Back to Journals » International Journal of Nanomedicine » Volume 13
Olaparib nanoparticles potentiated radiosensitization effects on lung cancer
Authors Wu M, Liu J, Hu C, Li D, Yang J, Wu Z, Yang L, Chen Y, Fu S ![]() , Wu J
, Wu J
Received 26 July 2018
Accepted for publication 22 October 2018
Published 11 December 2018 Volume 2018:13 Pages 8461—8472
DOI https://doi.org/10.2147/IJN.S181546
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Linlin Sun
Min Wu,1,* Jing Liu,1,* ChuanFei Hu,1 Dong Li,1 Juan Yang,1 ZhouXue Wu,1 LingLin Yang,1 Yue Chen,2 ShaoZhi Fu,1 JingBo Wu1
1Department of Oncology, The Affiliated Hospital of Southwest Medical University, Luzhou 646000, China; 2Department of Nuclear Medicine, The Affiliated Hospital of Southwest Medical University, Luzhou 646000, China
*These authors contributed equally to this work
Background: Poly (ADP-ribose) polymerase (PARP) is a key enzyme in the repair process of DNA strand breaks (DSBs). Olaparib (Ola) is a PARP inhibitor that is involved in arresting PARP release from radiotherapy (RT)-induced damaged DNA to potentiate the effect of RT. Although the underlying mechanisms for the radiosensitization effects of Ola are well understood in vitro, the radiosensitization effects in vivo are still unclear. Moreover, poor water solubility and severe toxicity are two major impediments for the clinical success of Ola.
Materials and methods: Here, we developed olaparib nanoparticles (Ola-NPs) and investigated their radiosensitization mechanisms and toxicity using human non-small-cell lung cancer xenograft models in mice.
Results: The prepared Ola-NPs showed a mean size of 31.96±1.54 nm and a lower polydispersity index of about 0.126±0.014. In addition, the sensitization enhancement ratio of Ola-NPs (3.81) was much higher than that of free Ola (1.66). The combination of Ola-NPs and RT (Ola-NPs + RT) significantly inhibited tumor growth and prolonged survival in mice. The mechanism of enhanced antitumor efficacy might be related to the inhibition of DSB repair and the promotion of cell apoptosis in vivo. No additional toxicity caused by Ola-NPs was observed.
Conclusion: This study demonstrated the principle of using Ola-NPs as a potent radiosensitizer to improve the therapeutic effect of RT relative to free Ola (P<0.05 in all cases).
Keywords: lung cancer, nanoparticles, olaparib, radiosensitization
Introduction
Lung cancer is a major disease that significantly endangers human life and health.1 Non-small-cell lung cancer (NSCLC) accounts for 80%–85% of all lung cancers worldwide.2,3 Radiotherapy (RT) is an effective treatment for local NSCLC. Indeed, RT has shown to improve local tumor control and the survival rate in patients. Nevertheless, local tumor control is still suboptimal because of radio-resistance.4 The repair of damaged DNA is the main reason for radio-resistance. Inhibiting DNA damage repair has represented an effective strategy to enhance the treatment effect of RT.5
DNA damage caused by RT mainly involves DNA strand breaks (DSBs). Poly (ADP-ribose) polymerase (PARP) is a DNA damage-sensing protein, which is the key enzyme in the repair of DSBs.6 PARP inhibitor (PARPi) has been known as sensitizers, which enhanced the therapeutic effect of chemotherapy or RT.5 PARPi arrested PARP release from damaged DNA.7 As a result, DNA repair proteins were unable to access the single-strand breaks (SSBs). Unrepaired SSBs then converted to DSBs, which cause lethal damage to tumor cells, thereby enhancing radiotoxicity. Olaparib (Ola), which specifically acted on the repair of SSBs of target cells, was the first PARPi approved by the Food and Drug Administration. Clinically, Ola has been approved for the treatment of advanced ovarian cancer in 2014.8 Nevertheless, the poor water solubility in gastric juice shortens the action time and decreases the bioavailability and therapeutic activity. Furthermore, due to the widespread distribution of Ola in normal tissues, treatment with Ola could cause side effects, including anemia, nausea, fatigue, headache, and diarrhea.9 Accordingly, it is of critical importance to identify a novel Ola delivery system to potentiate its water solubility and reduce biologic toxicity.
Novel nanoparticle delivery system brings a potential solution to the above-mentioned problems. In recent years, nanoparticles have been widely used as an antitumor drug delivery system because of their passive targeting ability on tumor tissue that has an enhanced permeation and retention effect (EPR).10,11 Degradable polymer nanomaterials play an important role in the transmission system of antitumor drugs.12,13 Among them, two block polymers methoxy poly (ethylene glycol)–poly (e-caprolactone) (MPEG-PCL) with a core-shell structure has become a promising carrier of nanoscale drugs due to its biodegradability and sustained drug release properties.14 MPEG-PCL has a hydrophobic core (PCL) for connecting with hydrophobic drugs, and the hydrophilic shell (PEG) contributes to the dissolution of the polymeric micelle. Accordingly, MPEG-PCL polymeric micelles are suitable for encapsulating hydrophobic drugs and stable drug release.15
Although numerous studies have reported the radiosensitization effects of Ola,5,16,17 no reports have been found on the selection of optimum RT dose. Previous studies on radiosensitization effects have shown that the similar drug dosages combined with different doses of RT produced different radiosensitization effects.18 The radiosensitization effects of radiosensitizer combined with RT resulted in an S-shaped curve. Once the optimum RT dose was achieved, the radiosensitization effect no longer increased with the RT dose.19 Moreover, no studies have been performed that demonstrated a radiosensitization effect or side effect of MPEG-PCL/Ola. Therefore, the objective of this study was to prepare olaparib nanoparticles (Ola-NPs) and investigate the radiosensitization effect and toxicity of Ola-NPs combined with RT on tumor-bearing BABL/c mice.
Materials and methods
Materials
Ola with 99.8% purity was purchased from Meilun Co., Ltd (Dalian, China). MPEG (Mn =2,000) was purchased from Sigma-Aldrich (St Louis, MO, USA), ε-caprolactone from Alfa Aesar (Reston, VA, USA), and stannous octoate (Sn(Oct)2) purchased from Sigma-Aldrich. Dimethylsulfoxide (DMSO), anhydrous ethanol, and methanol (HPLC grade) were purchased from KeLong Co., Ltd (Chengdu, China). γ-H2AX, CC3, Ki-67, and CD-31 polyclonal antibodies were purchased from Bioworld Technology Co. Ltd. (Nanjing, China).
Formulation and characterization of Ola-NPs
Ola-NPs were formulated as follows: the polymerization methods for the synthesis of MPEG-PCL (Mw =4,000) included open-ring polymerization, which was published previously.20 MPEG-PCL and Ola at various dosages of 6, 8, or 10 wt% were used. Briefly, for 8 wt% of Ola as an example, 92 mg MPEG–PCL and 8 mg Ola were co-dissolved in 10 mL acetone. A rotary evaporator was used to evaporate the mixed solution using a 37°C water bath with a revolving speed of 110±5 rpm until the organic solvent was totally removed. Next, the resulting harvest was washed using preheated (60°C) deionized water. Then, the solution was filtered through a 0.22 μm filter. The size and Zeta potential of Ola-NPs were measured using dynamic light scattering (NanoBrook 90Plus Zeta, Brookhaven, NY, USA) at 25°C. Morphologic characteristics of Ola-NPs were determined by transmission electron microscopy (TEM, Tecnai G2 F20, USA). The filtered Ola-NPs solution was lyophilized and redissolved in deionized water. HPLC was used to detect the drug loading rate (DL) and entrapment efficiency (EE) of Ola-NPs. The specific steps are as follows: the mobile phase A was deionized water and mobile phase B was methanol (40/60, v/v). The mobile phases were degassed 30 minutes before use, and then were applied at a flow rate of 1.0 mL/min through a reversed-phase C18 alkyl silane column (150 mm 4.6 mm, 5 μm, Agilent Santa Clara, CA, USA) at 25°C. The injection volume was 20 μL and the detection wavelength was set at 276 nm. The peak value appears around 2.8 minutes. The DL and EE were detected by HPLC according to the following formula:
|
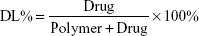
|
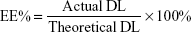
In vitro release of Ola and Ola-NPs
For in vitro studies, the release behavior was explored as follows: 1 mL (1 mg/mL) Ola or 1 mL Ola-NPs (the theoretical DL was 8%, equal to 1 mg of Ola) in distilled water was separately dispersed in DMSO and deionized water, then separately poured into dialysis bags (molecular weight cutoff, 3.5 kDa). The bags were incubated at 37°C in 30 mL PBS (pH =7.4) containing Tween-80 (0.5%, w/w) with gently shaking (100 rpm). The incubation medium was replaced by the same volume of freshly preheated medium (2 mL) at predetermined time points (0, 2, 4, 6, 8, 12, 24, 48 hours … until 8 days). Subsequently, the collected released drug was centrifuged at 12,000 rpm for 15 min at 4°C. Supernatant was collected and stored at −20°C for further analysis. Supernatant was analyzed by HPLC as described in the section “DL and EE of Ola-NPs”, and experiments were performed at least in triplicate. Data represented the mean of a triplicate and expressed as the mean±SD.
In vivo antitumor and radiosensitization effect
A549 lung cancer cells used in our experiment were purchased by the Experimental Research Center of the Affiliated Hospital of Southwest Medical University (Luzhou, China) from the Cell Bank of the Chinese Academy of Sciences. A549 cells were maintained in RPMI-1640 medium (1640, Gibco, USA) supplemented with 10% FBS and antibiotics (penicillin 100 U/mL and streptomycin 100 μg/mL) and incubated at 37°C in a humidified standard atmosphere (95% air and 5% CO2). Cells in the logarithmic phase were harvested and used in experiments.
BALB/c nude mice (female, 3 to 4 weeks old) were purchased from Chongqing TengXin Biotechnology Co., Ltd (Chongqing, China). Animal experiments were performed according to the animal care guidelines and were approved by the Institutional Animal Care and Treatment Committee of Southwest Medical University (Luzhou, China). In brief, mice were subcutaneously injected in the right thigh with 0.1 mL A549 cell suspension at a density of 107/mL. Mouse weights were monitored every other day and once palpable tumors were formed their volumes were calculated every other day using the following equation: volume = 1/2 × length × width.2 To select the optimal dose of RT, a total of 72 mice were randomly divided into 3 groups (n=24/group) and when tumor measured 100–150 mm3, subgroups were made as follows: 1) RT (a single dose ranges from 0 to 18 Gy); 2) Ola + RT (50 mg/kg/d, 3 days, intravenous injection [IT], combined with RT); 3) Ola-NPs + RT (equivalent to 50 mg/kg Ola). Each subgroup included six mice (n=6/subgroup) that received irradiation of 0, 2, 6, 10, 14, and 18 Gy, respectively. RT was given 30 minutes after the first time of using drug. The optimal dose of RT was calculated by monitoring tumor growth delay (TGD). Tumor growth curves were prepared in accordance with the Compertz model by using the following equation: y=V0*exp(k*(1−exp(−a*X))), where y represented the TGD; V0 represented the original tumor volume (100–150 mm3); a and k are coefficients, and X indicated the RT dose.
After calculating the optimal RT dose, the radiosensitization effect and toxicity of Ola-NPs drugs was further studied. In this study, a xenograft model of A549 lung cancer in nude mice was established. Tumor-bearing nude mice were randomly divided into the following six groups (n=12/group): 1) control (0.9% normal saline [NS]); 2) Ola; 3) Ola-NPs; 4) RT (a single dose of 10 Gy); 5) Ola + RT; (6) Ola-NPs + RT.
Micro 18F-FDG PET/CT imaging
To explore Ola-NPs therapy response and the metabolism of tumor tissue, micro 18F-FDG PET/CT imaging scans (Siemens, Munich, Germany) were performed. Briefly, mice were starved from eating and drinking for at least 8 hours prior to the scan, anesthetized with 1% pentobarbital sodium 5 mg/kg through intraperitoneal injection and intravenously injected with 150–250 μCi FDG (0.1–0.2 mL) for at least 30 minutes before scanning. After that, mice were placed in the center of the PET/CT imaging field and an image was obtained. The parameters used were as follows: 80 kV, 500 μA, 1.5 mm slice collimation, at 10 minutes per bed position.
Histopathology analysis
Tumor tissue was obtained 24–48 hours after the first treatment for immunohistochemical (IHC) detection of γ-H2AX. Tumor tissue from mice in different groups was harvested 10 days after treatment for other indicators of IHC (CC3, Ki-67, and CD-31). Specific steps included the following: the tumor was fixed in neutral formaldehyde for at least 24 hours, then tumor tissue was embedded in paraffin. Paraffin-embedded tissue was cut into 3–4 mm thick sections and stained with H&E. Paraffin sections were incubated with antihuman γ-H2AX, CC3, Ki-67, and CD-31 primary antibodies. And the secondary antibody was a biotinylated goat anti-mouse antibody. Detailed operation procedure was performed according to the manufacturer’s instruction (Bioworld Technology, Nanjing, China). Sections were stained with hematoxylin and observed using a microscope (Leica TE2000-S Microscope, Tokyo, Japan). γ-H2AX, Ki-67, and CC3 expressions were calculated in five randomly selected areas in each tumor sample at 400× magnification as the number of positive cells/total cells. In each tumor section, microvessel density (MVD) was calculated in five randomly selected areas (at 400× magnification) as the mean value of CD31-positive microvessels.
Flow cytometry
Flow cytometry (BD FACSVerse, San Diego, CA, USA) was used to explore the cell-cycle phase and apoptosis. In brief, tumor tissue was cut into small pieces and incubated at 37°C in the presence of 2 mL trypsinization buffer for 40 minutes when gently shaking. After 40 minutes of digestion, medium containing serum was added to terminate the digestion. Then, the mixture was filtered using a nylon gauze to remove undigested tumor tissue. The harvested cell suspension was washed twice with NS and centrifuged at 1,000 rpm for 2 minutes. Then, a total of 5 μL of fluorescein isothiocyanate and 5 μL of propidium iodide were added to each tube. Cells were incubated for 15 minutes at room temperature in the dark, then 400 μL of ice-cold 1× binding buffer was added and cells were gently mixed. Next, cell suspensions were analyzed using flow cytometry. Cell-cycle phage was also analyzed by flow cytometry using a BD cycle test plus DNA kit.
Evaluation of the in vivo toxicity of Ola-NPs
Mice were killed and the liver, heart, spleen, lungs, and kidneys of each mice were prepared for H&E staining. To assess the toxicity of different treatments and to evaluate the degree of damage of the different treatments to normal tissues, histologic changes of different organs were determined.
Statistical analysis
Statistical analysis was performed by SPSS 17.0 software using Student’s test for two-group comparison or one-way ANOVA for multiple-group comparisons. Data were expressed as the mean±SD. P<0.05 was considered statistically significant.
Results
The physicochemical characterization and release behavior of Ola-NPs
In our study, Ola-NPs containing different Ola concentrations (6%, 8%, and 10% wt/wt) were successfully established. Table 1 shows that an 8% theoretical DL in Ola-NPs resulted in a higher EE (almost 99.17%) and a smaller mean size (31.96±1.54 nm). As a result, we selected Ola-NPs containing 8% Ola for further studies. As shown in Figure 1A, Ola-NPs aqueous solution appeared as a faint blue color. In addition, Ola-NPs presented as spherical or approximately spherical with a smooth surface using TEM analysis (Figure 1B). Ola-NPs had a negative surface charge of −22.53±1.96 mV (Figure 1C). Ola-NPs showed a mean size of 31.96±1.54 nm (Figure 1D) and a lower polydispersity index of about 0.126±0.014. As presented in Figure 1E, free Ola demonstrated a rapid release, and more than 90% of Ola was released into the medium within 3 days. In addition, 75% of Ola was slowly released from Ola-NPs within 8 days.
 | Table 1 Characteristics of olaparib nanoparticles |
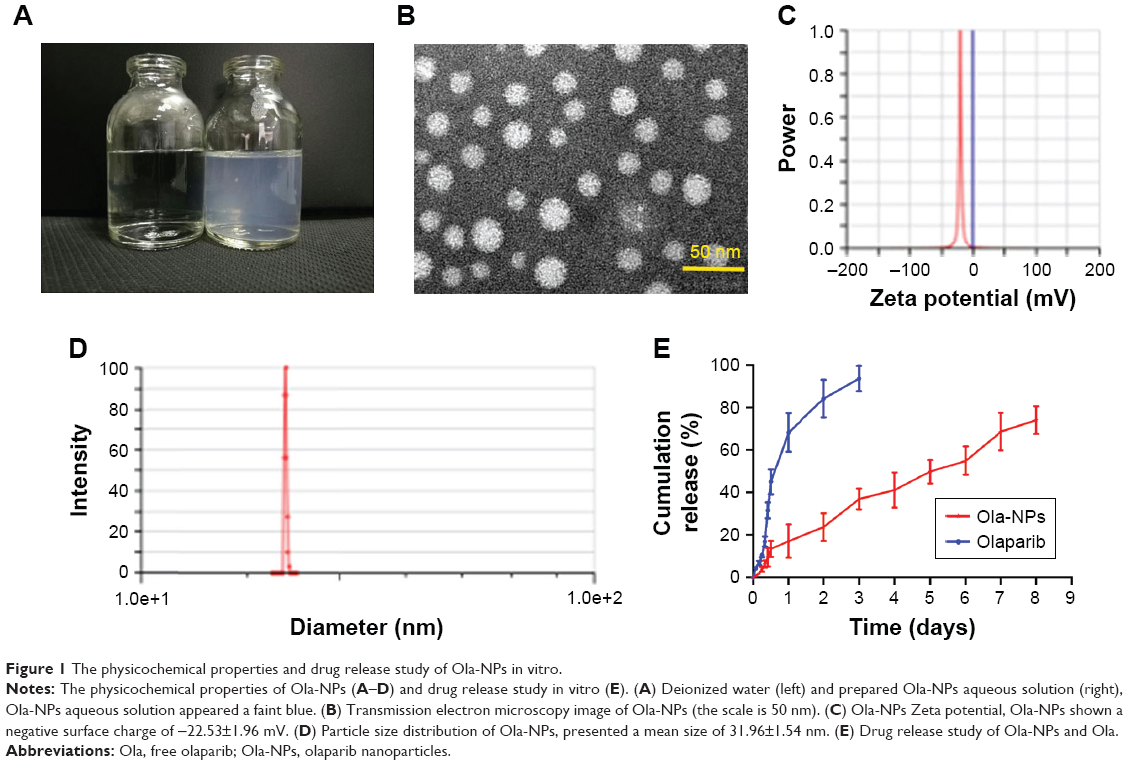 | Figure 1 The physicochemical properties and drug release study of Ola-NPs in vitro. |
In vivo radiosensitization testing
To assess the efficacy of Ola-NPs + RT on A549 xenograft mice, tumor volumes (Figure 2A–C) were calculated and expressed by TGD (Table 2). Dose–effect curves were prepared for RT, Ola + RT, and Ola-NPs + RT (Figure 2D). The curves were nearly linear when TGDs were between 8 and 12 days. Therefore, a TGD of 10 days was taken as a biologic endpoint. To achieve a TGD of 10 days, 7.28, 4.39, and 1.91 Gy were required for RT, Ola + RT, and Ola-NPs + RT groups, respectively. Sensitization enhancement ratio (SER) was 1.66 (7.28/4.39) for the Ola + RT group and 3.81 (7.28/1.91) for Ola-NPs + RT group. As shown in Figure 2D, by increasing the RT dose, tumor growth slowed down and the TGD was gradually extended in the RT, Ola + RT, and Ola-NPs + RT groups. Moreover, drug plus RT prolonged the TGD and was more pronounced for Ola-NPs + RT. The TGD reached a maximum at 10 Gy and then decreased. To explore the radiosensitization effect of Ola-NPs, a single dose of 10 Gy was selected as the optimum dose and was given to mice in the corresponding groups. The time needed for the tumor volume to reach 4 times of the initial volume (T4V0) was 6.3 days (Control), 9.8 days (Ola), 15.7 days (Ola-NPs), 19.9 days (RT), 26.2 days (Ola + RT), and 33.5 days (Ola-NPs + RT), respectively (Figure 3C). These data demonstrated that Ola-NPs + RT possessed a more powerful effect on delaying tumor growth (P<0.05 in all cases). At the same time, a similar effect was observed from the median survival time of mice (Figure 3E). The median survival time of mice in the Ola-NPs + RT group (69.5±11.8 days) was significantly longer when compared to that of mice treated with Ola + RT (53.5±8.0 days), RT (51.83±7.3 days), Ola-NPs (42.8±8.3 days), Ola (40.5±7.2 days), and Controls (31.8±6.7 days) (P<0.05). Body weight was recorded every 2 days but no significant differences were observed between groups throughout the entire study (Figure 3D, P>0.05 in all cases).
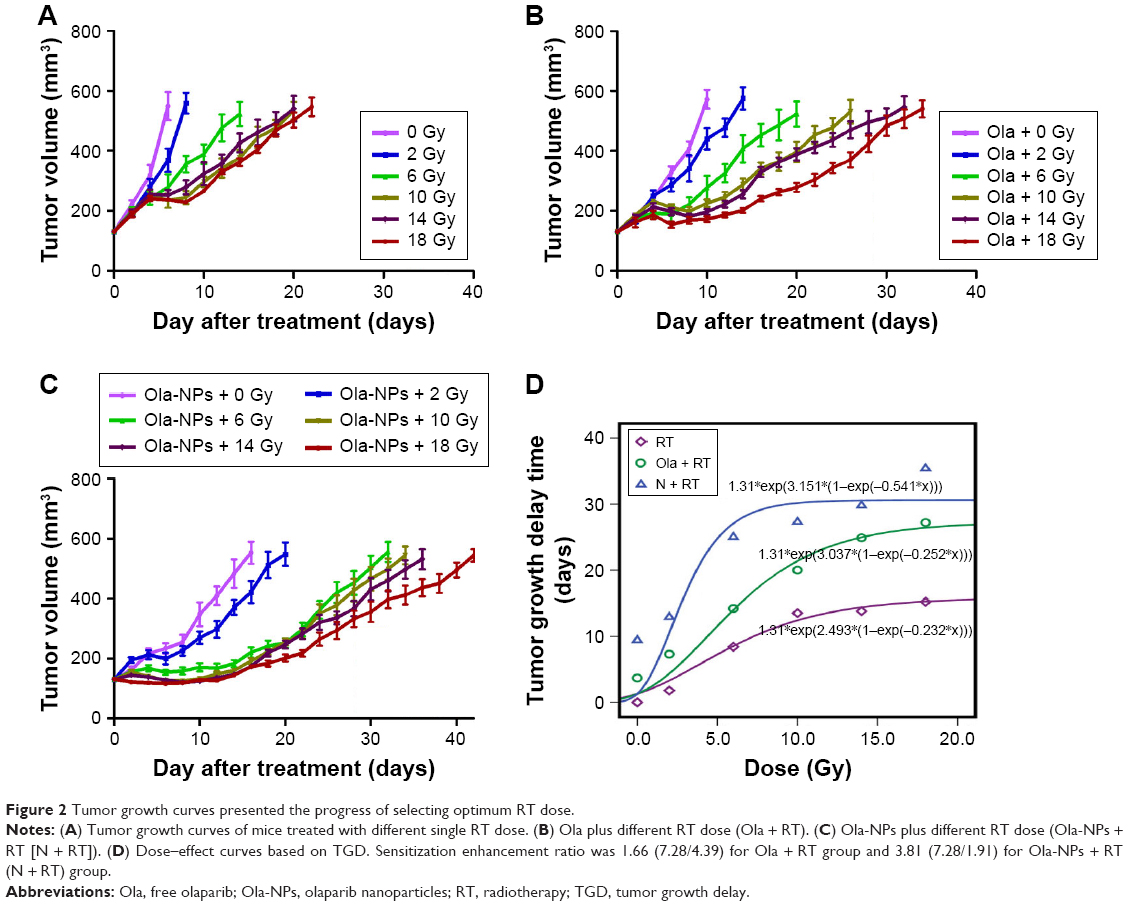 | Figure 2 Tumor growth curves presented the progress of selecting optimum RT dose. |
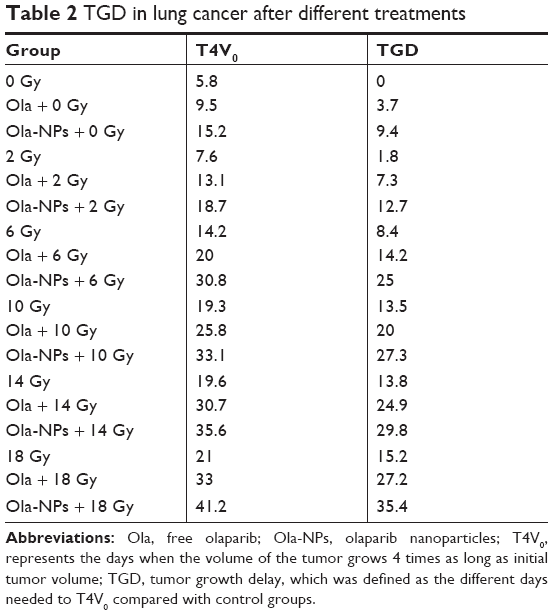 | Table 2 TGD in lung cancer after different treatments |
 | Figure 3 Evaluation of antitumor efficiency in vivo. |
Micro 18F-FDG PET/CT imaging
PET/CT imaging results of mice in each group are shown in Figure 3B, the mean SUVmax of the tumor in each group are as follows: 2.2±0.15 (control), 1.7±0.08 (Ola), 1.3±0.15 (Ola-NPs), 1.2±0.04 (RT), 0.93±0.05 (Ola + RT), and 0.82±0.07 (Ola-NPs + RT). When compared with other groups, mice in the Ola-NPs + RT group had the lowest SUVmax, suggesting a lower metabolism and thereby implying a more powerful antitumor effect of Ola-NPs + RT (P<0.05), which were consisted with the general view of tumor xenograft models (Figure 3A).
IHC analysis
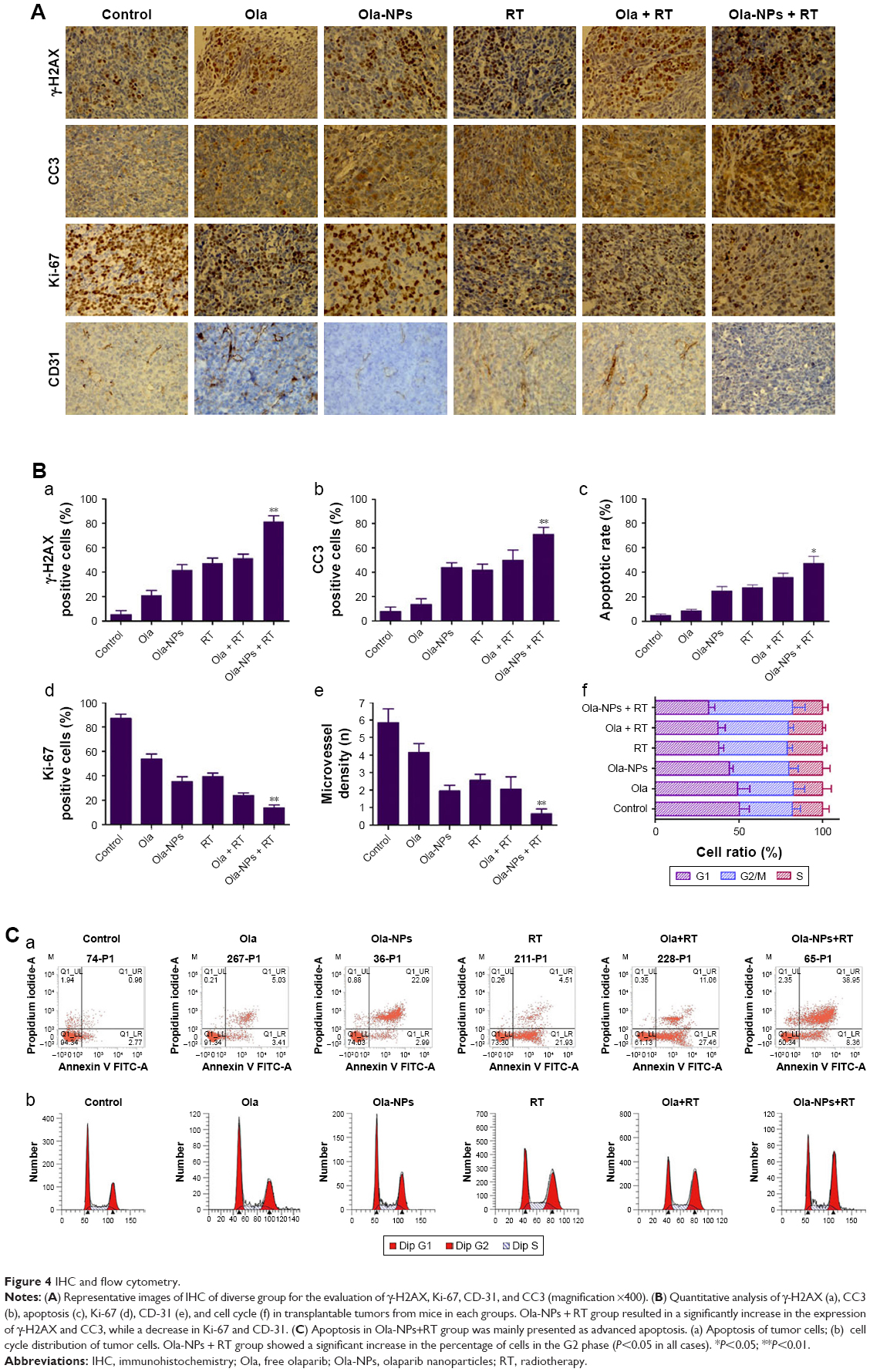
IHC results are depicted in Figure 4A. An increased number of γ-H2AX-positive cells was observed in the Ola-NPs + RT group (81.47%±4.65%) when compared to other groups (Figure 4B-a). The quantification of γ-H2AX-positive cells of other groups was as follows: Control (5.27%±3.35%), Ola (21.23%±3.85%), Ola-NPs (41.77%±4.31%), RT (47.47%±4.10%), and Ola + RT (51.4%±3.34%) (P<0.01).
 | Figure 4 IHC and flow cytometry. |
CC3 is a cellular marker of apoptosis, which participates in programmed cell death.21 As shown in Figure 4B-b, Ola-NPs + RT significantly increased apoptosis (71.03%±5.91%) when compared to control (7.63%±3.81%), RT (41.57%±5.03%), Ola-NPs (43.75%±4.14%), and other groups (P<0.01). These data indicated that Ola-NPs dramatically enhanced radiation-induced cellular killing when combined with RT.
The proliferation of tumor cell was accessed by Ki-67 assay. As shown in Figure 4B-d, the percentage of Ki-67-positive cells was 87.1%±3.48% in the control group and 39.27%±2.97% in the RT group. Moreover, in the Ola + RT (23.8%±2.33%) and Ola-NPs + RT (13.8%±2.35%) treatment groups, a significant decrease in Ki-67-positive cells was observed when compared to the control group (P<0.05).
The expression of CD-31 was used to evaluate MVD of tumor tissues. The MVD in each group ranged as follows: control > Ola > RT > Ola + RT > Ola-NPs > Ola-NPs + RT (Figure 4B-e). A dramatically lower MVD (0.65±0.27) was observed in Ola-NPs + RT-treated mice when compared to those of mice in the Ola + RT (2.05±0.71), RT (2.55±0.35), and other groups (P<0.01).
Flow cytometry
Flow cytometry was used to analyze the cell cycle phase (Figure 4C-d) as well as apoptosis (Figure 4C-a). As shown in Figure 4B-c, mice in the Ola-NPs + RT group showed an increased apoptotic rate (47.28%±5.67%) when compared to Ola + RT (35.88%±3.3%), RT (27.56%±2.12%), and other treatment groups (P<0.05 in all cases). The differences in cell-cycle phase between groups were mainly reflected in the percentage of cells in the G2/M phase. As shown in Figure 4B-f, mice in the Ola-NPs + RT group showed a significant increase in the percentage of cells in the G2/M phase (50.12%±7.52%), when compared to the Ola + RT (42.03%±3.21%), RT (40.93%±3.14%), and other groups (P<0.05). No significant differences were observed in the S phase (P>0.05 in all cases).
Evaluation the side effects of Ola-NPs in vivo
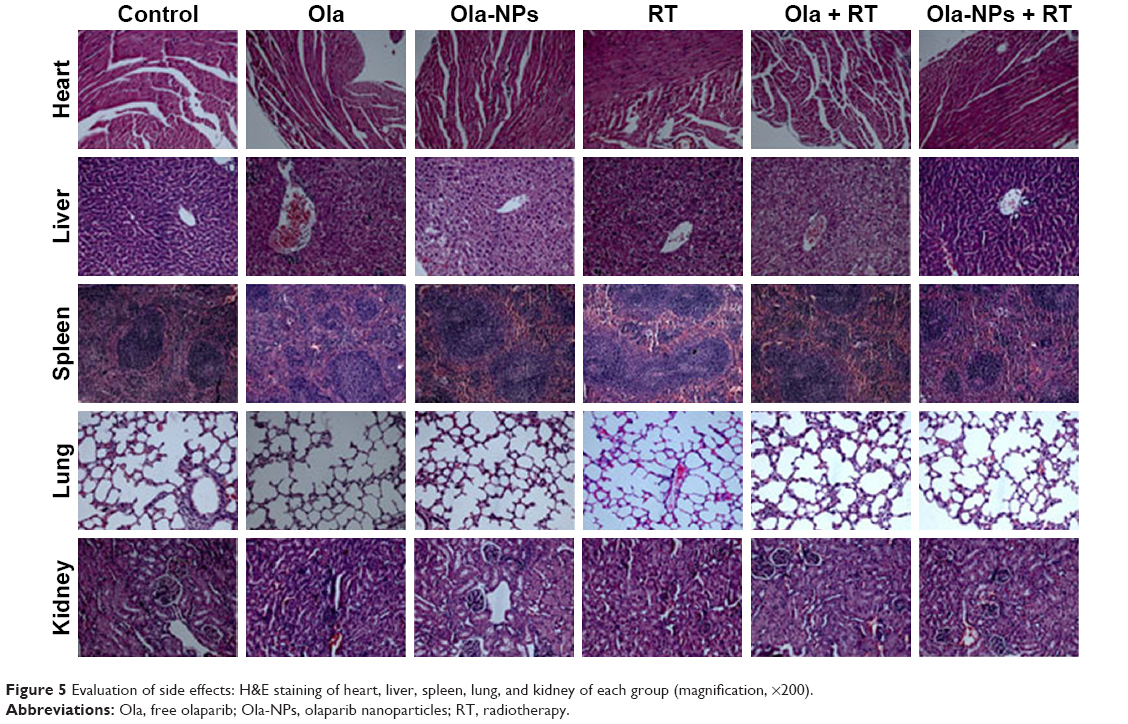
In this study, we investigated the side effect of Ola-NPs on changes in body weight and toxicity in organs. Throughout the entire study, no significant differences were observed in body weight between groups (Figure 3D, P>0.05 in all cases). After mice were euthanized, heart, liver, spleen, lungs, and kidneys were dissected and prepared for H&E staining to determine the toxic effect of Ola-NPs in organs. As shown in Figure 5, the primary pathologic changes involved the balloon degeneration in the liver of mice that were treated with Ola but not Ola-NPs. No obvious pathologic changes were observed in other organs.
 | Figure 5 Evaluation of side effects: H&E staining of heart, liver, spleen, lung, and kidney of each group (magnification, ×200). |
Discussion
Lung cancer is one of the most prevalent malignant tumors that threatens human health and life. RT is a frequently used treatment regimen for lung cancer as it not only improves tumor control but also the survival rate.22 Nevertheless, the radio-resistance caused by the repair of damaged DNA of the tumor tissue remains a daunting challenge.5 Inhibiting damaged DNA repair has been considered an effective strategy to enhance the treatment outcomes of RT.23,24
Ola is a DNA damage repair inhibitor, which can act as a radiosensitizer when combined with RT. Previous studies have shown that Ola + RT significantly enhanced antitumor effects.16,17 Unfortunately, no reports are available that focus on the radiosensitization effect or side effect of Ola-NPs. Ola presents with a series of physicochemical properties, such as poor solubility, low bioavailability, and an extensive range of tissue toxicity that largely offset the use and therapeutic benefits of Ola. In our previous study, we demonstrated that MPEG-PCL nanoparticles were outstanding carriers for hydrophobic drugs in virtue of the hydrophilic PEG segment being introduced into PCL backbones.25–28 Therefore, in our study, MPEG-PCL was used as a carrier to prepare Ola-NPs.
The main purpose of this study was to prepare Ola-NPs and confirm the radiosensitization of Ola-NPs. First, we use a rotary evaporation method to successfully prepare injectable Ola-NPs using an MPEG-PCL copolymer.29–31 These nanoparticles had stable particle size with a higher DL when compared with other methods reported in recent studies.25,32,33 The lyophilized nanoparticles were relatively narrowly distributed and could be stably dissolved in water to achieve the requirements for intravenous injection. Recent studies have shown that the importance of nanoparticle size will greatly affect the metabolism of nanoparticles: <200 nm of nanoparticles will result in a longer blood circulation time, favorable stability, lower cytotoxicity, and greater absorptive capacity due to EPR effect.13,34,35 In addition, the sustained slow drug release as shown by Ola-NPs could be attributed to the stable structure of MPEG-PCL polymeric micelles, which might be due to the drug diffusion in the PCL core, thereby indicating that Ola-NPs had an excellent DL and EE.
Free Ola released rapidly indicating that the membrane did not hinder drug release, and suggested that the mechanism of Ola-NPs drug release involved diffusion, swelling, and degradation of polymeric micelles. Previous studies have shown that the cumulative drug release of NPs is 30%–40% within 1 month, which was too slow to achieve the therapeutic needs.36,37 Therefore, the Ola-NPs prepared in our study had better advantages regarding drug release. Such advantages allowed for a strong synergistic effect of Ola-NPs + RT, which turned out to be of outstanding therapeutic efficacy.
Subsequently, we set up a lung cancer xenograft model in nude mice to explore the radiosensitization of Ola-NPs. A general linear model was used to assess the effect of drugs with different doses of RT, as previously reported.18,19 The data suggested that Ola-NPs possessed greater tumor growth inhibition and better radiosensitization effects when compared to Ola (SER of 3.81 vs 1.66). This might be attributed to the fact that Ola-NPs slowly release Ola. In vivo studies demonstrated that treatment with Ola-NPs + RT (10 Gy) showed the most significant radiosensitization when compared to other groups. Another reason for choosing 10 Gy was that when increasing RT doses, severe side effects appeared gradually, including emaciation, diarrhea, and radiodermatitis. During the study, a series of observations were used to assess the therapeutic effects of RT combined with Ola-NPs. Mice treated with Ola-NPs + RT showed the best therapeutic effects among other treatment groups whether in TGD or survival rate. This further indicated that Ola encapsulated in Ola-NPs was more effective than free Ola in a lung cancer xenograft model.
18F-FDG PET/CT was used to detect the therapy response in our experiment. Several studies have shown that 18F-FDG PET/CT recognized a very early decrease of glucose metabolism that is associated with progression-free survival and overall survival in many cancers.12,25,38 A higher 18F-FDG uptake may indicate a worse therapeutic response, while a lower uptake in tumors may suggest a better outcome. In this study, 18F-FDG uptake in mice in the Ola-NPs + RT group was the lowest compared to that in other groups, suggesting that Ola-NPs were more effective in inhibiting tumor metabolism.
Tumor tissue was used for IHC analysis, using antibodies directed against γ-H2AX, CC3, Ki-67, and CD-31. γ-H2AX is a biomarker of DSBs. It has been reported that radiosensitization by PARPi is more effective in the repair-defective DNA DSBs of tumor cells than repairable DNA.39 In our study, mice in the Ola-NPs + RT group presented a significantly higher expression of γ-H2AX compared to mice in other groups. These results were the synergistic effects of Ola-NPs combined with RT, which was consistent with previously published findings.5 The expression of CC3, which participated in programmed cell death, was investigated as a biomarker.21 Mice in the Ola-NPs + RT group showed the highest expression of CC3 among all groups, suggesting that Ola-NPs + RT played a significant role in the induction of apoptosis of tumor cells. It is the inhibition caused by Ola-NPs on the repair of damaged DNA that causes apoptosis of tumor cells. As previously reported, a lower MVD is usually related to slower tumor growth and better biologic and clinical behavior.40–43 Mice in the Ola-NPs group showed the lowest expression of CD-31 and Ki-67 when compared to mice in other groups. The results might indicate that Ola-NPs + RT inhibited the growth of tumor cells by inhibiting tumor angiogenesis and proliferation.
Flow cytometry was used to investigate the cell cycle and apoptosis of tumor tissue.
Previous studies have shown that cells in the G2/M phase presented the greatest sensitivity to PARPi + RT.5,44,45 The susceptibility of tumor cells to apoptosis may be increased by downregulating anti-apoptosis proteins.46,47 Our results also showed that Ola-NPs might increase the number of cells in the G2/M phase, thereby further accelerating apoptosis of tumor cells, which was consistent with the expression of CC3 in the tumor tissue.
The toxicity of Ola-NPs in vivo was also investigated. As shown in our study, the primary pathologic changes were caused by free Ola. Due to the lack of targeting of Ola, Ola was not only distributed in tumor tissues but also other normal tissues such as the liver, to increase toxicity. Nevertheless, Ola-NPs were well tolerated and no obviously toxic effects were observed. The findings might be attributed to the EPR effects of Ola-NPs. The stable structure of MPEG-PCL polymeric micelles contributed to the sustained slow drug release, so that Ola-NPs could be passively targeted and distributed in tumor tissues as needed, thereby reducing toxicity.
These results generally indicated the outstanding radiosensitization effect of Ola-NPs in an NSCLC xenograft model. Although the data are promising, we are aware of the limitations of our study. Therefore, a more detailed in vivo study on Ola-NPs including blood drug concentration will be further explored soon. Also, further studies containing additional cell lines are currently underway.
Conclusion
In this study, we have successfully prepared injectable Ola-NPs. Data presented that Ola-NPs indicated outstanding radiosensitization effects and were well tolerated in human xenograft tumor models. Ola-NPs + RT possessed powerful effects on delaying the growth of tumor cells and prolonging the median survival time in mice. Moreover, we have explored the mechanism of the radiosensitization and found that Ola-NPs inhibited DSBs repair and increased tumor cell apoptosis. Ola-NPs also enhanced the number of cells in G2/M phase and reduced angiogenesis of tumor tissue. In summary, Ola-NP is an excellent radiosensitization drug without extra toxicity for NSCLC.
Acknowledgment
This study was supported by the project from Health and Family Planning Commission of Sichuan Province (No 17PJ557), the research project from Office of Science & Technology and Intellectual Property of Luzhou (No 2017), and the Union Project of Luzhou and Southwest Medical University under Grant Nos 14JC0144 and 2013LZLY-J40.
Disclosure
The authors report no conflicts of interest in this work.
References
Jemal A, Bray F, Center MM, et al. Global cancer statistics. CA Cancer J Clin. 2011;61(2):69–90. | ||
Lee WH, Loo CY, Ong HX, et al. Synthesis and characterization of inhalable flavonoid nanoparticle for lung cancer cell targeting. J Biomed Nanotechnol. 2016;12(2):371–386. | ||
Herbst RS, Heymach JV, Lippman SM. Lung cancer. N Engl J Med. 2008;359(13):1367–1380. | ||
Klein C, Dokic I, Mairani A, et al. Overcoming hypoxia-induced tumor radioresistance in non-small cell lung cancer by targeting DNA-dependent protein kinase in combination with carbon ion irradiation. Radiat Oncol. 2017;12(1):208. | ||
Jiang Y, Verbiest T, Devery AM, et al. Hypoxia potentiates the radiation-sensitizing effect of olaparib in human non-small cell lung cancer xenografts by contextual synthetic lethality. Int J Radiat Oncol Biol Phys. 2016;95(2):772–781. | ||
Prasad CB, Prasad SB, Yadav SS. Olaparib modulates DNA repair efficiency, sensitizes cervical cancer cells to cisplatin and exhibits anti-metastatic property. Sci Rep. 2017;7:12876. | ||
Gravells P, Neale J, Grant E, et al. Radiosensitization with an inhibitor of poly(ADP-ribose) glycohydrolase: a comparison with the PARP1/2/3 inhibitor olaparib. DNA Repair. 2018;61:25–36. | ||
Kim G, Ison G, McKee AE, et al. FDA Approval summary: olaparib monotherapy in patients with deleterious germline BRCA-mutated advanced ovarian cancer treated with three or more lines of chemotherapy. Clin Cancer Res. 2015;21(19):4257–4261. | ||
Liu JF, Barry WT, Birrer M, et al. Combination cediranib and olaparib versus olaparib alone for women with recurrent platinum-sensitive ovarian cancer: a randomised phase 2 study. Lancet Oncol. 2014;15(11):1207–1214. | ||
Kobayashi H, Choyke PL. Super enhanced permeability and retention (SUPR) effects in tumors following near infrared photoimmunotherapy. Nanoscale. 2016;8(25):12504–12509. | ||
Chen L, Zang F, Wu H, et al. Using PEGylated magnetic nanoparticles to describe the EPR effect in tumor for predicting therapeutic efficacy of micelle drugs. Nanoscale. 2018;10(4):1788–1797. | ||
Ni XL, Chen LX, Zhang H, et al. In vitro and in vivo antitumor effect of gefitinib nanoparticles on human lung cancer. Drug Deliv. 2017;24(1):1501–1512. | ||
Zhang H, Jiang Y, Ni X, et al. Glycyrrhetinic acid-modified norcantharidin nanoparticles for active targeted therapy of hepatocellular carcinoma. J Biomed Nanotechnol. 2018;14(1):114–126. | ||
Gong C, Xie Y, Wu Q, et al. Improving anti-tumor activity with polymeric micelles entrapping paclitaxel in pulmonary carcinoma. Nanoscale. 2012;4(19):6004–6017. | ||
Gao X, Wang B, Wei X, et al. Anticancer effect and mechanism of polymer micelle-encapsulated quercetin on ovarian cancer. Nanoscale. 2012;4(22):7021–7030. | ||
Gani C, Coackley C, Kumareswaran R, et al. In vivo studies of the PARP inhibitor, AZD-2281, in combination with fractionated radiotherapy: an exploration of the therapeutic ratio. Radiother Oncol. 2015;116(3):486–494. | ||
Senra JM, Telfer BA, Cherry KE, et al. Inhibition of PARP-1 by olaparib (AZD2281) increases the radiosensitivity of a lung tumor xenograft. Mol Cancer Ther. 2011;10(10):1949–1958. | ||
Zhang X, Yang H, Gu K, et al. In vitro and in vivo study of a nanoliposomal cisplatin as a radiosensitizer. Int J Nanomedicine. 2011;6:437–444. | ||
Xu S, Tang YY, Yu YX, Yx Y, et al. Novel composite drug delivery system as a novel radio sensitizer for the local treatment of cervical carcinoma. Drug Deliv. 2017;24(1):1139–1147. | ||
Diab R, Hamoudeh M, Boyron O, Elaissari A, Fessi H. Microencapsulation of cytarabine using poly(ethylene glycol)-poly(epsilon-caprolactone) diblock copolymers as surfactant agents. Drug Dev Ind Pharm. 2010;36(4):456–469. | ||
Ito M, Jiang C, Krumm K, et al. TIP30 deficiency increases susceptibility to tumorigenesis. Cancer Res. 2003;63(24):8763–8767. | ||
Chen W, Zheng R, Baade PD, et al. Cancer statistics in China, 2015. CA Cancer J Clin. 2016;66(2):115–132. | ||
Helleday T, Lo J, van Gent DC, Engelward BP. DNA double-strand break repair: from mechanistic understanding to cancer treatment. DNA Repair. 2007;6(7):923–935. | ||
Hühn D, Bolck HA, Sartori AA. Targeting DNA double-strand break signalling and repair: recent advances in cancer therapy. Swiss Med Wkly. 2013;143:w13837. | ||
Yu Y, Xu S, You H, et al. In vivo synergistic anti-tumor effect of paclitaxel nanoparticles combined with radiotherapy on human cervical carcinoma. Drug Deliv. 2017;24(1):75–82. | ||
You H, Fu S, Qin X, et al. A study of the synergistic effect of folate-decorated polymeric micelles incorporating hydroxycamptothecin with radiotherapy on xenografted human cervical carcinoma. Colloids Surf B Biointerfaces. 2016;140:150–160. | ||
Wu Z, Zou X, Yang L, et al. Thermosensitive hydrogel used in dual drug delivery system with paclitaxel-loaded micelles for in situ treatment of lung cancer. Colloids Surf B Biointerfaces. 2014;122:90–98. | ||
Wang Y, Wang C, Wang Y, et al. Micelles of methoxy poly(ethylene glycol)-poly(epsilon-caprolactone) as a novel drug delivery vehicle for tacrolimus. J Biomed Nanotechnol. 2013;9(2):147–157. | ||
van de Ven AL, Tangutoori S, Baldwin P, et al. Nanoformulation of olaparib amplifies PARP inhibition and sensitizes PTEN/TP53-deficient prostate cancer to radiation. Mol Cancer Ther. 2017;16(7):1279–1289. | ||
Ullal AV, Reiner T, Yang KS, et al. Nanoparticle-mediated measurement of target-drug binding in cancer cells. ACS Nano. 2011;5(11):9216–9224. | ||
Yang Q, Liao J, Deng X, et al. Anti-tumor activity and safety evaluation of fisetin-loaded methoxy poly(ethylene glycol)-poly(epsilon-caprolactone) nanoparticles. J Biomed Nanotechnol. 2014;10(4):580–591. | ||
Wang Y, Wu M, Gu L, et al. Effective improvement of the neuroprotective activity after spinal cord injury by synergistic effect of glucocorticoid with biodegradable amphipathic nanomicelles. Drug Deliv. 2017;24(1):391–401. | ||
Chen LX, Ni XL, Zhang H, et al. Preparation, characterization, in vitro and in vivo anti-tumor effect of thalidomide nanoparticles on lung cancer. Int J Nanomedicine. 2018;13:2463–2476. | ||
Xia Y, Wu X, Zhao J, et al. Three dimensional plasmonic assemblies of AuNPs with an overall size of sub-200 nm for chemo-photothermal synergistic therapy of breast cancer. Nanoscale. 2016;8(44):18682–18692. | ||
Xu Z, Liu S, Kang Y, Wang M. Glutathione- and pH-responsive nonporous silica prodrug nanoparticles for controlled release and cancer therapy. Nanoscale. 2015;7(13):5859–5868. | ||
Zhang Z, Feng SS. Nanoparticles of poly(lactide)/vitamin E TPGS copolymer for cancer chemotherapy: synthesis, formulation, characterization and in vitro drug release. Biomaterials. 2006;27(2):262–270. | ||
Mu L, Feng SS. Vitamin E TPGS used as emulsifier in the solvent evaporation/extraction technique for fabrication of polymeric nanospheres for controlled release of paclitaxel (Taxol). J Control Release. 2002;80(1–3):129–144. | ||
Van den Abbeele AD. The lessons of GIST – PET and PET/CT: a new paradigm for imaging. Oncologist. 2008;13(Suppl 2):8–13. | ||
Löser DA, Shibata A, Shibata AK, et al. Sensitization to radiation and alkylating agents by inhibitors of poly(ADP-ribose) polymerase is enhanced in cells deficient in DNA double-strand break repair. Mol Cancer Ther. 2010;9(6):1775–1787. | ||
Glatzel-Plucinska N, Piotrowska A, Grzegrzolka J, et al. SATB1 level correlates with Ki-67 expression and is a positive prognostic factor in non-small cell lung carcinoma. Anticancer Res. 2018;38(2):723–736. | ||
Xie F, Ding RL, He WF, et al. In vivo antitumor effect of endostatin-loaded chitosan nanoparticles combined with paclitaxel on Lewis lung carcinoma. Drug Deliv. 2017;24(1):1410–1418. | ||
Sun CJ, Yang ZG, Zhou XP, et al. Non-small cell lung cancer evaluated by first pass dynamic contrast-enhanced 16-slice spiral CT: correlation of tumor vascularity with pathological characteristics. Zhonghua Zhong Liu Za Zhi. 2007;29(6):429–433. | ||
Ranjan AP, Mukerjee A, Gdowski A, et al. Curcumin-ER prolonged subcutaneous delivery for the treatment of non-small cell lung cancer. J Biomed Nanotechnol. 2016;12(4):679–688. | ||
Chalmers A, Johnston P, Woodcock M, Joiner M, Marples B. PARP-1, PARP-2, and the cellular response to low doses of ionizing radiation. Int J Radiat Oncol Biol Phys. 2004;58(2):410–419. | ||
Lin S, Yan Y, Liu Y, et al. Sensitisation of human lung adenocarcinoma A549 cells to radiotherapy by Nimotuzumab is associated with enhanced apoptosis and cell cycle arrest in the G2/M phase. Cell Biol Int. 2015;39(2):146–151. | ||
Deben C, Lardon F, Wouters A, et al. APR-246 (PRIMA-1(MET)) strongly synergizes with AZD2281 (olaparib) induced PARP inhibition to induce apoptosis in non-small cell lung cancer cell lines. Cancer Lett. 2016;375(2):313–322. | ||
Kumar CG, Poornachandra Y, Chandrasekhar C. Green synthesis of bacterial mediated anti-proliferative gold nanoparticles: inducing mitotic arrest (G2/M phase) and apoptosis (intrinsic pathway). Nanoscale. 2015;7(44):18738–18750. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.