41:56–59. | ||
dePaiva A, Meunier FA, Molgó J, Aoki KR, Dolly JO. Functional repair of motor endplates after botulinum neurotoxin A poisoning: biphasic switch of synaptic activity between nerve sprouts and their parent terminals. Proc Natl Acad Sci U S A. 1999;96:3200–3205. | ||
Aoki KR, Francis J. Updates on the antinociceptive mechanism hypothesis of botulinum toxin A. Parkinsonism Relat Disord. 2011;17(suppl 1): S28–S33. | ||
Durham PL, Cady R. Insights into the mechanism of onabotulinumtoxinA in chronic migraine. Headache. 2011;51(10):1573–1577. | ||
Aoki KR. Evidence for antinociceptive activity of botulinum toxin type A in pain management. Headache. 2003;43(suppl 1):S9–S15. | ||
Dolly O. Synaptic transmission: inhibition of neurotransmitter release by botulinum toxins. Headache. 2003;43(suppl 1):S16–S24. | ||
Cernuda-Morollon E, Ramon C, Martinez-Camblor P, Serrano-Pertierra E, Larrosa D, Pascual J. OnabotulinumtoxinA decreases interictal CGRP plasma levels in chronic migraine patients. Pain. 2015;156(5): 820–824. | ||
Matak I, Bach-Rojecky L, Filipovic B, Lackovic Z. Behavioral and immunohistochemical evidence for central antinociceptive activity of botulinum toxin A. Neuroscience. 2011;186:201–207. | ||
Binder WJ, Brin MF, Blitzer A, Schoenrock LD, Pogoda JM. Botulinum toxin type A (Botox) for treatment of migraine headaches: an open-label study. Otolaryngol Head Neck Surg. 2000;123:669–676. | ||
Silberstein S, Mathew N, Saper J, Jenkins S. Botulinum toxin type A as a migraine preventive treatment. For the BOTOX Migraine Clinical Research Group. Headache. 2000;40(6):445–450. | ||
Schmitt WJ, Slowey E, Fravi N, Weber S, Burgunder JM. Effect of botulinum toxin A injections in the treatment of chronic tension-type headache: a double-blind, placebo-controlled trial. Headache. 2001;41(7): 658–664. | ||
Rollnik JD, Tanneberger O, Schubert M, Schneider U, Dengler R. Treatment of tension-type headache with botulinum toxin type A: a double-blind, placebo-controlled study. Headache. 2000;40(4):300–305. | ||
Rollnik JD, Karst M, Fink M, Dengler R. Botulinum toxin type A and EMG: a key to the understanding of chronic tension-type headaches? Headache. 2001;41(10):985–989. | ||
Ondo WG, Vuong KD, Derman HS. Botulinum toxin A for chronic daily headache: a randomized, placebo-controlled, parallel design study. Cephalalgia. 2004;24(1):60–65. | ||
Mathew NT, Frishberg BM, Gawel M, et al; BOTOX CDH Study Group. Botulinum toxin type A (BOTOX) for the prophylactic treatment of chronic daily headache: a randomized, double-blind, placebo-controlled trial. Headache. 2005;45(4):293–307. | ||
Silberstein SD, Stark SR, Lucas SM, et al; BoNTA-039 Study Group. Botulinum toxin type A for the prophylactic treatment of chronic daily headache: a randomized, double-blind, placebo-controlled trial. Mayo Clin Proc. 2005;80(9):1126–1137. | ||
Silberstein SD, Göbel H, Jensen R, et al. Botulinum toxin type A in the prophylactic treatment of chronic tension-type headache: a multicentre, double-blind, randomized, placebo-controlled, parallel-group study. Cephalalgia. 2006;26:790–800. | ||
Evers S, Olesen J. Botulinum toxin in headache treatment: the end of the road. Cephalalgia. 2006;26:769–771. | ||
Freitag FG, Diamond S, Diamond M, Urban G. Botulinum toxin type A in the treatment of chronic migraine without medication overuse. Headache. 2008;48(2):201–209. | ||
Aurora S, Dodick D, Turkel C, et al. OnabotulinumtoxinA for treatment of chronic migraine: results from the double-blind, randomized, placebo-controlled phase of the PREEMPT 1 trial. Cephalalgia. 2010;30(7):793–803. | ||
Diener HC, Dodick DW, Aurora SK, et al; PREEMPT 2 Chronic Migraine Study Group. OnabotulinumtoxinA for treatment of chronic migraine: results from the double-blind, randomized, placebo-controlled phase of the PREEMPT 2 trial. Cephalalgia. 2010;30(7):804–814. | ||
Dodick DW, Turkel CC, DeGryse RE, et al; PREEMPT Chronic Migraine Study Group. OnabotulinumtoxinA for treatment of chronic migraine: pooled results from the double-blind, randomized, placebo-controlled phases of the PREEMPT clinical program. Headache. 2010;50(6):921–936. | ||
Aurora SK, Winner P, Freeman MC, et al. OnabotulinumtoxinA for treatment of chronic migraine: pooled analyses of the 56-week PREEMPT clinical program. Headache. 2011;51:1358–1373. | ||
Silberstein S, Tfelt-Hansen P, Dodick DW, et al; Task Force of the International Headache Society Clinical Trials Subcommittee. Guidelines for controlled trials of prophylactic treatment of chronic migraine in adults. Cephalagia. 2008;28:484–495. | ||
Tfelt-Hansen P, Pascual J, Ramadan N, et al; International Headache Society Clinical Trials Subcommittee. Guidelines for controlled trials of drugs in migraine: third edition. A guide for investigators. Cephalalgia. 2012;32:6–38. | ||
Schwedt TJ, Hentz JG, Dodick DW. Factors associated with the prophylactic effect of placebo injections in subjects enrolled in a study of botulinum toxin for migraine. Cephalalgia. 2007;27:528–534. | ||
Manack A, Turkel C, Silberstein S. The evolution of chronic migraine: classification and nomenclature. Headache. 2009;49:1206–1213. | ||
Diener HC, Schorn CF, Bingel U, Dodick DW. The importance of placebo in headache research. Cephalalgia. 2008;28:1003–1011. | ||
Geraud G, Olesen J, Pfaffenrath V, et al. Comparison of the efficacy of zolmitriptan and sumatriptan: issues in migraine trial design. Cephalalgia. 2000;20:30–38. | ||
Mathew NT, Jaffri SF. A double-blind comparison of onabotulinumtoxina (BOTOX®) and topiramate (TOPAMAX®) for the prophylactic treatment of chronic migraine: a pilot study. Headache. 2009;29: 1466–1478. | ||
Cady RK, Schreiber CP, Porter JA, Blumenfeld AM, Farmer KU. A multi-center double-blind pilot comparison of onabotulinumtoxinA and topiramate for the prophylactic treatment of chronic migraine. Headache. 2011;51(1):21–32. | ||
Magalhaes E, Menezes C, Cardeal M, Melo A. Botulinum toxin type A versus amitriptyline for the treatment of chronic daily migraine. Clin Neurol Neurosurg. 2010;112(6):463–466. | ||
Aurora SK, Dodick DW, Diener HC, et al. OnabotulinumtoxinA for chronic migraine: efficacy, safety, and tolerability in patients who received all five treatment cycles in the PREEMPT clinical program. Acta Neurol Scand. 2014;129(1):61–70. | ||
Rothrock JF, Bloudek LM, Houle TT, Andress-Rothrock D, Varon SF. Real-world economic impact of onabotulinumtoxinA in patients with chronic migraine. Headache. 2014;54:1565–1573. | ||
Batty AJ, Hansen RN, Bloudek LM, et al. The cost-effectiveness of onabotulinumtoxinA for the prophylaxis of headache in adults with chronic migraine in the UK. J Med Econ. 2013;16(7):877–887. | ||
Khalil M, Zafar H, Ahmed F. Hull prospective analysis of onabotulinumtoxinA (Botox) in the treatment of chronic migraine; real-life data in 359 patients: an update. In: 4th European Headache Migraine Trust International Congress; 2014; Copenhagen. Abstract. | ||
Khalil M, Zafar H, Quarshie V, Ahmed F. Prospective analysis of the use of onabotulinumtoxinA (BOTOX) in the treatment of chronic migraine; real-life data in 254 patients from Hull, UK. J Headache Pain. 2014;15:54. | ||
Zafar H, Khalil M, Ahmed F. OnabotulinumtoxinA (Botox) in the prevention of chronic migraine: comparing NICE criteria versus Hull criteria for evaluating responder rates. In: 4th European Headache Migraine Trust International Congress; 2014; Copenhagen. Abstract. | ||
Zafar H, Khalil M, Ahmed F. How long to continue Botox in Chronic Migraine patients? A two year follow up of 85 patients treated in Hull, UK. In: 4th European Headache Migraine Trust International Congress; 2014; Copenhagen. Abstract. | ||
Rothrock JK, Scanlon C, Weibelt S. OnabotulinumtoxinA for the treatment of chronic migraine: long-term outcome. In: American Headache Association Meeting; 2011; Washington DC, USA. Abstract. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.