Back to Journals » Therapeutics and Clinical Risk Management » Volume 17
Ocrelizumab for the Treatment of Multiple Sclerosis: Safety, Efficacy, and Pharmacology
Authors Mancinelli CR, De Rossi N, Capra R
Received 6 April 2021
Accepted for publication 18 July 2021
Published 30 July 2021 Volume 2021:17 Pages 765—776
DOI https://doi.org/10.2147/TCRM.S282390
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Editor who approved publication: Professor Garry Walsh
Chiara Rosa Mancinelli, Nicola De Rossi, Ruggero Capra
Multiple Sclerosis Centre, Spedali Civili di Brescia, Brescia, Italy
Correspondence: Chiara Rosa Mancinelli
Multiple Sclerosis Centre - Spedali Civili di Brescia, Via Ciotti, 154, Montichiari (BS), Brescia, 25018, Italy
Tel +39 0309963474
Fax +39 0309963475
Email [email protected]
Abstract: The success of selective B-cells depleting therapies, as the anti-CD20 antibodies, in patients with multiple sclerosis (MS) has confirmed that B-cells are critical in the immune pathogenesis of the disease. Ocrelizumab, a humanized monoclonal antibody that selectively targets CD20+ B-cells, profoundly suppresses acute inflammatory disease activity, representing a highly effective therapy for relapsing-remitting multiple sclerosis (RRMS). It is also the first proven therapy able to slow disability progression in primary progressive multiple sclerosis (PPMS), particularly in patients with signs of acute radiological activity before being enrolled. Effectiveness has widely been demonstrated in randomized clinical trials (RCTs), and recently confirmed in open-label extension trials. Here, we review the role of B-cells in MS, the mechanism of action of ocrelizumab, its pharmacokinetics and pharmacodynamics, and the clinical data supporting its use, as well as safety data. We focus on issues related to the maintenance of immunocompetence, essential to ensure an immune response to either a primary infection or a vaccination. Lastly, we discuss about the possible role of ocrelizumab as an exit strategy from natalizumab-treated patients at risk of developing multifocal progressive leukoencephalopathy. In view of using ocrelizumab chronically, collecting long-term safety data and finding strategies to minimize adverse events will be extremely relevant.
Keywords: ocrelizumab, anti-CD20 therapies, relapsing-remitting multiple sclerosis, primary progressive multiple sclerosis
Introduction
Multiple Sclerosis (MS) is a chronic autoimmune and neurodegenerative disease of the central nervous system (CNS) that typically affects young adults, with an onset between 20 and 40 years of age in most cases. Most patients have a relapsing form characterized by episodes of neurological deficits corresponding to an acute demyelination process (relapsing-remitting multiple sclerosis, RRMS), but over time a progressive course could develop with accumulation of clinical disability (secondary progressive multiple sclerosis, SPMS). Approximately 15% of patients have a progressive disease from onset (primary progressive multiple sclerosis, PPMS).
The pathological hallmark of disease in the acute phase is the accumulation of focal demyelinating lesions in the white and grey matter of the brain and spinal cord. Active inflammatory lesions are sustained by macrophages, reactive astrocytes, and an infiltrate of T and B lymphocytes, which enter the CNS and are histologically characterized by the predominance of CD8+ T-cells, B-lymphocytes, and plasma cells, whereas CD4+ T-cells are relatively scarce.1,2 This lymphocytic infiltrate is associated with profound blood–brain barrier damage, while in the progressive stage, inflammation becomes more compartmentalized within the leptomeninges and the CNS parenchyma.3 Chronically activated microglia and macrophages, as well as B-cells aggregated in meninges, could sustain this compartmentalized inflammation4,5 and maintain the chronic injury in MS.
Although MS was traditionally considered a T-cell-mediated autoimmune disease, during the last years, evidence on the implication of B-cells in MS pathophysiology has accumulated both in the early stage and with the progression of the disease. B-cells have now emerged as an important target for several established MS therapies, including interferon-β (IFN-β), fingolimod, and cladribine. A more selective depletion of B-cells is obtained with anti-CD20 monoclonal antibodies (mAbs) such as rituximab, ocrelizumab, and ofatumumab.6 These drugs deplete circulating B-cells by targeting the CD20 antigen, a surface marker expressed throughout the majority of the B-cell lineage, including pre-B-cells and mature B-cells, and results in very effective treatment for MS. The exact mechanism by which B-cell depletion is effective in treating MS remains unproven. Antibody production is one of the important B-cells functions, however B-cells are also antigen-presenting cells (APCs) and have immunomodulator activity through cytokine secretion.
Rituximab, a chimeric anti-CD20 mAb, whose effectiveness was demonstrated in two randomized placebo-controlled Phase II trials (HERMES in RRMS and OLYMPUS in PPMS, respectively)7,8 and in several observational studies, can now be administrated only off-label in MS patients. Ofatumumab, a fully human monoclonal antibody, has been evaluated in phase II and III trials,9–11 and just approved by the Food and Drug Administration (FDA) and by the European Medicines Agency (EMA).
In 2017, ocrelizumab, a recombinant humanized anti-CD20 mAb, was approved for relapsing MS (RMS) and was the first ever approved therapy for PPMS, based on its ability to drastically reduce inflammatory disease activity and to slow disability progression in Phase III randomized controlled trials (RCTs).12,13
In this review we analyze the efficacy, safety, pharmacokinetics, and pharmacodynamics of ocrelizumab, summarizing relevant adverse events and proposing a strategy to minimize them.
B-Cells in MS: Implications for Clinical Use of Anti-CD20 mAbs
The success of B-cell-depleting therapies in MS has highlighted the important role that B lymphocytes have in the pathogenesis of the disease. The evidence of the involvement of B-cells in MS has steadily increased through time and several examples are here presented. Despite the experimental murine model of MS requirs only the involvement of activated myelin-specific T-cells, in other species, such as the marmoset, B-cells depletion prevents the development of clinical and pathological signs of experimental autoimmune encephalomyelitis (EAE).14,15 Anatomopathological studies have also shown that B-cells are prominent cell populations together with CD8+ T-cells in active MS lesions,2 with B-lymphocytes accumulating in perivascular lesions and within the subarachnoid space of leptomeninges rather than in the CNS parenchyma.2,16,17 Interestingly, B cell-enriched ectopic lymphoid follicles, particularly evident in patients with Secondary Progressive MS (SPMS), can be found in the proximity of cortical brain lesions and may contribute to the propagation of subpial cortical injury.17–19
A deficit of tolerance in autoreactive B-cells is found in MS such as in many autoimmune diseases. Two major checkpoints, along the B-cell development pathway, contribute to the normal elimination of autoreactive B-cells in healthy humans: central tolerance in the bone marrow and peripheral tolerance in secondary lymphoid organs.20 While in Neuromyelitis Optica Spectrum Disorders (NMOSD),21 systemic lupus erythematosus,22 and rheumatoid arthritis,23 central and peripheral tolerance are both involved, in MS only the peripheral checkpoint seems to be dysregulated by deficit of the regulatory T-cells (Treg) function.24 Indeed, in MS some individuals treated with selective B-cell depletion experience durable quiescence of disease activity, even as their B-cells (mainly naive) reemerge, thus suggesting that a form of tolerance may have been achieved.
Accumulating evidence has highlighted the importance of the antigen-presenting function of B-cells for T-cells activation in MS. This process can occur in the periphery, in fact antigen-experienced B-cells are clearly present in the CNS, but maturation and activation seem to occur in the deep cervical lymph nodes.25,26 Jelcic et al27 have provided a model for how B- and T-cell interactions, outside the CNS, might generate and maintain autoreactive T-cells able to enter the brain. Proliferation of these brain-homing autoreactive CD4+ T-cells, that occurred without any cell stimuli in patients who express the HLA-DR15 haplotype, the stronger genetic risk factor for MS, was sustained by the presence of B lymphocytes. These were substantially memory B-cells and could activate autoreactive CD4+ T-cells that recognize antigens expressed in B-cells and in brain lesions. The elimination of B-cells via anti-CD20 therapies inhibited T-cell activation and proliferation, providing further evidence that interruption of the crosstalk between T- and B-cells could be clinically relevant.27 Interesting scenarios are opening up on the nature of the autoantigen presented by B-cells to autoreactive T-cells and on the underlying mechanism of antigen presentation, finding links with the most important genetic and environmental risk factors for MS,28,29 already previously investigated.30
Anti-CD20 mAbs have been shown to also deplete a subset of T-cells that express CD20 at a low level.31 Despite CD20+, T lymphocytes represent a small percentage of total circulating T-cells, approximately 6%, they have a highly pro-inflammatory activity and a fast kinetic of proliferation in MS patients, suggesting another possible mechanism of action of anti-CD20-depleting therapies.32 Moreover, beyond B-cell depletion, the involvement of T-cells is emerging specifically in ocrelizumab-treated patients.33,34
In summary, these findings may explain why B-cell-depleting agents are so effective in controlling clinical relapses and focal inflammation in RMS, but the mechanisms underlying the benefits on PPMS are less clear and investigated. Of note, in the ORATORIO trial, a minority of patients with PPMS, especially younger and closer to disease onset, have signs of acute radiological activity before being enrolled,13 suggesting an action of the drug on relapse biology that may have occurred subclinically in some of these patients.35 However, it remains difficult to explain how B- cells’ accumulation as part of CNS-compartmentalized inflammation, may be limited by therapies as anti-CD20 antibodies that do not cross the blood–brain barrier (BBB).36
Pharmacodynamics and Pharmacokinetics
Anti-CD20 mAbs can rapidly and profoundly deplete circulating B-cells through three main different mechanisms: cell-mediated antibody-dependent cellular cytotoxicity (ADCC), complement-dependent cytotoxicity (CDC), and antibody-triggered apoptosis. The physiological ligand and the precise function of CD20 are currently unknown; CD20 is a transmembrane protein involved in B-cell activation and proliferation thought to function as a calcium channel.37 CD20 is a B-cell marker expressed during cell differentiation by late pre-B, naïve and memory B-cells. B-cells that do not express CD20 – pro-B-cells, late plasma blasts, and terminally differentiated antibody-producing plasma cells – are spared from this selective depletion. B-cells precursor on hematopoietic bone marrow are continuously repopulated, enabling peripheral reconstitution after depletion, while antibody-producing plasma cells guarantee antibodies production and preexisting humoral immunity, at least, in the short-term.38
The understanding about the kinetics of depletion and reconstitution of B-cells after anti-CD20 mAbs has come almost exclusively from phenotyping analysis of circulation B-cells subsets. The proportion of circulating B lymphocytes (2% of the entire population) is almost totally depleted of the expose to the anti-CD20 antibodies where they persist in the bone marrow and in various secondary lymphoid organs. There are limited data on the degree of depletion of B-cells in secondary lymphoid organs and these derive mainly from preclinical studies and observations on humans with other autoimmune diseases treated with rituximab. In murine MS models, B-cell-rich compartments, such as bone marrow, spleen, and lymph nodes, are depleted to varying degrees, though CD20 cells expressing the maturation marker CD27 remain detectable in the spleen.39 In patients with rheumatoid arthritis, switched memory B-cells (CD27+ IgD-) persisted in lymph nodes biopsies after rituximab treatment,40,41 suggesting that memory B-cells can escape depletion in peripheral organs more than in blood.
The very small numbers of circulating B cells that can be detected during periods of depletion usually show a phenotype of plasma cell precursor but memory cells have also been reported.38,42 Following depletion, B reconstitution starts from naïve and immature B-cells, while memory B-cells and plasma blasts show a slow and delayed repopulation.43–45
Ocrelizumab (Ocrevus®), an intravenously administered glycosylated IgG1, is a recombinant, humanized anti-CD20 mAb, that targets the extracellular loop of the CD20 epitope on B-cells, a different but overlapping binding site than that of rituximab.46 Due to its mostly human origin, ocrelizumab is expected to be less immunogenic compared with rituximab, induces fewer anti-drug antibodies, as well as milder infusion reactions. In a rituximab phase II trial,7 a higher proportion of anti-drug antibodies was found compared to the ocrelizumab pivotal trial,12 suggesting a greater immunogenicity of rituximab. However, caution is warranted considering that different assay methods were used between studies and the association with the development of anti-drug antibodies and infusion reactions are not completely demonstrated. Furthermore, compared with rituximab, the mechanism of action of ocrelizumab in more dependent on ADCC and less on CDC.47
The initial dose of ocrelizumab is administrated as two separate 300 mg infusions 2 weeks apart; subsequent doses are administrated as a single 600 mg infusion every 24 weeks. The half-life is 26 days and the pharmacokinetic parameters are typical for an IgG1 monoclonal antibody.48 Because there is a substantial overlap of CD20 and CD19 expression from pre-B to memory B lymphocytes, CD19 cell counts are used to evaluate B-cells depletion and repopulation.49 Ocrelizumab administration leads to a rapid and complete depletion of CD19 peripheral cells by 2 weeks,12 which was sustained over time. Slow repopulation began about 6 months after the last infusion with a median-repletion of over 15 months.50
The scheduled dose of ocrelizumab is fixed, but ongoing trials are investigating if a higher dose of ocrelizumab every 24 weeks could be more effective than the approved 600 mg dose. Post-hoc analysis of the pharmacodynamics and pharmacokinetics of ocrelizumab in the Phase II–III clinical trials suggested that higher exposure to ocrelizumab is associated with greater immune B-cell depletion in the blood and lower risk of disability progression. No increase of adverse side-effects has been reported.51
Ocrelizumab for Relapsing–Remitting Multiple Sclerosis: Efficacy Data
Two identically designed Phase III, randomized, controlled trials (OPERA I and OPERA II) enrolling 821 and 835 adults with RMS, compared, in a 1:1 ratio, ocrelizumab (600 mg every 24 weeks) with subcutaneous IFNβ-1a (IFNβ-1a; 44 µg 3-times weekly) for 96 weeks.12 The primary endpoint, ie, annualized relapse rate (ARR) at 96 weeks, and other ten secondary endpoints were analyzed specifically for OPERA I and II; a pooled data analysis was obtained to evaluate disability progression at 12 and 24 weeks. Both studies showed a significant decrease in the ARR in patients treated with ocrelizumab compared to IFNβ-1a at 96 weeks (0.16 vs 0.29; p<0.001), that is a 46% lower relapse rate in OPERA I and 47% in OPERA II. Regarding the radiological endpoints, the mean number of gadolinium-enhancing lesions was reduced by 94% and 95%, the number of new and/or enlarging T2 lesions was reduced by 77% and 83%, and rate of brain volume loss was reduced by 22.8% and 14.9% in OPERA I and OPERA II, respectively. The proportion of subjects with confirmed disability progression was lower with ocrelizumab than with IFNβ-1a at both 12 (9.1 vs 13.6%; p<0.001) and 24 weeks (6.9 vs 10.5% p=0.003). Recently, the results of the open-label extension phase of the pooled OPERA I and II studies showed that early initiation and continuous treatment with ocrelizumab for up to 5 years was associated with a reduction of confirmed disability progression compared to switching to ocrelizumab after 2 years of IFNβ-1a. Furthermore, patients switching from IFNβ-1a to ocrelizumab had a near complete suppression of MRI disease activity (gadolinium-enhanced and new/enlarging T2 lesions) from year 3 to year 5.52
Ocrelizumab for Primary Progressive Multiple Sclerosis: Efficacy Data
The double-blind, placebo-controlled phase III ORATORIO trial, randomly assigned 732 adult patients with PPMS to receive 600 mg of ocrelizumab or placebo every 24 weeks for at least 120 weeks, in a 2:1 ratio.13 Patients receiving ocrelizumab had lower 12-week confirmed disability progression (32.9% vs 39.3%; p=0.03) and 24-week confirmed disability progression (29.6% vs 35.7%, p=0.04), compared to placebo. Ocrelizumab also reduced walking impairment vs placebo, as assessed by the mean percent change from baseline in the timed 25-foot walk test (T25FW) at week 120 (mean decline of 38.9% vs 55.1% with placebo; p=0.04) and led to significant improvements on other radiological endpoints, including change in MRI T2 lesion volume and whole brain volume loss. More in detail, the total volume of T2 lesions decreased in patients treated with ocrelizumab and increased in placebo recipients at week 120 (−3.4% vs 7.4%; p<0.001) and in the ocrelizumab group, brain volume loss from week 24 to 120 was smaller than in the placebo group (−0.90 vs −1.09%; p=0.02). Based on these results, ocrelizumab was the first DMT approved for PPMS.
The post hoc analysis of the open-label extension of the ORATORIO trial showed sustained benefits on measures of disease progression over a 6.5 years follow-up period. The proportion of patients with progression was lower in those who started ocrelizumab early compared to those who started, in the double-blind period, placebo in most measures of 24-weeks confirmed disability progression.53
Safety and Tolerability Data
Infusion-Related Reactions
Infusion-related reactions (IRRs) occur commonly with ocrelizumab during or within a few hours of infusions, although symptoms may be delayed for up to 24 hours. They are due to a type 2 hypersensitive reaction sustained by cytokine release and are reduced with appropriate premedication (ie, with intravenous glucocorticoids). In clinical trials, IRRs were the most frequently reported adverse events (AEs), which occurred in 34.3% and 39.9% of ocrelizumab-treated patients in the pooled analysis of OPERA and ORATORIO trials, respectively.54 The frequency of IRRs was highest with the first dose and decreased with subsequent dosing,54 supporting for a shorter infusion period than the conventional 3.5-hour infusion, after the first dose. Based on the results of the randomized, double-blind ENSAMBLE PLUS study,55 the Food and Drugs Administration (FDA) and the European Medicines Agency (EMA) approved a shorter two-hour infusion time for ocrelizumab in patients with relapsing and progressive MS who have not experienced any previous serious IRRs.
Infections and Laboratory Findings
In the phase III clinical trials, the percentage of ocrelizumab-treated patients reporting any infection was 59.9% (vs 54.3% in IFNβ-1a) in OPERA I, 60.2% (vs 52.5% in IFNβ-1a) in OPERA II, and 71.4% (vs 69.9% in placebo) in ORATORIO. The most frequent infections observed in the ocrelizumab group were nasopharyngitis, upper respiratory tract, and urinary tract infections.12,13 In large open-label extension trials, infective adverse events were consistent with the past reports of the double-blind phases52,53 and, to date, no particular new safety signals emerged regarding infections. Although, compared to IFNβ and placebo, ocrelizumab treatment is associated with a slight increase in the risk of infections, they are generally mild-to-moderate. In fact, the rate of serious infection remains low over 7 years of treatment and in line with the rate of infection-related hospitalizations reported in real-world MS cohorts.56 Also, opportunistic infections are rare and the rates remain stable year-on-year with the rate observed during phase III studies.56 The occurrence of invasive fungal infections was in line with the occurrence reported among patients treated with other monoclonal antibodies and fingolimod.57 The risk of Progressive Multifocal Leukoencephalopathy (PML) will be discussed in the following sections.
Hepatitis B virus reactivation is a possible complication in ocrelizumab-treated patients who are positive for HBsAg or in those who are negative for HBsAg but positive for hepatitis B core antibody (HBcAb+).12,13,58 Data referring to hepatitis C reactivation are more limited56 and mainly concern patients treated with rituximab for rheumatoid arthritis.59 Anyway, prior to starting ocrelizumab, screening for hepatitis B is mandatory, while screening for hepatitis C should be taken into account. Patients with evidence of infection should consult liver disease experts before treatment and be managed in accordance with current practice guidelines.60,61
Late-onset neutropenia (LON) is a rare complication of anti-CD20 therapies that occurs after 4 weeks following the last dose in less than 6.5% of patients with rheumatoid arthritis receiving rituximab, as observed in various studies.62,63 Most patients with severe neutropenia recover quickly, and there is no evidence that the condition worsens with further treatment cycles.64 The cause of LON has been poorly investigated and many theories have been formulated; the most credited hypotheses is that homeostasis of granulocytes may be disturbed by chemokine stromal-derived factor-1 interacting with B-lymphocyte recovery.65 In the PPMS clinical trial with ocrelizumab, decreased neutrophil counts were transiently found in 13% of patients compared to 10% in placebo. Only 1% of patients displayed neutrophil counts less than 1.0 x 109/L, and none of these cases was associated with infections.66 Association with serious infections leading to significant adverse outcomes and the necessity of treatment with granulocyte colony-stimulating factor (G-CSF) are debated.67
While not expressing the CD20 antigen, the plasma cells population is based on the evolution of the B lymphocyte of the germinal centers and repeated cycles of treatment can lead to reduced plasma cell production, which might result in a reduction of immunoglobulin levels. Rituximab use can be complicated by decreases in IgG, IgM, or IgA levels, a condition known as hypogammaglobulinemia, that has been reported in people receiving long-term rituximab treatment.64 The true frequency of hypogammaglobulinemia is unknown and the clinical significance, ie, the possible association with recurrent or serious infections remains controversial.68 In OPERA I and II the proportion of ocrelizumab-treated patients with immunoglobulin levels below the normal limits at week 96 were 1.5% for IgG, 2.4% for IgA, and 16.5% for IgM.12 In the ORATORIO trial only IgM levels differed between groups; treatment with ocrelizumab resulted in a higher proportion of patients with IgG levels below the lower limit (15.5% for ocrelizumab vs 1.2% for placebo) at week 120.13 Over 5.5 years of ocrelizumab treatment, a reduction in immunoglobulin levels and an apparent association with increased rates of serious infections, especially for IgG deficits, were observed.69
Estimation of infectious risk in ocrelizumab-treated patients is challenging. Generally, DMTs impact on the immune response and may carry an increased probability of infections; data from RCTs70 and large observational studies have been consistent with an increase of serious infections in patients taking natalizumab and rituximab.71,72 As for any other DMTs, it would be desirable to perform a baseline evaluation of infectious disease history and of the vaccinal immunization status. A strict monitoring to detect infections during treatment is just as important to consider.73 A screening before starting ocrelizumab is reported in Table 1. Post-marketing-surveillance initiatives and pharmacovigilance studies will be essential to determinate the safety of the use of ocrelizumab.
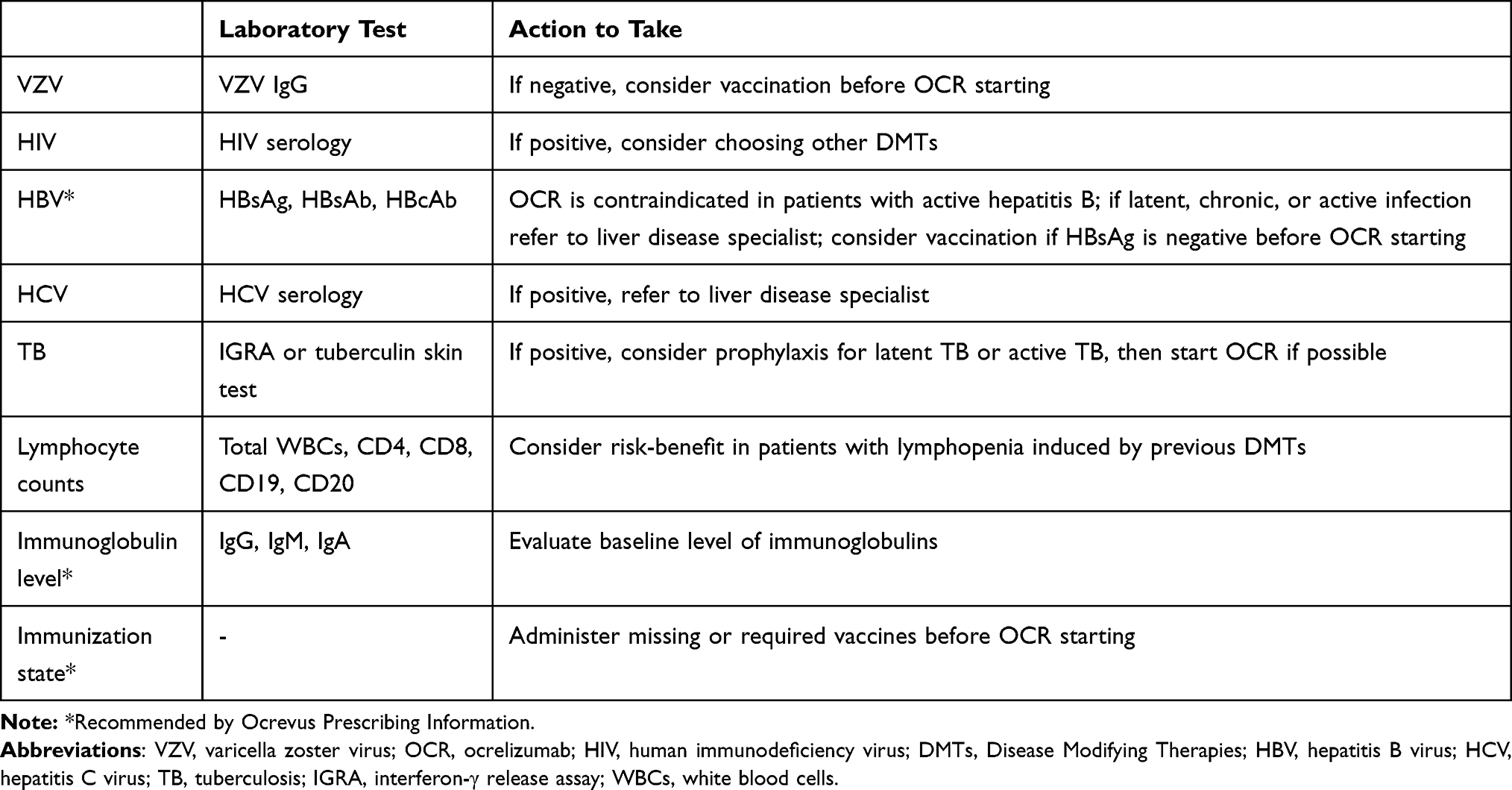 |
Table 1 Suggested Screening Before Ocrelizumab Starting |
The COVID-19 pandemic has raised many questions about risks of infection in patients with MS and treated with immunotherapies. In non-MS patients who have recovered from COVID-19, CD4+ T-cells, CD8+ T-cells, and neutralizing antibodies all contribute, with different roles, to control the primary SARS-CoV-2 infection,74 as well as the innate immune response.75 Anti-viral neutralizing antibodies levels decrease over time76,77 and could not be essential to recover from the disease, such as shown in two adults with X-linked agammaglobulinemia that fully recovered from COVID-19 despite the absence of B lymphocytes.78 On the contrary, the development of SARS-CoV-2-specific T and B memory cells may confer durable protective immunity against the virus.79,80 In view of this immunological knowledge, the possible impact of MS therapies on the SARS-CoV-2 infection should be evaluated.
Giving the emerging data, MS patients appear to respond to SARS-CoV-2 in a similar way to non-MS patients; in fact the risk of severe COVID-19 disease increases with older age, greater disability, and comorbidity such as cardiovascular disease and obesity.81–83 DMTs have been shown to be generally safe, while concerns regarding anti-CD20 therapies have emerged. Indeed, as reported by an Italian MS Registry, anti-CD20 mAbs (rituximab and ocrelizumab) may increase the risk of severe COVID-19, defined as infection requiring hospitalization, intensive care, or death.82 However, this does not seem to be confirmed in two other multicenter retrospective studies,81,83 and further evidence collecting more data is required. Recently, a study investigating the characteristic of COVID-19 severity in the largest cohort of people with MS and COVID-19 available showed that use of rituximab and ocrelizumab is associated with increased risk of hospitalization, Intensive Care Unit (ICU) admission, and for rituximab also risk for requiring artificial ventilation.84 In the context of the ongoing pandemic, the evaluation of the risk-vs-benefit of the use of ocrelizumab should be evaluated case by case.
Malignancies
Among patients treated with ocrelizumab in the phase III RCTs, 15 patients with neoplasms were identified (1.1%), compared to four cases in the placebo or IFN-β arms (0.38%). Six cases of breast cancer were diagnosed in women who received ocrelizumab, while no such cases were observed in the comparator group.12,13 Updated data from ongoing ocrelizumab clinical trials shows that rates of all malignancies, including breast cancer, remain stable over time and within the range reported in epidemiological studies.56 Furthermore, long-term safety studies have not revealed an increased risk of malignancy in patients treated with rituximab than in patients treated with other DMTs, either in MS or in rheumatoid arthritis.64,85 Although these data are encouraging, it is important to continue monitoring the long-term safety of ocrelizumab to identify potential cumulative risks associated with prolonged and repeated peripheral blood B-cell depletion, such as immunosuppression-associated malignancy.
PML Risk and Possible Use in JCV-Positive Patients
DMTs for MS are associated with various degrees of risk of PML, a rare opportunistic infection caused by the John Cunningham virus (JCV). While natalizumab is associated with the highest overall incidence, lesser degrees of risk exist for other drugs.86 As of December 2020, ten confirmed cases of PML were reported in MS patients treated with ocrelizumab, but nine of these cases are carry-over PML from previous treatment with either natalizumab (n=8) or fingolimod (n=1).87 An additional two case reports, which occurred 78 and 97 days after natalizumab withdrawal, were recently described by Toorop et al.88 Therefore, to date only one PML case was considered to be directly associated with ocrelizumab. This case occurred in a 78-year-old patient treated with ocrelizumab for approximately 2 years, without history of a previous DMT, but notably with lymphopenia (Grade 1 before treatment and Grade 2 during treatment).87,89 The occurrence of PML is very rare among rituximab-treated patients with autoimmune systemic disorders, despite the increasing use of this therapy.90
Because the risk of PML related to anti-CD20 therapies is much lower than natalizumab, the possibility of using ocrelizumab as an effective exit strategy from natalizumab is under investigation. In patients at high risk, even though discontinuation of natalizumab can lead to MS disease recurrence, the suspension of treatment and the shifting to other highly efficacious DMTs represents a possible strategy to limit the occurrence of PML. Recently, results from a study on 42 JCV-positive patients suggests that switching to ocrelizumab could be considered a possible choice to mitigate the risk of MS reactivation in patients who were previously treated with natalizumab and at risk of PML.91 Other studies, where switching to ocrelizumab was analyzed in 28, 39, and 64 natalizumab-treated patients, respectively,92–94 come to the same conclusions. Overall, in these observations, no case of carry-over PML has been described. The small sample size and the short follow-up warrant cautious interpretation of these data; a clinical trial is currently ongoing, evaluating the efficacy and safety of switching patients from natalizumab to ocrelizumab, with estimated completion in June 2022 (Clinicaltrial.gov Identifier: NCT03157830).
Another issue that must be considered is the washout time interval for the switch, that should be as short as possible to reduce disease reactivation,95 but sufficient to exclude a carry-over PML. In fact, a possible diagnosis of asymptomatic PML should be ruled out in patients who discontinue natalizumab before starting ocrelizumab. Because it is now known that PML lesion volume is related with viral load and smaller natalizumab-PML lesions are associated with a higher likelihood of undetectable JCV DNA in CSF,96 the strategy to perform CSF analysis after natalizumab interruption and before ocrelizumab starting could be insufficient to detect asymptomatic PML.88 A stringent MRI surveillance plan for JCV-positive patients remains a chance to potentially lead to an early diagnosis and detect asymptomatic PML,97 before starting high effective DMT like ocrelizumab.
Whether the anti JCV antibody index, determined by the two-step second-generation STRATIFY JCVTM enzyme-linked immunosorbent assay (ELISA), changes after natalizumab interruption and during ocrelizumab treatment remains uncertain and uninvestigated, as well as its link with PML risk in these patients. Anyway, screening for anti-JCV antibody status is not recommended for ocrelizumab treatment.
Maintenance of Immunocompetence: Possible Implication in Vaccination Strategy
In MS patients using DMTs, the entity of the immune response to either a primary infection or a vaccination must be considered. Immunization for vaccine-preventable infections is an important aspect of MS disease management, because infections can exacerbate MS symptoms and can represent relevant adverse events during treatment.71,72,98
Generally, vaccines confer protection through the induction of neutralizing antibodies, but the role of T-cell response, although less characterized, seems to be also important.99 Immunocompetence is essential for the maintenance of the preexisting specific humoral immunity to common pathogens and to allow the valid immune response to neoantigens, therefore vaccination strategies in people with altered immunocompetence may not confer a complete protection.99 However, the level of immunosuppression is difficult to define and, in patients receiving immunosuppressive drugs, could vary according to doses, duration of therapy, and concomitant use of other drugs.100
Memory B-cells are crucial in the development of an effective active immunization through differentiation in 1) long-lived antibody-secreting plasma cells that do not express CD20 and produce protective antibodies and 2) long-lived memory CD20+ B-cells that are able to respond on reinfection to pathogens.101 Regarding the possible impact of ocrelizumab on the immunization after vaccination, the same considerations can be drawn. First, long-lived plasma cells are not the target of anti-CD20 therapies and peripheral B-cell depletion is not linked to the same degree of depletion in secondary lymphoid organs.39–41 Second, long-lived plasma cells survive in absence of memory B-cells.102,103 However, upon antigen re-exposure memory B-cells can re-enter the germinal center and differentiate in plasma cells, ensuring a response to reinfection and providing a second layer of defense against variants of the original pathogens that escape neutralization by serum antibodies.101,104
In the VELOCE trial, a randomized open-label study, the population treated with ocrelizumab was exposed to four different antigens: tetanoid, pneumococcal, and influenza vaccine, as well as a new antigen, keyhole limpet hemocyanin (KLH). In patients receiving ocrelizumab, pre-existing humoral immunity is not affected and patients mount humoral responses, although attenuated, to the inactivated vaccines studied and to the neoantigen KLH.105 Inactivated vaccines, and in particular inactivate seasonal influenza vaccine,105,106 are recommended in ocrelizumab-treated patients, whereas live and live-attenuated vaccines were not used during and after discontinuation of anti-CD20 therapies until B-cell repletion has occurred.98,107 Correct timing for vaccine administration is also paramount; waiting at least 4 weeks before starting ocrelizumab for live vaccines and at least 2 weeks or more for non-live vaccines should be considered.98,107 During the course of the treatment, vaccinations with inactivate vaccines should not overlap with the infusion and, recently, a 3-months’ delay was proposed,106 but it is unknown what degree of B-cell repopulation needs to make vaccine responses effective. The different repopulation kinetics of memory B-cells and naive B-cells should be exploited to try to personalize the timeframe to vaccinate these patients, and these elements should be taken into account in the course of anti-SARS-CoV2 vaccination. In fact, first data by Achiron et al.108 on the immune response after vaccination with BNT162b2 provided evidence that humoral vaccine responses were attenuated in most patients under treatment with ocrelizumab.
Conclusions
The effectiveness of ocrelizumab in patients with multiple sclerosis has now been widely demonstrated in RCTs, profoundly suppressing disease activity in RRMS, and mildly slowing disability progression in PPMS. Infusion-related reactions are the most common adverse events reported in RCTs, whereas, even if in the absence of new safety signals, the risk of infections should be carefully monitored, given the profound and sustained depletion of circulating B-cells that ocrelizumab leads to. However, there are several critical points that need further elucidation. Among these, the best degree and duration of B-cell depletion is still not entirely known, particularly considering that anti-CD20 therapies do not fully deplete B-cells in peripheral lymphoid tissue, thus providing a possible source of peripherally maintained disease activity. Moreover, the best time window to allow vaccinations and to schedule a pregnancy safely has yet to be defined. Different dosing and administration of anti-CD20 therapies are being evaluated to identify the best approach balancing efficacy with safety.
Taken together, ocrelizumab has been a great step forward in the treatment of MS, however collecting long-term safety data and finding strategies to minimize adverse events will be extremely relevant in the future.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising, or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Disclosure
CRM: fees as invited speaker and travel grants for attending meeting from Biogen Idec, Novartis and Roche; NDR: speaker honoraria from Biogen Idec, Genzyme, Novartis, Sanofi-Aventis; funding for participation in advisory board to Novartis and Genzyme-sanofi and for travel to scientific meetings from Biogen Idec, Teva, Sanofi-genzyme, Roche, Almirall and Novartis; RC: lecture fees and/or travel grants from Novartis, Biogen, Roche, Celgene and Merck. These relationships are not related to the content in the manuscript. The authors report no other conflicts of interest in this work.
References
1. Frischer JM, Weigand SD, Guo Y, et al. Clinical and pathological insights into the dynamic nature of the white matter multiple sclerosis plaque. Ann Neurol. 2015;78(5):710–721. doi:10.1002/ana.24497
2. Machado-Santos J, Saji E, Tröscher AR, et al. The compartmentalized inflammatory response in the multiple sclerosis brain is composed of tissue-resident CD8+ T lymphocytes and B cells. Brain. 2018;141(7):2066–2082. doi:10.1093/brain/awy151
3. Absinta M, Lassmann H, Trapp BD. Mechanisms underlying progression in multiple sclerosis. Curr Opin Neurol. 2020;33(3):277–285. doi:10.1097/WCO.0000000000000818
4. Magliozzi R, Howell OW, Reeves C, et al. A gradient of neuronal loss and meningeal inflammation in multiple sclerosis. Ann Neurol. 2010;68(4):477–493. doi:10.1002/ana.22230
5. Zrzavy T, Hametner S, Wimmer I, Butovsky O, Weiner HL, Lassmann H. Loss of “homeostatic” microglia and patterns of their activation in active multiple sclerosis. Brain. 2017;140(7):1900–1913. doi:10.1093/brain/awx113
6. Longbrake EE, Cross AH. Effect of multiple sclerosis disease-modifying therapies on B cells and humoral immunity. JAMA Neurol. 2016;73(2):219–225. doi:10.1001/jamaneurol.2015.3977
7. Hauser SL, Waubant E, Arnold DL, et al. B-cell depletion with rituximab in relapsing–remitting multiple sclerosis. N Engl J Med. 2008;358(7):676–688. doi:10.1056/nejmoa0706383
8. Hawker K, O’Connor P, Freedman MS, et al. Rituximab in patients with primary progressive multiple sclerosis: results of a randomized double-blind placebo-controlled multicenter trial. Ann Neurol. 2009;66(4):460–471. doi:10.1002/ana.21867
9. Sorensen PS, Lisby S, Grove R, et al. Safety and efficacy of ofatumumab in relapsing-remitting multiple sclerosis: a Phase 2 study. Neurology. 2014;82(7):573–581. doi:10.1212/WNL.0000000000000125
10. Bar-Or A, Grove RA, Austin DJ, et al. Subcutaneous ofatumumab in patients with relapsing-remitting multiple sclerosis: the MIRROR study. Neurology. 2018;90(20):E1805–E1814. doi:10.1212/WNL.0000000000005516
11. Hauser SL, Bar-Or A, Cohen JA, et al. Ofatumumab versus teriflunomide in multiple sclerosis. N Engl J Med. 2020;383(6):546–557. doi:10.1056/nejmoa1917246
12. Hauser SL, Bar-Or A, Comi G, et al. Ocrelizumab versus interferon beta-1a in relapsing multiple sclerosis. N Engl J Med. 2017;376(3):221–234. doi:10.1056/nejmoa1601277
13. Montalban X, Hauser SL, Kappos L, et al. Ocrelizumab versus placebo in primary progressive multiple sclerosis. N Engl J Med. 2017;376(3):209–220. doi:10.1056/NEJMoa1606468
14. Kap YS, Bauer J, Driel N, et al. B-cell depletion attenuates white and gray matter pathology in marmoset experimental autoimmune encephalomyelitis. J Neuropathol Exp Neurol. 2011;70(11):992–1005. doi:10.1097/NEN.0b013e318234d421
15. Kap YS, van Driel N, Blezer E, et al. Late B Cell Depletion with a Human Anti-Human CD20 IgG1κ Monoclonal Antibody Halts the Development of Experimental Autoimmune Encephalomyelitis in Marmosets. J Immunol. 2010;185(7):3990–4003. doi:10.4049/jimmunol.1001393
16. Lucchinetti C, Brück W, Parisi J, Scheithauer B, Rodriguez M, Lassmann H. Heterogeneity of multiple sclerosis lesions: implications for the pathogenesis of demyelination. Ann Neurol. 2000;47(6):707–717. doi:10.1002/1531-8249(200006)47:6<707::AID-ANA3>3.0.CO;2-Q
17. Serafini B, Rosicarelli B, Magliozzi R, Stigliano E, Aloisi F. Detection of ectopic B-cell follicles with germinal centers in the meninges of patients with secondary progressive multiple sclerosis. Brain Pathol. 2004;14(2):164–174. doi:10.1111/j.1750-3639.2004.tb00049.x
18. Magliozzi R, Howell O, Vora A, et al. Meningeal B-cell follicles in secondary progressive multiple sclerosis associate with early onset of disease and severe cortical pathology. Brain. 2007;130(4):1089–1104. doi:10.1093/brain/awm038
19. Howell OW, Reeves CA, Nicholas R, et al. Meningeal inflammation is widespread and linked to cortical pathology in multiple sclerosis. Brain. 2011;134(9):2755–2771. doi:10.1093/brain/awr182
20. Meffre E. The establishment of early B cell tolerance in humans: lessons from primary immunodeficiency diseases. Ann N Y Acad Sci. 2011;1246(1):1–10. doi:10.1111/j.1749-6632.2011.06347.x
21. Cotzomi E, Stathopoulos P, Lee CS, et al. Early B cell tolerance defects in neuromyelitis optica favour anti-AQP4 autoantibody production. Brain. 2019;142(6):1598–1615. doi:10.1093/brain/awz106
22. Yurasov S, Wardemann H, Hammersen J, et al. Defective B cell tolerance checkpoints in systemic lupus erythematosus. J Exp Med. 2005;201(5):703–711. doi:10.1084/jem.20042251
23. Samuels J, Ng YS, Coupillaud C, Paget D, Meffre E. Impaired early B cell tolerance in patients with rheumatoid arthritis. J Exp Med. 2005;201(10):1659–1667. doi:10.1084/jem.20042321
24. Kinnunen T, Chamberlain N, Morbach H, et al. Specific peripheral B cell tolerance defects in patients with multiple sclerosis. J Clin Invest. 2013;123(6):2737–2741. doi:10.1172/JCI68775
25. Stern JNH, Yaari G, Vander Heiden JA, et al. B cells populating the multiple sclerosis brain mature in the draining cervical lymph nodes. Sci Transl Med. 2014;6(248):1–11. doi:10.1126/scitranslmed.3008879
26. Palanichamy A, Apeltsin L, Kuo TC, et al. Immunoglobulin class-switched B cells form an active immune axis between CNS and periphery in multiple sclerosis. Sci Transl Med. 2014;6:248. doi:10.1126/scitranslmed.3008930
27. Jelcic I, Al Nimer F, Wang J, et al. Memory B Cells Activate Brain-Homing, Autoreactive CD4+ T Cells in Multiple Sclerosis. Cell. 2018;175(1):85–100.e23. doi:10.1016/j.cell.2018.08.011
28. Wang J, Jelcic I, Mühlenbruch L, et al. HLA-DR15 molecules jointly shape an autoreactive T cell repertoire in multiple sclerosis. Cell. 2020;183(5):1264–1281.e20. doi:10.1016/j.cell.2020.09.054
29. Zamvil SS, Ph D, Hauser SL. Antigen Presentation by B Cells in Multiple Sclerosis. N Engl J Med. 2021;2:2–5.
30. van Sechel AC, Bajramovic JJ, van Stipdonk MJ, Persoon-Deen C, Geutskens SB, van Noort JM. EBV-induced expression and HLA-DR-restricted presentation by human B cells of alpha B-crystallin, a candidate autoantigen in multiple sclerosis. J Immunol. 1999;162(1):129–135.
31. Palanichamy A, Jahn S, Nickles D, et al. Rituximab Efficiently Depletes Increased CD20-Expressing T Cells in Multiple Sclerosis Patients. J Immunol. 2014;193(2):580–586. doi:10.4049/jimmunol.1400118
32. Von Essen MR, Ammitzbøll C, Hansen RH, et al. Proinflammatory CD20+ T cells in the pathogenesis of multiple sclerosis. Brain. 2019;142(1):120–132. doi:10.1093/brain/awy301
33. Capasso N, Nozzolillo A, Scalia G, et al. Ocrelizumab depletes T-lymphocytes more than rituximab in multiple sclerosis. Mult Scler Relat Disord. 2021;49(July):2020. doi:10.1016/j.msard.2021.102802
34. Howlett-Prieto Q, Feng X, Kramer JF, Kramer KJ, Houston TW, Reder AT. Anti-CD20 therapy corrects a CD8 regulatory T cell deficit in multiple sclerosis. Mult Scler J. 2021;2:1–10. doi:10.1177/13524585211003301
35. Li R, Patterson KR, Bar-Or A. Reassessing B cell contributions in multiple sclerosis. Nat Immunol. 2018;19(7):696–707. doi:10.1038/s41590-018-0135-x
36. Comi G, Bar-Or A, Lassmann H, et al. Role of B cells in multiple sclerosis and related disorders. Ann Neurol. 2021;89(1):13–23. doi:10.1002/ana.25927
37. Parolini D, Cassinelli L, Razini P, et al. Expression of CD20 reveals a new store-operated calcium entry modulator in skeletal muscle. Int J Biochem Cell Biol. 2012;44(12):2095–2105. doi:10.1016/j.biocel.2012.09.001
38. Leandro MJ, Cambridge G, Ehrenstein MR, Edwards JCW. Reconstitution of peripheral blood B cells after depletion with rituximab in patients with rheumatoid arthritis. Arthritis Rheum. 2006;54(2):613–620. doi:10.1002/art.21617
39. Häusler D, Häusser-Kinzel S, Feldmann L, et al. Functional characterization of reappearing B cells after anti-CD20 treatment of CNS autoimmune disease. Proc Natl Acad Sci U S A. 2018;115(39):9773–9778. doi:10.1073/pnas.1810470115
40. Ramwadhdoebe TH, Van Baarsen LGM, Boumans MJH, et al. Effect of rituximab treatment on T and B cell subsets in lymph node biopsies of patients with rheumatoid arthritis. Rheumatology. 2019;58(6):1075–1085. doi:10.1093/rheumatology/key428
41. Kamburova EG, Koenen HJPM, Borgman KJE, Ten Berge IJ, Joosten I, Hilbrands LB. A single dose of rituximab does not deplete b cells in secondary lymphoid organs but alters phenotype and function. Am J Transplant. 2013;13(6):1503–1511. doi:10.1111/ajt.12220
42. Vital EM, Dass S, Buch MH, et al. B cell biomarkers of rituximab responses in systemic lupus erythematosus. Arthritis Rheum. 2011;63(10):3038–3047. doi:10.1002/art.30466
43. Roll P, Palanichamy A, Kneitz C, Dorner T, Tony HP. Regeneration of B cell subsets after transient B cell depletion using anti-CD20 antibodies in rheumatoid arthritis. Arthritis Rheum. 2006;54(8):2377–2386. doi:10.1002/art.22019
44. Leandro MJ. B-cell subpopulations in humans and their differential susceptibility to depletion with anti-CD20 monoclonal antibodies. Arthritis Res Ther. 2013;15(Suppl 1):1–8. doi:10.1186/ar3908
45. Nissimov N, Hajiyeva Z, Torke S, et al. B cells reappear less mature and more activated after their anti-CD20-mediated depletion in multiple sclerosis. Proc Natl Acad Sci U S A. 2020;117(41):25690–25699. doi:10.1073/pnas.2012249117
46. Klein C, Lammens A, Schäfer W, et al. Epitope interactions of monoclonal antibodies targeting CD20 and their relationship to functional properties. MAbs. 2013;5(1):22–33. doi:10.4161/mabs.22771
47. Kappos L, Li D, Calabresi PA, et al. Ocrelizumab in relapsing-remitting multiple sclerosis: a phase 2, randomised, placebo-controlled, multicentre trial. Lancet. 2011;378(9805):1779–1787. doi:10.1016/S0140-6736(11)61649-8
48. Gibiansky E, Petry C, Mercier F, et al. Ocrelizumab in relapsing and primary progressive multiple sclerosis: pharmacokinetic and pharmacodynamic analyses of OPERA I, OPERA II and ORATORIO. Br J Clin Pharmacol. 2020:1:1–10. doi:10.1111/bcp.14658
49. Edwards JCW, Szczepański L, Szechiński J, et al. Efficacy of B-cell–targeted therapy with rituximab in patients with rheumatoid arthritis. N Engl J Med. 2004;350(25):2572–2581. doi:10.1056/nejmoa032534
50. Baker D, Pryce G, James LK, Marta M, Schmierer K. The ocrelizumab phase II extension trial suggests the potential to improve the risk: benefit balance in multiple sclerosis. Mult Scler Relat Disord. 2020;44(March):102279. doi:10.1016/j.msard.2020.102279
51. Kletzl H, Gibiansky E, Petry C, et al. Pharmacokinetics, pharmacodynamics and exposure-response analyses of ocrelizumab in patients with multiple sclerosis (N4.001). ANN 2019. 2019.
52. Hauser SL, Kappos L, Arnold DL, et al. Five years of ocrelizumab in relapsing multiple sclerosis: OPERA studies open-label extension. Neurology. 2020;95(13):e1854–e1867. doi:10.1212/WNL.0000000000010376
53. Wolinsky JS, Arnold DL, Brochet B, et al. Long-term follow-up from the ORATORIO trial of ocrelizumab for primary progressive multiple sclerosis: a post-hoc analysis from the ongoing open-label extension of the randomised, placebo-controlled, Phase 3 trial. Lancet Neurol. 2020;19(12):998–1009. doi:10.1016/S1474-4422(20)30342-2
54. Mayer L, Kappos L, Racke MK, et al. Ocrelizumab infusion experience in patients with relapsing and primary progressive multiple sclerosis: results from the phase 3 randomized OPERA I, OPERA II, and ORATORIO studies. Mult Scler Relat Disord. 2019;30(January):236–243. doi:10.1016/j.msard.2019.01.044
55. Hartung HP. Ocrelizumab shorter infusion: primary results from the ENSEMBLE PLUS substudy in patients with MS. Neurol Neuroimmunol Neuroinflammation. 2020;7(5):e807. doi:10.1212/NXI.0000000000000807
56. Hauser S, Kappos L, Montalban X, et al. Safety of ocrelizumab in multiple sclerosis: updated analysis in patients with relapsing and primary progressive multiple sclerosis.
57. Scotto L, Reia A, Buonomo A. et al. Risk of invasive fungal infections among patients treated with disease modifying treatments for multiple sclerosis: a comprehensive review. Expert Opin Drug Saf;2021. 1–12. doi:10.1080/14740338.2021.1918673
58. Ciardi MR, Iannetta M, Zingaropoli MA, et al. Reactivation of hepatitis B virus with immune-escape mutations after ocrelizumab treatment for multiple sclerosis. Open Forum Infect Dis. 2019;6(1):1–3. doi:10.1093/ofid/ofy356
59. Chen YM, Chen HH, Chen YH, et al. A comparison of safety profiles of tumour necrosis factor α inhibitors and rituximab therapy in patients with rheumatoid arthritis and chronic hepatitis C. Ann Rheum Dis. 2015;74(3):626–627. doi:10.1136/annrheumdis-2014-206711
60. Lampertico P, Agarwal K, Berg T, et al. EASL 2017 clinical practice guidelines on the management of hepatitis B virus infection. J Hepatol. 2017;67(2):370–398. doi:10.1016/j.jhep.2017.03.021
61. Pawlotsky JM, Negro F, Aghemo A, et al. EASL Recommendations on Treatment of Hepatitis C 2018. J Hepatol. 2018;69(2):461–511. doi:10.1016/j.jhep.2018.03.026
62. Monaco WE, Jones JD, Rigby WFC. Rituximab associated late-onset neutropenia—a rheumatology case series and review of the literature. Clin Rheumatol. 2016;35(10):2457–2462. doi:10.1007/s10067-016-3313-y
63. Cohen BA. Late-onset neutropenia following ocrelizumab therapy for multiple sclerosis. Neurology. 2019;92(9):435–436. doi:10.1212/WNL.0000000000006924
64. Garcia-Montoya L, Villota-Eraso C, Yusof MYM, Vital EM, Emery P. Lessons for rituximab therapy in patients with rheumatoid arthritis. Lancet Rheumatol. 2020;2(8):e497–e509. doi:10.1016/S2665-9913(20)30033-3
65. Boison D, Patel D, Silberstein P. Drug induced neutropenia: a Focus on Rituximab-Induced Late-Onset Neutropenia. P T. 2016;41(12):765–768. doi:10.1016/B978-008055232-3.60622-1
66. Ocrevus full prescribing information. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/761053lbl.pdf.
67. Wolach O, Bairey O, Lahav M. Late-onset neutropenia after rituximab treatment: case series and comprehensive review of the literature. Med. 2010;89(5):308–318. doi:10.1097/MD.0b013e3181f2caef
68. Evangelatos G, Fragoulis GE, Klavdianou K, Moschopoulou M, Vassilopoulos D, Iliopoulos A. Hypogammaglobulinemia after rituximab for rheumatoid arthritis is not rare and is related with good response: 13 years real-life experience. Rheumatology. 2020;60:1–8. doi:10.1093/rheumatology/keaa617
69. Derfuss T, Weber M, Hughes R, et al. Serum immunoglobulin levels and risk of serious infections in the pivotal Phase III trials of ocrelizumab in multiple sclerosis and their open-label extensions.
70. Winkelmann A, Loebermann M, Reisinger EC, Hartung HP, Zettl UK. Disease-modifying therapies and infectious risks in multiple sclerosis. Nat Rev Neurol. 2016;12(4):217–233. doi:10.1038/nrneurol.2016.21
71. Wijnands JMA, Zhu F, Kingwell E, et al. Disease-modifying drugs for multiple sclerosis and infection risk: a cohort study. J Neurol Neurosurg Psychiatry. 2018;89(10):1050–1056. doi:10.1136/jnnp-2017-317493
72. Luna G, Alping P, Burman J, et al. Infection risks among patients with multiple sclerosis treated with fingolimod, natalizumab, rituximab, and injectable therapies. JAMA Neurol. 2020;77(2):184–191. doi:10.1001/jamaneurol.2019.3365
73. Moiola L, Barcella V, Benatti S, et al. The risk of infection in patients with multiple sclerosis treated with disease-modifying therapies: a Delphi consensus statement. Mult Scler J. 2020:52:331–346. doi:10.1177/1352458520952311
74. Sette A, Crotty S. Adaptive immunity to SARS-CoV-2 and COVID-19. Cell. 2021;184(4):861–880. doi:10.1016/j.cell.2021.01.007
75. Bastard P, Rosen LB, Zhang Q, et al. Autoantibodies against type I IFNs in patients with life-threatening COVID-19. Science. 2020;370:6515. doi:10.1126/science.abd4585
76. Gudbjartsson DF, Norddahl GL, Melsted P, et al. Humoral Immune Response to SARS-CoV-2 in Iceland. N Engl J Med. 2020;383(18):1724–1734. doi:10.1056/nejmoa2026116
77. Stubblefield WB, Talbot HK, Feldstein LR, et al. Change in Antibodies to SARS-CoV-2 Over 60 Days Among Health Care Personnel in Nashville, Tennessee. JAMA. 2020;324(17):2020–2021. doi:10.1093/cid/ciaa936
78. Soresina A, Moratto D, Chiarini M, et al. Two X-linked agammaglobulinemia patients develop pneumonia as COVID-19 manifestation but recover. Pediatr Allergy Immunol. 2020;31(5):565–569. doi:10.1111/pai.13263
79. Dan JM, Mateus J, Kato Y, et al. Immunological memory to SARS-CoV-2 assessed for up to 8 months after infection. Science. 2021;371:6529. doi:10.1126/science.abf4063
80. Hartley GE, Edwards ESJ, Aui PM, et al. Rapid generation of durable B cell memory to SARS-CoV-2 spike and nucleocapsid proteins in COVID-19 and convalescence. Sci Immunol. 2020;5(54):1–15. doi:10.1126/sciimmunol.abf8891
81. Louapre C, Collongues N, Stankoff B, et al. Clinical characteristics and outcomes in patients with coronavirus disease 2019 and multiple sclerosis. JAMA Neurol. 2020;77(9):1079–1088. doi:10.1001/jamaneurol.2020.2581
82. Sormani MP, De Rossi N, Schiavetti I, et al. Disease modifying therapies and coronavirus disease 2019 severity in multiple sclerosis. Ann Neurol. 2021;89(4):780–789. doi:10.1002/ana.26028
83. Salter A, Fox RJ, Newsome SD, et al. Outcomes and Risk Factors Associated With SARS-CoV-2 Infection in a North American Registry of Patients With Multiple Sclerosis. JAMA Neurol. 2021;1093:1–10. doi:10.1001/jamaneurol.2021.0688
84. Simpson-Yap S, de Brouwer E, Kalincik T, et al. Associations of DMT therapies with COVID-19 severity in multiple sclerosis. medRxiv. 2021;2021:455. doi:10.1101/2021.02.08.21251316
85. Alping P, Askling J, Burman J, et al. Cancer risk for fingolimod, natalizumab, and rituximab in multiple sclerosis patients. Ann Neurol. 2020;87(5):688–699. doi:10.1002/ana.25701
86. Major EO, Yousry TA, Clifford DB. Pathogenesis of progressive multifocal leukoencephalopathy and risks associated with treatments for multiple sclerosis: a decade of lessons learned. Lancet Neurol. 2018;17(5):467–480. doi:10.1016/S1474-4422(18)30040-1
87. Roche data on file 2020. Available from: https://www.ocrelizumabinfo.global/en/homepage/additional-topics-of-interest/progressive-multifocal.html..html. Accessed July 25, 2021.
88. Toorop AA, van Lierop ZYG, Strijbis EEM, et al. Mild progressive multifocal leukoencephalopathy after switching from natalizumab to ocrelizumab. Neurol Neuroimmunol Neuroinflammation. 2021;8(1):1–6. doi:10.1212/NXI.0000000000000904
89. Sul J, Patel A, Gordon ML, et al. Progressive multifocal leukoencephalopathy in a patient with progressive multiple sclerosis treated with ocrelizumab monotherapy. JAMA Neurol. 2021;78:736. doi:10.1001/jamaneurol.2021.0627
90. Berger JR, Malik V, Lacey S, Brunetta P, Lehane PB. Progressive multifocal leukoencephalopathy in rituximab-treated rheumatic diseases: a rare event. J Neurovirol. 2018;24(3):323–331. doi:10.1007/s13365-018-0615-7
91. Mancinelli CR, Scarpazza C, Cordioli C, et al. Switching to ocrelizumab in RRMS patients at risk of PML previously treated with extended interval dosing of natalizumab. Mult Scler J. 2020;58:1–5. doi:10.1177/1352458520946017
92. Levin SN, Ezuma C, Levine L, et al. Switching from natalizumab to ocrelizumab in patients with multiple sclerosis. Mult Scler J. 2020;26(14):1964–1965. doi:10.1177/1352458520927631
93. Ellwardt E, Rolfes L, Klein J, et al. Ocrelizumab initiation in patients with MS: a multicenter observational study. Neurol Neuroimmunol Neuroinflammation. 2020;7(4):1–8. doi:10.1212/NXI.0000000000000719
94. Zanghì A, Gallo A, Avolio C, et al. Exit Strategies in Natalizumab-Treated RRMS at High Risk of Progressive Multifocal Leukoencephalopathy: a Multicentre Comparison Study. Neurotherapeutics. 2021. doi:10.1007/s13311-021-01037-2
95. Kappos L, Radue EW, Comi G, et al. Switching from natalizumab to fingolimod: a randomized, placebo-controlled study in RRMS. Neurology. 2015;85(1):29–39. doi:10.1212/WNL.0000000000001706
96. Wijburg MT, Kleerekooper I, Lissenberg-Witte BI, et al. Association of progressive multifocal leukoencephalopathy lesion volume with jc virus polymerase chain reaction results in cerebrospinal fluid of natalizumab-treated patients with multiple sclerosis. JAMA Neurol. 2018;75(7):827–833. doi:10.1001/jamaneurol.2018.0094
97. Scarpazza C, Signori A, Cosottini M, Sormani MP, Gerevini S, Capra R. Should frequent MRI monitoring be performed in natalizumab-treated MS patients? A contribution to a recent debate. Mult Scler J. 2020;26(10):1227–1236. doi:10.1177/1352458519854162
98. Farez MF, Correale J, Armstrong MJ, Rae-grant A. Practice guideline update summary: vaccine- preventable infections and immunization in multiple sclerosis report of the guideline development, dissemination, and implementation. Neurology. 2019;93(13):584–594. doi:10.1212/WNL.0000000000008157
99. Pollard AJ, Bijker EM. A guide to vaccinology: from basic principles to new developments. Nat Rev Immunol. 2021;21(2):83–100. doi:10.1038/s41577-020-00479-7
100. Chinen J, Shearer WT. Secondary immunodeficiencies, including HIV infection. J Allergy Clin Immunol. 2010;125(2suppl. 2):S195–S203. doi:10.1016/j.jaci.2009.08.040
101. Akkaya M, Kwak K, Pierce SK. B cell memory: building two walls of protection against pathogens. Nat Rev Immunol. 2020;20(4):229–238. doi:10.1038/s41577-019-0244-2
102. DiLillo DJ, Hamaguchi Y, Ueda Y, et al. Maintenance of Long-Lived Plasma Cells and Serological Memory Despite Mature and Memory B Cell Depletion during CD20 Immunotherapy in Mice. J Immunol. 2008;180(1):361–371. doi:10.4049/jimmunol.180.1.361
103. Hammarlund E, Thomas A, Amanna IJ, et al. Plasma cell survival in the absence of B cell memory. Nat Commun. 2017;8(1). doi:10.1038/s41467-017-01901-w
104. Kurosaki T, Kometani K, Ise W. Memory B cells. Nat Rev Immunol. 2015;15(3):149–159. doi:10.1038/nri3802
105. Bar-or A, Calkwood JC, Chognot C, Evershed J. Effect of ocrelizumab on vaccine responses in patients with multiple sclerosis The VELOCE study. Neurology. 2020;95(14):1999–2008. doi:10.1212/WNL.0000000000010380
106. Filippi M, Capra R, Centonze D, et al. Therapeutic recommendations and seasonal influenza vaccine for multiple sclerosis patients in treatment with ocrelizumab: an expert consensus. J Neurol. 2021;268:1540. doi:10.1007/s00415-021-10466-0
107. Riva A, Barcella V, Benatti SV, et al. Vaccinations in patients with multiple sclerosis: a Delphi consensus statement. Mult Scler J. 2020;25:347–359. doi:10.1177/1352458520952310
108. Achiron A, Mandel M, Dreyer-Alster S, et al. Humoral immune response to COVID-19 mRNA vaccine in patients with multiple sclerosis treated with high-efficacy disease-modifying therapies. Ther Adv Neurol Disord. 2021;14:1–8. doi:10.1177/17562864211012835
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.