Back to Journals » Nature and Science of Sleep » Volume 10
Obstructive sleep apnea: current perspectives
Authors Osman AM, Carter SG, Carberry JC, Eckert DJ ![]()
Received 14 September 2017
Accepted for publication 6 December 2017
Published 23 January 2018 Volume 2018:10 Pages 21—34
DOI https://doi.org/10.2147/NSS.S124657
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Steven Shea
Amal M Osman,1,2 Sophie G Carter,1,2 Jayne C Carberry,1,2 Danny J Eckert1,2
1Neuroscience Research Australia (NeuRA), 2School of Medical Sciences, University of New South Wales, Sydney, NSW, Australia
Abstract: The prevalence of obstructive sleep apnea (OSA) continues to rise. So too do the health, safety, and economic consequences. On an individual level, the causes and consequences of OSA can vary substantially between patients. In recent years, four key contributors to OSA pathogenesis or “phenotypes” have been characterized. These include a narrow, crowded, or collapsible upper airway “anatomical compromise” and “non-anatomical” contributors such as ineffective pharyngeal dilator muscle function during sleep, a low threshold for arousal to airway narrowing during sleep, and unstable control of breathing (high loop gain). Each of these phenotypes is a target for therapy. This review summarizes the latest knowledge on the different contributors to OSA with a focus on measurement techniques including emerging clinical tools designed to facilitate translation of new cause-driven targeted approaches to treat OSA. The potential for some of the specific pathophysiological causes of OSA to drive some of the key symptoms and consequences of OSA is also highlighted.
Keywords: pathophysiology, sleep-disordered breathing, arousal, upper airway physiology, control of breathing, precision medicine
Introduction
Obstructive sleep apnea (OSA) is an increasingly common, chronic, sleep-related breathing disorder.1–3 OSA is characterized by periodic narrowing and obstruction of the pharyngeal airway during sleep. Untreated OSA is associated with long-term health consequences including cardiovascular disease,4,5 metabolic disorders,6 cognitive impairment,7 and depression.8 Common symptoms include excessive daytime sleepiness, fatigue, non-refreshing sleep, nocturia, morning headache, irritability, and memory loss.9,10 Untreated OSA is also associated with lost productivity and workplace and motor vehicle accidents resulting in injury and fatality.11–13 The costs of untreated OSA and sleep loss are substantial.14,15 Recommended therapy can relieve symptoms16,17 and reduce some of the associated sequelae.18,19 However, many people with OSA struggle with the first-line therapy, continuous positive airway pressure (CPAP), for which adherence rates remain unacceptably low.20,21 Non-CPAP therapies (e.g., oral appliance therapy and upper airway surgery) are beneficial in many cases but have variable and unpredictable efficacy.22–25 Thus, new approaches to treat OSA are required.
Indeed, most people with OSA are undiagnosed and untreated.26–28 In some cases, this may be attributed to, at least in part, a lack of awareness of the disorder.29 Other potential barriers to seek treatment include stigma related to some of the features of the disease such as snoring, access to polysomnography (PSG) and diagnostic services (particularly in remote communities and in the developing world),27,29 perceived lack of enthusiasm with existing treatment options, and, in some cases, concern that driving licenses will be revoked. In addition, primary care physicians may not be prompted to explore an early diagnosis of OSA. This is especially true if patients do not present with subjective sleepiness and the classic characteristics of a high body mass index. Symptoms such as fatigue or sleepiness may also be attributed to comorbid disease that is common in people with OSA.30 However, we know that absence of subjective sleepiness does not rule out substantial sleep-disordered breathing and up to 50% of people with OSA are not obese.2,31 Indeed, 25% of individuals with moderate OSA have neither subjective nor objective sleepiness.32,33 Nonetheless, given the burden of disease, the shortcomings of existing diagnostic and treatment approaches, and the substantial health, safety, and economic consequences of untreated OSA, there is a pressing need to continue to raise awareness and develop new strategies to manage and treat this common chronic health condition.
There are multiple contributors to OSA.34,35 Each contributor represents a therapeutic target.34,35 While these new research findings offer hope for new therapies, identification of these new targets has not yet translated to new models of care for OSA.36 However, there has been recent progress toward achieving this goal. For example, strategies to extract information from existing clinical PSG studies to help inform treatment decisions according to a cause-driven targeted therapy model for OSA have been developed.37–41 In accordance with this objective, simple wakefulness upper airway and respiratory physiology tests may also be useful.42–45 These concepts are the focus of the current review.
Existing clinical measures of OSA
The gold standard method used to diagnose sleep-disordered breathing is a comprehensive in-laboratory PSG. The main outcome used to define OSA severity is the apnea-hypopnea index (AHI). This index represents the number of breathing stoppages (apneas) and periods of reduced airflow (hypopneas) lasting greater than 10 seconds that result in a brief awakening (arousal) or reduced oxygenation that occur per hour of sleep. While severity cutoffs vary, mild sleep apnea is typically defined as 5–15, moderate 15–30, and severe more than 30 respiratory events/h sleep.
While in-laboratory PSG is comprehensive, it is also labor intensive, time-consuming, and costly (see the study by Edwards et al46 for a review). To facilitate the diagnosis process, home-monitoring technologies have emerged. These range from a replication of the same measurements used in the laboratory (a level 2 unattended study) to limited channel devices that focus on a few core signals (e.g., oxygen and an airflow sensor). These tend to be most useful for the detection of severe disease, provided patients do not have severe comorbidity.47–49
Despite the quantity of neurophysiological signals obtained during an overnight PSG, most of the data collected is ignored and treatment decisions rely heavily on the AHI. While the AHI remains a widely used measure of OSA severity clinically and for research purposes, it has several limitations. For example, a patient with very long respiratory events may experience substantial hypoxemia but have a relatively low AHI. Conversely, another patient may have more frequent events and therefore a much higher AHI, but minimal exposure to hypoxemia.50 Thus, the effects of hypoxia from OSA and its adverse impact on the cardiovascular system in the patient with a low AHI may be more pronounced.51,52 In addition, non-apneic respiratory events that do not meet the scoring criteria for a hypopnea are associated with heart rate changes and increased expiratory pharyngeal resistance.53 Breathing disruptions that do not cause major hypoxemia are also associated with objective daytime sleepiness.54 Furthermore, the total AHI correlates poorly with the key causes and consequences of the disease.35,55 Conversely, recent studies indicate that REM sleep apnea may be more important in mediating insulin resistance and the cardiovascular consequences of OSA.56–59 Thus, these examples highlight the heterogeneity in the various clinical manifestations of OSA and its consequences and some of the limitations with currently used diagnostic methods.
OSA pathophysiology
Similar to the clinical heterogeneity, OSA pathogenesis is also multifactorial. There are “anatomical” and “non-anatomical” causes34,35,60 (Figure 1). In recent years, the potential role that factors beyond pharyngeal anatomy and craniofacial structure play in OSA pathophysiology has been recognized. Indeed, OSA can develop due to multiple contributors, the combination of which likely varies substantially between patients. These concepts have been the focus of several recent review articles (e.g., studies by Eckert,34 Carberry et al,36 Edwards et al,61 and Eckert and Wellman62) and are therefore only briefly summarized here.
 | Figure 1 Schematic of the anatomical and non-anatomical causes of OSA. Notes: Some degree of anatomical vulnerability is present in OSA. However, the extent of impairment varies widely between patients. The non-anatomical contributors, which are present in approximately 70% of OSA patients, play a key role in mediating the absence or presence of OSA. In the schematic, the gray tracings indicate the desired response, whereas the black tracings represent impairment in the non-anatomical trait. Refer to the text for further detail. Reprinted from Chest, Carberry JC, Amatoury J, Eckert DJ, Personalized management approach for OSA, Epub 2017 June 16, Copyright (2017), with permission from Elsevier.36 Abbreviations: EEG, electroencephalography; EMG, genioglossus electromyography; MTA, 100 ms moving time average of the rectified raw EMG signal; OSA, obstructive sleep apnea. |
Non-anatomical contributors include impaired pharyngeal dilator muscle function, premature awakening to mild airway narrowing (low respiratory arousal threshold), and unstable control of breathing (high loop gain3,34,35) (Figure 1). As highlighted in later sections, when combined with a pharyngeal airway that is susceptible to closure during sleep, impairment in one or more of these non-anatomical contributors can perpetuate OSA severity. Given that airway obstruction in OSA only occurs during sleep, the combination of an anatomical predisposition combined with state-dependent changes in non-anatomical contributors is crucial in driving this common disorder.34,35,63
OSA is largely a sleep-dependent anatomical problem
As highlighted, OSA is a multifactorial disorder. However, some level of upper airway anatomical impairment is essential.34,35 Thus, it is logical that most existing therapies for OSA aim to correct the anatomical problem. Imaging studies64–66 have identified key pharyngeal anatomical abnormalities in people with OSA. For example, a narrow pharyngeal airway, increased airway length, and certain pharyngeal lumen shapes are all associated with the propensity for pharyngeal collapse during sleep.67–69 The upper airway can collapse at one or multiple sites.70 The pharyngeal structures that can contribute to airway crowding and collapse include the dilator muscles such as genioglossus, soft palate, lateral pharyngeal walls, and the epiglottis. Obesity is an important risk factor.71 Neck circumference is routinely measured in the clinic and has been used to help predict OSA risk.72,73 Craniofacial morphology,74,75 position of the hyoid bone,76 airway surface tension,77 tongue scalloping,78 and tongue fat79 are some of the factors associated with OSA risk and its severity. While these approaches have provided insight into OSA pathogenesis, limitations for clinical use include 1) high cost of imaging procedures and 2) awake static imaging provides limited insight into the properties of a dynamic structure that closes involuntarily during sleep.
Upper airway collapsibility
Pharyngeal critical closing pressure (Pcrit) is a well-established technique used to quantify upper airway collapsibility during sleep.80–82 Pcrit has been used to describe differences in upper airway collapsibility across the sleep-disordered breathing spectrum (from snoring to OSA80). It is considered as the gold standard approach to quantify “functional anatomy” during sleep.34 The Pcrit technique allows the pharyngeal airway to be examined under conditions of reduced,82 although not absent,83 neuromuscular input compared to wakefulness. Once a therapeutic CPAP level that prevents airway obstruction or narrowing is established, brief reductions (5 breaths) in the holding pressure are applied during stable sleep.70 This procedure is repeated at different levels of mask pressure until airflow limitation and closure occurs. The pressure-flow relationship between peak inspiratory flow for flow-limited breaths and the corresponding mask pressure is compared. These values are then extrapolated to determine the “Pcrit”, the mask pressure at zero airflow. Within an individual, Pcrit measurements are stable over time (days to months).84 However, factors such as weight gain over a longer period would be expected to increase airway collapsibility (Pcrit).85,86
On average, OSA patients tend to have Pcrit values near atmospheric pressure. This indicates that their airway closes at or near 0 cmH2O during sleep.35,76,86 However, there is substantial variability in Pcrit in OSA and therefore anatomical vulnerability to pharyngeal collapse. Indeed, Pcrit can range from approximately −5 to greater than +5 cmH2O in OSA. A Pcrit at or near +5 cmH2O indicates a highly collapsible airway, whereas a sub-atmospheric Pcrit indicates a relatively stable upper airway as suction pressure is required to close the upper airway during sleep. OSA is very rare in people with Pcrit values less than −5 cmH2O.35,86 However, within the sub-atmospheric range (0 to −5 cmH2O), there is considerable overlap in Pcrit between people with and without OSA. Indeed, approximately 20% of OSA patients have similar pharyngeal collapsibility during sleep compared to people without OSA.35 In this group, the interaction between mild anatomical susceptibility and impairment in one or more of the non-anatomical causes of OSA is crucial in driving OSA pathogenesis.34,35 These patients are more likely to benefit from targeted non-CPAP therapies compared to those with very high Pcrits.34,36 Thus, given the key role that upper airway anatomy/collapsibility plays in driving OSA pathogenesis, a simple measure of airway collapsibility would be invaluable to inform targeted treatment decisions. The problem, however, is that the Pcrit technique is not clinically viable as the protocol is technically challenging, somewhat invasive (requires CPAP and ideally a pharyngeal pressure catheter), time-consuming, and requires skilled personnel to collect and analyze the data.
New simplified methods to estimate upper airway collapsibility
There has been recent progress toward development of simple and reliable methods to estimate the extent of anatomical/airway collapsibility contribution to OSA. The first technique involves an existing tool traditionally used to assess expiratory flow limitation in patients with chronic obstructive pulmonary disease.42 Participants are fitted with a nasal breathing mask and brief (2 second) periods of negative pressure (−5 cmH2O) are delivered during early expiration.42 This elicits a transient increase in expiratory airflow the extent to which is mediated, at least in part, by upper airway collapsibility/anatomy. The average response is quantified as the ratio between the exhaled volumes (during the first 0.2 seconds) for at least 4 breaths prior to the expiratory pressure application versus the expiratory pressure breaths for 10 replicate trials. An increase in this ratio suggests a collapsible airway. A modest relationship with Pcrit and other important anatomical components that contribute to OSA was detected.42 Thus, this technique alone is unlikely to be helpful in informing treatment decisions, but if combined with other simple measures it may play a role.
Preliminary data from our group indicate that a 10–15 minute protocol in which brief pulses of suction are applied during early inspiration through a nasal mask during wakefulness correlates well with Pcrit.43 The prescribed CPAP level from a routine CPAP titration study is also associated with passive Pcrit.37 Thus, the therapeutic CPAP level may be useful in distinguishing patients with mildly versus highly collapsible upper airways.37 Genta et al39 have also recently demonstrated that analysis of the shape of the inspiratory flow curve during airflow limitation during sleep and the degree of negative effort dependence (extent to which the airway narrows during inspiration) can inform the site of upper airway collapse. This was determined using endoscopy to locate the site of collapse while simultaneously monitoring nasal airflow and pharyngeal pressures. Averaging multiple flow-limited breaths revealed characteristic flow patterns that were associated with different sites of airway narrowing/collapse.39 In addition, simply quantifying peak flow during routine polysomnography has recently been shown to be associated with active Pcrit (a measure that encompasses upper airway collapsibility and neuromuscular compensation40). Thus, there are several new promising approaches to estimate the extent of anatomical impairment in people with OSA. Given their relative simplicity, one or more of these approaches may be preferable compared to more invasive procedures such as drug-induced endoscopy, which is becoming increasingly used to help inform patient selection for upper airway surgery.87
The upper airway muscles
The human pharynx is unique in that it lacks rigid bony support. Its predominant soft tissue structure enables it to change cross-sectional area with varying intraluminal pressures. However, depending on the dynamic balance of intraluminal pressure and neural drive to the upper airway dilator muscles, the human pharynx is vulnerable to collapse during sleep.88 There are over 20 muscles in the upper airway. These are involved in respiratory and non-respiratory tasks (speech, mastication, swallowing, and breathing). A subset of these muscles plays a predominant role in airway stability during breathing.89 In healthy individuals and people with OSA during wakefulness, activation of the upper airway dilator muscles is effective in opposing the collapsing pressures generated during inspiration. However, during sleep, state-dependent reductions in muscle activity when combined with anatomical susceptibility (narrow/crowded/collapsible airway) can induce airway collapse.88 Thus, understanding the neural control of the airway muscles and their mechanical consequences is important for development of new treatments and preventative measures to improve upper airway function in OSA.
The upper airway muscles have complex patterns of neural activation that differ between muscles. For example, the genioglossus, the largest pharyngeal dilator muscle located at the base of the tongue, receives up to six different patterns of drive.90 It receives central input from the brainstem (respiratory pattern generator neurons) and reflex input from pharyngeal mechanoreceptors and chemoreceptors. The summation of drive to genioglossus typically results in a phasic pattern of activation (i.e., more activity during inspiration and less during expiration, Figures 1–4). Genioglossus activity is reduced at sleep onset91 and varies between sleep stages.83 However the tensor palatini muscle (a palatal muscle) displays predominantly tonic (constant throughout the breathing cycle) patterns of activation.92 It is less sensitive to small changes in pharyngeal pressure compared to genioglossus but can be activated in a similar manner with larger transient pressure swings93 and is sensitive to sleep state but has minimal change across sleep stages in the absence of upper airway resistance.83 The combinations of a loss in central drive and reflex input to the upper airway muscles during sleep are thought to be important contributors to OSA pathogenesis.94,95 Similarly, the ability to increase reflex drive to airway narrowing during sleep is important in OSA pathogenesis.35,96,97 Indeed, approximately 30% of OSA patients have poor genioglossus muscle responsiveness to airway narrowing during sleep35 (Figure 2). Many patients have a high recruitment threshold to respiratory stimuli during sleep that is not reached without awakening from sleep (arousal).96 Conversely, others are able to restore airflow during sleep via pharyngeal muscle recruitment without arousal (Figure 3). In addition, enhanced muscle responsiveness can protect certain obese individuals from developing OSA despite their anatomical compromise.98 Thus, the combination of non-anatomical and anatomical compromise is crucial in preventing or promoting OSA.
 | Figure 2 Example of minimal genioglossus muscle responsiveness. Notes: In this example of a naturally occurring apnea, despite clear phasic activation of the genioglossus muscle (as shown in the raw and MTA genioglossus EMG channels), there is minimal activation of genioglossus during the respiratory event. This is despite substantial increasing negative epiglottic pressure (Pepi) swings from the first to last effort (last effort nadir epiglottic pressure = arousal threshold). It is only when cortical arousal occurs (as shown in the EEG channel) that major genioglossus activation occurs (signal clipped in this example) and airflow is restored. Abbreviations: EEG, electroencephalography; EMG, genioglossus electromyography; MTA, 100 ms moving time average of the rectified raw EMG signal. |
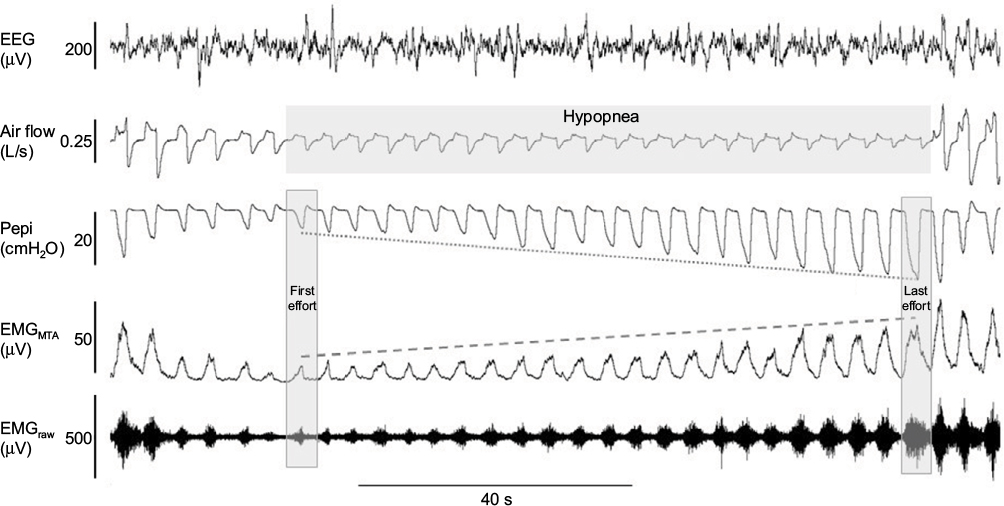 | Figure 3 Example of robust genioglossus muscle responsiveness and restoration of airflow without cortical arousal. Notes: In contrast to the example in Figure 2, in this example of a naturally occurring hypopnea, there is robust activation of the genioglossus muscle (as shown in the raw and MTA genioglossus EMG channels), to increasing negative epiglottic pressure (Pepi) swings from the first to last effort which ultimately results in recovery of airflow without cortical arousal. Abbreviations: EEG, electroencephalogram; EMG, genioglossus electromyography; MTA, 100 ms moving time average of the rectified raw EMG signal. |
In addition, recent findings suggest a mismatch between central neural drive to the genioglossus muscle and the mechanical response of the muscles in OSA patients.99 Indeed, in healthy individuals, dynamic magnetic resonance imaging shows anterior movement of this fan-shaped muscle during inspiration and increased cross-sectional area (CSA) of the pharynx.100 However, tongue movement patterns during quiet breathing vary in people with OSA.101 Some patients have counterproductive motion characterized by anterior motion at the base of the tongue followed by airway narrowing at the level of the soft palate, while others have little to no movement during inspiration.101 These patterns of movement are dependent, at least in part, on OSA severity whereby minimal movement is most common in severe OSA.101 Compensatory mechanisms in healthy individuals who have a narrow airway CSA compared to controls display larger anterior movement of the tongue during inspiration.102 Accordingly, breathing stability is associated with greater genioglossus activity.103 However, increased genioglossus activity is sometimes insufficient to re-open the airway (Figure 4). Thus, the contribution of the other upper airway muscles may also play a contributing role.104 How the various components of upper airway muscle function change over time is unknown. However, increased weight gain over time and fat accumulation in the tongue are predicted to worsen upper airway motion.79
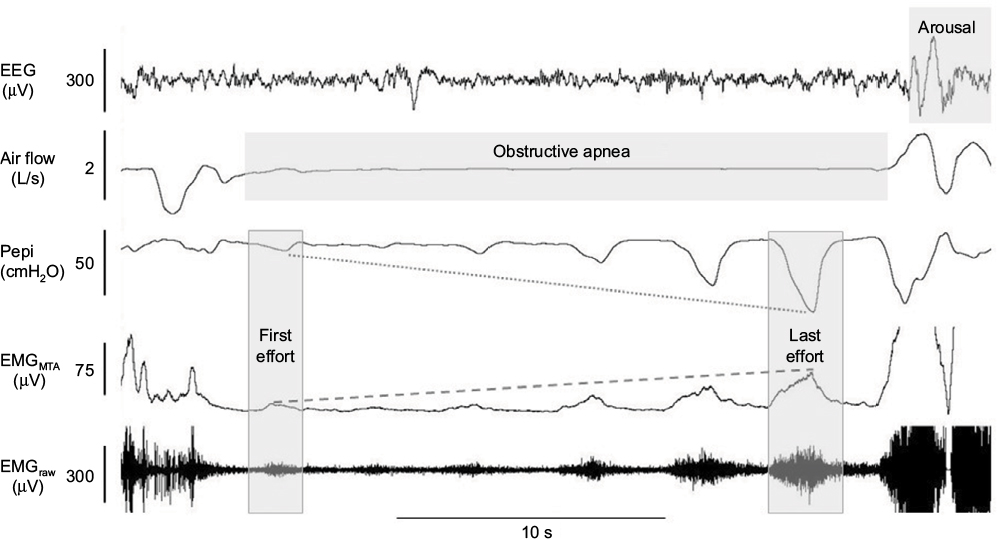 | Figure 4 Example of robust genioglossus muscle responsiveness without restoration of airflow. Notes: In contrast to the example in Figure 3, in this example of a naturally occurring apnea, there is robust activation of the genioglossus muscle (as shown in the raw and MTA genioglossus EMG channels) to increasing negative epiglottic pressure (Pepi) swings from the first to last effort (last effort nadir epiglottic pressure = arousal threshold). However, despite substantial genioglossus muscle activation during the apnea, it is insufficient to restore airflow, which only occurs with cortical arousal (as shown in the EEG channel). The genioglossus signal is clipped in this example when airflow is restored with arousal. Abbreviations: EEG, electroencephalogram; EMG, genioglossus electromyography; MTA, 100 ms moving time average of the rectified raw EMG signal. |
Treatments to target the upper airway muscles
One approach to activate the upper airway muscles during sleep is to deliver current to the muscles via direct stimulation or via stimulation of the hypoglossal nerve. Clinically, this is achieved via surgical implantation of a stimulation device connected to a cuff placed around the nerve.105 Mechanistic studies have also used fine-wire electrodes or non-invasive methods such as transcutaneous electrodes (see recent review to compare different methods106). Improvements in inspiratory airflow, AHI, apnea duration, oxygen saturation, arousal index, sleep architecture, and excessive daytime sleepiness have all been reported to varying degrees using these methods.107–111 Clinical trial follow-up studies show long-term sustained reductions in AHI (~50–66%).111–114 However, it is difficult to predict responders. The STAR trial used endoscopy for pre-screening to eliminate patients with concentric airway collapse, which may have increased the response rate to therapy.105 However, one-third of patients were still classified as non-responders.105 Thus, other factors such as airway shape and Pcrit are also likely to be important considerations to optimize treatment response rates.105,110
To date, attempts to develop pharmacotherapy to increase upper airway muscle activity to treat OSA have not been successful. Targets have included serotonergic, noradrenergic and GABAergic systems, as well as potassium channels (for reviews see).115,116 However, recent studies with the tricyclic antidepressant desipramine which has strong noradrenergic, mild antimuscarinic, and mild serotonergic effects, have shown preservation of sleep-related reductions in genioglossus activity.117 Desipramine also yields improvements in airway collapsibility and OSA severity in patients with poor muscle responsiveness.118 Thus, this combined approach may be superior to single system targets.115,119–121 Interestingly, the hypnotic zolpidem (which acts on the GABAergic system) shows potential to increase pharyngeal muscle responsiveness during airway narrowing without impairing the other key causes of OSA.122 Designer Receptors Exclusively Activated by Designer Drugs that allow for selective targeting of a group of neurons via introduction of an engineered macromolecule (designer receptor) with viral vectors that can be activated with a specific drug (designer drug) have also recently been tested in animal models with the objective to increase pharyngeal muscle activity during sleep.123,124 These exciting findings demonstrate lasting increases in genioglossus activity and offer promise that these new concepts will ultimately translate to humans.
Other strategies to improve pharyngeal muscle function include training modalities. Indeed, regular didgeridoo playing and oropharyngeal exercises can reduce snoring and OSA severity (~50% reduction in AHI) and daytime sleepiness.125–128 However, the mechanisms are largely unknown. No studies have investigated upper airway muscle tone or muscle properties pre-training versus post-training. Longitudinal follow-up studies are also lacking. Understanding the mechanisms may help to inform which OSA phenotypes are most likely to respond to this form of therapy.
Prediction tools and simplified methods to estimate pharyngeal muscle function
Identification of patients who have poor pharyngeal muscle function may facilitate development of targeted therapies directed toward this trait118 and improve treatment success rates with existing therapies (e.g., hypoglossal nerve stimulation). However, gold standard methodology to quantify pharyngeal muscle activity is complex, invasive (fine-wire electrodes inserted directly into the muscle), requires specialized personnel and equipment, and is time-consuming.34 There are no simplified tools to estimate pharyngeal muscle function to identify people with this clinical phenotype accurately. However, while multiple variables impact inspiratory flow during sleep, mean peak inspiratory airflow during airflow limitation appears to be a good surrogate for active Pcrit, a measure that incorporates both anatomical and neuromuscular components.40 Thus, if this approach proves useful to predict treatment outcomes, automated signal processing algorithms based on routine PSG data could be implemented.40 Nonetheless, additional practical tools to determine pharyngeal muscle function are urgently required in order to advance personalized treatment approaches that target the upper airway muscles.
Respiratory arousal threshold
The role of the respiratory arousal threshold in OSA pathogenesis has been described in detail.129 Accordingly, these concepts are outlined only briefly here. Historically, given that most respiratory events are associated with a cortical arousal, arousals were considered crucial to reopen the upper airway following a respiratory event in OSA.130,131 However, approximately 20% of respiratory events cease without cortical arousal and an additional 20% occur after the upper airway has already reopened and airflow has been restored.129,132–134 Indeed, 75% of adults with OSA have respiratory events that terminate without an arousal or the arousal occurs following airway reopening at some stage of the night.132 Thus, airway reopening can occur without arousal. Rather, continual unnecessary arousals can worsen OSA and contribute to OSA pathophysiolgy.129,132 Specifically, repetitive arousals can perpetuate blood-gas disturbances and cause sleep fragmentation to promote cyclical breathing and prevent establishment and maintenance of more stable, deeper stages of sleep.135–138
Numerous respiratory stimuli can contribute to arousal from sleep during a respiratory event.129,139–142 Increased respiratory effort due to a narrowed pharyngeal airway increases negative intrathoracic pressure. Although the amount of negative intrathoracic pressure generated can vary greatly between individuals and in different stages of sleep,35,132,143–147 the magnitude of negative pressure required to cause an arousal from sleep is relatively constant within an individual.147–149 This is the case regardless of whether the respiratory disturbance is caused by hypoxia, hypercapnia, or respiratory loading.147
Accordingly, the gold standard method to quantify the threshold for arousal to respiratory stimuli requires an epiglottic or esophageal pressure catheter combined with PSG recording equipment. Specifically, the respiratory arousal threshold is the nadir pressure immediately prior to cortical arousal (Figures 1, 2, and 4). The respiratory arousal threshold is quantified by averaging multiple pressure values throughout the night to an experimental intervention designed to cause airway narrowing or activate respiratory afferents35,147 or during naturally occurring respiratory events.144,145
In people with OSA who require large intrathoracic pressure swings to cause an arousal (i.e., patients with high respiratory arousal thresholds ≤25cmH2O), respiratory events are often prolonged, particularly if these patients also have poor upper airway muscle responsiveness.129 Thus, in the absence of neuromuscular compensation, arousal from sleep and reintroduction of wakefulness drive can act as a last line of defense to facilitate rapid reopening of the airway to re-establish airflow and normalize blood-gas levels in these individuals.129,130,150 The consequences of OSA such as sleep deprivation increase, while CPAP therapy decreases the respiratory arousal threshold.151,152 Nonetheless, approximately 30–50% of OSA patients wake to relatively small intrathoracic/epiglottic pressure swings (i.e., patients with low respiratory arousal thresholds ≥−15 cmH2O).35,38,129,145 In these patients, given that the stimuli for arousal are the same as the stimuli required to recruit the pharyngeal dilator muscles (i.e., blood gas changes and negative pressure swings), premature arousal limits the opportunity for neuromuscular compensation mechanisms to overcome airway narrowing and stabilize breathing.132 Frequent arousals can also cause sleep fragmentation and sleep instability, prevent deeper stages of sleep, and perpetuate unstable breathing.132,142 Thus, strategies to reduce arousals in these patients may allow for more stable breathing during sleep.
Treatments to target the respiratory arousal threshold
Given the abovementioned rationale, the potential therapeutic role of hypnotics to treat OSA in patients with a low respiratory arousal threshold phenotype has been an area of recent research focus.150 This strategy requires a careful targeted approach to optimize benefit in those with the desired phenotype and avoid harm in those with high arousal thresholds. In particular, the selected agent to increase the threshold for arousal to respiratory stimuli must do so without impairment in pharyngeal muscle activity. Apart from an early wakefulness benzodiazepine study,22 subsequent studies during sleep have not shown systematic reductions in pharyngeal muscle activity or responsiveness to negative pharyngeal pressure with common doses of zopiclone,96,144 trazodone,153 temazepam,122 or zolpidem.122 While a recent study found more variable effects on genioglossus muscle responsiveness with temazepam, paradoxically, on average, zolpidem increased muscle responsiveness 3-fold in people with and without OSA.122
The other concern with hypnotic use in OSA is prolongation of respiratory events and worsening hypoxemia due to blunted arousal responses in those individuals with a high threshold for respiratory arousal. Indeed, this can occur with high doses or in obese patients with very severe disease.129 By contrast, the hypnotics eszopiclone,145 zopiclone,22 and trazodone154 can reduce OSA severity as measured by the AHI without worsening hypoxemia. In the eszopiclone study,145 reductions in AHI occurred invariably in those with a low arousal threshold phenotype. The number of arousals per hour of sleep also decreased.145 Given the contrasting effects of hypnotics in OSA, screening tools to distinguish between patients with low and high respiratory arousal thresholds and to determine who will benefit versus be susceptible to harm are important.
Prediction tools and simplified methods to estimate the respiratory arousal threshold
The gold standard approach to quantify the respiratory arousal threshold is impractical for routine clinical use as it is time-consuming, costly, and somewhat invasive (requires an airway pressure catheter). Preliminarily findings indicate that respiratory sensation to inspiratory loading during wakefulness is related to the respiratory arousal threshold during sleep.155 Edwards et al have developed a simple tool to estimate the respiratory arousal threshold with high sensitivity and specificity based on three measures from a standard overnight PSG (AHI, nadir oxygen saturation, and the apnea to hypopnea ratio).38 Thus, while prospective intervention studies are required, this simple approach could easily be implemented in the clinical setting to inform treatment decisions. Given that over 40% of OSA patients may also have insomnia,156–158 simple accurate tools to determine which OSA patients will benefit versus those at risk of harm with hypnotics would be invaluable.
Loop gain
Loop gain is a term used to describe the stability of a feedback control system. In the context of respiratory physiology, loop gain is the ventilatory response to ventilatory disturbance ratio. It comprises three principal components: 1) plant gain (i.e., tissues, blood, and lungs where CO2 is stored) and 2) delays in circulation (i.e., time it takes for a change in CO2 to mix with the existing blood to arrive and be detected by the chemoreceptors) and 3) controller gain (i.e., chemosensitivity). Any medical condition that modifies one or more of these components (e.g., heart failure) will alter loop gain.34 Components of OSA such as intermittent hypoxia can also alter respiratory control.159 People with high loop gain have exaggerated ventilatory responses to minimal changes in CO2. This is a marker of an unstable control system. This can be reduced with CPAP therapy.160 On the other hand, those with extremely low loop gain often experience hypoventilation during sleep, as is the case in people with obesity hypoventilation syndrome.34
In their landmark study using proportional assist ventilation to induce breathing oscillations during sleep after the airway was stabilized with CPAP, Younes et al showed that severe OSA patients have high loop gain.138 Subsequent studies confirmed that many people with OSA have high loop gain.35,161 When combined with even a modest impairment in upper airway anatomy, high loop gain can drive OSA pathogenesis.35,161 Similar to the other phenotypic traits, loop gain can be quantified using transient reductions in CPAP during sleep to create a disturbance to breathing.161,162 Rapid reintroduction of CPAP is then applied so that the ventilatory response (overshoot) can be quantified (using a breathing mask and pneumotachograph) to calculate loop gain. This procedure is repeated as many times as possible throughout the night. Loop gain is then calculated as the ventilatory response divided by the ventilatory disturbance ratio after scaling for the different levels of ventilatory disturbances presented.161,162 This technique results in a negative number such that more negative numbers reflect higher loop gain.162 Approximately one-third of OSA patients have high loop gain (<−5) which indicates a >5 L/min increase in minute ventilation in response to 1 L/min reduction in minute ventilation.35
Treatments to target loop gain and simplified methods to estimate loop gain
O2 therapy reduces loop gain and OSA severity in people with high loop gain.163 Carbonic anhydrase inhibitors such as acetazolamide also reduce loop gain by approximately 40% and OSA severity.164 Similarly, zonisamide reduces OSA severity.24 In addition, stabilization of CO2 and hypercapnia can prevent hypoventilation and unstable respiratory control during sleep in OSA.165 Strategies that combine O2 therapy to reduce loop gain with a hypnotic to increase the arousal threshold can yield major reductions in OSA severity.146 In addition, recent studies from the Mateika group indicate that several of the key contributors to OSA such as airway collapsibility, arousal threshold, and respiratory control are influenced by circadian phase.166–168 Thus, novel strategies that target the circadian system may have therapeutic potential in OSA.
The current approaches to quantify loop gain during sleep require experienced personnel to perform CPAP manipulations and analyze the data. However, Terrill et al have developed an analysis approach that uses the nasal pressure signal from a standard PSG to estimate loop gain.41 This approach has been used to explain and predict changes to a range of interventions in OSA.30,47,146 Wakefulness tests of respiratory control may also be helpful in estimating responses to pharmacotherapies.44,45
Potential links between phenotypic traits, treatment, and health consequences
In addition to the role that the phenotypic traits play in contributing to OSA pathogenesis, the traits may also provide insight into disease consequences and physiological reasons for treatment failure. For example, a low arousal threshold trait may be a physiological contributor to poor CPAP adherence and compliance.31 A low arousal threshold trait and its consequences such as sleep fragmentation and frequent hypopneas with minimal changes in oxygenation may be a marker for cognitive impairment and daytime sleepiness.54,169 Similarly, patients who tend to have more “intense” arousals, which appear to be an inherent trait,47,170 may experience more daytime sleepiness than those who do not. People who have OSA that is driven by high loop gain may be more vulnerable to the cardiovascular consequences of OSA. Conversely, strategies that target certain components of respiratory control such as mild intermittent hypoxia to elicit respiratory plasticity may help improve CPAP compliance and potentially also directly target the autonomic, cardiovascular, neurocognitive, and metabolic systems.171,172 While possible links between the phenotypic traits and specific disease consequences have not yet been investigated, emergence of simplified tools to estimate the traits will enable these theoretical concepts to be tested systematically in cohort studies.
Conclusion
Characterization of the different causes or phenotypes of OSA in recent years has provided new pathways for targeted therapy. New simplified approaches to estimate each of the key causes of OSA have recently been developed. While more work is required, particularly directed toward the impaired pharyngeal muscle trait, these new tools offer promise for the translation of detailed phenotyping concepts to the clinic. Identification of the traits may also provide insight in to which patients are more likely to develop specific disease consequences.
Disclosure
DJE is supported by a National Health and Medical Research Council of Australia Senior Research Fellowship (1116942), has a Commonwealth Government of Australia Cooperative Research Centre grant (industry partner: Oventus Medical), and serves as a consultant for Bayer. The other authors report no conflicts of interest in this work.
References
Punjabi NM. The epidemiology of adult obstructive sleep apnea. Proc Am Thorac Soc. 2008;5(2):136–143. | ||
Peppard PE, Young T, Barnet JH, Palta M, Hagen EW, Hla KM. Increased prevalence of sleep-disordered breathing in adults. Am J Epidemiol. 2013;177(9):1006–1014. | ||
Heinzer R, Vat S, Marques-Vidal P, et al. Prevalence of sleep-disordered breathing in the general population: the HypnoLaus study. Lancet Respir Med. 2015;3(4):310–318. | ||
Kapur VK, Resnick HE, Gottlieb DJ; Sleep Heart Health Study Group. Sleep disordered breathing and hypertension: does self-reported sleepiness modify the association? Sleep. 2008;31(8):1127–1132. | ||
Walia HK, Li H, Rueschman M, et al. Association of severe obstructive sleep apnea and elevated blood pressure despite antihypertensive medication use. J Clin Sleep Med. 2014;10(8):835–843. | ||
Drager LF, Togeiro SM, Polotsky VY, Lorenzi-Filho G. Obstructive sleep apnea: a cardiometabolic risk in obesity and the metabolic syndrome. J Am Coll Cardiol. 2013;62(7):569–576. | ||
Olaithe M, Bucks RS, Hillman DR, Eastwood PR. Cognitive deficits in obstructive sleep apnea: insights from a meta-review and comparison with deficits observed in COPD, insomnia, and sleep deprivation. Sleep Med Rev. Epub 2017 Mar 30. | ||
Wheaton AG, Perry GS, Chapman DP, Croft JB. Sleep disordered breathing and depression among U.S. adults: National Health and Nutrition Examination Survey, 2005–2008. Sleep. 2012;35(4):461–467. | ||
Antic NA, Catcheside P, Buchan C, et al. The effect of CPAP in normalizing daytime sleepiness, quality of life, and neurocognitive function in patients with moderate to severe OSA. Sleep. 2011;34(1):111–119. | ||
Romero E, Krakow B, Haynes P, Ulibarri V. Nocturia and snoring: predictive symptoms for obstructive sleep apnea. Sleep Breath. 2010;14(4):337–343. | ||
Mulgrew AT, Ryan CF, Fleetham JA, et al. The impact of obstructive sleep apnea and daytime sleepiness on work limitation. Sleep Med. 2007;9(1):42–53. | ||
Howard ME, Desai AV, Grunstein RR, et al. Sleepiness, sleep-disordered breathing, and accident risk factors in commercial vehicle drivers. Am J Respir Crit Care Med. 2004;170(9):1014–1021. | ||
Stoohs RA, Guilleminault C, Itoi A, Dement WC. Traffic accidents in commercial long-haul truck drivers: the influence of sleep-disordered breathing and obesity. Sleep. 1994;17(7):619–623. | ||
Hillman DR, Murphy AS, Pezzullo L. The economic cost of sleep disorders. Sleep. 2006;29(3):299–305. | ||
Sleep Health Foundation. Asleep on the job: Costs of inadequate sleep in Australia; 2017:1–112. | ||
Montserrat JM, Ferrer M, Hernandez L, et al. Effectiveness of CPAP treatment in daytime function in sleep apnea syndrome: a randomized controlled study with an optimized placebo. Am J Resp Crit Care Med. 2001;164(4):608–613. | ||
Knudsen TB, Laulund AS, Ingerslev J, Homøe P, Pinholt EM. Improved apnea-hypopnea index and lowest oxygen saturation after maxillomandibular advancement with or without counterclockwise rotation in patients with obstructive sleep apnea: a meta-analysis. J Oral Maxillofac Surg 2015;73(4):719–726. | ||
George CF. Reduction in motor vehicle collisions following treatment of sleep apnoea with nasal CPAP. Thorax. 2001;56(7):508–512. | ||
Dong Y, Dai Y, Wei G, Cha L, Li X. Effect of continuous positive airway pressure on blood pressure in hypertensive patients with coronary artery bypass grafting and obstructive sleep apnea. Int J Clin Exp Med. 2014;7(11):4308–4315. eCollection 2014. | ||
Olsen S, Smith S, Oei T, Douglas J. Health belief model predicts adherence to CPAP before experience with CPAP. Eur Respir J. 2008; 32(3):710–717. | ||
Libman E, Bailes S, Fichten CS, et al. CPAP treatment adherence in women with obstructive sleep apnea. Sleep Disord. 2017; 2017:2760650. | ||
Ferguson KA, Ono T, Lowe AA, Keenan SP, Fleetham JA. A randomized crossover study of an oral appliance vs nasal-continuous positive airway pressure in the treatment of mild-moderate obstructive sleep apnea. Chest. 1996;109(5):1269–1275. | ||
Noller MW, Guilleminault C, Gouveia CJ, et al. Mandibular advancement for adult obstructive sleep apnea: a systematic review and meta-analysis. J Craniomaxillofac Surg. Epub 2017 Oct 13. | ||
Choi JH, Lee JY, Cha J, Kim K, Hong SN, Lee SH. Predictive models of objective oropharyngeal OSA surgery outcomes: success rate and AHI reduction ratio. PLoS One. 2017;12(9):e0185201. | ||
Phillips CL, Grunstein RR, Darendeliler MA, et al. Health outcomes of continuous positive airway pressure versus oral appliance treatment for obstructive sleep apnea: a randomized controlled trial. Am J Respir Crit Care Med. 2013;187(8):879–887. | ||
Appleton SL, Vakulin A, McEvoy RD, et al. Undiagnosed obstructive sleep apnea is independently associated with reductions in quality of life in middle-aged, but not elderly men of a population cohort. Sleep Breath. 2015;19(4):1309–1316. | ||
Simpson L, Hillman DR, Cooper MN, et al. High prevalence of undiagnosed obstructive sleep apnoea in the general population and methods for screening for representative controls. Sleep Breath. 2013;17(3):967–973. | ||
Kapur V, Strohl KP, Redline S, Iber C, O’Connor G, Nieto J. Underdiagnosis of sleep apnea syndrome in U.S. communities. Sleep Breath. 2002;6(2):49–54. | ||
Jaiswal SJ, Owens RL, Malhotra A. Raising awareness about sleep disorders. Lung India. 2017;34(3):262–268. | ||
Bixler EO, Vgontzas AN, Lin HM, Calhoun SL, Vela-Bueno A, Kales A. Excessive daytime sleepiness in a general population sample: the role of sleep apnea, age, obesity, diabetes, and depression. J Clin Endocrinol Metab. 2005;90(8):4510–4515. | ||
Gray EL, McKenzie DK, Eckert DJ. Obstructive sleep apnea without obesity is common and difficult to treat: evidence for a distinct pathophysiological phenotype. J Clin Sleep Med. 2017;13(1):81–88. | ||
Ye L, Pien GW, Ratcliffe SJ, et al. The different clinical faces of obstructive sleep apnoea: a cluster analysis. Eur Respir J. 2014;44(6):1600–1607. | ||
Arnardottir ES, Bjornsdottir E, Olafsdottir KA, Benediktsdottir B, Gislason T. Obstructive sleep apnoea in the general population: highly prevalent but minimal symptoms. Eur Respir J. 2016;47(1):194–202. | ||
Eckert DJ. Phenotypic approaches to obstructive sleep apnoea – new pathways for targeted therapy. Sleep Med Rev. 2018;37:45–59. | ||
Eckert DJ, White DP, Jordan AS, Malhotra A, Wellman A. Defining phenotypic causes of obstructive sleep apnea. Identification of novel therapeutic targets. Am J Respir Crit Care Med. 2013;188(8):996–1004. | ||
Carberry JC, Amatoury J, Eckert DJ. Personalized management approach for OSA. Chest. Epub 2017 June 16. | ||
Landry SA, Joosten SA, Eckert DJ, et al. Therapeutic CPAP level predicts upper airway collapsibility in patients with obstructive sleep apnea. Sleep. 2017;40(6). | ||
Edwards BA, Eckert DJ, McSharry DG, et al. Clinical predictors of the respiratory arousal threshold in patients with obstructive sleep apnea. Am J Respir Crit Care Med. 2014;190(11):1293–1300. | ||
Genta PR, Sands SA, Butler JP, et al. Airflow shape is associated with the pharyngeal structure causing OSA. Chest. 2017;152(3):537–546. | ||
Azarbarzin A, Sands SA, Taranto-Montemurro L, et al. Estimation of pharyngeal collapsibility during sleep by peak inspiratory airflow. Sleep. 2017;40(1). | ||
Terrill PI, Edwards BA, Nemati S, et al. Quantifying the ventilatory control contribution to sleep apnoea using polysomnography. Eur Respir J. 2015;45(2):408–418. | ||
Hirata RP, Schorr F, Kayamori F, et al. Upper airway collapsibility assessed by negative expiratory pressure while awake is associated with upper airway anatomy. J Clin Sleep Med. 2016;12(10):1339–1346. | ||
Osman A, Nguyen C, Carberry JC, et al. Awake upper airway collapsibility is related to airway collapsibility during sleep (Pcrit) in obstructive sleep apnea. J Sleep Res. 2016;25(2):A57. | ||
Wang D, Marshall NS, Duffin J, et al. Phenotyping interindividual variability in obstructive sleep apnoea response to temazepam using ventilatory chemoreflexes during wakefulness. J Sleep Res. 2011;20(4):526–532. | ||
Wang D, Somogyi AA, Yee BJ, et al. The effects of a single mild dose of morphine on chemoreflexes and breathing in obstructive sleep apnea. Respir Physiol Neurobiol. 2013;185(3):526–532. | ||
Edwards BA, Wellman A, Owens RL. PSGs: more than just the AHI. J Clin Sleep Med. 2013;9(6):527–528. | ||
Cairns A, Poulos G, Bogan R. Who is getting tested for obstructive sleep apnea using a portable recording system? Test results from 193,221 patients. J Clin Sleep Med. 2014;10(11):1193–1198. | ||
Donovan LM, Patel SR. Making the most of simplified sleep apnea testing. Ann Intern Med. 2017;166(5):366–367. | ||
Collop NA, Anderson WM, Boehlecke B, et al; Portable Monitoring Task Force of the American Academy of Sleep Medicine. Clinical guidelines for the use of unattended portable monitors in the diagnosis of obstructive sleep apnea in adult patients. Portable Monitoring Task Force of the American Academy of Sleep Medicine. J Clin Sleep Med. 2007;3(7):737–747. | ||
Muraja-Murro A, Nurkkala J, Tiihonen P, et al. Total duration of apnea and hypopnea events and average desaturation show significant variation in patients with a similar apnea-hypopnea index. J Med Eng Technol. 2012;36(8):393–398. | ||
Alex R, Manchikatla S, Machiraju K, et al. Effect of apnea duration on apnea induced variations in cerebral blood flow velocity and arterial blood pressure. Conf Proc IEEE Eng Med Biol Soc. 2014;2014:270–273. | ||
Wu H, Zhan X, Zhao M, Wei Y. Mean apnea-hypopnea duration (but not apnea-hypopnea index) is associated with worse hypertension in patients with obstructive sleep apnea. Medicine (Baltimore). 2016;95(48):e5493. | ||
Sankari A, Pranathiageswaran S, Maresh S, Hosni AM, Badr MS. Characteristics and consequences of non-apneic respiratory events during sleep. Sleep. 2017;40(1). | ||
Koch H, Schneider LD, Finn LA, et al. Breathing disturbances without hypoxia are associated with objective sleepiness in sleep apnea. Sleep. 2017;40(11). | ||
Weaver EM, Woodson BT, Steward DL. Polysomnography indexes are discordant with quality of life, symptoms, and reaction times in sleep apnea patients. Otolaryngol Head Neck Surg. 2005;132(2):255–262. | ||
Alzoubaidi M, Mokhlesi B. Obstructive sleep apnea during rapid eye movement sleep: clinical relevance and therapeutic implications. Curr Opin Pulm Med. 2016;22(6):545–554. | ||
Appleton SL, Vakulin A, Martin SA, et al. Hypertension is associated with undiagnosed OSA during rapid eye movement sleep. Chest. 2016;150(3):495–505. | ||
Aurora RN, Crainiceanu C, Gottlieb DJ, Kim JS, Punjabi NM. Obstructive sleep apnea during rapid eye movement sleep and cardiovascular disease. Am J Respir Crit Care Med. Epub 2017 Nov 7. | ||
Chami HA, Gottlieb DJ, Redline S, Punjabi NM. Association between glucose metabolism and sleep-disordered breathing during REM sleep. Am J Respir Crit Care Med. 2015;192(9):1118–1126. | ||
Neelapu BC, Kharbanda OP, Sardana HK, et al. Craniofacial and upper airway morphology in adult obstructive sleep apnea patients: a systematic review and meta-analysis of cephalometric studies. Sleep Med Rev. 2017;31:79–90. | ||
Edwards BA, Eckert DJ, Jordan AS. Obstructive sleep apnoea pathogenesis from mild to severe: is it all the same? Respirology. 2017;22(1):33–42. | ||
Eckert DJ, Wellman A. Physiological phenotypes. In: Barbé F, Pépin JL, editors. European Respiratory Monograph: Obstructive Sleep Apnoea. Plymouth, UK: European Respiratory Society; 2015:9–23. | ||
Dempsey JA, Xie A, Patz DS, Wang D. Physiology in medicine: obstructive sleep apnea pathogenesis and treatment--considerations beyond airway anatomy. J Appl Physiol (1985). 2014;116(1):3–12. | ||
Carlisle T, Carthy ER, Glasser M, et al. Upper airway factors that protect against obstructive sleep apnoea in healthy older males. Eur Respir J. 2014;44(3):685–693. | ||
Safiruddin F, Koutsourelakis I, de Vries N. Upper airway collapse during drug induced sleep endoscopy: head rotation in supine position compared with lateral head and trunk position. Eur Arch Otorhinolaryngol. 2015;272(2):485–488. | ||
Coxson HO, Eastwood PR, Williamson JP, Sin DD. Phenotyping airway disease with optical coherence tomography. Respirology. 2011;16(1):34–43. | ||
Ito E, Tsuiki S, Maeda K, Okajima I, Inoue Y. Oropharyngeal crowding closely relates to aggravation of OSA. Chest. 2016;150(2):346–352. | ||
Schwab RJ, Pasirstein M, Pierson R, et al. Identification of upper airway anatomic risk factors for obstructive sleep apnea with volumetric magnetic resonance imaging. Am J Respir Crit Care Med. 2003;168(5):522–530. | ||
Segal Y, Malhotra A, Pillar G. Upper airway length may be associated with the severity of obstructive sleep apnea syndrome. Sleep Breath. 2008;12(4):311–316. | ||
Morrison DL, Launois SH, Isono S, Feroah TR, Whitelaw WA, Remmers JE. Pharyngeal narrowing and closing pressures in patients with obstructive sleep apnea. Am Rev Respir Dis. 1993;148(3):606–611. | ||
Young T, Peppard PE, Taheri S. Excess weight and sleep-disordered breathing. J Appl Physiol (1985). 2005;99(4):1592–1599. | ||
Lai CC, Friedman M, Lin HC, et al. Clinical predictors of effective continuous positive airway pressure in patients with obstructive sleep apnea/hypopnea syndrome. Laryngoscope. 2015;125(8):1983–1987. | ||
Pinto JA, Godoy LB, Marquis VW, Sonego TB, Leal Cde F, Artico MS. Anthropometric data as predictors of Obstructive Sleep Apnea Severity. Braz J Otorhinolaryngol. 2011;77(4):516–521. | ||
Caron CJJM, Pluijmers BI, Maas BDPJ, et al. Obstructive sleep apnoea in craniofacial microsomia: analysis of 755 patients. Int J Oral Maxillofac Surg. 2017;46(10):1330–1337. | ||
Schorr F, Kayamori F, Hirata RP, et al. Different craniofacial characteristics predict upper airway collapsibility in Japanese-Brazilian and white men. Chest. 2016;149(3):737–746. | ||
Sforza E, Bacon W, Weiss T, Thibault A, Petiau C, Krieger J. Upper airway collapsibility and cephalometric variables in patients with obstructive sleep apnea. Am J Respir Crit Care Med. 2000;161(2 Pt 1):347–352. | ||
Verma M, Seto-Poon M, Wheatley JR, Amis TC, Kirkness JP. Influence of breathing route on upper airway lining liquid surface tension in humans. J Physiol. 2006;574(Pt 3):859–866. | ||
Weiss TM, Atanasov S, Calhoun KH. The association of tongue scalloping with obstructive sleep apnea and related sleep pathology. Otolaryngol Head Neck Surg. 2005;133(6):966–971. | ||
Kim AM, Keenan BT, Jackson N, et al. Tongue fat and its relationship to obstructive sleep apnea. Sleep. 2014;37(10):1639–1648. | ||
Gleadhill IC, Schwartz AR, Schubert N, Wise RA, Permutt S, Smith PL. Upper airway collapsibility in snorers and in patients with obstructive hypopnea and apnea. Am Rev Respir Dis. 1991;143(6):1300–1303. | ||
Eastwood PR, Szollosi I, Platt PR, Hillman DR. Collapsibility of the upper airway during anesthesia with isoflurane. Anesthesiology. 2002;97(4):786–793. | ||
Schwartz AR, O’Donnell CP, Baron J, et al. The hypotonic upper airway in obstructive sleep apnea: role of structures and neuromuscular activity. Am J Respir Crit Care Med. 1998;157(4 Pt 1):1051–1057. | ||
Carberry JC, Jordan AS, White DP, Wellman A, Eckert DJ. Upper airway collapsibility (Pcrit) and pharyngeal dilator muscle activity are sleep stage dependent. Sleep. 2016;39(3):511–521. | ||
Kirkness JP, Peterson LA, Squier SB, et al. Performance characteristics of upper airway critical collapsing pressure measurements during sleep. Sleep. 2011;34(4):459–467. | ||
Genta PR, Schorr F, Eckert DJ, et al. Upper airway collapsibility is associated with obesity and hyoid position. Sleep. 2014;37(10):1673–1678. | ||
Kirkness JP, Schwartz AR, Schneider H, et al. Contribution of male sex, age, and obesity to mechanical instability of the upper airway during sleep. J Appl Physiol (1985). 2008;104(6):1618–1624. | ||
Bachar G, Feinmesser R, Shpitzer T, Yaniv E, Nageris B, Eidelman L. Laryngeal and hypopharyngeal obstruction in sleep disordered breathing patients, evaluated by sleep endoscopy. Eur Arch Otorhinolaryngol. 2008;265(11):1397–1402. | ||
Horner RL, Hughes SW, Malhotra A. State-dependent and reflex drives to the upper airway: basic physiology with clinical implications. J Appl Physiol (1985). 2014;116(3):325–336. | ||
Kubin L. Neural control of the upper airway: respiratory and state-dependent mechanisms. Compr Physiol. 2016;6(4):1801–1850. | ||
Saboisky JP, Butler JE, Fogel RB, et al. Tonic and phasic respiratory drives to human genioglossus motoneurons during breathing. J Neurophysiol. 2006;95(4):2213–2221. | ||
Wilkinson V, Malhotra A, Nicholas CL, et al. Discharge patterns of human genioglossus motor units during sleep onset. Sleep. 2008;31(4):525–533. | ||
Nicholas CL, Jordan AS, Heckel L, et al. Discharge patterns of human tensor palatini motor units during sleep onset. Sleep. 2012;35(5):699–707. | ||
Eckert DJ, Saboisky JP, Jordan AS, White DP, Malhotra A. A secondary reflex suppression phase is present in genioglossus but not tensor palatini in response to negative upper airway pressure. J Appl Physiol (1985). 2010;108(6):1619–1624. | ||
Eckert DJ, Malhotra A, Lo YL, White DP, Jordan AS. The influence of obstructive sleep apnea and gender on genioglossus activity during rapid eye movement sleep. Chest. 2009;135(4):957–964. | ||
Eckert DJ, McEvoy RD, George KE, Thomson KJ, Catcheside PG. Genioglossus reflex inhibition to upper-airway negative-pressure stimuli during wakefulness and sleep in healthy males. J Physiol. 2007;581(Pt 3):1193–1205. | ||
Loewen AH, Ostrowski M, Laprairie J, Maturino F, Hanly PJ, Younes M. Response of genioglossus muscle to increasing chemical drive in sleeping obstructive apnea patients. Sleep. 2011;34(8):1061–1073. | ||
Jordan AS, Wellman A, Heinzer RC, et al. Mechanisms used to restore ventilation after partial upper airway collapse during sleep in humans. Thorax. 2007;62(10):861–867. | ||
Sands SA, Eckert DJ, Jordan AS, et al. Enhanced upper-airway muscle responsiveness is a distinct feature of overweight/obese individuals without sleep apnea. Am J Respir Crit Care Med. 2014;190(8):930–937. | ||
Dotan Y, Pillar G, Tov N, et al. Dissociation of electromyogram and mechanical response in sleep apnoea during propofol anaesthesia. Eur Respir J. 2013;41(1):74–84. | ||
Cheng S, Butler JE, Gandevia SC, Bilston LE. Movement of the tongue during normal breathing in awake healthy humans. J Physiol. 2008;586(17):4283–4294. | ||
Brown EC, Cheng S, McKenzie DK, Butler JE, Gandevia SC, Bilston LE. Respiratory movement of upper airway tissue in obstructive sleep apnea. Sleep. 2013;36(7):1069–1076. | ||
Cheng S, Brown EC, Hatt A, Butler JE, Gandevia SC, Bilston LE. Healthy humans with a narrow upper airway maintain patency during quiet breathing by dilating the airway during inspiration. J Physiol. 2014;592(21):4763–4774. | ||
Jordan AS, White DP, Owens RL, et al. The effect of increased genioglossus activity and end-expiratory lung volume on pharyngeal collapse. J Appl Physiol (1985). 2010;109(2):469–475. | ||
Dotan Y, Pillar G, Schwartz AR, Oliven A. Asynchrony of lingual muscle recruitment during sleep in obstructive sleep apnea. J Appl Physiol (1985). 2015;118(12):1516–1524. | ||
Strollo PJ Jr, Soose RJ, Maurer JT, et al; STAR Trial Group. Upper-airway stimulation for obstructive sleep apnea. N Eng J Med. 2014;370(2):139–149. | ||
Bisogni V, Pengo MF, De Vito A, et al. Electrical stimulation for the treatment of obstructive sleep apnoea: a review of the evidence. Expert Rev Respir Med. 2017;11(9):711–720. | ||
Hida W, Okabe S, Miki H, et al. Effects of submental stimulation for several consecutive nights in patients with obstructive sleep apnoea. Thorax. 1994;49(5):446–452. | ||
Hofauer B, Philip P, Wirth M, Knopf A, Heiser C. Effects of upper-airway stimulation on sleep architecture in patients with obstructive sleep apnea. Sleep Breath. 2017;21(4):901–908. | ||
Schwartz AR, Barnes M, Hillman D, et al. Acute upper airway responses to hypoglossal nerve stimulation during sleep in obstructive sleep apnea. Am J Respir Crit Care Med. 2012;185(4):420–426. | ||
Schwartz AR, Smith PL, Oliven A. Electrical stimulation of the hypoglossal nerve: a potential therapy. J Appl Physiol (1985). 2014;116(3):337–344. | ||
Eastwood PR, Barnes M, Walsh JH, et al. Treating obstructive sleep apnea with hypoglossal nerve stimulation. Sleep. 2011;34(11):1479–1486. | ||
Steffen A, Sommer JU, Hofauer B, Maurer JT, Hasselbacher K, Heiser C. Outcome after one year of upper airway stimulation for obstructive sleep apnea in a multicenter German post-market study. Laryngoscope. Epub 2017 May 31. | ||
Gillespie MB, Soose RJ, Woodson BT, et al; STAR Trial Investigators. Upper airway stimulation for obstructive sleep apnea: patient-reported outcomes after 48 months of follow-up. Otolaryngol Head Neck Surg. 2017;156(4):765–771. | ||
Woodson BT, Soose RJ, Gillespie MB, et al; STAR Trial Investigators. Three-year outcomes of cranial nerve stimulation for obstructive sleep apnea: the STAR Trial. Otolaryngol Head Neck Surg. 2016;154(1):181–188. | ||
White DP. Pharmacologic approaches to the treatment of obstructive sleep apnea. Sleep Med Clin. 2016;11(2):203–212. | ||
Horner RL, Grace KP, Wellman A. A resource of potential drug targets and strategic decision-making for obstructive sleep apnoea pharmacotherapy. Respirology. 2017;22(5):861–873. | ||
Taranto-Montemurro L, Edwards BA, Sands SA, et al. Desipramine increases genioglossus activity and reduces upper airway collapsibility during Non-REM sleep in healthy subjects. Am J Respir Crit Care Med. 2016;194(7):878–885. | ||
Taranto-Montemurro L, Sands SA, Edwards BA, et al. Desipramine improves upper airway collapsibility and reduces OSA severity in patients with minimal muscle compensation. Eur Respir J. 2016;48(5):1340–1350. | ||
Marshall NS, Yee BJ, Desai AV, et al. Two randomized placebo-controlled trials to evaluate the efficacy and tolerability of mirtazapine for the treatment of obstructive sleep apnea. Sleep. 2008;31(6):824–831. | ||
Kraiczi H, Hedner J, Dahlöf P, Ejnell H, Carlson J. Effect of serotonin uptake inhibition on breathing during sleep and daytime symptoms in obstructive sleep apnea. Sleep. 1999;22(1):61–67. | ||
Taranto-Montemurro L, Sands SA, Azarbarzin A, et al. Effect of 4-aminopyridine on genioglossus muscle activity during sleep in healthy adults. Ann Am Thorac Soc. 2017;14(7):1177–1183. | ||
Carberry JC, Fisher LP, Grunstein RR, et al. Role of common hypnotics on the phenotypic causes of OSA: paradoxical effects of zolpidem. Eur Respir J. 2017;50(6):pii:1701344. | ||
Horton GA, Fraigne JJ, Torontali ZA, et al. Activation of the hypoglossal to tongue musculature motor pathway by remote control. Sci Rep. 2017;7:45860. | ||
Fleury Curado T, Fishbein K, Pho H, et al. Chemogenetic stimulation of the hypoglossal neurons improves upper airway patency. Sci Rep. 2017;7:44392. | ||
Ieto V, Kayamori F, Montes MI, et al. Effects of oropharyngeal exercises on snoring: a randomized trial. Chest. 2015;148(3):683–691. | ||
Guimarães KC, Drager LF, Genta PR, Marcondes BF, Lorenzi-Filho G. Effects of oropharyngeal exercises on patients with moderate obstructive sleep apnea syndrome. Am J Respir Crit Care Med. 2009;179(10):962–966. | ||
Puhan MA, Suarez A, Lo Cascio C, Zahn A, Heitz M, Braendli O. Didgeridoo playing as alternative treatment for obstructive sleep apnoea syndrome: randomised controlled trial. BMJ. 2006;332(7536):266–270. | ||
Camacho M, Certal V, Abdullatif J, et al. Myofunctional therapy to treat obstructive sleep apnea: a systematic review and meta-analysis. Sleep. 2015;38(5):669–675. | ||
Eckert DJ, Younes MK. Arousal from sleep: implications for obstructive sleep apnea pathogenesis and treatment. J Appl Physiol (1985). 2014;116(3):302–313. | ||
Phillipson EA, Sullivan CE. Arousal: the forgotten response to respiratory stimuli. Am Rev Respir Dis. 1978;118(5):807–809. | ||
Remmers JE, deGroot WJ, Sauerland EK, Anch AM. Pathogenesis of upper airway occlusion during sleep. J Appl Physiol Respir Environ Exerc Physiol. 1978;44(6):931–938. | ||
Younes M. Role of arousals in the pathogenesis of obstructive sleep apnea. Am J Respir Crit Care Med. 2004;169(5):623–633. | ||
Rees K, Spence DP, Earis JE, Calverley PM. Arousal responses from apneic events during non-rapid-eye-movement sleep. Am J Respir Crit Care Med. 1995;152(3):1016–1021. | ||
Younes M, Ostrowski M, Atkar R, Laprairie J, Siemens A, Hanly P. Mechanisms of breathing instability in patients with obstructive sleep apnea. J Appl Physiol (1985). 2007;103(6):1929–1941. | ||
Ratnavadivel R, Chau N, Stadler D, Yeo A, McEvoy RD, Catcheside PG. Marked reduction in obstructive sleep apnea severity in slow wave sleep. J Clin Sleep Med. 2009;5(6):519–524. | ||
White DP, Younes MK. Obstructive sleep apnea. Compr Physiol. 2012;2(4):2541–2594. | ||
Younes M. Role of respiratory control mechanisms in the pathogenesis of obstructive sleep disorders. J Appl Physiol (1985). 2008;105(5):1389–1405. | ||
Younes M, Ostrowski M, Thompson W, Leslie C, Shewchuk W. Chemical control stability in patients with obstructive sleep apnea. Am J Respir Crit Care Med. 2001;163(5):1181–1190. | ||
Puddy A, Giesbrecht G, Sanii R, Younes M. Mechanism of detection of resistive loads in conscious humans. J Appl Physiol (1985). 1992;72(6):2267–2270. | ||
Younes M, Jung D, Puddy A, Giesbrecht G, Sanii R. Role of the chest wall in detection of added elastic loads. J Appl Physiol (1985). 1990;68(5):2241–2245. | ||
Banzett RB, Lansing RW, Brown R, et al. ‘Air hunger’ from increased PCO2 persists after complete neuromuscular block in humans. Respir Physiol. 1990;81(1):1–17. | ||
Chamberlin NL. Brain circuitry mediating arousal from obstructive sleep apnea. Curr Opin Neurobiol. 2013;23(5):774–779. | ||
Berry RB, Gleeson K. Respiratory arousal from sleep: mechanisms and significance. Sleep. 1997;20(8):654–675. | ||
Carter SG, Berger MS, Carberry JC, et al. Zopiclone increases the arousal threshold without impairing genioglossus activity in obstructive sleep apnea. Sleep. 2016;39(4):757–766. | ||
Eckert DJ, Owens RL, Kehlmann GB, et al. Eszopiclone increases the respiratory arousal threshold and lowers the apnoea/hypopnoea index in obstructive sleep apnoea patients with a low arousal threshold. Clin Sci (Lond). 2011;120(12):505–514. | ||
Edwards BA, Sands SA, Owens RL, et al. The combination of supplemental oxygen and a hypnotic markedly improves obstructive sleep apnea in patients with a mild to moderate upper airway collapsibility. Sleep. 2016;39(11):1973–1983. | ||
Gleeson K, Zwillich CW, White DP. The influence of increasing ventilatory effort on arousal from sleep. Am Rev Respir Dis. 1990;142(2):295–300. | ||
Sforza E, Krieger J, Petiau C. Arousal threshold to respiratory stimuli in OSA patients: evidence for a sleep-dependent temporal rhythm. Sleep. 1999;22(1):69–75. | ||
Berry RB, Asyali MA, McNellis MI, Khoo MC. Within-night variation in respiratory effort preceding apnea termination and EEG delta power in sleep apnea. J Appl Physiol (1985). 1998;85(4):1434–1441. | ||
Jordan AS, O’Donoghue FJ, Cori JM, Trinder J. Physiology of arousal in obstructive sleep apnea and potential impacts for sedative treatment. Am J Respir Crit Care Med. 2017;196(7):814–821. | ||
Berry RB, Kouchi KG, Der DE, Dickel MJ, Light RW. Sleep apnea impairs the arousal response to airway occlusion. Chest. 1996;109(6):1490–1496. | ||
Haba-Rubio J, Sforza E, Weiss T, Schröder C, Krieger J. Effect of CPAP treatment on inspiratory arousal threshold during NREM sleep in OSAS. Sleep Breath. 2005;9(1):12–19. | ||
Eckert DJ, Malhotra A, Wellman A, White DP. Trazodone increases the respiratory arousal threshold in patients with obstructive sleep apnea and a low arousal threshold. Sleep. 2014;37(4):811–819. | ||
Smales ET, Edwards BA, Deyoung PN, et al. Trazodone effects on obstructive sleep apnea and non-REM arousal threshold. Ann Am Thorac Soc. 2015;12(5):758–764. | ||
Carberry JC, Fisher LP, Carter SG, et al. Respiratory sensation during wakefulness is related to the respiratory arousal threshold during sleep in obstructive sleep apnoea. Sleep Biol Rhythms. 2015;13:58. | ||
Benetó A, Gomez-Siurana E, Rubio-Sanchez P. Comorbidity between sleep apnea and insomnia. Sleep Med Rev. 2009;13(4):287–293. | ||
Krakow B, Romero E, Ulibarri VA, Kikta S. Prospective assessment of nocturnal awakenings in a case series of treatment-seeking chronic insomnia patients: a pilot study of subjective and objective causes. Sleep. 2012;35(12):1685–1692. | ||
Krakow B, Melendrez D, Ferreira E, et al. Prevalence of insomnia symptoms in patients with sleep-disordered breathing. Chest. 2001;120(6):1923–1929. | ||
Mateika JH, Panza G, Alex R, El-Chami M. The impact of intermittent or sustained carbon dioxide on intermittent hypoxia initiated respiratory plasticity. What is the effect of these combined stimuli on apnea severity? Respir Physiol Neurobiol. Epub 2017 Oct 31. | ||
Loewen A, Ostrowski M, Laprairie J, et al. Determinants of ventilatory instability in obstructive sleep apnea: inherent or acquired? Sleep. 2009;32(10):1355–1365. | ||
Wellman A, Edwards BA, Sands SA, et al. A simplified method for determining phenotypic traits in patients with obstructive sleep apnea. J Appl Physiol (1985). 2013;114(7):911–922. | ||
Wellman A, Eckert DJ, Jordan AS, et al. A method for measuring and modeling the physiological traits causing obstructive sleep apnea. J Appl Physiol (1985). 2011;110(6):1627–1637. | ||
Wellman A, Malhotra A, Jordan AS, Stevenson KE, Gautam S, White DP. Effect of oxygen in obstructive sleep apnea: role of loop gain. Respir Physiol Neurobiol. 2008;162(2):144–151. | ||
Edwards BA, Sands SA, Eckert DJ, et al. Acetazolamide improves loop gain but not the other physiological traits causing obstructive sleep apnoea. J Physiol. 2012;590(5):1199–1211. | ||
Xie A, Teodorescu M, Pegelow DF, et al. Effects of stabilizing or increasing respiratory motor outputs on obstructive sleep apnea. J Appl Physiol (1985). 2013;115(1):22–33. | ||
El-Chami M, Shaheen D, Ivers B, et al. Time of day affects chemoreflex sensitivity and the carbon dioxide reserve during NREM sleep in participants with sleep apnea. J Appl Physiol (1985). 2014;117(10):1149–1156. | ||
El-Chami M, Shaheen D, Ivers B, et al. Time of day affects the frequency and duration of breathing events and the critical closing pressure during NREM sleep in participants with sleep apnea. J Appl Physiol (1985). 2015;119(6):617–626. | ||
Wains SA, El-Chami M, Lin HS, Mateika JH. Impact of arousal threshold and respiratory effort on the duration of breathing events across sleep stage and time of night. Respir Physiol Neurobiol. 2017;237:35–41. | ||
Djonlagic I, Saboisky J, Carusona A, Stickgold R, Malhotra A. Increased sleep fragmentation leads to impaired off-line consolidation of motor memories in humans. PLoS One. 2012;7(3):e34106. | ||
Amatoury J, Azarbarzin A, Younes M, Jordan AS, Wellman A, Eckert DJ. Arousal intensity is a distinct pathophysiological trait in obstructive sleep apnea. Sleep. 2016;39(12):2091–2100. | ||
Mateika JH, El-Chami M, Shaheen D, Ivers B. Intermittent hypoxia: a low-risk research tool with therapeutic value in humans. J Appl Physiol (1985). 2015;118(5):520–532. | ||
Mateika JH, Komnenov D. Intermittent hypoxia initiated plasticity in humans: a multipronged therapeutic approach to treat sleep apnea and overlapping co-morbidities. Exp Neurol. 2017;287(Pt 2):113–129. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.