")
Back to Journals » Clinical and Experimental Gastroenterology » Volume 17
Obstacles to Early Diagnosis of Acute Hepatic Porphyria: Current Perspectives on Improving Early Diagnosis and Clinical Management
Authors Thapar M , Singh A, Robinson KM , Bonkovsky HL
Received 11 September 2023
Accepted for publication 28 November 2023
Published 5 January 2024 Volume 2024:17 Pages 1—8
DOI https://doi.org/10.2147/CEG.S348507
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Everson L.A. Artifon
Manish Thapar,1 Akash Singh,2 Kevin M Robinson,3 Herbert L Bonkovsky4
1Division of Hepatology, Jefferson- Einstein Medical Center, Philadelphia, PA, USA; 2Department of Medicine, Jefferson- Einstein Medical Center Montgomery, East Norriton, PA, USA; 3Department of Medicine Jefferson- Einstein Medical Center, Philadelphia, PA, USA; 4Division of Gastroenterology and Hepatology, Wake Forest University School of Medicine/NC Baptist Hospital, Winston-Salem, NC, USA
Correspondence: Manish Thapar, Division of Hepatology, Jefferson-Einstein Medical Center, 5401 Old York Road, Philadelphia, PA, USA, Email [email protected]
Abstract: Porphyrias are, for the most part, inherited disorders of the heme biosynthetic pathway which lead to accumulation of specific intermediates responsible for most of the symptoms and signs of biochemically active disease. Acute hepatic porphyrias usually come to clinical attention primarily in women in their reproductive years who present with episodic, severe, generalized abdominal pain. Such acute attacks may also be associated with tachycardia, systemic arterial hypertension, hyponatremia, recent history of dark reddish to brownish urine, and anxiety, delirium, and sensory or motor neuropathies. Diagnosing AHPs is often challenging, requiring a high index of suspicion and the appropriate testing showing elevated ALA and/or PBG in a random urine specimen. Obstacles to diagnosis include inappropriate testing for porphyrins only, inadequate sample handling, and ordering genetic testing as the initial diagnostic test. While some of these pitfalls in diagnosis are surmountable with current knowledge, others are in need of more research.
Keywords: porphyria, diagnosis, porphyrins, ALA, PBG, misdiagnosis, obstacle to diagnosis, acute intermittent porphyria, secondary coproporphyrinuria
Introduction
Porphyrias are, for the most part, inherited disorders of the heme biosynthetic pathway. Heme is synthesized from glycine and succinyl-coA through a pathway (Figure 1) that involves eight enzymatic steps, and mutations along the pathway leading to reduced enzymatic activity result in inherited porphyrias, except X-linked protoporphyria (XLP) which is due to gain in function mutation.1 Accumulation of specific intermediates in the heme biosynthetic pathway is responsible for most of the symptoms and signs of biochemically active and clinically manifest porphyrias. Porphyrias are often classified as hepatic or erythropoietic, depending on the site of major over production and accumulation of the precursors of heme. Hepatic porphyrias are classified as acute or chronic, depending on clinical presentation. Acute hepatic porphyrias have an acute neurovisceral presentation. There are four acute hepatic porphyrias (AHP): acute intermittent porphyria (AIP); variegate porphyria (VP); hereditary coproporphyria (HCP); and 5-aminolevulinic acid dehydratase deficiency porphyria (ALAD). Approximately 90% of symptomatic AHP patients are women in their reproductive years, and the most common presentation is episodic severe, generalized abdominal pain. The diagnosis of AHPs is often missed, with a delay as long as 15 years from onset of symptomatic AHP to diagnosis.2 Fortunately, acute porphyrias causing such attacks can be diagnosed with a simple urine test that is sensitive and specific, namely, urinary measurement of 5-Aminolevulinic acid (ALA) and Porphobilinogen (PBG), normalized to urinary creatinine concentration, and effective treatments are available.
 |
Figure 1 The pathway of heme synthesis, showing pathway intermediates and end-product regulation by heme. The eight steps of heme synthesis (left columns) are shown with the enzyme (middle column) that catalyzes each step. The enzymes in bold face are the clinically most prevalent porphyrias. Acute intermittent porphyria is the most common acute hepatic porphyria. Note: Reprinted from New England Journal of Medicine, Bissell DM, Anderson KE, Bonkovsky HL. Porphyria. volume 377(9), pages 862–872. Copyright © (2017) Massachusetts Medical Society. Reprinted with permission from Massachusetts Medical Society.3 |
Pathophysiology
All human cells have the capacity to synthesize heme, although it is predominantly produced in the bone marrow (75–80%) and a smaller amount in the hepatocytes (15–20%).4 The heme biosynthesis pathway is controlled by the first and rate-limiting enzyme, 5-aminolevulinic acid (ALA) synthase which exists in two isoforms; ALA Synthase 1 and ALA Synthase 2. ALA Synthase 1, the isoform in the non-erythroid cells is subject to feedback regulation by heme. In AHP’s, ALA Synthase 1 is the-rate limiting enzyme despite other partial enzyme defects downstream. 5-Aminolevulinic acid (ALA) accumulates during an acute attack and is thought to play a role in both acute and chronic pain.5 Other porphyrin intermediaries, which can accumulate in HCP or VP, absorb light from the visible spectrum and cause skin damage.
AIP, HCP, and VP are inherited as autosomal dominant disorders, while ALAD porphyria is an extremely rare (~10 cases in the world) autosomal recessive disorder. Symptomatic AHP is seen in approximately 1:100,000 persons even though the genetic prevalence of its pathogenic variants is between 1 in 1300 and 1 in 1785 among Europeans and Americans.4,6 The vast majority of carriers of AHP mutations do not experience symptomatic acute attacks in their lifetime. HCP and VP are more often latent [without symptoms or signs] than AIP. These latent, asymptomatic HCP and VP patients have normal ALA and PBG levels. The low penetrance of symptomatic AHP underscores the role external factors play in precipitating an acute attack. Medications, especially those metabolized by the cytochrome P-450 system are the most common precipitants, along with other precipitants such as stress in the form of infection, surgery, starvation or crash dieting; excess alcohol intake, and progesterone, the latter accounting for the cyclical monthly acute attacks suffered by some women during the luteal phase of their menstrual cycle.
Clinical Presentation
AHPs have low clinical penetrance with the exception of ADP. Most subjects with HCP, VP and even AIP likely never experience an acute neurovisceral attack. Of the AHP’s HCP and VP can present with both neurovisceral and photocutaneous symptoms (due to accumulation of porphyrins).7 Neurovisceral attacks are characterized by severe, generalized and poorly localized abdominal pain (cardinal symptom) often in association with nausea, vomiting, and constipation; central nervous system symptoms such as seizures and altered mental status; and, peripheral nervous system symptoms including limb paresis and peripheral neuropathy.8 The rate of progression of symptoms can be variable and progressive. Untreated patients can develop flaccid tetraplegia and rapidly develop respiratory paralysis requiring mechanical ventilation. Despite severe abdominal pain, physical examination typically reveals a non-focal abdominal exam since the pain is neuropathic in nature, guarding and rigidity are usually absent. These patients may present with an SIADH-like syndrome with hyponatremia. VP often and HCP rarely also manifest with cutaneous symptoms, typically blistering lesions seen primarily on sun-exposed areas.9,10 The non-specific nature of symptoms and physical examination findings as well as the overall rarity of AHP require a high index of suspicion by clinicians to think of the possibility of AHP and to obtain appropriate diagnostic testing.
The majority of symptomatic AHP patients typically have one or a few attacks throughout their lifetime. Less than 5% of AHP patients experience frequent recurrent attacks with resultant chronic neurological symptoms, impaired quality of life and are at a higher risk of long-term complications of AHP. AHP patients are at risk of hepatocellular cancer, hypertension and chronic renal failure.
Diagnosis of Acute Hepatic Porphyrias
Early and accurate diagnosis of acute hepatic porphyrias starts with an astute clinician obtaining a focused and pertinent history, ordering the appropriate first-line screening biochemical tests, followed by confirmatory genetic testing if first-line testing is abnormal.
The symptoms, signs, and complications of AHP are thought to be due mainly to the accumulation of porphyrin precursors of the heme synthesis pathway; namely, ALA and PBG.7 Elevated ALA is the hallmark of all acute attacks of AHP. Therefore, the initial screening test of choice involves random urinary testing for ALA, PBG, and creatinine. Significant elevation of urinary PBG/creatinine is typically observed in AIP, HCP and VP. However, urinary PBG is normal in ALAD because the enzymatic defect in this extremely rare porphyria is upstream to the formation of PBG.11 ALA and PBG levels are elevated 5–50 fold during an acute attack. All urine samples should also have creatinine concentrations measured so that concentrations of analytes can be normalized to creatinine concentrations, to adjust for variations in the degree of urinary dilution, such as with large doses of oral or IV fluids, which may have been administered.
Elevated urinary porphyrins can be seen in a variety of hepatobiliary or hematological diseases, recent alcohol use, concomitant drug therapy, infections, and others. It is also important to note that urine samples for porphyrins must be protected from light in order to yield accurate results.10,12 Urinary porphyrin level should be normalized per gram or per mmol of urine creatinine to help prevent false negatives in a dilute sample, and samples should best be collected during an acute attack and prior to treatment with IV heme or givosiran when ALA and PBG levels/ g creatinine are typically greater than ten times the upper limit of normal.9
In patients with positive screening results, confirmatory genetic testing to ascertain the type of AHP should be performed. Sequencing of the ALAD, HMBS, CPOX and PPOX genes identifies the proband mutation which can be used to screen family members.
A diagnosis of VP is suspected when screening urinary PBG is elevated, indicating the presence of AHP, in a patient with typical neurovisceral symptoms of AHP associated with blistering skin lesions. The diagnosis of VP would further be supported by analysis of plasma and fecal porphyrin, usually done using high performance liquid chromatography with fluorescence detection or mass spectrometry followed by genetic testing for a mutation in the protoporphyrinogen oxidase gene.13
Like VP, HCP diagnosis is suspected in patients with neurovisceral symptoms and photocutaneous lesions who have positive urinary ALA and PBG. The diagnosis of HCP can be distinguished from VP based on plasma and fecal porphyrin analysis. Typically, in VP, both plasma and stool porphyrins will be markedly elevated. In contrast, HCP is typically associated with markedly elevated fecal porphyrins, but normal or only slightly increased plasma porphyrins.14 Subsequently, genetic testing for a mutation in coproporphyrinogen oxidase gene will confirm the diagnosis of HCP.
Diagnostic Challenges and Clinical Perspectives
Early and definitive diagnosis of acute hepatic porphyrias (AHP) poses a diagnostic challenge due to their diverse and non-specific clinical presentation and the rarity of the disorders. Clinicians need to consider the diagnosis in the appropriate clinical setting and order appropriate diagnostic testing accordingly.
Diagnosis of AHP is based on high index of suspicion in patients presenting with unexplained recurrent generalized, poorly localized abdominal pain, especially in women in the age range of 15–50 years. While the vast majority of symptomatic AHP is seen in young females, men can also present with symptoms. Male gender alone is not a reason to exclude the diagnosis, especially when other work up has been unrevealing. Often clinicians disregard the diagnosis for lack of an identifiable trigger or precipitant. It is important to remember that such a trigger may not always be readily apparent in clinical practice. A positive family history of AHP is helpful in making a diagnosis, but most patients do not have a family history of porphyria. The lack of family history does not exclude the diagnosis of AIP, HCP and VP as they have autosomal dominant inheritance and the point mutation might have arisen de novo in the subject.
ALA and PBG are not porphyrins, but rather porphyrin precursors. Clinicians need to seek out and order the right test in the EMR when looking to make the diagnosis of AHP. Ordering only plasma or urinary porphyrins as a screening test is a common error. The test of choice is urinary ALA, PBG, and creatinine levels.
Measurement of urinary ALA, PBG and creatinine is most useful during an acute attack. A rapid urine PBG is the initial screening test whenever acute porphyria is suspected.15,16 To improve the bedside diagnosis of porphyria, recently a new rapid test for PBG (Teco Diagnostics) has been approved in the USA although it is not yet widely available. The lack of an easy and widely available, rapid point of care test for ALA and PBG is a barrier to diagnosis in a timely manner.
A quantitative test of the urine sample for PBG level and ALA level is confirmatory. A PBG level of >10 mg/g creatinine (normal <2–4) and ALA >10 mg/g creatinine is highly specific for acute porphyria.17 In AIP, Urine PBG and ALA levels can remain elevated for weeks to months after an acute attack and increase further during recurrent acute attacks.18 However, in HCP and VP, the ALA and PBG levels typically drop to normal soon after an attack abates. Such patients may require a confirmatory testing during an acute attack and the timing of testing can pose a diagnostic challenge in patients with HCP and VP. Plasma emission fluorescence testing showing a peak at 626 nm is very specific in diagnosing VP, even in the absence of an attack.
The turnaround time for ALA and PBG is usually about 1–2 weeks as the test is only performed at large reference laboratories. This delay in obtaining the results in a timely manner is both a diagnostic challenge and a source of frustration for patients and physicians alike causing a delay in treatment at times.
Testing symptomatic family members may identify asymptomatic high excreters (ASHE), who have chronically elevated levels of ALA and PBG, in the absence of any symptoms. This subgroup of ASHE patients are often started on treatment as patients with symptomatic AHP. This does pose a unique therapeutic dilemma as data is lacking about efficacy or need of treating patients who are ASHE. There is consensus that ASHE patients might be at an increased risk of developing chronic complications of AHP such as hepatocellular cancer and chronic renal failure.
PBG in urine exposed to air and/or light and at room temperature may be converted non-enzymatically to uroporphyrin 1. Therefore, in biochemically active AIP there may be an increase in urinary uro and copro porphyrin levels.19–21
In variegate porphyria (VP) there are elevated plasma porphyrins with peak emission fluorescence at 626 nm following excitation at 410nm along with raised fecal protoporphyrin and coproporphyrin.22,23 Unfortunately, in the USA, at least, many commercial or other reference laboratories do not perform this simple and useful plasma fluorescence test. It is performed routinely in the academic laboratory of Karl Anderson, MD, UTMB, Galveston, TX. Laboratory personnel are often not fully trained on sample handling and the importance of protection from sunlight, which leads to erroneous results. Urinary samples for ALA, PBG and porphyrins should be protected from light, wrapped in foil and refrigerated. Care should also be taken to transport the samples frozen.
In hereditary coproporphyria (HCP) fecal porphyrin, particularly coproporphyrin, is markedly elevated.23 The elevated urinary coproporphyrin may suggest HCP which should be confirmed with elevated plasma porphyrins and fecal coproporphyrin III. Patients find it tedious and unpleasant to collect a 24- or 72-hour stool sample. This can be overcome by laboratories accepting a random stool sample of at least 30 g. Patients with HCP and VP presenting with blistering skin lesions are sometimes misdiagnosed as porphyria cutanea tarda (PCT) in clinical practice.
If urinary ALA excretion in mg/g creatinine is elevated while that of PBG/creatinine is normal, ADP or an alternative diagnosis such as lead poisoning or hereditary tyrosinemia should be considered. Alcohol ingestion and P-450 inducing drugs may also increase urinary porphyrin excretion. Fecal porphyrins may be increased in patients with gastrointestinal bleeding or those who consume large amount of red meat.
Porphobilinogen deaminase (PBGD) activity is reduced in AIP patients and measurement of PBGD in red blood cells has been used as a diagnostic test. About 5–10% of bona fide AIP patients have normal RBC PBGD, because the mutation is found in the exon that is not expressed in RBC, but only in hepatic PBGD. Thus, RBC PBGD as a confirmatory test has now largely been superseded by genetic testing. Yet, there are instances in which genetic testing has not revealed a pathogenic mutation, RBC PBGD is low and patients clearly do have AIP.
Genetic testing is used to confirm the diagnosis of AHP in patients with biochemical evidence of disease and identify the specific type of AHP. Genetic sequencing of the 4 genes ALAD, HMBS, CPOX, and PPOX helps diagnose ALAD, AIP, HCP and VP, respectively. Unveiling of known pathogenic mutations can be used to identify carriers among family members of the proband case.24 The recent easy availability of genetic testing and its use in diagnosing patients and family members with symptoms suggestive of AHP raises a new set of diagnostic dilemmas. Genetic sequencing of the culprit genes often reports variants of uncertain significance (VUS) or lack of reported pathogenic mutations even in patients with biochemical evidence of AHP. Such cases need to be referred to centers of excellence for confirmatory testing. The finding of a VUS in a patient with lack of biochemical evidence of disease raises more questions than it answers. There is no consensus on how to manage patients with VUS or pathogenic mutations with no biochemical evidence of disease. Genetic testing should not be used as first-line testing for suspected AHP.
Secondary porphyrinuria: In secondary porphyrinuria, conditions unrelated to porphyria may result in raised urinary levels of porphyrins. In some hepatobiliary disorders, etc, as described above, coproporphyrins are raised in urine as there is a decrease in the biliary excretion of porphyrins. Alcohol and P-450 inducing drugs may cause increase in urine porphyrin level by inhibiting organic anion transporters which normally transport porphyrins specially coproporphyrins into bile.25,26 These disorders show normal or only minimally elevated levels of ALA-PBG which helps distinguish secondary porphyrinuria from acute porphyrias.
Improving Clinical Management
Hemin is currently approved treatment for acute attacks, and givosiran is approved for patients with recurrent attacks of AHP.27 Both are highly effective at preventing acute clinical deterioration and recurrent attacks, if used early on in the disease course. It behooves the treating clinician to diagnose patients in a timely manner to prevent complications both acute and chronic. Patients should be tested with any suspicion of disease and the importance of early and accurate testing cannot be over-emphasized. Treating providers need to be educated about appropriate testing to order, but also in the correct interpretation of tests. This would avoid not only a delay in diagnosis but also inaccurate diagnosis. Health insurance companies and health systems need to work on ensuring easy, widespread availability and coverage of both point of care testing and genetic testing. Laboratory medical directors on the other hand need to standardize testing, adopt best practices and ensure that their personnel are adequately trained on handling samples and processing. EMR vendors can also help in improving clinical outcomes by optimizing the ease of ordering the appropriate testing and also promote the use of artificial intelligence in diagnosis. The high cost of currently available treatment along with the onerous prior authorization requirements are also a barrier to treating patients. Pharmaceutical manufacturers and healthcare plans also owe it to the patients to ensure accessibility of life saving and altering treatment. These diagnostic and therapeutic challenges are summarized in Table 1 and can be viewed as action items which can be worked on with currently available knowledge and as future areas of additional research.
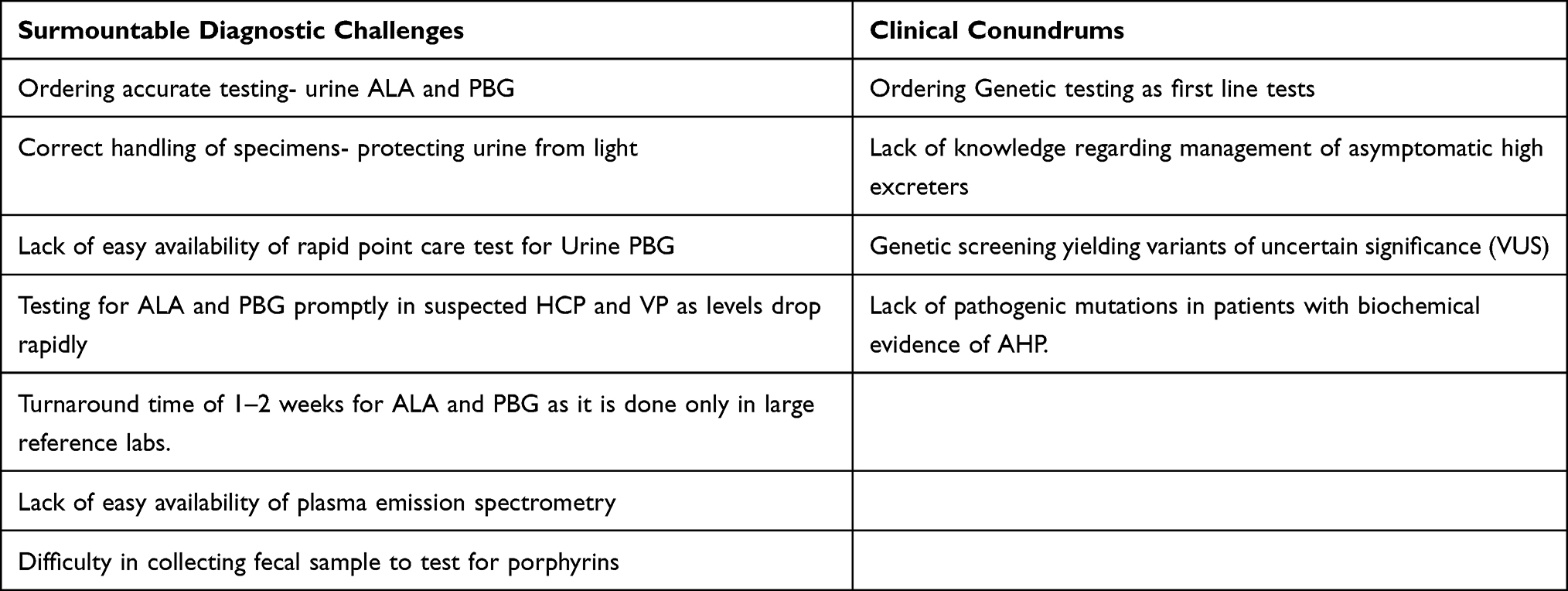 |
Table 1 Obstacles to Early Diagnosis and Clinical Conundrums |
In conclusion clinicians, geneticists, researchers and industry need to work together keeping the patients front and center. There are several unanswered questions such as the role and utility of treating ASHE, interpretation of variants of unknown significance, and when to order genetic testing. We are still unclear on how often to follow patients with known pathogenic genetic mutations but no clinical or biochemical evidence of disease. While a meeting of minds with a consensus conference to move the needle might be need of the hour, it would be presumptuous to propose it as a panacea. AHP is a poster child on how all stakeholders involved in health care need to work together to improve diagnosis, ensure access to treatment and answer outstanding clinical questions.
Disclosure
Dr Manish Thapar is a consultant for Alnylam Pharmaceuticals, Disc Medicine, and Recordati Rare Diseases, outside the submitted work. Dr Herbert Bonkovsky reports grants/personal fees from Alnylam Pharma, Disc Medicine, Mitsubishi-Tanabe, NA, Calliditas, SA, Gilead Sciences, and Recordati Rare Chemicals, outside the submitted work. The authors report no other conflicts of interest in this work.
References
1. Bonkovsky HL, Guo JT, Hou W, Li T, Narang T, Thapar M. Porphyrin and heme metabolism and the porphyrias. Compr Physiol. 2013;3(1):365–401. PMID: 23720291. doi:10.1002/cphy.c120006
2. Bonkovsky HL, Maddukuri VC, Yazici C, et al. Acute porphyrias in the USA: features of 108 subjects from porphyrias consortium. Am J Med. 2014;127(12):1233–1241. doi:10.1016/j.amjmed.2014.06.036
3. Bissell DM, Anderson KE, Bonkovsky HL Porphyria. N Engl J Med. 2017;377(9):862–872.
4. Chen B, Solis-Villa C, Hakenberg J, et al. Acute intermittent porphyria: predicted pathogenicity of HMBS variants indicates extremely low penetrance of the autosomal dominant disease. Hum Mutat. 2016;37:1215–1222. doi:10.1002/humu.23067
5. Kazamel M, Pischik E, Desnick RJ. Pain in acute hepatic porphyrias: updates on pathophysiology and management. Front Neurol. 2022;13:1004125. doi:10.3389/fneur.2022.1004125
6. Lenglet H, Schmitt C, Grange T, et al. From a dominant to an oligogenic model of inheritance with environmental modifiers in acute intermittent porphyria. Hum Mol Genet. 2018;27(7):1164–1173. doi:10.1093/hmg/ddy030
7. Simon A, Pompilus F, Querbes W, et al. Patient perspectives on acute intermittent porphyria with frequent attacks: a disease with intermittent and chronic manifestations. Patient. 2018;11(5):527–537. doi:10.1007/s40271-018-0319-3
8. Ventura P, Cappellini MD, Biolcati G, Guida CC, Rocchi E. Gruppo Italiano Porfiria (GrIP). A challenging diagnosis for potential fatal diseases: recommendations for diagnosing acute porphyrias. Eur J Intern Med. 2014;25(6):497–505. doi:10.1016/j.ejim.2014.03.011
9. Anderson KE, Bloomer JR, Bonkovsky HL, et al. Recommendations for the diagnosis and treatment of the acute porphyrias. Ann Intern Med. 2005;142(6):439–450. doi:10.7326/0003-4819-142-6-200503150-00010
10. Balwani M, Wang B, Anderson KE, et al. Porphyrias consortium of the rare diseases clinical research network. Acute hepatic porphyrias: recommendations for evaluation and long-term management. Hepatology. 2017;66(4):1314–1322. doi:10.1002/hep.29313
11. Szlendak U, Bykowska K, Lipniacka A. Clinical, biochemical and molecular characteristics of the main types of porphyria. Adv Clin Exp Med. 2016;25:361–368.
12. Woolf J, Marsden JT, Degg T, et al. Best practice guidelines on first-line laboratory testing for porphyria. Ann Clin Biochem. 2017;54(2):188–198. doi:10.1177/0004563216667965
13. Anderson KE, Lobo R, Salazar D, et al. Biochemical diagnosis of acute hepatic porphyria: updated expert recommendations for primary care physicians. Am J Med Sci. 2021;362(2):113–121. doi:10.1016/j.amjms.2021.03.004
14. Rigor J, Pinto SA, Martins-Mendes D. Porphyrias: a clinically based approach. Eur J Intern Med. 2019;67:24–29. doi:10.1016/j.ejim.2019.06.014
15. Sardh E, Wahlin S, Björnstedt M, Harper P, Andersson DE. High risk of primary liver cancer in a cohort of 179 patients with acute hepatic porphyria. J Inherit Metab Dis. 2013;36:1063–1071. doi:10.1007/s10545-012-9576-9
16. Andant C, Puy H, Bogard C, et al. Hepatocellular carcinoma in patients with acute hepatic porphyria: frequency of occurrence and related factors. J Hepatol. 2000;32:933–939. doi:10.1016/S0168-8278(00)80097-5
17. Bissell DM, Wang B. Acute hepatic porphyria. J Clin Transl Hepatol. 2015;3:17–26.
18. Marsden JT, Rees DC. Urinary excretion of porphyrins, porphobilinogen and delta-aminolaevulinic acid following an attack of acute intermittent porphyria. J Clin Pathol. 2014;67:60–65. doi:10.1136/jclinpath-2012-201367
19. Bonkovsky HL. Porphyrin and heme metabolism and the porphyrias. In: Zakim D, Boyer T, editors. Hepatology: A Textbook of Liver Disease.
20. Bonkovsky HL, Barnard GF. Diagnosis of porphyric syndromes: a practical approach in the era of molecular biology. Semin Liver Dis. 1998;18(01):57–65. doi:10.1055/s-2007-1007141
21. Hahn M, Bonkovsky HL. Disorders of porphyrin metabolism. In: Wu G, Israel J, editors. Diseases of the Liver and Bile Ducts: A Practical Guide to Diagnosis and Treatment.
22. Singal AK, Anderson KE. Variegate porphyria. In: Pagon RA, Adam MP, Ardinger HH, editors. Gene Reviews. Seattle (WA): University of Washington; 1993.
23. Ramanujam VM, Anderson KE. Porphyria diagnostics-part 1: a brief overview of the porphyrias. Curr Protoc Hum Genet. 2015;86:17.20.1–26. doi:10.1002/0471142905.hg1720s86
24. Whatley SD, Mason NG, Woolf JR, et al. Diagnostic strategies for autosomal dominant acute porphyrias: retrospective analysis of 467 unrelated patients referred for mutational analysis of the HMBS, CPOX, or PPOX gene. Clin Chem. 2009;55(7):1406–1414. doi:10.1373/clinchem.2008.122564
25. An G, Wang X, Morris ME. Flavonoids are inhibitors of human organic anion transporter 1 (OAT1)-mediated transport. Drug Metab Dispos. 2014;42(9):1357–1366. doi:10.1124/dmd.114.059337
26. Duan P, Li S, Ai N, Hu L, Welsh WJ, You G. Potent inhibitors of human organic anion transporters 1 and 3 from clinical drug libraries: discovery and molecular characterization. Mol Pharm. 2014;9(11):3340–3346, 2012. doi:10.1021/mp300365t
27. Thapar M, Rudnick S, Bonkovsky HL. Givosiran, a novel treatment for acute hepatic porphyrias. Expert Rev Precis Med Drug Dev. 2021;6(1):9–18. doi:10.1080/23808993.2021.1838275
 © 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.