Back to Journals » International Journal of Nanomedicine » Volume 12
Octanoyl galactose ester-modified microemulsion system self-assembled by coix seed components to enhance tumor targeting and hepatoma therapy
Authors Qu D ![]() , Liu M, Huang M, Wang L, Chen Y
, Liu M, Huang M, Wang L, Chen Y ![]() , Liu C, Liu Y
, Liu C, Liu Y
Received 20 October 2016
Accepted for publication 27 December 2016
Published 14 March 2017 Volume 2017:12 Pages 2045—2059
DOI https://doi.org/10.2147/IJN.S125293
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Lei Yang
Ding Qu,1,2 Mingjian Liu,1 Mengmeng Huang,1,2 Lixiang Wang,1 Yan Chen,1,2 Congyan Liu,1,2 Yuping Liu1,2
1Research Center for Multicomponent Traditional Medicine and Microecology, Affiliated Hospital of Integrated Traditional Chinese and Western Medicine, Nanjing University of Chinese Medicine, 2Research Center for Multicomponent Traditional Medicine and Microecology, Jiangsu Provincial Academy of Traditional Chinese Medicine, Nanjing, People’s Republic of China
Abstract: A nanosized drug delivery platform with a combination of rational components and tumor targeting is significant for enhancement of anticancer therapy and reduction of side effects. In this study, we developed a octanoyl galactose ester-modified microemulsion system self-assembled by coix seed components (Gal(oct)-C-MEs), which improved the tumor accumulation through asialoglycoprotein receptor-mediated endocytosis and promoted the antitumor efficacy through multicomponent-mediated synergistic effect. Octanoyl galactose ester (Gal(oct)) with a yield of 82.3% was synthesized through a green enzymatic reaction and multidimensional characterization. Gal(oct)-C-MEs with a spherical shape had a small and uniform particle size (58.49±1.03 nm), narrow polydispersity index (0.09±0.01) and neutral surface charge (-5.82±0.57 mV). In the cellular uptake studies, the internalized Gal(oct)-C-ME was 2.28-fold higher relative to that of coix seed component-based microemulsions (C-MEs). The half-maximal inhibitory concentration of Gal(oct)-C-MEs against HepG2 cells was 46.5±2.4 µg/mL, which was notably higher than that of C-MEs. Importantly, the intratumor fluorescence of HepG2 xenograft-bearing nude mice treated with Cy5/Gal(oct)-C-MEs was 1.9-fold higher relative to treatment with Cy5/C-MEs. In the study of antitumor efficacy in vivo, HepG2 xenograft-bearing nude mice intragastrically administered Gal(oct)-C-MEs for 14 days exhibited the strongest inhibition of tumor growth and the lowest toxicity against liver and kidney among all the treatments. In summary, Gal(oct)-C-ME, as a highly effective and safe anticancer drug delivery system, showed promising potential for hepatoma therapy.
Keywords: coix seed oil, coixan, multicomponent-based microemulsion, hepatic targeting, antihepatoma, oral drug delivery system
Introduction
Hepatocellular carcinoma (HCC) is the fifth most frequently diagnosed cancer worldwide and the second and sixth leading cause of cancer death in men and women, respectively.1 Higher incidence of HCC is reported in East Asia and sub-Saharan Africa due to the dietary habits and public hygiene.2 Although chemotherapy is a most commonly used and effective approach for treatment of HCC, the severe systemic side effects resulting from nonselective biodistribution still enormously restrict the clinical applications.3,4 Recently, nano-sized drug delivery system (DDS) showed a promising potentiality in tumor-specific accumulation by enhanced permeability and retention (EPR) effect. For example, there are several clinically applied nanomedicines for cancer therapy, such as Doxil® (PEGylated liposomal doxorubicin [DOX], Johnson & Johnson, NJ, USA), micellular paclitaxel (PTX/NK-105) and Lipusu® (liposomal paclitaxel [PTX], Luye Pharma, Shandong, China).5,6 Until now, however, few commercial formulations have been modified with well-known tumor ligands (folic acid, cholic acid, transferrin, hyaluronic acid, etc) or exclusively designed for active tumor targeting, although the nanomedicines improved the therapeutic efficacy and reduced the adverse effects of the anticancer drugs.7–9 In fact, tumor-targeted, ligand-modified nanomedicines have to overcome a serious problem of the exogenous interference to tumor-specific recognition. Besides, the modification method also influences tumor targeting, because it decides the exposure destiny of tumor ligands on the surface of the nanoparticle.
As we know, galactose- and N-acetylgalactosamine-terminated glycoproteins have high affinity for asialoglycoprotein receptor, which is highly expressed on the surface of the hepatoma cells. Galactose-modified nanosized DDSs have increasingly become one of the most promising hepatoma-targeted nanomedicines for treatment of liver cancer.10–13 Previous publications demonstrated that the galactose-containing, fluorescein isothiocyanate (FITC)-labeled dextran-g-PCL micelles had an apparent targeting to HepG2 cells and liver tissue.14 However, such a modification method of galactose usually needs chemical catalyzer and harsh reaction conditions. Recently, our group has developed butyryl galactose ester (Gal(but)) and stearic galactose ester (Gal(ste)) by a green enzymatic reaction method. In addition, a multicomponent microemulsion system was prepared through self-assembly of coix seed oil, coixan, RH40 and polyethylene glycol 400 (PEG400) using Gal(but) or Gal(ste) as the targeting ligand.15,16 Interestingly, we found that galactose ester with different lengths of chain significantly influenced the targeting on the liver and liver tumor. Sparked by this strategy, we were interested to determine whether galactose ester with medium lengths of chain probably had a better liver and hepatoma targeting.
Herein, octanoyl galactose ester with a medium length chain was synthesized and octanoyl galactose ester-modified coix seed component microemulsion (Gal(oct)-C-ME) based on coix seed oil and coixan was prepared by using a previously reported method (Figure 1). After crossing the intestinal tract, Gal(oct)-C-ME was capable of accumulating at the tumor sites by the EPR-mediated passive targeting and galactose ester-mediated active targeting, thereby enhancing the cellular uptake and anticancer efficacy. The enhanced tumor targeting was characterized by fluorescent probe techniques and near-infrared imaging in vivo. The advantages of such a design were investigated through evaluation of tumor cell killing, cell apoptosis induction, in vivo anticancer efficacy and safety.
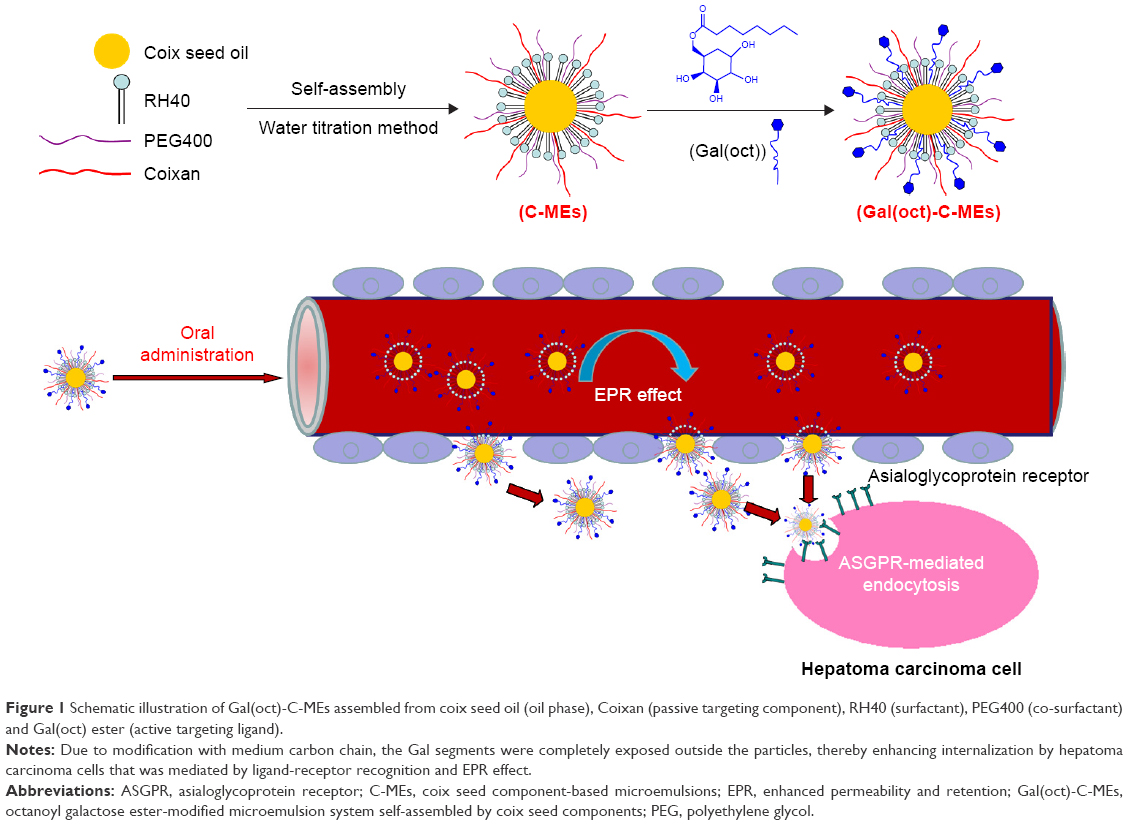 | Figure 1 Schematic illustration of Gal(oct)-C-MEs assembled from coix seed oil (oil phase), Coixan (passive targeting component), RH40 (surfactant), PEG400 (co-surfactant) and Gal(oct) ester (active targeting ligand). |
Materials and methods
Materials
Coix seed oil was extracted by our group using a supercritical CO2 fluid extraction technology (purity of glycerin trilaurate >85%, determined by ultraviolet spectroscopy). Coix seed polysaccharides (Coixan) were purified using a classical water extraction and alcohol precipitation method by our group (purity >75%, determined by ultraviolet spectroscopy). Novozym 435 and 4A molecular sieve were purchased from Nanjing Cheng-Na Chemical Co., Ltd (Nanjing, People’s Republic of China). Vinyl n-octanoate, D-galactose and PEG400 were obtained from Sigma-Aldrich (St Louis, MO, USA). Cremophor® RH40 was obtained from BASF Co., Ltd (Ludwigshafen, Germany). Roswell Park Memorial Institute 1640 medium, fetal bovine serum (FBS), penicillin–streptomycin solution and phosphate-buffered saline (PBS) were provided by Thermo Fisher Scientific Inc. (Waltham, MA, USA). 3-[4,5-Dimethylthiahiazol-2-yl]-2,5-diphenyltetrazolium bromide (MTT) assay and Annexin V-PE assay kit were both provided by KeyGen Biotech Co., Ltd (Jiangsu, People’s Republic of China). FITC and Cy5 were purchased from Tianjin Biolite Biotech Co., Ltd (Tianjin, People’s Republic of China). Human hepatoma (HepG2) cells were bought from Shanghai Institute of Biochemistry and Cell Biology. Male Balb/c mice (6–8 weeks old) weighing 22–25 g were purchased from SLAC Co., Ltd (Shanghai, People’s Republic of China). Water in this study was prepared by the Milli-Q-water purification system (Merck Millipore, Billerica, MA, USA). All other chemicals and solvents were of analytical grade, unless otherwise stated.
Synthesis and characterization of octanoyl galactose ester
Octanoyl galactose ester (Gal(oct)), a hepatic-targeted ligand, was synthesized by a enzymatic reaction between vinyl n-octanoate and D-galactose using Novozym 435 immobilized lipase as a catalyzator. Briefly, vinyl n-octanoate (3.0 mL, 15.5 mmol) and D-galactose (2.8 g, 15.6 mmol) were dissolved in 50 mL of dehydrated acetone in the presence of 4A molecular sieve, which was followed by addition of Novozym 435 immobilized lipase slowly. After vigorous stirring for 15 h at 60°C, the mixture was filtered to remove the enzyme and molecular sieve, and consequently, the crude product was obtained through evaporating acetone under reduced pressure. Highly purified octanoyl galactose ester was prepared with a yield of 82.3% by using a silica gel column separation technology (100–200 mesh) with an eluent of methanol/dichloromethane (1/10, v/v). The chemical structure of the product was multidimensionally characterized, including Fourier transform infrared (FTIR) spectroscopy, hydrogen spectrum nuclear magnetic resonance (1H NMR) and high-resolution mass spectrum (HRMS) analysis. 1H NMR spectra were recorded on a Bruker AVANCE-300 spectrometer (300 MHz; Bruker Optik GmbH, Ettlingen, Germany) in deuterated chloroform (CDCl3) at room temperature. Chemical shifts are reported in ppm with the internal tetramethylsilane signal at 0.0 ppm as a standard. FTIR spectra were recorded with KBr pellets on an FTIR spectrometer (Nicolet 2000; Magna IR 550; Madison, Nicolet Inc., WI, USA) at room temperature. HRMS analysis was performed using Agilent Q-TOF system (6545; Agilent Technologies, Santa Clara, CA, USA) using electrospray ionization (ESI+) model; prior to the experiment, the sample was dissolved in mass grade methanol at a concentration of 20 ng/mL.
Preparation and characterization of microemulsions
The Gal(oct)-C-MEs were prepared using “one-step” emulsion method described previously.16 Briefly, octanoyl galactose ester (32 mg), coixan (240 mg), Cremophor RH40 (150 mg) and PEG400 (50 mg) were mixed with vigorous magnetic stirring at room temperature until a completely homogeneous mixture was obtained. Afterward, coix seed oil (200 mg) was added in batches to the above-mentioned mixture, followed by stirring for another 2 h. A faint yellow transparent Gal(oct)-C-MEs solution with low viscosity was obtained on adding 2.5 mL of deionized water slowly into the mixture. Coix seed components microemulsions (C-MEs) were prepared by a similar method, but without adding octanoyl galactose ester.
The average size, polydispersity index and zeta potential of various types of microemulsions were measured in triplicate by dynamic light scattering analyzer (Nano ZS; Malvern Instruments Ltd, Malvern, UK). For the morphologic study, 30 μL of Gal(oct)-C-MEs was deposited onto a film-coated copper grid, followed by staining with 30 μL of 1.5% aqueous solution of phosphotungstic acid and observing immediately by transmission electron microscopy (JEM-200CX; JEOL, Tokyo, Japan) after air drying.
Cell culture
HepG2 cells were cultured in Roswell Park Memorial Institute 1640 medium containing 10% FBS (v/v) at 37°C under an atmosphere of 5% CO2 and 90% relative humidity. The cells were passaged using trypsin at a split ratio of 1:8, after they reached 80% confluence.
Cellular uptake and internalization mechanism
For cellular uptake study, FITC was employed as a fluorescence probe to visualize the intracellular Gal(oct)-C-MEs and other formulations. Various types of FITC-labeled formulations were prepared using the above-mentioned method, but by adding FITC and adjusting the concentration to 2 μM. The exponential phase HepG2 cells were cultured in 24-well plates at a density of 1.0×105 cells per well for 24 h. After reaching 60% overspread, the culture medium was replaced with FITC-labeled formulations, followed by incubation for another 2 h. Then the cells were washed with precooling PBS thoroughly to remove the remaining fluorescence. At the end of this time, the adherent cells were immediately observed by fluorescence inverse microscope (IX73; Olympus Corporation, Tokyo, Japan). Likewise, the cell suspension harvested in 0.3 mL of PBS was also analyzed by flow cytometry (Guava 6HT, Merck Millipore).
The mechanism of cellular uptake was evaluated by pretreatment with specific inhibitors.17,18 When the cells grew up to 60% confluence, 200 μL of FBS-free culture medium containing 100 mM galactose, 0.1% NaN3, 10 nM NH4Cl, 0.8 μM sucrose, 0.2 nM genistein and 0.4 nM amiloride was incubated with the cells for 2 h respectively, which was followed by adding various FITC-labeled formulations in the presence of different inhibitors. After another 2 h, the cells were again treated in the above-mentioned process and analyzed by flow cytometry.
In vitro cytotoxicity and cell apoptosis
The cytotoxicity of Gal(oct)-C-MEs against HepG2 cells was investigated using conventional MTT method. C-MEs (without galactose ligand) and coix seed oil-MEs (without coixan and tumor ligand) were also used as control groups. Briefly, HepG2 cells were planted in 96-well plates at 5.0×103 per well. After incubation for 24 h, the medium was removed, which was followed by addition of various formulations. After treatment for another 24 h, the cells in each well were stained with 10 μL of MTT (0.5 mg/mL) PBS solution at 37°C for 4 h. The culture medium was replaced with 150 μL of dimethyl sulfoxide and the absorbance of the dissolved blue formazan crystals in each well was measured at 492 nm using an enzyme-linked immunosorbent assay (Thermo Fisher Scientific Inc.). The cell viability was calculated as an absorbance ratio of the tested groups to the negative control group. The half-maximal inhibitory concentration (IC50) was calculated using Statistical Package for the Social Sciences 18.0 (SPSS, Inc., Chicago, IL, USA) software.
We have previously validated the strong apoptosis-inducing ability of coix seed oil-based MEs in human lung tumor (A-549) cell model.17 To investigate the combined effect of galactose ligand modification and coix polysaccharide incorporation, the cell apoptosis after treatment with various formulations was studied by flow cytometry using Annexin V-PE apoptosis detection kit. In brief, 1.0×105 HepG2 cells/well were seeded into 24-well plates and allowed to grow until they reached 80% confluence. Next, the coix seed oil concentration of various formulations including Gal(oct)-C-MEs, C-MEs and coix seed oil-MEs were adjusted as 160 μg/mL, respectively. After incubation for 12 h, the cells were trypsinized by ethylenediaminetetraacetic acid-free pancreatin, rinsed with ice-cold PBS thrice and resuspended in 500 μL of binding buffer successively, which was followed by adding 5 μL of Annexin V-PE and equivalent 7-AAD. At the end of the staining for 15 min, the apoptotic induction was evaluated immediately by flow cytometry (Guava 6HT, Merck Millipore).
In vivo imaging
In our previous studies, stearate galactose-modified C-MEs were capable of accumulation in tumor sites in HepG2 xenograft nude mice models.15 In this study, to validate that galactose ester with the medium length of carbon chain had a better tumor-targeting ability than stearate galactose ligand, we investigated the biodistribution of Gal(oct)-C-MEs through encapsulating Cy5 (a near-infrared dye) using in vivo near-infrared imaging system (Caliper, Hopkinton, MA, USA). HepG2 xenograft nude mice models were built as described in our previous paper.15 When the tumor volume reached about 300 mm3, the mice were intragastrically administered with different formulations, including free Cy5, Cy5-labeled C-MEs (Cy5/C-MEs) and Cy5-labeled Gal(oct)-C-MEs (Cy5/Gal(oct)-C-MEs) at a Cy5 dose of 250 μg/kg. The NIR images were observed at determined intervals post-intragastric administration by in vivo imaging system equipped with an excitation band pass filter at 630 nm and an emission filter at 700 nm. At 48 h posttreatment, the mice were sacrificed and tumors were excised. The ex vivo fluorescent images of the tumors were also detected qualitatively and quantitatively. Besides, Cy5 formulation-treated rats (220–250 g) were also used to investigate the preliminary pharmacokinetic property. The blood samples were collected at determined intervals, and the plasma was obtained by low temperature centrifugation (1,000 rpm ×3 min). The fluorescence was measured by microplate reader (Varioskan Flash, Thermo Fisher Scientific Inc.).
Antitumor efficacy in vivo
Fifty male BALB/c mice (22–25 g) bearing HepG2 tumor xenograft were used in antitumor efficacy study. The animal experiments were carried out in accordance with the protocol approved by the animal ethics committee of Nanjing University of Chinese Medicine (Nanjing, People’s Republic of China). When the tumor volume reached about 80 mm3, the mice were randomly separated into five groups and treated with Gal(oct)-C-MEs, C-MEs and coix seed oil-MEs intragastrically each day for 14 days at a coix seed oil dose of 2.5 g/kg/day. Saline and Kanglaite injection (commercial formulation of coix seed oil, Kanglaite Pharmacy Inc., Zhejiang, China) were used as a negative and positive control, respectively. The volumes of the tumor and the body weight of each mouse were monitored every day. The tumor volume (V) was calculated using the formula: V = longitudinal (L) × transverse (W)2/2. All mice were sacrificed at the end of the treatment, which was followed by weighing the tumor tissues and harvesting various organs. Hematoxylin and eosin staining was used to evaluate the toxicity of treatment against organs through embedding the sections of 5 μm thickness in paraffin and observing by optical microscope.
Safety evaluation
Hematologic toxicity, acute liver–kidney damage and immune suppression are commonly found in anticancer therapy. In this study, we measured the above-mentioned indices through collecting the blood of mice treated with various formulations. Specifically, 300 μL of blood sample was collected from the tail vein at 24 h after the last administration. Thereafter, 50 μL of whole blood was used for blood routine analysis and the remaining sample was allowed to coagulate to obtain serum by centrifuging the clotted blood. Several indices representing the functions of liver and kidney, such as aspartate aminotransferase, alanine aminotransferase and blood urea nitrogen, were detected by blood biochemical analyzer (LH 750, Beckman Coulter). Likewise, the serum immune indices were determined by using enzyme-linked immunosorbent assay kits for interleukin-6 and tumor necrosis factor-α (enzyme linked Biological Technology Co., Ltd., Shanghai, People’s Republic of China).
Statistical analysis
Data are presented as the mean ± standard deviation (SD). Statistical significance of data was determined using Student’s t-test with *P<0.05 as statistical significance and **P<0.01 as extreme statistical significance.
Results and discussion
Characterization of octanoyl galactose ester
Gal(oct) was synthesized by conjugation of galactose (hydroxyl group) and vinyl n-octanoate (acyl group) in the presence of Novozym 435 immobilized lipase (illustrated in Figure 2A). Note that an aqueous environment could result in a change in the conformation of Novozym 435 lipase, and thereby reduce the yield of product. Therefore, the solvent and reaction system were kept away from any trace of moisture.19 As shown in Figure 2B, the molecular weight of Gal(oct) was detected as 307.2941 by HRMS. According to the results of FTIR (Figure 2C), specific carbonyl stretch of ester was observed at 1,705.94 cm−1, which was ascribed to the newly generated bond of Gal(oct). Besides, a broad absorption band at 3,366.61 cm−1 was also found, which represented the O–H stretch of substantial hydroxyl group in galactose. Likewise, the obvious absorption peaks at 2,923 and 2,849.96 cm−1 could be attributed to the aliphatic C–H stretching vibrations of octanoate. The chemical structure of Gal(oct) was further confirmed through the proton nuclear magnetic resonance spectra (1H NMR). As shown in Figure 2D, the characteristic peaks around 4.85 ppm representing the double bond hydrogen of vinyl group were not found, suggesting that the vinyl groups were substituted by galactose. In addition, the aliphatic chain (δ=0.89–2.32 ppm) and the galactose segment (δ=3.51–4.49 and 5.41 ppm) were observed, validating the chemical structure of Gal(oct). In our previous studies, we also synthesized a series of galactose esters with different carbon chain lengths, such as stearoyl, butyryl and acetyl galactose ester.
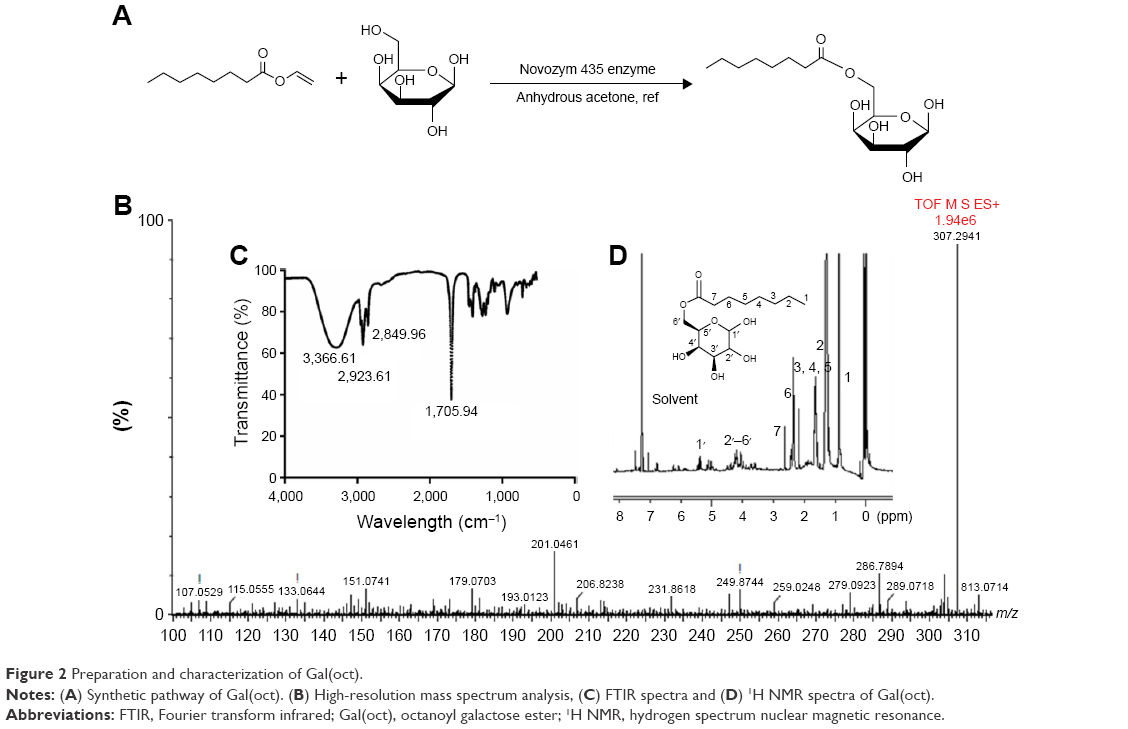 | Figure 2 Preparation and characterization of Gal(oct). |
Previously, we synthesized a series of galactose esters with different aliphatic chains, including stearoyl, butyryl and acetyl segments. The preliminary findings suggested that galactose ester with a medium carbon chain length (Gal(oct)) displayed the most obvious HepG2 cell/tumor affinity among all the synthesized galactose ester in cellular uptake or in vivo tumor targeting all tumor-targeting ligands. The potential explanation for such a phenomenon is that the length of the carbon chain determines the hydrophilic–lipophilic balance (HLB) of galactose ester. Stearoyl galacotse (long chain) might be covered by the coixan shell outside the microemulsion because of the strong hydrophobicity. Conversely, butyryl or acetyl galacotse (short chain) is difficult to coat stably on the surface of the microemulsions due to the relatively strong hydrophilicity. In view of this, Gal(oct) with medium chain length was used in further studies.
Preparation and characterization of microemulsions
Gal(oct)-C-MEs and C-MEs were prepared by simple operation and a reliable technology according to our previous reported method.15,16 As shown in Figure 3A, the average particle size and zeta potential of Gal(oct)-C-MEs at pH 7.4 were approximately 55 nm and −5.8 mV, respectively. With a decrease in pH, no significant change was found in the size and surface potential, which suggests that Gal(oct)-C-MEs have acceptable stability in different pH environments. There was no obvious alternation in the size and potential of microemulsion before and after modification with galactose ester. However, the particle size and zeta potential of coix oil-MEs reduced significantly than those of C-MEs, indicating that coixan coated outside the particles increased the size of microemulsions and shielded the partial negative charge, which might be helpful to decrease the adsorption by serum protein and prolong the blood circulation time.20 In order to investigate the influence of coixan and Gal(oct), the morphology of various microemulsions was also determined (Figure 3B). All test samples displayed as spherical particles with around 50 nm, which was essentially in agreement with the results of dynamic light scattering analysis. In addition, we also investigated the in vitro release profile of microemulsions using DiI as a fluorescence probe. As shown in Figure 3C, approximately 30% of DiI was released from DiI-labeled Gal(oct)-C-MEs (DiI/Gal(oct)-C-MEs) and DiI-labeled C-MEs (DiI/C-MEs) at 24 h post-incubation by using dialysis method. In contrast, the accumulative release amount of DiI was detected as only 15% in DiI-labeled coix seed oil-MEs (DiI/coix seed oil-MEs). It suggests that the modification of Gal(oct) had no influence on the release behavior on Gal(oct)-C-MEs, but incorporation of coixan seemed to promote the release rate of drugs. The storage stability and pH tolerance tests were performed through observing the transmittance. As displayed in Figure 3D and E, the clarity of the numbers written behind the bottles represented the stability of Gal(oct)-C-MEs. The results suggest that the Gal(oct)-C-MEs was stable within 5 days or under pH environment of >5.0.
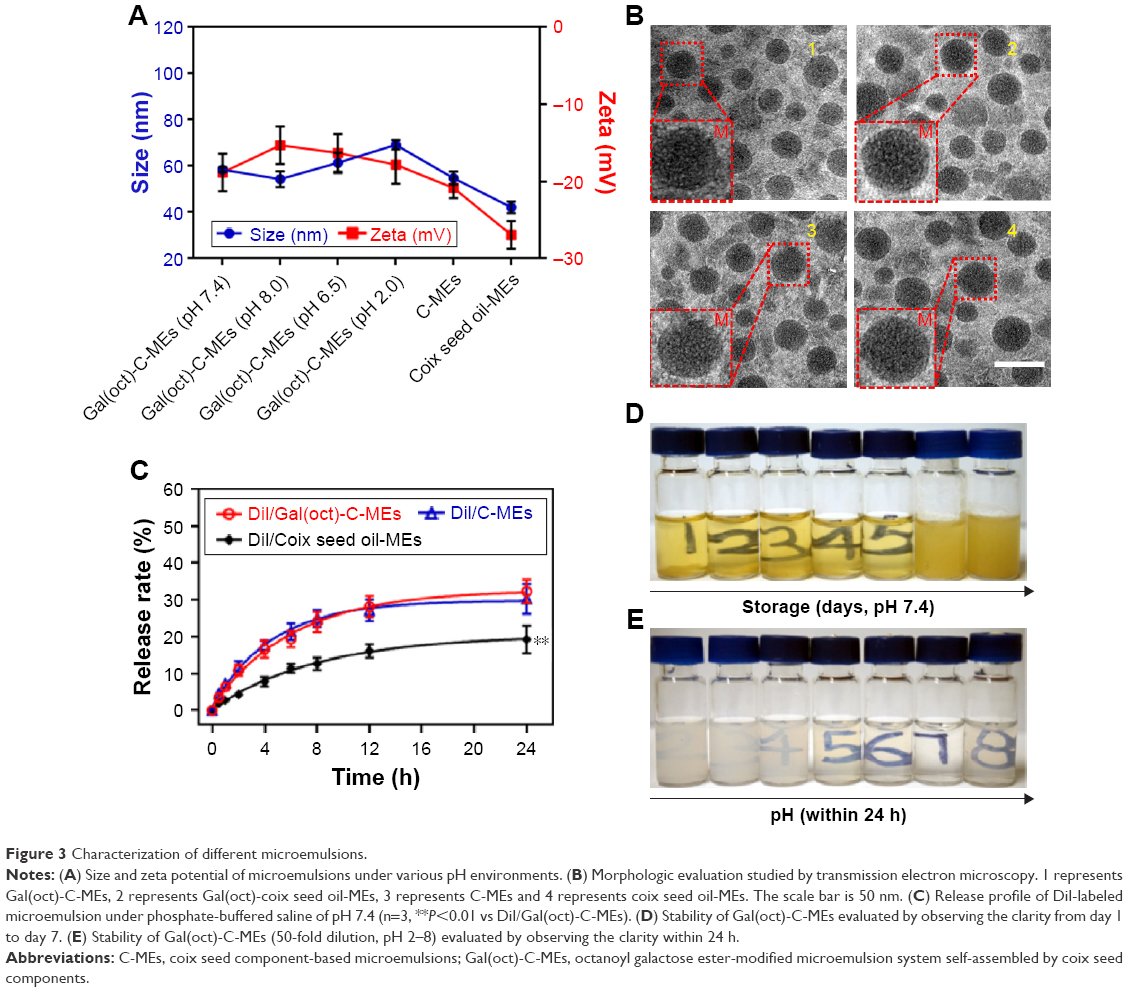 | Figure 3 Characterization of different microemulsions. |
Cellular uptake
We have previously validated that C-MEs modified with stearate galactose ester could significantly increase the endocytosis of HepG2 cells.15 In the synthesis of various galactose esters, we found that different aliphatic chains were closely related to the HLB, and thereby determined the compatibility of the tumor-targeted ligand with the microemulsions. Due to the appropriate HLB, Gal(oct) with a medium length chain was assumed to further enhance the cellular uptake of microemulsions. To validate the hypothesis, the internalization of various microemulsions was investigated qualitatively and quantitatively using FITC as a fluorescence probe. As depicted in Figure 4A, the cells treated with FITC-Gal(oct)-C-MEs displayed overwhelming green fluorescence among all the groups. Comparison between FITC-Gal(oct)-C-MEs and FITC-Gal(oct)-coix oil-MEs showed that the introduction of coixan also synergistically increased the endocytosis. Notably, the cellular uptake of FITC-Gal(oct)-C-MEs sharply decreased when the cells were pretreated with 100 mM galactose, suggesting that the promotion of internalization was mainly contributed by modification of galactose ester.21
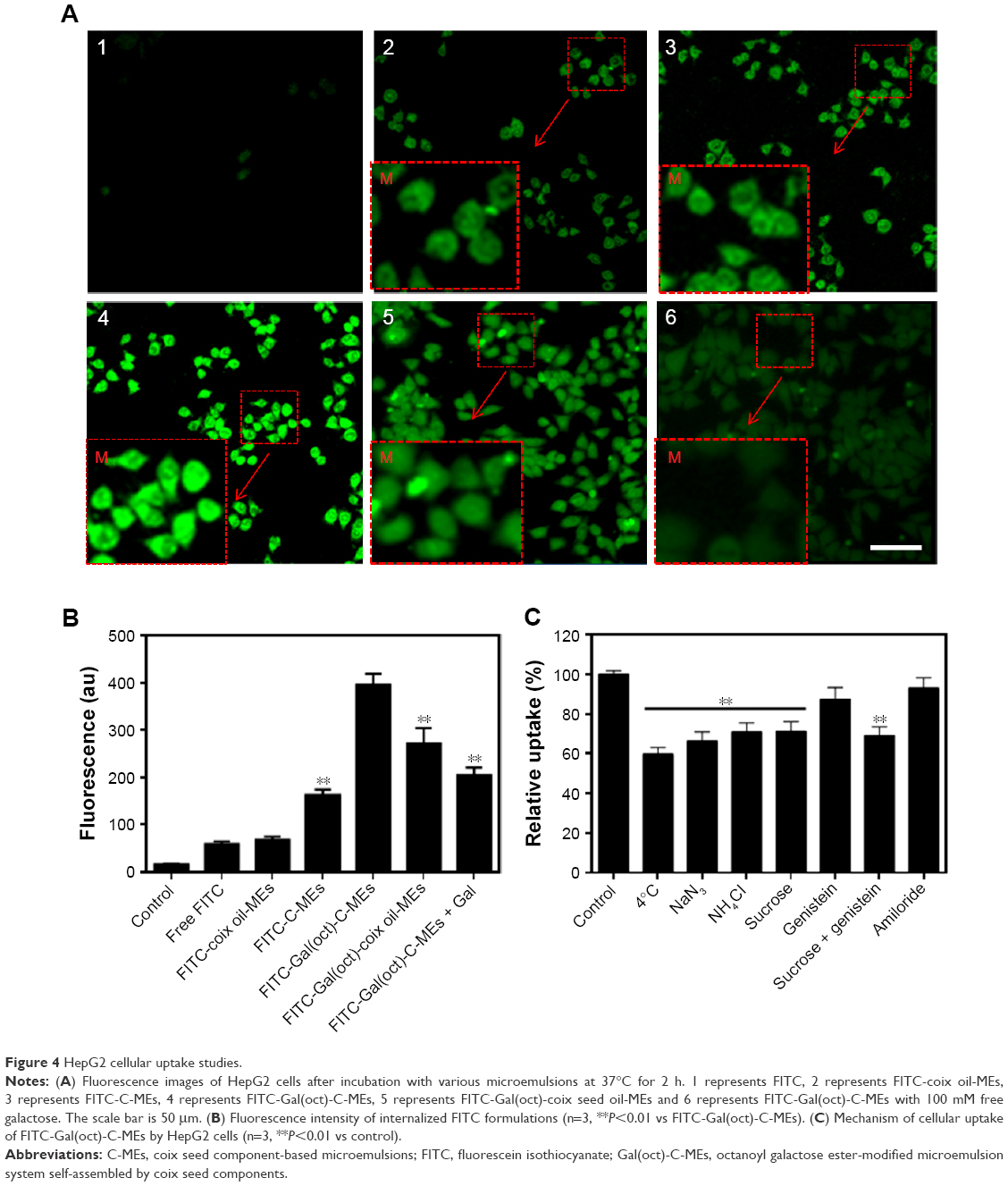 | Figure 4 HepG2 cellular uptake studies. |
The fluorescence signal of HepG2 cells treated with various formulations was analyzed by flow cytometry. As shown in Figure 4B, the fluorescence intensity of tumor cells was approximately 400 after treatment with FITC-Gal(oct)-C-MEs, which was 2.28-fold higher than FITC-C-MEs. Compared with FITC-Gal(ste)-C-MEs,15 modification of octanoyl galactose ester showed an obvious improvement in the cellular uptake of C-MEs, indicating that galactose ester with medium length chain had significant advantage over that with long length chain in cellular uptake. Besides, the fluorescence intensity of FITC-Gal(oct)-coix oil-MEs was 1.4-fold lower than that of FITC-Gal(oct)-C-MEs, further validating that coixan and galactose ester synergistically enhanced internalization. The internalization mechanism of Gal(oct)-C-MEs was studied using various specific inhibitors or treatment, such as 4°C/NaN3 (energy inhibitor), NH4Cl (lyso-endosomers endocytosis inhibitor), sucrose (clathrin-mediated endocytosis inhibitor), genistein (caveolae-mediated endocytosis inhibitor) and amiloride (macropinocytosis inhibitor).22 As displayed in Figure 4C, the cellular uptake greatly reduced when the cells were cultured under low temperature, as well as when treated with NaN3, suggesting that Gal(oct)-C-MEs entered the tumor cells in a energy-dependent manner. In addition, the cellular uptake was also inhibited after treatment with NH4Cl and sucrose, suggesting that Gal(oct)-C-MEs crossed the cell membrane by clathrin-mediated pathway and was consequently entrapped by lyso-endosomers.
In vitro antitumor efficacy
In the cellular uptake studies, Gal(oct)-C-MEs were capable of carrying more active components than C-MEs and Gal(oct)-coix oil-MEs into tumor cells. In this experiment, the cytotoxicity of different formulations was investigated to figure out whether Gal(oct) could further enhance the cytotoxicity of Gal(oct)-C-MEs against HepG2 cells. As shown in Figure 5A, the IC50 of C-MEs was 132.5±7.3 μg/mL, which was a 1.5-fold decrease compared with coix oil-MEs, suggesting that coixan-mediated enhancement of cellular uptake promoted killing of tumor cells. With the modification of Gal(oct), the IC50 of Gal(oct)-C-MEs reduced up to 46.5±2.4 μg/mL, which was a 2.8-fold decrease in comparison with that of C-MEs. In our previous studies, we had verified that C-MEs show 1.8-fold enhancement in cytotoxicity against HepG2 cells due to modification with stearate galactose ester. Obviously, galactose ester with chains of different length notably influenced the cellular behavior of microemulsions, further validating our assumption. Besides, the cytotoxicity of Gal(oct) was also tested to eliminate the effects of galactose ester. As depicted in Figure 5B, no significant inhibition of cell growth was observed after incubation with Gal(oct) at the concentrations ranging from 1.5 to 48 μg/mL.
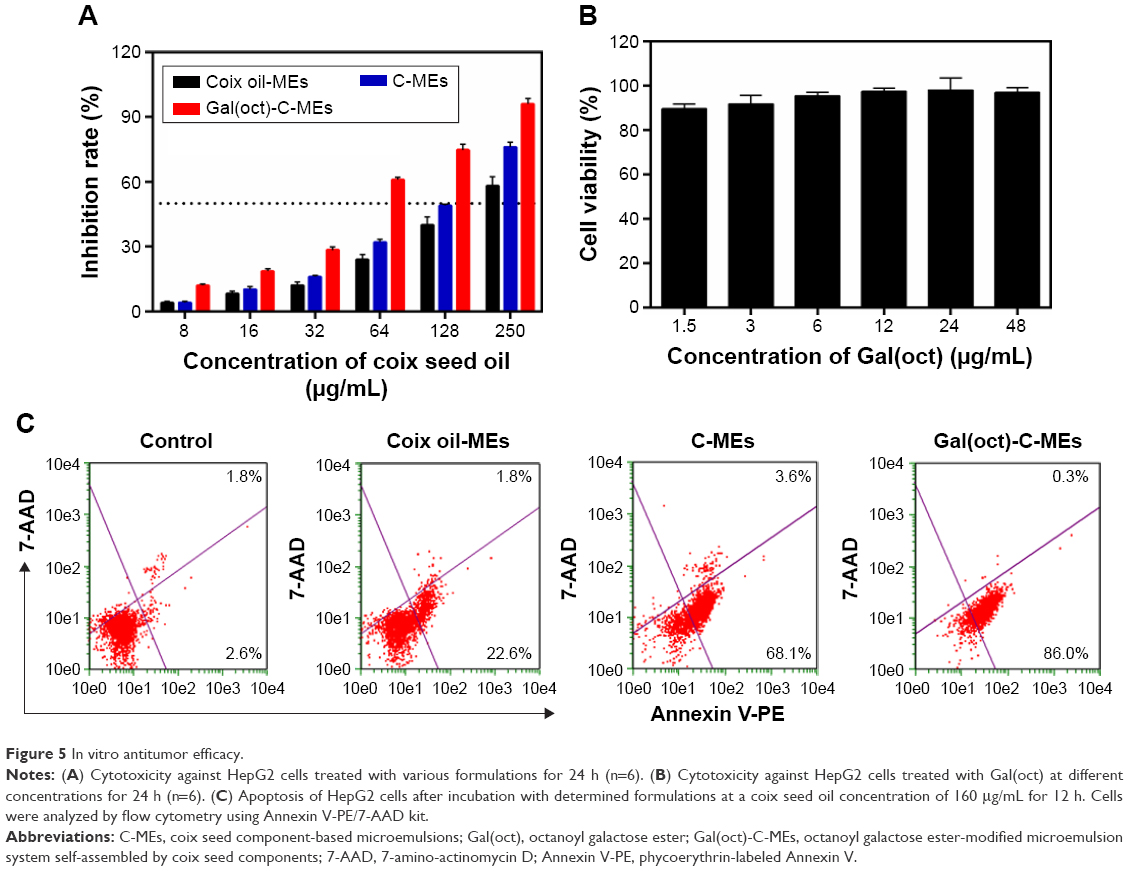 | Figure 5 In vitro antitumor efficacy. |
Generally, cellular uptake and drug release of nanosize DDS would determine the cell apoptosis within a certain period of treatment.23 For evaluating the promotion in apoptosis-inducing effect of Gal(oct)-C-MEs by modification with Gal(oct), the cells treated with various microemulsions were analyzed using an Annexin V-PE/7-AAD kit.24 As shown in Figure 5C, both C-MEs and Gal(oct)-C-MEs produced a significantly enhanced apoptosis effect compared with coix oil-MEs, suggesting that coixan and coix oil were capable of apoptosis induction synergistically using such coix component-based microemulsion.15–18 Note that 86.3% of HepG2 cells treated with Gal(oct)-C-MEs induced apoptosis, which was 1.2-fold higher than that in the C-MEs group. These findings indicated that the synergistic anticancer effect by rational drug combination and the improved intracellular accumulation by the rational design of tumor-targeted ligand were of great importance in cell apoptosis induction.
Tumor targeting in vivo and pharmacokinetics
To validate our assumption that modification of galactose ester with medium carbon chain was favorable for improving tumor targeting of microemulsions, we evaluated the biodistribution of Gal(oct)-C-MEs using the in vivo imaging technique with C-MEs as the control group. Cy5, a commonly used near-infrared dye, was encapsulated into various microemulsions for visualization of distribution in vivo.25,26 At 8 h post-intragastric administration of free Cy5, the fluorescence signal of HepG2 tumor-bearing mice mainly accumulated in the intestinal tract and was eliminated rapidly from 8 to 24 h. Due to nanoparticle-mediated EPR effect, Cy5/C-MEs presented a potential of tumor targeting after crossing the enterocytes, especially at the initial 4 h. In sharp contrast, Cy5/Gal(oct)-C-MEs displayed the most obvious fluorescence signal in the tumor site, which was retained for 48 h (Figure 6A). For further confirming the accumulation of the tumor, ex vivo imaging was observed immediately after harvest of the tumor tissues from HepG2 tumor-bearing mice. As expected, the fluorescence intensity of the tumor tissue excised from Cy5/Gal(oct)-C-MEs–treated mice was 1.9-fold higher than that of Cy5/C-MEs–treated mice (Figure 6B and C), presenting a concrete proof of rationality of Gal(oct) modification.
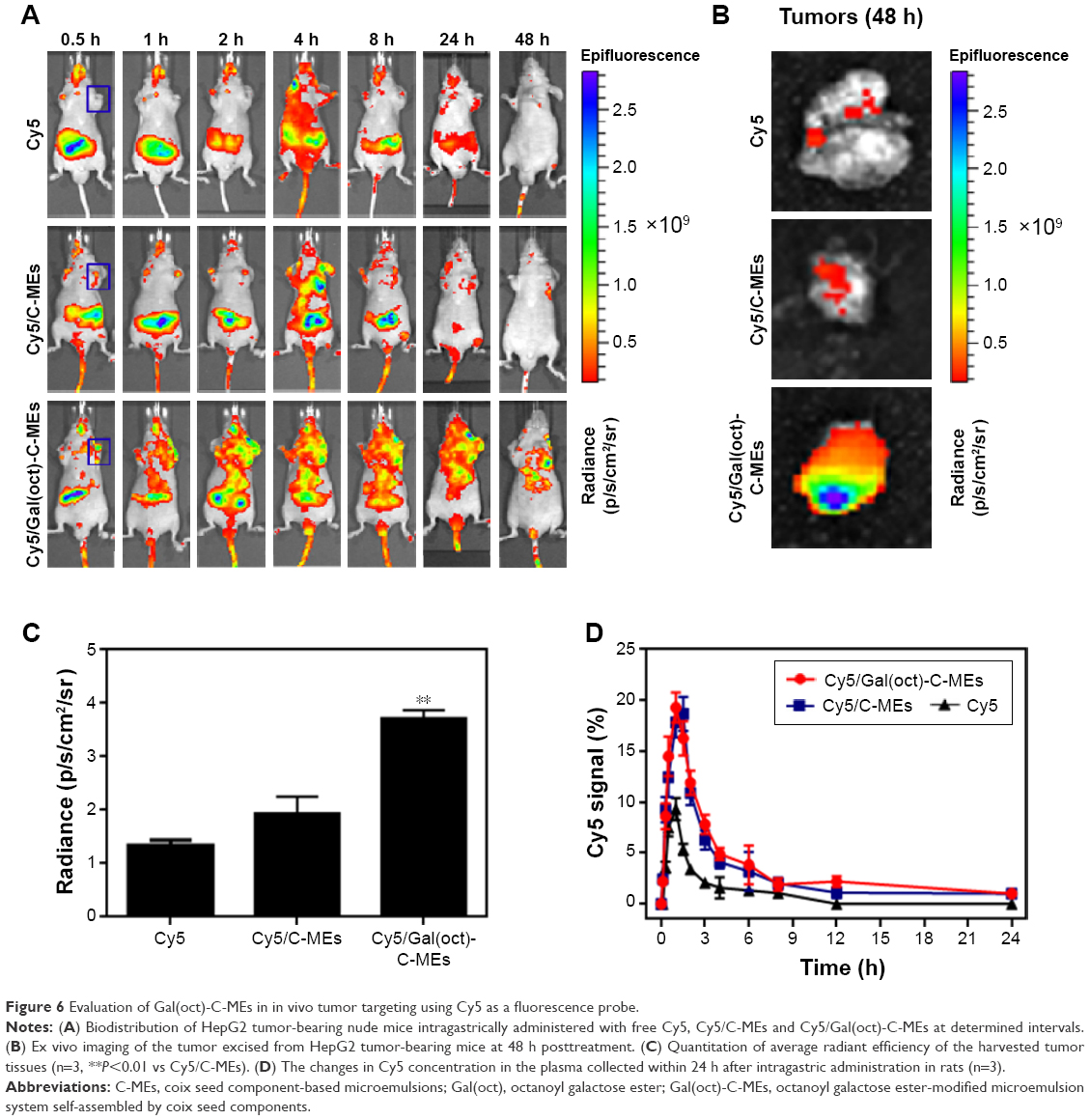 | Figure 6 Evaluation of Gal(oct)-C-MEs in in vivo tumor targeting using Cy5 as a fluorescence probe. |
Prolonged circulation in blood is a critical and inherent merit for orally administrated nanosized vehicle, because particles have more time to accumulate in the tumor region.27 In this experiment, we also evaluated the pharmacokinetics of Cy5/Gal(oct)-C-MEs after intragastric administration by quantifying the Cy5 intensity in plasma. Free Cy5 and Cy5/C-MEs were taken as references. As shown in Figure 6D, the concentration of Cy5 in all test groups reached the maximum at 1 h post-administration. However, the absorption peak of the two microemulsion groups was 2-fold higher than that of free Cy5, which exhibited an enhancement of microemulsion on oral bioavailability. Logically, there was no obvious difference in the pharmacokinetic behavior between Cy5/Gal(oct)-C-MEs and Cy5/C-MEs, suggesting that modification of Gal(oct) did not influence the oral absorption. These results indicated that the combination strategy of multicomponent microemulsion design and Gal(oct) modification offered a potential for tumor targeting in vivo and improved the pharmacokinetic characteristics.
In vivo antitumor efficacy
Oral delivery of coix seed oil and coixan using Gal(oct)-C-MEs could lead to significant accumulation in the tumor sites through the synergistic strategy of Gal(oct) modification and microemulsion formulation. Generally, such a characteristic could also promote the in vivo anticancer efficacy and reduce the side effects. In this experiment, the antitumor activity of Gal(oct)-C-MEs was evaluated in BALB/c nude mice bearing HepG2 xenografts, with C-MEs, coix oil-MEs and Kanglaite used as controls.28,29 After intragastric administration of various microemulsions for 14 days, inhibition of tumor growth significantly increased compared with the saline-treated group and the coix oil-treated group.30 In this, Gal(oct)-C-MEs presented a remarkable enhancement of retardation of tumor growth in comparison with C-MEs (Figure 7A) and even had similar potential to intraperitoneally administrated Kanglaite. According to our previously reported formula,15 the inhibition rates of tumor growth by Gal(oct)-C-MEs and Kanglaite were both >74%, which achieved a further improvement in comparison with Gal(ste)-C-MEs. As a orally administrated microemulsion assembled by the bioactive components of coix seed, such a strong anticancer efficacy was obviously connected with Gal(oct) modification, multicomponent combination and microemulsion-based passive tumor targeting. In addition, the time from the end of therapy to reach double the volume of tumor tissues was also used to evaluate the effect after anticancer therapy. As shown in Figure 7B, the tumor of mice treated with C-MEs grew to 2-fold volume within 11 days, which was the longest among all the test groups. It suggests that C-MEs could sustain the effect of tumor growth delay even after ending the treatment. As expected, the weight of the tumor tissues harvested from mice treated with Gal(oct)-C-MEs was obviously lighter than in other treatments (Figure 7C), which was consistent with the curve of tumor growth. Furthermore, the survival time of mice after different treatments was also recorded. As depicted in Figure 7D, Gal(oct)-C-MEs–treated group exhibited the longest survival time in HepG2 tumor-bearing mice model among all the formulations (54 days), further offering a concrete proof of our hypothesis.
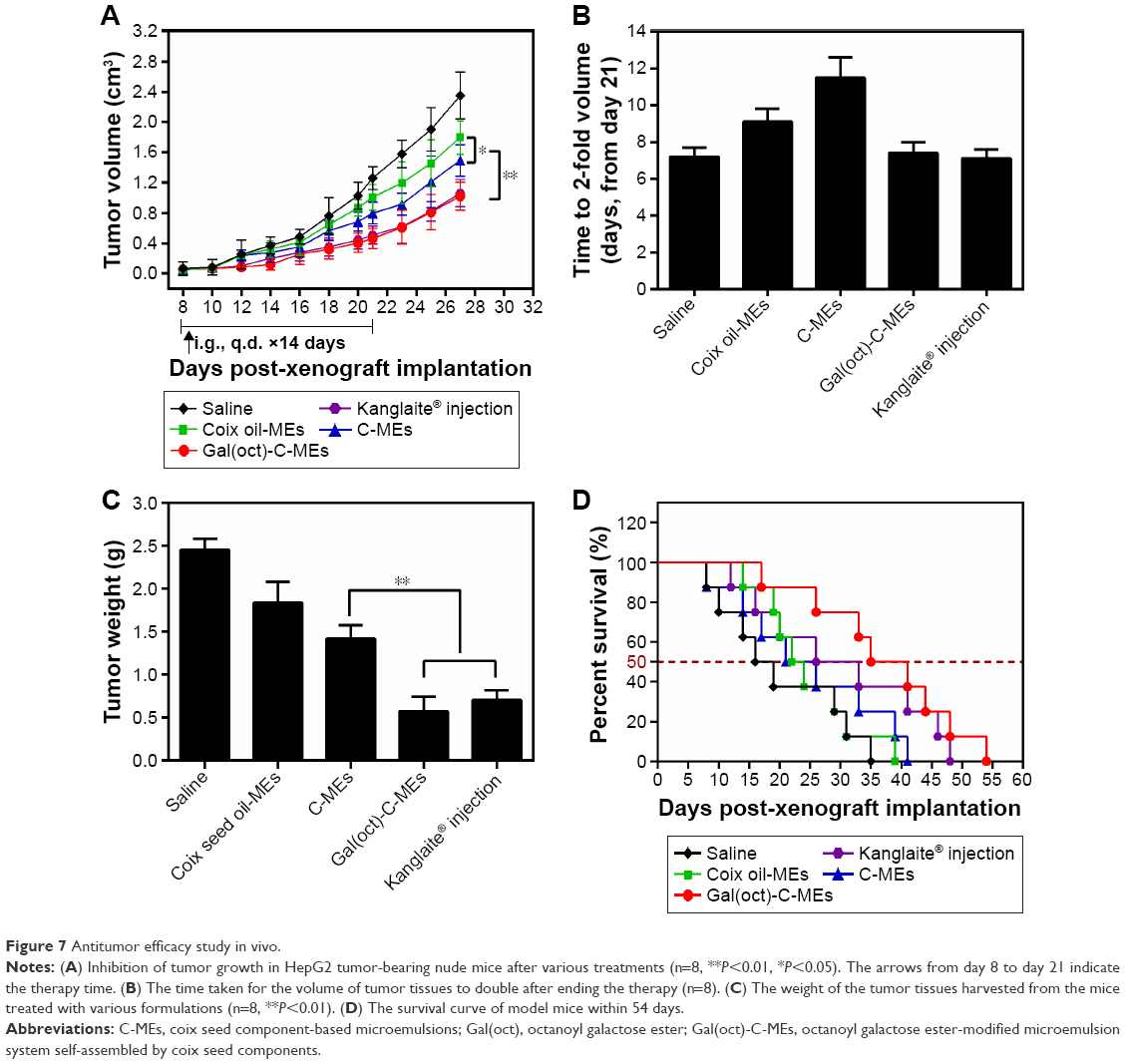 | Figure 7 Antitumor efficacy study in vivo. |
Safety evaluation
The main pharmacologically active components of Gal(oct)-C-MEs were extracted from coix seed, which did not exert antitumor effects powerfully like several other chemotherapeutics such as paclitaxol and doxorubicin. In order to evaluate the systemic toxicity against mice treated with Gal(oct)-C-MEs, various important indices of safety including body weight, liver/spleen index, the function of liver and kidney, pathology of normal tissues, hematology and preliminary immune function were thoroughly investigated during and after the anticancer treatment. As shown in Figure 8A, the body weight of mice did not significantly decrease after different therapies, indicating a low systemic side effect. The liver and spleen were collected at 72 h posttreatment to evaluate the influence of treatment on the important immune organs. The liver and spleen index showed no obvious difference among all the groups (Figure 8B), suggesting low toxicity of Gal(oct)-C-MEs against the immune organs. Acute damage of liver and kidney commonly occurred in the conventional chemotherapy.24,31 However, treatment with Gal(oct)-C-MEs did not bring about any alternation in the liver enzymes (alanine aminotransferase and aspartate aminotransferase) and acute renal lesion indicators (blood urea nitrogen; Figure 8C and D). The histopathology of kidney, liver and tumor tissues was evaluated through hematoxylin and eosin staining. Oral administration of Gal(oct)-C-MEs did not result in solid lesion or abnormality in the liver and kidney, but led to significant remission in the section of tumor tissues (Figure 8E).
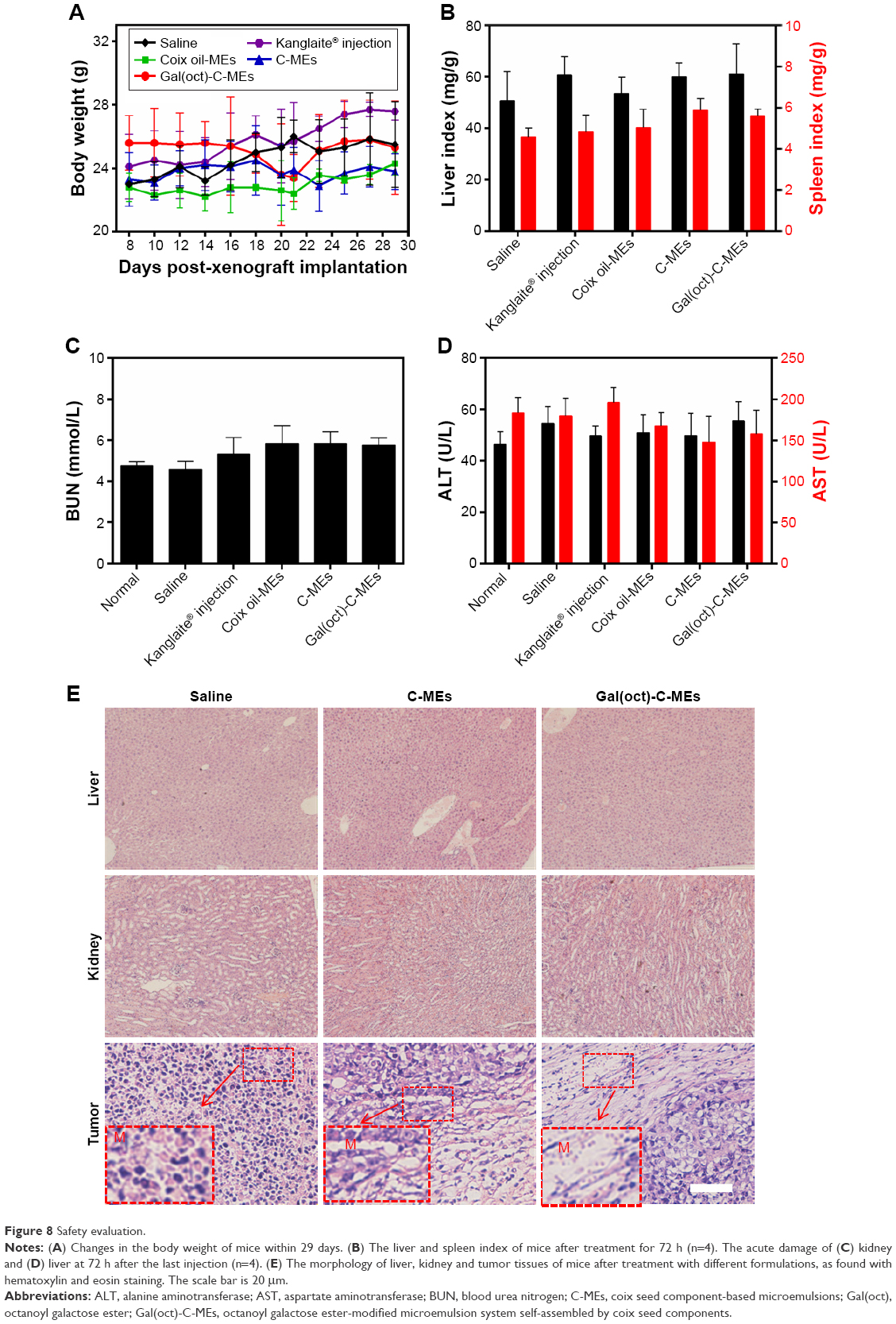 | Figure 8 Safety evaluation. |
In addition, the hematology of mice after therapy for 24 h was investigated through routine blood analysis. As shown in Table 1, Gal(oct)-C-MEs administration brought about a significant increase in white blood cells and platelets, offering a feasibility of combination therapy with chemotherapeutics.17,32,33 As for the influence on the immune function, the concentrations of cytokines were also detected. As shown in Table 2, treatment with microemulsion did not reduce the serum concentration of tumor necrosis factor-α and interleukin-6 significantly. All the data suggest that Gal(oct)-C-ME could be used as a safe nanosized DDS in vivo which acts by improved tumor targeting and as a highly effective anticancer vehicle through rational drug combination.
 | Table 1 Evaluation of WBC, RBC, HGB and PLT in the serum of mice treated with various formulations (n=5) |
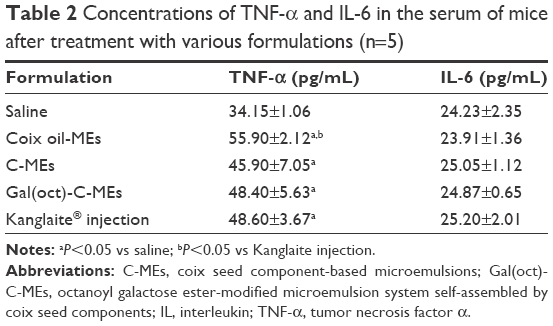 | Table 2 Concentrations of TNF-α and IL-6 in the serum of mice after treatment with various formulations (n=5) |
Conclusion
We developed a octanoyl galactose ester-modified coix seed multicomponent microemulsion system (Gal(oct)-C-MEs), which achieved significant enhancement of HepG2 cellular uptake, remarkable improvement in the cytotoxicity and obvious promotion of the induction of cell apoptosis. In addition, orally administrated Gal(oct)-C-MEs could prolong the intestinal retention, elevate the oral bioavailability and realize tumor-specific distribution. Importantly, Gal(oct)-C-MEs displayed an overwhelming antihepatic efficacy in the xenograft tumor mice models among all the test groups. These findings suggested that Gal(oct)-C-MEs could be used as an efficient oral delivery system for hepatoma therapy.
Acknowledgments
This work was carried out with the financial support of the National Natural Science Foundation (81373979 and 81503264), the key projects of Jiangsu Provincial Administration of Traditional Chinese Medicine (ZD201509), and the Jiangsu Provincial Natural Science Foundation (BK20141038 and BK 20141058).
Disclosure
The authors report no conflicts of interest in this work.
References
Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61(2):69–90. | ||
Mittal S, El-Serag HB. Epidemiology of hepatocellular carcinoma: consider the population. J Clin Gastroenterol. 2013;47 Suppl:S2–S6. | ||
Shah SM, Goel PN, Jain AS, et al. Liposomes for targeting hepatocellular carcinoma: use of conjugated arabinogalactan as targeting ligand. Int J Pharm. 2014;477(1–2):128–139. | ||
Wang J, Zhang Z, Wang X, Wu W, Jiang X. Size- and pathotropism-driven targeting and washout-resistant effects of boronic acid-rich proteinnanoparticles for liver cancer regression. J Control Release. 2013;168(1):1–9. | ||
Mo R, Jiang T, Gu Z. Recent progress in multidrug delivery to cancer cells by liposomes. Nanomedicine (Lond). 2014;9(8):1117–1120. | ||
Deshpande PP, Biswas S, Torchilin VP. Current trends in the use of liposomes for tumor targeting. Nanomedicine (Lond). 2013;8(9):1509–1528. | ||
Krais A, Wortmann L, Hermanns L, et al. Targeted uptake of folic acid-functionalized iron oxide nanoparticles by ovarian cancer cells in the presence but not in the absence of serum. Nanomedicine. 2014;10(7):1421–1431. | ||
Song S, Chen F, Qi H, et al. Multifunctional tumor-targeting nanocarriers based on hyaluronic acid-mediated and pH-sensitive properties for efficient delivery of docetaxel. Pharm Res. 2014;31(4):1032–1045. | ||
Zeng X, Tao W, Mei L, Huang L, Tan C, Feng SS. Cholic acid-functionalized nanoparticles of star-shaped PLGA-vitamin E TPGS copolymer for docetaxel delivery to cervical cancer. Biomaterials. 2013;34(25):6058–6067. | ||
Ding J, Xiao C, Li Y, et al. Efficacious hepatoma-targeted nanomedicine self-assembled from galactopeptide and doxorubicin driven by two-stage physical interactions. J Control Release. 2013;169(3):193–203. | ||
Chen W, Zou Y, Meng F, et al. Glyco-nanoparticles with sheddable saccharide shells: a unique and potent platform for hepatoma-targeting delivery of anticancer drugs. Biomacromolecules. 2014;15(3):900–907. | ||
Yu CY, Wang YM, Li NM, et al. In vitro and in vivo evaluation of pectin-based nanoparticles for hepatocellular carcinoma drug chemotherapy. Mol Pharm. 2014;11(2):638–644. | ||
Zhu D, Tao W, Zhang H, et al. Docetaxel (DTX)-loaded polydopamine-modified TPGS-PLA nanoparticles as a targeted drug delivery system for the treatment of liver cancer. Acta Biomater. 2016;30:144–154. | ||
Wu DQ, Lu B, Chang C, et al. Galactosylated fluorescent labeled micelles as a liver targeting drug carrier. Biomaterials. 2009;30(7):1363–1371. | ||
Qu D, Sun W, Liu M, Liu Y, Zhou J, Chen Y. Bitargeted microemulsions based on coix seed ingredients for enhanced hepatic tumor delivery and synergistic therapy. Int J Pharm. 2016;503(1–2):90–101. | ||
Liu MJ, Qu D, Chen Y, Liu CY, Liu YP, Ding XF. Preparation of novel butyryl galactose ester-modified coix component microemulsions and evaluation on hepatoma-targeting in vitro and in vivo. Drug Deliv. 2016;23(9):3444–3451. | ||
Qu D, Ma Y, Sun W, et al. Microemulsion-based synergistic dual-drug co-delivery system for enhanced apoptosis of tumor cells. Int J Nanomed. 2015;10:1173–1187. | ||
Qu D, Lin H, Zhang C, Xue J, Zhang C. In vitro evaluation on novel modified chitosan for targeted antitumor drug delivery. Carbohydr Polym. 2013;92(1):545–554. | ||
Lee MY, Dordick JS. Enzyme activation for nonaqueous media. Curr Opin Biotechnol. 2002;13(4):376–384. | ||
De Luca MA, Lai F, Corrias F, et al. Lactoferrin- and antitransferrin-modified liposomes for brain targeting of the NK3 receptor agonist senktide: preparation and in vivo evaluation. Int J Pharm. 2015;479(1):129–137. | ||
Mou Q, Ma Y, Zhu X, Yan D. A small molecule nanodrug consisting of amphiphilic targeting ligand-chemotherapy drug conjugate for targeted cancer therapy. J Control Release. 2016;230:33–34. | ||
Su Z, Xing L, Chen Y, et al. Lactoferrin-modified poly(ethylene glycol)-grafted BSA nanoparticles as dual-targeting carrier for treating brain gliomas. Mol Pharm. 2014;11(6):1823–1834. | ||
Mo R, Jiang T, DiSanto R, Tai W, Gu Z. ATP-triggered anticancer drug delivery. Nat Commun. 2014;5:3364. | ||
Huang X, Li L, Liu T, et al. The shape effect of mesoporous silica nanoparticles on biodistribution, clearance, and biocompatibility in vivo. ACS Nano. 2011;5(7):5390–5399. | ||
Chen G, Jaskula-Sztul R, Harrison A, et al. KE108-conjugated unimolecular micelles loaded with a novel HDAC inhibitor thailandepsin-A for targeted neuroendocrine cancer therapy. Biomaterials. 2016;97:22–33. | ||
Saurabh S, Perez AM, Comerci CJ, et al. Super-resolution imaging of live bacteria cells using a genetically directed, highly photostable fluoromodule. J Am Chem Soc. 2016;138(33):10398–10401. | ||
Pridgen EM, Alexis F, Kuo TT, et al. Transepithelial transport of Fc-targeted nanoparticles by the neonatal fc receptor for oral delivery. Sci Transl Med. 2013;5(213):213ra167. | ||
Wang Y, Dou L, He H, Zhang Y, Shen Q. Multifunctional nanoparticles as nano-carrier for vincristine sulfate delivery to overcome tumor multidrug resistanc. Mol Pharm. 2014;11(3):885–894. | ||
Ju C, Mo R, Xue J, et al. Sequential intra-intercellular nanoparticle delivery system for deep tumor penetration. Angew Chem Int Ed Engl. 2014;53(24):6253–6258. | ||
Qu D, He J, Liu C, Zhou J, Chen Y. Triterpene-loaded microemulsion using Coix lacryma-jobi seed extract as oil phase for enhanced antitumor efficacy: preparation and in vivo evaluation. Int J Nanomed. 2014;9:109–119. | ||
Charoensuk L, Pinlaor P, Wanichwecharungruang S, et al. Nanoencapsulated curcumin and praziquantel treatment reduces periductal fibrosis and attenuates bile canalicular abnormalities in Opisthorchis viverrini-infected hamsters. Nanomedicine. 2016;12(1):21–32. | ||
Zhou M, Zhang X, Yu C, Nan X, Chen X, Zhang X. Shape regulated anticancer activities and systematic toxicities of drug nanocrystals in vivo. Nanomedicine. 2016;12(1):181–189. | ||
Kretzer IF, Maria DA, Guido MC, Contente TC, Maranhão RC. Simvastatin increases the antineoplastic actions of paclitaxel carried in lipid nanoemulsions in melanoma-bearing mice. Int J Nanomedicine. 2016;11:885–904. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.