")
Back to Journals » Therapeutics and Clinical Risk Management » Volume 15
Obstacles to early treatment of idiopathic pulmonary fibrosis: current perspectives
Received 1 October 2018
Accepted for publication 13 December 2018
Published 3 January 2019 Volume 2019:15 Pages 73—81
DOI https://doi.org/10.2147/TCRM.S160248
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Garry Walsh
Teng Moua, Jay H Ryu
Department of Medicine, Division of Pulmonary and Critical Care Medicine, Mayo Clinic, Rochester MN, USA
Abstract: Idiopathic pulmonary fibrosis is a progressive and fatal fibrotic lung disease which has seen new opportunity for drug treatment in the last several years with the approval of nintedanib and pirfenidone, two antifibrotic agents aimed at slowing decline in lung function as defined by FVC on pulmonary function testing. Despite these promising effects, delays in drug initiation have been reported undermining the premise that earlier drug initiation may sustain lung function and prolong survival. This review explores obstacles to earlier treatment, inclusive of defining so-called early idiopathic pulmonary fibrosis, difficulties in achieving a confident diagnosis in that setting, and uncertainties regarding drug-related benefits among specific patient subgroups such as those with no symptoms or advanced disease at presentation. Goals of therapy balanced with the burdens associated with antifibrotic drug therapy are negotiated on an individual basis. We review the evidence for and against earlier initiation of antifibrotic drug therapy along with its role in patient-centered outcomes.
Keywords: idiopathic pulmonary fibrosis, nintedanib, pirfenidone, antifibrotics
Introduction
Idiopathic pulmonary fibrosis (IPF) is a progressive fibrotic lung disease associated with worsening lung function and commonly results in death over the course of several years. A pathologic pattern of usual interstitial pneumonia (UIP) on lung biopsy, characteristic radiologic findings, and absence of an identifiable etiology support the diagnosis.1,2 A recent algorithmic estimate of disease prevalence based on insurance claims data suggested 58.7 cases per 100,000 persons,3 while prevalence in patients <65 years was 18.2 cases per 100,000 persons.4 Though the disease characteristically progresses over time, rate of progression may vary among patients and sometimes abruptly worsen due to acute exacerbation.5 Often-cited median survival from time of diagnosis ranges from 2.5 to 3.8 years.1,6
Historical treatments have ranged from corticosteroids and steroid-sparing agents such as azathioprine, to colchicine and N-acetylcysteine. The past decade and a half has seen multiple unsuccessful therapeutic trials, including the use of pulmonary vasodilators7,8 and even warfarin anticoagulation.9 Percent predicted FVC was previously shown to be predictive of survival10 and has been the main outcome measure of therapeutic trials and currently available therapy.11,12 This was until 2014 when two antifibrotic agents, aimed at slowing the rate of FVC% decline, were approved by the Food and Drug Administration for the treatment of IPF in the United States.13,14
The exact biologic mechanism of pirfenidone as an antifibrotic agent is unknown, while nintedanib is a triple tyrosine kinase inhibitor with antifibrotic properties.15 Clinical trials for both drugs demonstrated a statistically significant reduction in the rate of FVC% decline over a 52-week study period. Patients involved in both studies had baseline pulmonary function test (PFT) results considered mild to moderate in terms of impairment (FVC% >50% with percent predicted diffusion capacity of the lungs for carbon monoxide [DLCO%] >30%).13,14 Commonly associated adverse effects include diarrhea and nausea with nintedanib, and nausea and rash with pirfenidone.
As both drugs demonstrated similar efficacy in slowing but not stopping disease progression, a theoretical advantage of initiating drug therapy earlier to preserve remaining lung function and perhaps improve survival appears reasonable. A recent survey of European physicians noted only 60% of confidently diagnosed IPF patients were being prescribed antifibrotic therapy, primarily due to a “watch and wait” approach taken by both clinicians and patients in the presence of mild or stable disease.16 Pros–cons debates in the literature illustrate contentious issues of when and in whom antifibrotic therapy should be provided.17–19 This article highlights current perspectives on potential obstacles to earlier drug initiation, and arguments for or against this therapeutic maneuver (see Figure 1). Our focus will be on the antifibrotic medications, excluding a review of other interventions including antacid therapy or fundoplication, pulmonary vasodilators for those with pulmonary hypertension, and lung transplantation.
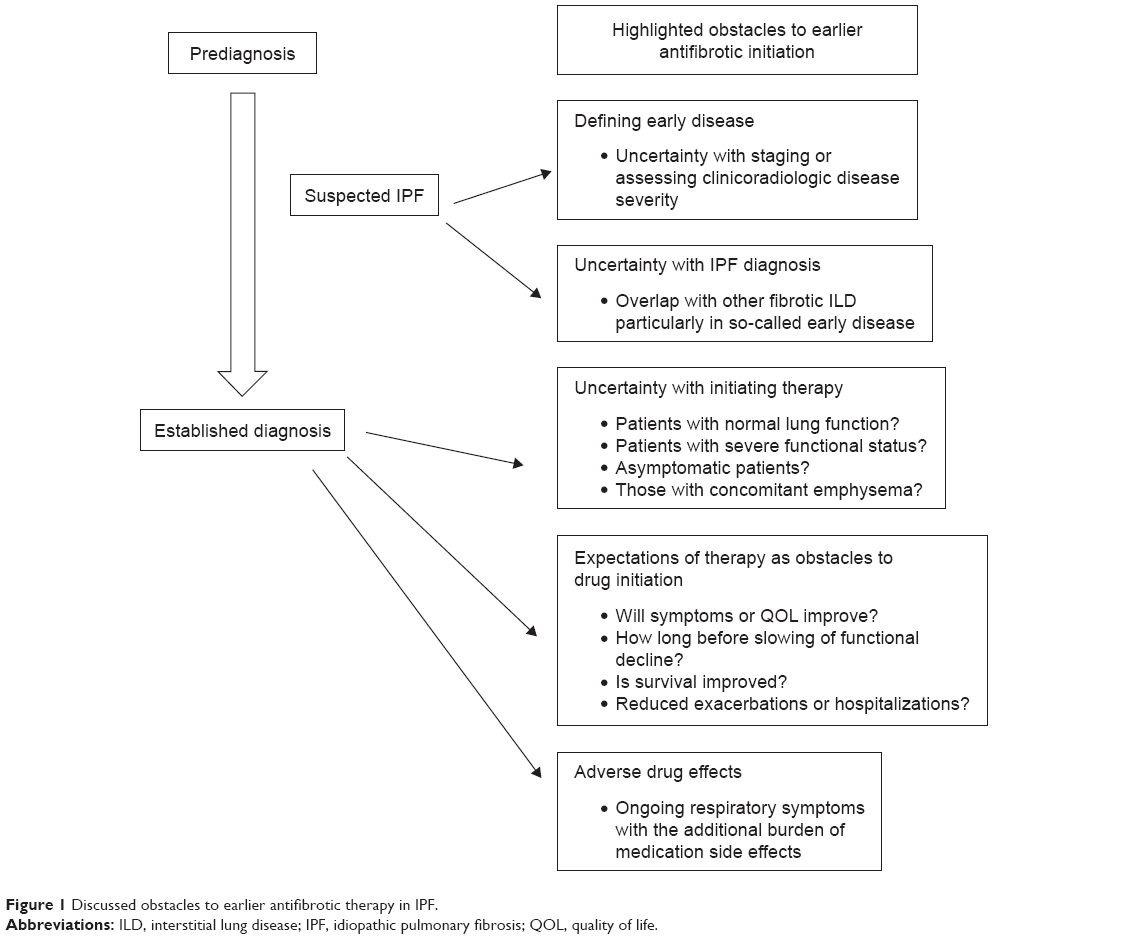 | Figure 1 Discussed obstacles to earlier antifibrotic therapy in IPF. |
Defining “early” IPF
For the purposes of this discussion, earlier antifibrotic treatment in IPF may be thought of in two ways. The first refers to treatment of disease earlier in its natural course or evolution, while the second emphasizes earlier treatment relative to the time of diagnosis, no matter the severity of presentation. For the latter, starting therapy sooner after diagnosis may reflect less the severity of findings but more the balance of benefits vs harms. Treating so-called earlier stage disease instead may mean attempting to treat when diagnosis is not definitively established, or when disease progression or symptoms have not manifested.
As there is no widely accepted staging criteria for IPF, definitions of early disease remain nebulous and confounded by discrepancies between patient-reported symptom burden, severity of radiologic findings, and PFT. FVC% has been recognized as predictive of death in earlier, large, single-center cohort studies,10,20 and remains the primary end point for assessing therapeutic efficacy.21 A recent multi-center review suggested up to 29% of IPF patients may have significant FVC decline (defined as >10%) in the first 6 months after diagnosis.22 PFT criteria for inclusion in the international antifibrotic clinical trials required FVC% to be >50% and DLCO to be >30%.13,14 Applying such trial-specific PFT criteria for treatment of patients in the real world would mean precluding many from receiving therapy, which has been the focus of many posttrial subgroup analyses. It is also recognized that FVC% may not correlate with severity of symptom burden or radiologic disease pattern at diagnosis,23 the former of which is often not well characterized in daily practice.
Radiologic staging based on computed tomography (CT) findings or patterns has also not been well established in IPF. While anecdotally, patients may proceed from so-called earlier or atypical (nondefinitive) UIP to more consistent or definite UIP CT patterns, systematic analysis of this progression is incomplete and may not reflect the extent to which radiologic severity affects survival or morbidity. Prior studies have suggested patients with “possible” UIP CT pattern at diagnosis have improved survival (suggesting perhaps less severe or “earlier” disease),24,25 while a recent study suggested no difference in long-term survival among various UIP CT pattern types.26 Two studies have confirmed radiologic progression from “possible” or “inconsistent” UIP CT pattern to more “consistent UIP” in about third to half of patients.26,27 What is more interesting is the observation that relative pattern stability may be seen in some over many years of follow-up, making the supposition of so-called earlier radiologic disease based on the absence of honeycombing or more extensive fibrotic changes less reliable for delineating how long disease has been present. Delay in the recognition of IPF even when fibrosis is present was recently demonstrated in a large multicenter registry of IPF patients, with a quarter being diagnosed >13 months after an initial scan suggesting fibrotic disease.28
The GAP Index (Gender, Age, and Physiology) is a prognostic prediction tool whose baseline criteria have been used to stage disease severity into three categories.29 Stages I through III are associated with increasing mortality risk. A post-hoc analysis of clinical trial data by GAP staging recently demonstrated its possible role as a research end point.30 It was suggested though that two of its main components, age and gender, which are not influenced by drug therapy, may confound results.
In summary, current staging based on PFT criteria may not completely take into account other measures of disease severity, or predict future disease course. The majority of clinical trials have continued to focus on PFT criteria as end points.31 Criteria such as acute exacerbation or respiratory-related hospitalization have been recently considered as applicable and clinically salient.32 Other measures that may be theoretically relevant for assessing disease severity include quantitative CT assessment,33 6-minute walk distance,34,35 or patient-reported outcomes as assessed on solicited surveys.36 A discussion of disease severity or staging in regards to timing of drug therapy remains challenging. So-called earlier disease is not yet well defined; therefore, treatment initiation may not be so much a matter of when, but in whom and by which parameters.
Getting the right diagnosis – equipoise in the diagnosis of early IPF
The current high cost of antifibrotic therapy and its related adverse effects support the need for an accurate diagnosis of IPF. Accurate diagnosis not only benefits and avoids harm in patients but justifies cost of drug therapy for payers. Antifibrotic therapy is currently approved only for IPF, with the treatment of other fibrotic interstitial lung diseases (ILDs) still undergoing trial investigation. The described clinical, radiologic, and pathologic patterns associated with IPF are not unique and may overlap with autoimmune or exposure-related causes of fibrotic lung disease.
A significant proportion of IPF patients present without honeycombing on CT, which is believed to perhaps represent earlier stage disease. Such CT findings, though, may be difficult to distinguish from other fibrotic ILDs. Advances in radiologic characterization have reduced the need for surgical lung biopsy for diagnosing IPF,37 particularly in those with older age and male gender.38 There is a known radiologic overlap between IPF and idiopathic nonspecific interstitial pneumonia (NSIP),25,39 where the presence of honeycombing while pointing toward UIP may also be seen in up to 32% of pathologically confirmed NSIP.40 Chronic hypersensitivity pneumonitis, which currently does not have widely accepted diagnostic criteria,41,42 may manifest overlapping pathologic and radiologic findings with IPF.43 Serologic findings associated with autoimmune or rheumatologic diseases are not uncommonly found in patients with IPF.44,45 A recent working group delineated more standardized criteria for ILD presenting with autoimmune features (“interstitial pneumonia with autoimmune features”),46 though the presence of UIP among these patients either radiologically or pathologically portended similar outcomes to IPF,47,48 suggesting such patients may simply represent atypical IPF more than true autoimmune-related disease.
Recent interest in IPF patients presenting without honeycombing has led to the identification of additional findings to support a reliable IPF diagnosis in those with nondefinitive radiologic findings and no lung biopsy. Post-hoc analysis of a large clinical trial suggested the accurate diagnosis of IPF in the absence of radiologic honeycombing can be achieved in the presence of typical clinical presentation and demographics.49 A prediction model involving high pretest suspicion of IPF also increased positive predictive value.41 Surgical lung biopsy has been performed less in recent years, despite recent evidence that when performed in the appropriate clinical context, procedure-related mortality rate may be lower than feared.50 The advancement of bronchoscopic cryobiopsy may allow for a less invasive approach to confirming UIP pathology,51 such as in those patients with progressive ILD unresponsive to initial empiric anti-inflammatory or immunosuppressant therapy. The role of bronchoscopic cryobiopsy in the evaluation of diffuse parenchymal lung disease needs further exploration. Finally, the gold standard of multidisciplinary discussion (MDD) to achieve accurate ILD diagnosis may not be widely available or feasible in busy clinical centers, particularly in nonacademic settings. Decline in surgical biopsy may in essence limit MDD to only the clinician and radiologist.
In summary, the absence of radiologic honeycombing in patients with fibrotic ILD may still represent IPF, though significant clinical and radiologic overlap occurs with other fibrotic ILDs, leading to uncertainty in the earlier initiation of antifibrotic therapy. A “watch and wait” approach is often taken under such circumstances to monitor for disease progression over time.
Is IPF diagnosis alone indication for treatment? (When and who should be treated?)
While “early” IPF may be difficult to define and uncertainty is more common in “earlier” or atypically presenting disease, additional questions remain even after a confident diagnosis is made. Such questions may delay initiation of antifibrotic therapy and include the following:
- Should patients with advanced disease or low FVC (<50%) be treated?
- Should patients with normal or preserved lung function be treated?
- Should asymptomatic patients (no matter the FVC severity) be treated?
- Should patients with concomitant emphysema (combined pulmonary fibrosis and emphysema [CPFE]) be treated?
Many reports have appeared in the literature over the past several years describing single or multi-center, real-world experiences in the treatment of IPF with antifibrotic agents, as well as non-peer reviewed conference abstracts attempting to address these issues. For the purposes of this discussion, we will include primarily peer-reviewed data (many conference abstracts did not result in peer-reviewed publications).
Should patients with advanced disease or low FVC (<50%) be treated?
Patients presenting with severely impaired pulmonary function are common in IPF and are often associated with substantial symptom burden and radiologic abnormalities. The major clinical trials published to date excluded patients with severe pulmonary impairment as characterized by FVC% <50% or DLCO <30%. This has led to post-hoc and subgroup analyses reviewing drug efficacy across the spectrum of disease severity as defined by pulmonary function results.
Efficacy in severe disease has not been prospectively assessed or reported in a systematic manner. While patients with FVC <50% were excluded from the INPULSIS-R nintedanib trial, some were allowed access to drug therapy in the subsequent open label study INPULSIS-ON, which found a similar 48-week absolute decline in FVC (−62.3 mL) in such patients compared to those with FVC >50% (−87.9 mL).52 Although these data suggest drug efficacy is similar in those with advanced or severe disease, care should be taken in noting the analysis included only 24 patients with severe disease. A single-center retrospective assessment of patients with FVC <50% or DLCO <30% treated with pirfenidone for at least 6 months noted a trend toward slower decline in FVC.53 Such an effect on FVC decline was lost after treating for 1 year. Another series of 18 patients with advanced-stage disease (based on clinical and radiologic criteria) and relative decline in FVC% of 10% or more in the prior 6 months achieved prespecified FVC stability in 8 (44%) while on pirfenidone.54 This particular study also highlighted the additional benefit of oral N-acetylcysteine in combination with pirfenidone for achieving FVC% stability and improved survival.
These reports would suggest response to therapy may be possible even in those with advanced disease. However, there may be reluctance to initiate therapy in such patients as the risk of additional burden related to adverse drug reactions and economic costs are judged to outweigh potential benefits in the presence of advanced lung disease and its associated manifestations.
Should patients with normal or preserved lung function be treated?
A “watch and wait” approach is often taken when patients present with normal or near-normal lung function, particularly if clinical and radiologic findings also reflect mild disease. Such patients may want to avoid unnecessary adverse drug effects. Unfortunately, disease progression may sometimes be abrupt or precipitous and unpredictable (acute exacerbation), leading to irreversible decline prior to drug initiation. For that reason, some clinicians may offer treatment for subclinical disease even when lung function is normal or only mildly affected.
More data for disease response in those with preserved lung function have become available compared to those presenting with advanced disease. Extended analysis of a Phase III pirfenidone trial results found better therapeutic effect in milder disease compared with those with moderate or severe disease.55 In a pooled analysis of the ASCEND and CAPACITY trials as stratified by those with FVC% > or <80% or GAP staging (I vs II/III), pirfenidone was found to have similar positive effects on end points of FVC% decline, 6-minute walk distance, and dyspnea scores, when compared with placebo. All groups, irrespective of baseline lung function, experienced disease progression at 12 months, though pirfenidone demonstrated similar efficacy in improving measured outcomes in these subgroups. Adverse events (AEs) were similar between those with or without preserved lung function.30 Patients with more rapidly progressive disease (rapid progressors) as defined by rate of pretreatment FVC decline appeared to gain greater beneficial effect from pirfenidone within 6–12 months of drug initiation when compared with those with slower progression.56 Such declines were not described as attributable to AE or other causes of rapid decline, suggesting pirfenidone may have an immediate observed effect in those with abrupt disease progression. Similarly, post-hoc analysis of the INPULSIS trials stratified by FVC > or <90% found placebo and treated patients had a similar decline in FVC regardless of baseline lung function.57 Additional post-hoc analysis of nintedanib effect in prespecified subgroups analyzed by age, sex, race, FVC (>70% or <70%), smoking history, prior use of corticosteroids, and severity of quality of life, found no statistical difference in its effect among all subgroups. Treatment effect among those with FVC <70% appeared more pronounced, perhaps supported by greater deterioration and worse quality of life in the placebo group. Such patients appeared to gain more in terms of their measurable effect than those with higher FVC.58 Radiologic honeycombing or UIP CT pattern was also assessed recently in terms of response to antifibrotic therapy. In a post-hoc subgroup analysis of patients from INPULSIS with or without honeycombing, nintedanib demonstrated a similar efficacy in slowing FVC decline, suggesting radiologic severity (presumptively more severe in those with honeycombing) did not affect likelihood of treatment response.59
Should asymptomatic patients (no matter the FVC severity) be treated?
The question of whether to treat asymptomatic patients with or without evidence of disease progression as measured by FVC or radiologic worsening is one of balancing the potential of future respiratory symptoms against immediate adverse drug reactions. Whether antifibrotics have a role in directly attenuating or improving respiratory symptoms remains dubious. In the ASCEND study, those on pirfenidone appeared to have statistically improved 6-minute walk distances compared with placebo, while there was no difference in measured symptom burden or quality of life assessments.13 This lack of benefit for dyspnea or other quality of life measures was similarly found with nintedanib.14 As FVC decline is slowed but not stopped or reversed with therapy, it should be clarified with patients that symptomatic improvement may not be achieved if functional limitations are already present. No studies have reported delay of symptom onset with drug therapy when initiated in asymptomatic patients. Indeed, the ability to tolerate adverse drug reactions in symptomatic or asymptomatic settings may influence long-term use of antifibrotic drugs and attenuate the possibility of any functional effect. Improving exercise tolerance and dyspnea with pulmonary rehabilitation may improve drug adherence, which has become the focus of future prospective investigations.60
Should patients with concomitant emphysema (CPFE) be treated?
Emphysema is common in patients with IPF and, when extensive, is often recognized as CPFE, highlighted by upper lobe predominant emphysema and lower lobe fibrosis.61 While CPFE has been reported in various clinical contexts,62–64 it is often idiopathic in terms of etiology for the underlying fibrosis, and carries with it significant morbidity and mortality including an increased risk of pulmonary hypertension. The question of whether to pursue antifibrotic treatment for such patients is often encountered as radiologic findings may include honeycombing or reticulation with architectural distortions suggestive of a UIP pattern. Unfortunately, CPFE patients often have normal or near-normal spirometry and lung volumes with severely decreased DLCO.65,66 Under such circumstances, assessment of response to therapy may be less reliable when following FVC change as a therapeutic end point. For this reason, patients with radiologic emphysema were specifically excluded from the pirfenidone trials. Cottin et al recently analyzed serial PFTs in IPF patients with emphysema from a previous Phase III IPF trial and found emphysema involvement of 15% or more of the lung was associated with reduced FVC decline for the 48-week study period.67 Whether that reduced FVC decline may, in part, be attributable to emphysema or represent response to antifibrotic therapy makes assessment of treatment response in IPF patients with emphysema challenging. Following alternative measures of disease progression such as scoring radiologic fibrosis or changes in DLCO or 6-minute walk distance in the context of CPFE may be reasonable, though unproven. A decision to treat fibrosis in this setting will likely depend on patient preference and the extent of emphysema, where predominantly fibrotic disease may justify therapy, particularly if there is documented progression.
Expectations for therapy: targeting symptoms, function, or survival?
Antifibrotic therapy may be delayed or simply not initiated if goals or outcomes of treatment do not align with the goals or expectations of patients. Clarifying realistic treatment outcomes and what is clinically and objectively expected is important for informed decision-making, and may become obstacles to treatment initiation or drug adherence if there is confusion or misunderstanding.
As respiratory symptoms in IPF often do not improve with antifibrotic therapy, sustaining lung function and improving survival may be emphasized as possible goals of therapy during treatment discussions. Recent follow-up data from INPULSIS-ON which assessed 430 continuing and 304 patients initiating nintedanib after a trial of placebo found clinical stability for a long term as well as reduced rate of FVC decline. Long-term therapy appeared to support relative maintenance of function up to 63 months with an acceptable safety profile.68 While evidence of FVC improvement or stability has not been reported with pirfenidone, a recent post-hoc analysis of the INPULSIS-R and INPULSIS-ON trials found a quarter of treated trial participants had improved or unchanged FVC at 52 weeks, compared with 9% in the placebo group.69 Whether this is drug effect or varied disease course in terms of improvement or stability from recent decline is unknown. Proposing actual functional improvement as an expectation with current antifibrotic therapy should probably be avoided given the weight of evidence against it.
Another benefit from antifibrotic therapy may be reduction in respiratory-related hospitalizations. Ley et al assessed pooled data of the three pirfenidone trials (CAPACITY I, II, and ASCEND) involving 1,247 treated patients and noted pirfenidone statistically reduced the risk of nonelective respiratory-related hospitalizations (7% vs 12%, HR of 0.52, P=0.001). All other causes of hospitalization were not reduced. Risk of death after hospitalization also appeared to be reduced with the use of pirfenidone, though this effect appeared lost or inconclusive after the study period.70 Only one of the INPULSIS trials found statistically prolonged time to acute exacerbation,71 as defined by local study investigators. Whether reduced hospitalizations for acute exacerbation offset the cost of drug therapy has not been formally assessed.
Mortality has not been a sufficiently powered study end point in recent clinical trials as background rates of death have traditionally been low during trial study periods, requiring larger study populations or a longer duration of study to assess. A post-hoc pooled analysis of the three multinational pirfenidone trials along with additional meta-analysis of two prior Japanese trials found improved survival for treatment-emergent all-cause and IPF-related mortality at 1 year, compared with placebo.72
The natural course of future treatment will likely involve combination therapy, an approach commonly pursued in other chronic diseases such as chronic obstructive pulmonary disease or diabetes. Safety and tolerance of combination therapy with nintedanib and pirfenidone was explored in a small, single-center trial recruiting patients with FVC >50% to assess a primary end point of combined gastrointestinal side effects.73 Patients were treated for only 3 months, with those on combination therapy noting greater incidence of GI-related symptoms. An exploratory end point of FVC decline suggested more stable disease in the combined group compared with nintedanib alone (−13.3 vs −40.9, respectively). Another small Phase IV study highlighted the tolerability of combination therapy over a 24-week period, where only 2 out of 73 completing the study suffered serious treatment-related emergent AEs.74 Larger trials powered toward assessing functional response are needed to determine synergistic or combined efficacy.
The cost of therapy: burden of adverse drug reactions as an obstacle to earlier treatment
Patients with mild functional limitation or related symptoms may find it difficult to initiate or maintain therapy due to frequent drug-related adverse effects. Long-term pirfenidone therapy was recently reviewed for safety outcomes from five clinical trials noting 37.6% had nausea, 28.1% had diarrhea, and 25% had rash, which were described as generally mild to moderate without severe sequelae.75 Liver enzyme elevation (greater than three times upper limit of normal) was noted in only 3.1% and reversed with dose modification or discontinuation. Patients with total drug exposure as high as 9.9 years still appeared to tolerate pirfenidone therapy without clinical sequelae.
An international expert panel recently provided a review of the extent and challenges of pirfenidone-related adverse effects with specific recommendations for management. The most common causes of drug discontinuation were gastrointestinal upset and photosensitivity, with suggested recommendations of taking pirfenidone during or after meals, and avoiding sun exposure or wearing sunscreen along with protective clothing, respectively.76 Frequency and severity of pirfenidone-related photosensitivity appears to diminish over time as reported in a large analysis of the pirfenidone trials.77
Patients treated with nintedanib may experience a similar efficacy even at reduced doses as described recently in a subgroup analysis of the INPULSIS-ON trial.78 Its most commonly reported adverse effects are nausea and diarrhea, the latter of which is usually manageable with the use of loperamide. Both nintedanib and pirfenidone can cause hepatic injury and require an initial phase of monthly monitoring of liver enzyme levels.
Conclusion
The antifibrotic drugs offer an opportunity to treat a progressive and commonly fatal lung disease, though obstacles to earlier drug initiation remain. Defining “early” IPF in terms of disease duration or evolution remains difficult. Commonly accepted parameters used in this designation include symptoms, PFT findings, and extent of radiologic involvement, though none alone are predictive of progression or outcome on an individual basis. Clinicoradiologic features associated with early IPF, such as nondefinitive UIP pattern on CT, may confound IPF diagnosis, as surgical lung biopsy is performed less and clinical overlap with other fibrotic ILDs is common. Once IPF diagnosis is established, additional concerns regarding the balance of drug efficacy vs drug-related burdens in those with minimal or no symptoms, normal or near-normal pulmonary function, advanced or severe disease, or those with concomitant emphysema, may further delay initiation of antifibrotic drug therapy. Discussions regarding treatment-related adverse effects and goals of therapy are essential as disease progression is likely while respiratory symptoms tend not to improve.
Disclosure
The authors report no conflicts of interest in this work.
References
Raghu G, Collard HR, Egan JJ, et al. An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med. 2011;183(6):788–824. | ||
Lynch DA, Sverzellati N, Travis WD, et al. Diagnostic criteria for idiopathic pulmonary fibrosis: a Fleischner Society White Paper. Lancet Respir Med. 2018;6(2):138–153. | ||
Esposito DB, Lanes S, Donneyong M, et al. Idiopathic pulmonary fibrosis in United States automated claims. Incidence, prevalence, and algorithm validation. Am J Respir Crit Care Med. 2015;192(10):1200–1207. | ||
Raghu G, Chen SY, Hou Q, Yeh WS, Collard HR. Incidence and prevalence of idiopathic pulmonary fibrosis in US adults 18–64 years old. Eur Respir J. 2016;48(1):179–186. | ||
Collard HR, Ryerson CJ, Corte TJ, et al. Acute exacerbation of idiopathic pulmonary fibrosis. An international working group report. Am J Respir Crit Care Med. 2016;194(3):265–275. | ||
Raghu G, Chen SY, Yeh WS, et al. Idiopathic pulmonary fibrosis in US Medicare beneficiaries aged 65 years and older: incidence, prevalence, and survival, 2001–11. Lancet Respir Med. 2014;2(7):566–572. | ||
King TE, Behr J, Brown KK, et al. BUILD-1: a randomized placebo-controlled trial of bosentan in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2008;177(1):75–81. | ||
Raghu G, Behr J, Brown KK, et al. Treatment of idiopathic pulmonary fibrosis with ambrisentan: a parallel, randomized trial. Ann Intern Med. 2013;158(9):641–649. | ||
Noth I, Anstrom KJ, Calvert SB, et al. A placebo-controlled randomized trial of warfarin in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2012;186(1):88–95. | ||
Collard HR, King TE, Bartelson BB, Vourlekis JS, Schwarz MI, Brown KK. Changes in clinical and physiologic variables predict survival in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2003;168(5):538–542. | ||
du Bois RM, Weycker D, Albera C, et al. Forced vital capacity in patients with idiopathic pulmonary fibrosis: test properties and minimal clinically important difference. Am J Respir Crit Care Med. 2011;184(12):1382–1389. | ||
Behr J, Bonella F, Bonnet R, et al. Position paper: significance of the forced vital capacity in idiopathic pulmonary fibrosis. Pneumologie. 2015;69(8):455–458. | ||
King TE, Bradford WZ, Castro-Bernardini S, et al. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N Engl J Med. 2014;370(22):2083–2092. | ||
Richeldi L, du Bois RM, Raghu G, et al. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med. 2014;370(22):2071–2082. | ||
Karimi-Shah BA, Chowdhury BA. Forced vital capacity in idiopathic pulmonary fibrosis – FDA review of pirfenidone and nintedanib. N Engl J Med. 2015;372(13):1189–1191. | ||
Maher TM, Molina-Molina M, Russell AM, et al. Unmet needs in the treatment of idiopathic pulmonary fibrosis-insights from patient chart review in five European countries. BMC Pulm Med. 2017;17(1):124. | ||
Moodley Y, Corte T, Richeldi L, King TE. Do all patients with idiopathic pulmonary fibrosis warrant a trial of therapeutic intervention? A pro-con perspective. Respirology. 2015;20(3):389–394. | ||
King CS, Nathan SD. POINT: should all patients with idiopathic pulmonary fibrosis, even those with more than moderate impairment, be treated with nintedanib or pirfenidone? Yes. Chest. 2016;150(2):273–275. | ||
Brown KK. COUNTERPOINT: should all patients with idiopathic pulmonary fibrosis, even those with more than moderate impairment, be treated with nintedanib or pirfenidone? No. Chest. 2016;150(2):276–278. | ||
Flaherty KR, Mumford JA, Murray S, et al. Prognostic implications of physiologic and radiographic changes in idiopathic interstitial pneumonia. Am J Respir Crit Care Med. 2003;168(5):543–548. | ||
Nathan SD, Reichmann WM, Macaulay D, Yf Y. Change in forced vital capacity and clinical outcomes in newly diagnosed idiopathic pulmonary fibrosis patients. Am J Respir Crit Care Med. 2015;191. | ||
Reichmann WM, Yu YF, Macaulay D, Wu EQ, Nathan SD. Change in forced vital capacity and associated subsequent outcomes in patients with newly diagnosed idiopathic pulmonary fibrosis. BMC Pulm Med. 2015;15(1):167. | ||
Arcadu A, Byrne SC, Pirina P, Hartman TE, Bartholmai BJ, Moua T. Correlation of pulmonary function and usual interstitial pneumonia computed tomography patterns in idiopathic pulmonary fibrosis. Respir Med. 2017;129:152–157. | ||
Salisbury ML, Tolle LB, Xia M, et al. Possible UIP pattern on high-resolution computed tomography is associated with better survival than definite UIP in IPF patients. Respir Med. 2017;131:229–235. | ||
Sumikawa H, Johkoh T, Colby TV. Computed tomography findings in pathological usual interstitial pneumonia – relationship to survival. Am J Respir Crit Care Med. 2008;177(4):433–439. | ||
de Giacomi F, White D, Cox CW, Moua T. Evolution of diagnostic UIP computed tomography patterns in idiopathic pulmonary fibrosis: disease spectrum and implications for survival. Respir Med. 2018;142:53–59. | ||
Lee HY, Lee KS, Jeong YJ, et al. High-resolution CT findings in fibrotic idiopathic interstitial pneumonias with little honeycombing: serial changes and prognostic implications. Am J Roentgenol. 2012;199(5):982–989. | ||
Deandrade J, Conoscenti C, Flaherty K, et al. Time from first imaging showing pulmonary fibrosis to diagnosis of idiopathic pulmonary fibrosis: data from the IPF-PRO registry. Chest. 2017;152(4):A445. | ||
Ley B, Ryerson CJ, Vittinghoff E, et al. A multidimensional index and staging system for idiopathic pulmonary fibrosis. Ann Intern Med. 2012;156(10):684–691. | ||
Albera C, Costabel U, Fagan EA, et al. Efficacy of pirfenidone in patients with idiopathic pulmonary fibrosis with more preserved lung function. Eur Respir J. 2016;48(3):843–851. | ||
Wells AU. Forced vital capacity as a primary end point in idiopathic pulmonary fibrosis treatment trials: making a silk purse from a sow’s ear. Thorax. 2013;68(4):309–310. | ||
Collard HR, Brown KK, Martinez FJ, Raghu G, Roberts RS, Anstrom KJ. Study design implications of death and hospitalization as end points in idiopathic pulmonary fibrosis. Chest. 2014;146(5):1256–1262. | ||
Ash SY, Vallejo DLL, Villalba JA. Quantitative computed tomography analysis predicts outcomes in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2016;193:A2603. | ||
Lederer DJ, Arcasoy SM, Wilt JS, D’Ovidio F, Sonett JR, Kawut SM. Six-minute-walk distance predicts waiting list survival in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2006;174(6):659–664. | ||
Morino A, Takahashi H, Ishiai S. Six-minute walk distance is a surrogate parameter of physical activity in patients with idiopathic pulmonary fibrosis. Eur Respir J. 2013;42:P1840. | ||
Swigris JJ, Fairclough D. Patient-reported outcomes in idiopathic pulmonary fibrosis research. Chest. 2012;142(2):291–297. | ||
Hunninghake GW, Lynch DA, Galvin JR, et al. Radiologic findings are strongly associated with a pathologic diagnosis of usual interstitial pneumonia. Chest. 2003;124(4):1215–1223. | ||
Fell CD, Martinez FJ, Liu LX, et al. Clinical predictors of a diagnosis of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2010;181(8):832–837. | ||
Macdonald SL, Rubens MB, Hansell DM, et al. Nonspecific interstitial pneumonia and usual interstitial pneumonia: comparative appearances at and diagnostic accuracy of thin-section CT. Radiology. 2001;221(3):600–605. | ||
Hartman TE, Swensen SJ, Hansell DM, et al. Nonspecific interstitial pneumonia: variable appearance at high-resolution chest CT. Radiology. 2000;217(3):701–705. | ||
Brownell R, Moua T, Henry TS, et al. The use of pretest probability increases the value of high-resolution CT in diagnosing usual interstitial pneumonia. Thorax. 2017;72(5):424–429. | ||
Morisset J, Johannson KA, Jones KD, et al. Identification of diagnostic criteria for chronic hypersensitivity pneumonitis. An International Modified Delphi Survey. Am J Respir Crit Care Med. 2018;197(8):1036–1044. | ||
Morell F, Villar A, Montero MÁ, et al. Chronic hypersensitivity pneumonitis in patients diagnosed with idiopathic pulmonary fibrosis: a prospective case-cohort study. Lancet Respir Med. 2013;1(9):685–694. | ||
Moua T, Maldonado F, Decker PA, Daniels CE, Ryu JH. Frequency and implication of autoimmune serologies in idiopathic pulmonary fibrosis. Mayo Clin Proc. 2014;89(3):319–326. | ||
Lee JS, Kim EJ, Lynch KL, et al. Prevalence and clinical significance of circulating autoantibodies in idiopathic pulmonary fibrosis. Respir Med. 2013;107(2):249–255. | ||
Fischer A, Antoniou KM, Brown KK, et al. An official European Respiratory Society/American Thoracic Society research statement: interstitial pneumonia with autoimmune features. Eur Respir J. 2015;46(4):976–987. | ||
Oldham JM, Adegunsoye A, Valenzi E, et al. Characterisation of patients with interstitial pneumonia with autoimmune features. Eur Respir J. 2016;47(6):1767–1775. | ||
Chartrand S, Swigris JJ, Stanchev L, Lee JS, Brown KK, Fischer A. Clinical features and natural history of interstitial pneumonia with autoimmune features: a single center experience. Respir Med. 2016;119:150–154. | ||
Raghu G, Lynch D, Godwin JD, et al. Diagnosis of idiopathic pulmonary fibrosis with high-resolution CT in patients with little or no radiological evidence of honeycombing: secondary analysis of a randomised, controlled trial. Lancet Respir Med. 2014;2(4):277–284. | ||
Hutchinson JP, Fogarty AW, Mckeever TM, Hubbard RB. In-hospital mortality after surgical lung biopsy for interstitial lung disease in the United States. 2000 to 2011. Am J Respir Crit Care Med. 2016;193(10):1161–1167. | ||
Tomassetti S, Wells AU, Costabel U. The diagnostic role of cryobiopsy in the multidisciplinary diagnosis of idiopathic pulmonary fibrosis. Eur Respir J. 2013;42(Suppl 57):469s. | ||
Wuyts WA, Kolb M, Stowasser S, Stansen W, Huggins JT, Raghu G. First data on efficacy and safety of nintedanib in patients with idiopathic pulmonary fibrosis and forced vital capacity of ≤50% of predicted value. Lung. 2016;194(5):739–743. | ||
Tzouvelekis A, Ntolios P, Karampitsakos T, et al. Safety and efficacy of pirfenidone in severe idiopathic pulmonary fibrosis: a real-world observational study. Pulm Pharmacol Ther. 2017;46:48–53. | ||
Sakamoto S, Itoh T, Muramatsu Y, et al. Efficacy of pirfenidone in patients with advanced-stage idiopathic pulmonary fibrosis. Intern Med. 2013;52(22):2495–2501. | ||
Taguchi Y, Ebina M, Hashimoto S, et al. Efficacy of pirfenidone and disease severity of idiopathic pulmonary fibrosis: extended analysis of phase III trial in Japan. Respir Investig. 2015;53(6):279–287. | ||
Biondini D, Balestro E, Cocconcelli E, et al. Effect of 24-month Pirfenidone treatment on functional decline in Idiopathic Pulmonary Fibrosis (IPF) patients with rapid and slow disease progression. Eur Respir J. 2017;50:PA2966. | ||
Kolb M, Richeldi L, Behr J, et al. Nintedanib in patients with idiopathic pulmonary fibrosis and preserved lung volume. Thorax. 2017;72(4):340–346. | ||
Costabel U, Inoue Y, Richeldi L, et al. Efficacy of nintedanib in idiopathic pulmonary fibrosis across prespecified subgroups in INPULSIS. Am J Respir Crit Care Med. 2016;193(2):178–185. | ||
Raghu G, Wells AU, Nicholson AG, et al. Effect of nintedanib in subgroups of idiopathic pulmonary fibrosis by diagnostic criteria. Am J Respir Crit Care Med. 2017;195(1):78–85. | ||
Kondoh Y, Taniguchi H, Kataoka K. Long-term effect of pulmonary rehabilitation under nintedanib treatment in idiopathic pulmonary fibrosis: methodology of the fitness study. Am J Respir Crit Care Med. 2017;195:A4280. | ||
Cottin V, Nunes H, Brillet PY, et al. Combined pulmonary fibrosis and emphysema: a distinct underrecognised entity. Eur Respir J. 2005;26(4):586–593. | ||
Cottin V, Nunes H, Mouthon L, et al. Combined pulmonary fibrosis and emphysema syndrome in connective tissue disease. Arthritis Rheum. 2011;63(1):295–304. | ||
Fernández Casares M, Fielli M, Cristaldo L, Zárate L, Nieves Capozzi M. Rheumatoid arthritis and combined pulmonary fibrosis and emphysema. Medicina. 2015;75(4):225–228. | ||
Soumagne T, Pana-Katatali H, Degano B, Dalphin JC. Combined pulmonary fibrosis and emphysema in hypersensitivity pneumonitis. BMJ Case Rep. 2015;2015:bcr2015211560. | ||
Kitaguchi Y, Fujimoto K, Hanaoka M, Honda T, Hotta J, Hirayama J. Pulmonary function impairment in patients with combined pulmonary fibrosis and emphysema with and without airflow obstruction. Int J Chron Obstruct Pulmon Dis. 2014;9:805–811. | ||
Kitaguchi Y, Fujimoto K, Hayashi R, Hanaoka M, Honda T, Kubo K. Annual changes in pulmonary function in combined pulmonary fibrosis and emphysema: over a 5-year follow-up. Respir Med. 2013;107(12):1986–1992. | ||
Cottin V, Hansell DM, Sverzellati N, et al. Effect of emphysema extent on serial lung function in patients with idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2017;196(9):1162–1171. | ||
Crestani B, Quaresma M, Kaye M, Stansen W, Stowasser S, Kreuter M. Long-term nintedanib treatment in idiopathic pulmonary fibrosis (IPF): new data from INPULSIS-ON. Eur Respir J. 2017;50:OA3402. | ||
Flaherty KR, Kolb M, Vancheri C, Tang W, Conoscenti CS, Richeldi L. Stability or improvement in forced vital capacity with nintedanib in patients with idiopathic pulmonary fibrosis. Eur Respir J. 2018;52(2):1702593. | ||
Ley B, Swigris J, Day BM, et al. Pirfenidone reduces respiratory-related hospitalizations in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2017;196(6):756–761. | ||
Richeldi L, du Bois R, Raghu G. Efficacy and safety of nintedanib in patients with idiopathic pulmonary fibrosis: results of two 52-week, Phase Iii, randomized, placebo-controlled trials (INPULSIS (TM)). Am J Respir Crit Care Med. 2014;19:28. | ||
Nathan SD, Albera C, Bradford WZ, et al. Effect of pirfenidone on mortality: pooled analyses and meta-analyses of clinical trials in idiopathic pulmonary fibrosis. Lancet Respir Med. 2017;5(1):33–41. | ||
Vancheri C, Kreuter M, Richeldi L, et al. Nintedanib with add-on pirfenidone in idiopathic pulmonary fibrosis. Results of the INJOURNEY Trial. Am J Respir Crit Care Med. 2018;197(3):356–363. | ||
Flaherty KR, Fell CD, Huggins JT, et al. Safety of nintedanib added to pirfenidone treatment for idiopathic pulmonary fibrosis. Eur Respir J. 2018;52(2):1800230. | ||
Lancaster L, Albera C, Bradford WZ, et al. Safety of pirfenidone in patients with idiopathic pulmonary fibrosis: integrated analysis of cumulative data from 5 clinical trials. BMJ Open Respir Res. 2016;3(1):e000105. | ||
Costabel U, Bendstrup E, Cottin V, et al. Pirfenidone in idiopathic pulmonary fibrosis: expert panel discussion on the management of drug-related adverse events. Adv Ther. 2014;31(4):375–391. | ||
Valeyre D, Albera C, Bradford WZ, et al. Comprehensive assessment of the long-term safety of pirfenidone in patients with idiopathic pulmonary fibrosis. Respirology. 2014;19(5):740–747. | ||
Crestani B, Kolb M, Wallaert B, Quaresma M, Stansen W, Richeldi L. Long-term efficacy of nintedanib is maintained in patients with idiopathic pulmonary fibrosis (IPF) irrespective of dose: subgroup analysis of INPULSIS-ON. Am J Respir Crit Care Med. 2017;195:A5408. |
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.