Back to Journals » International Journal of Nephrology and Renovascular Disease » Volume 7
Obesity, hypertension, and chronic kidney disease
Authors Hall M, do Carmo J, da Silva A, Juncos L, Wang Z, Hall J
Received 25 October 2013
Accepted for publication 4 December 2013
Published 18 February 2014 Volume 2014:7 Pages 75—88
DOI https://doi.org/10.2147/IJNRD.S39739
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Michael E Hall,1,2 Jussara M do Carmo,2 Alexandre A da Silva,2 Luis A Juncos,1,2 Zhen Wang,2 John E Hall2
1Department of Medicine, 2Department of Physiology and Biophysics, Mississippi Center for Obesity Research, University of Mississippi Medical Center, Jackson, MS, USA
Abstract: Obesity is a major risk factor for essential hypertension, diabetes, and other comorbid conditions that contribute to development of chronic kidney disease. Obesity raises blood pressure by increasing renal tubular sodium reabsorption, impairing pressure natriuresis, and causing volume expansion via activation of the sympathetic nervous system and renin-angiotensin-aldosterone system and by physical compression of the kidneys, especially when there is increased visceral adiposity. Other factors such as inflammation, oxidative stress, and lipotoxicity may also contribute to obesity-mediated hypertension and renal dysfunction. Initially, obesity causes renal vasodilation and glomerular hyperfiltration, which act as compensatory mechanisms to maintain sodium balance despite increased tubular reabsorption. However, these compensations, along with increased arterial pressure and metabolic abnormalities, may ultimately lead to glomerular injury and initiate a slowly developing vicious cycle that exacerbates hypertension and worsens renal injury. Body weight reduction, via caloric restriction and increased physical activity, is an important first step for management of obesity, hypertension, and chronic kidney disease. However, this strategy may not be effective in producing long-term weight loss or in preventing cardiorenal and metabolic consequences in many obese patients. The majority of obese patients require medical therapy for obesity-associated hypertension, metabolic disorders, and renal disease, and morbidly obese patients may require surgical interventions to produce sustained weight loss.
Keywords: visceral adiposity, type II diabetes, sodium reabsorption, glomerular filtration rate, sympathetic nervous system, renin-angiotensin-aldosterone system
Introduction
Obesity has rapidly become a major challenge to health care systems worldwide. According to the World Health Organization, more than 1.4 billion people worldwide were overweight and another 500 million were obese in 2008.1 In the United States, approximately two-thirds of the adult population is overweight and nearly one-third is obese.2
Obesity is a widely recognized risk factor for cardiovascular disease and for various metabolic disorders such as type II diabetes. Less attention has been paid to the link between increased body weight and chronic kidney disease (CKD), although there is evidence that the steady rise in CKD prevalence may be closely associated with increasing obesity.3 Obesity causes cardiovascular and renal diseases through several mechanisms including hypertension, hyperglycemia, dyslipidemia, inflammation, and atherosclerosis. These disorders often coexist, especially when there is excess visceral fat, and have often been referred to as the “metabolic syndrome.” However, there is substantial evidence that excess visceral fat is the main driving force for almost all of the disorders associated with the metabolic syndrome, including CKD.3,4
Increases in the rates of CKD have paralleled increases in overweight and obesity.3,5 In the United States, more than 10% of people over 20 years of age (or >20 million) have some degree of CKD, and the prevalence of CKD in those over age 60 is approximately 25%.6 In 2009, almost 900,000 people in the United States were being treated for end-stage renal disease (ESRD), with an estimated cost of nearly $40 billion.7 In addition to causing extremely inflated health care-related costs, CKD is associated with increased morbidity and mortality. After adjustments for age, sex, race, comorbidity, and prior hospitalizations, CKD patients experience a 59% higher mortality rate compared with patients without CKD.8
Chronic renal dysfunction is an important consequence of obesity, at least in part, because of the strong links between visceral adiposity and the two leading causes of CKD: hypertension and diabetes. Current evidence suggests that overweight and obesity account for 65%–75% of the risk for essential hypertension.9 Type II diabetes, which accounts for over 90% of diabetes, is almost always associated with increased visceral adiposity. Hypertension and diabetes, along with other disorders associated with the metabolic syndrome, may interact synergistically to increase the risk of CKD and progression to ESRD. However, there is also evidence that obesity may cause renal dysfunction and increased risk for CKD independent of diabetes and hypertension.3,10
Some of the major questions related to treatment of chronic kidney disorders are whether weight loss interventions by diet and lifestyle changes, pharmacological therapy, or surgical interventions, such as bariatric surgery, are effective in preventing and/or slowing development and progression of CKD. Also, the most effective therapies for hypertension and diabetes in obese subjects have not been fully elucidated with appropriate clinical trials.
In this review, we compare the onset of CKD in normal weight and overweight/obese individuals and discuss the mechanisms by which obesity may contribute to hypertension, renal dysfunction, and CKD. We also discuss weight loss interventions and their efficacy in preventing and reversing kidney dysfunction associated with obesity.
High body mass, visceral obesity, and renal dysfunction
The World Health Organization defines overweight as a body mass index (BMI) of 25 kg/m2 or greater and obesity as a BMI >30 kg/m2.1 Comparisons of BMI with outcomes such as CKD have produced conflicting results and can be confounded by differences in regional or ectopic fat deposition or differences in muscle mass. As BMI does not distinguish between lean and fat body mass, other markers of adiposity have been evaluated, including waist circumference and imaging of visceral fat and specific ectopic fat depots.
Despite the shortcomings of BMI as a measure of obesity, there is a clear association between BMI and visceral obesity as well as renal dysfunction. In a retrospective analysis of a cohort of over 320,000 individuals followed for at least 15 years, the adjusted relative risk for ESRD increased in a stepwise manner as BMI increased (Figure 1).11 This relationship persisted even after adjustment for blood pressure, diabetes, age, sex, race, education level, smoking status, history of myocardial infarction, cholesterol level, proteinuria, hematuria, and serum creatinine.
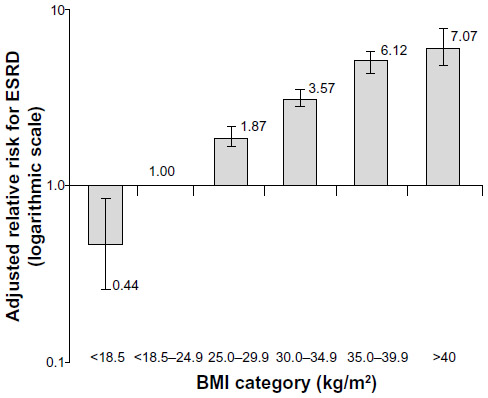 | Figure 1 Adjusted relative risk for end-stage renal disease (ESRD) by body mass index (BMI).Notes: In this retrospective cohort study of 320,252 adults who were followed for 15 to 35 years, the rate of ESRD increased in a stepwise manner as BMI increased. This relationship was not affected by blood pressure levels or diabetes. The model was adjusted for age, sex, race, education level, smoking status, history of myocardial infarction, serum cholesterol level, proteinuria, hematuria, and serum creatinine level. Copyright ©] 2006. Adapted from Hsu CY, McCulloch CE, Iribarren C, Darbinian J, Go AS. Body mass index and risk for end-stage renal disease. Ann Intern Med. 2006;144(1):21–28.11 |
Data from the Framingham Heart Study Offspring Cohort demonstrated a 68% relative increase in the odds of developing stage III CKD in individuals with a high BMI (>30 kg/m2) compared with those of normal weight.12 However, after adjustment for variables such as hypertension, diabetes, and smoking, this relationship was no longer significant. In a cohort of over 11,000 apparently healthy men followed for 14 years, higher baseline BMI was associated with an increased risk of incident CKD.13 Compared to participants with a BMI <22.7 kg/m2, those with BMI>26.6 kg/m2 had an odds ratio (OR) of 1.45 for developing CKD, and those who experienced a BMI increase of >10% had increased risk for CKD (OR 1.27).
Abdominal obesity is associated with CKD independently of overall adiposity or increased BMI, although there is generally a good association between BMI and visceral obesity. In a study of almost 6,500 nondiabetic participants, increasing BMI and waist circumference were associated with reduced estimated glomerular filtration rate (GFR) and increased CKD.14 Abdominal obesity, defined as a waist circumference >102 cm in men or >88 cm in women, was associated with higher risk of renal insufficiency (OR 1.40), even after adjustment for other components of the metabolic syndrome, such as dyslipidemia, hyperglycemia, hypertension, and BMI in patients with essential hypertension.15
There is also an association of ectopic kidney fat depots with renal dysfunction and hypertension. Renal sinus fat is associated with stage II hypertension and the number of antihypertensive medications required to control blood pressure.16 Furthermore, in the Framingham heart study, individuals with “fatty kidneys” (high perinephric fat levels) had a higher risk for hypertension (OR 2.12), which persisted after adjustment for BMI and visceral fat.17 Fatty kidney was also associated with increased risk for CKD (OR 2.30) even after adjustment for BMI and visceral adiposity.
It is clear that excess weight gain, especially when accompanied by increased visceral fat, is associated with many features of the metabolic syndrome which increase the risk for the development of CKD. Ectopic fat accumulation in and around the kidney may also have adverse consequences on renal function. Markers of visceral adiposity such as waist circumference are easily obtained in the clinic setting and may provide valuable prognostic information. More detailed examinations of specific fat depots evaluated with magnetic resonance and computed tomography imaging may also provide useful information related to the risk for development of CKD.
Obesity and susceptibility to other risk factors for kidney injury
There is evidence that obesity exacerbates the risk for developing CKD in response to other insults. Berthoux et al evaluated the absolute renal risk (ARR) of progression to dialysis or death in 331 patients with primary immunoglobulin A (IgA) nephropathy.18 The ARR was based on the presence of hypertension, ≥1 g/day of proteinuria, and histopathological renal lesions. Increased BMI at diagnosis was associated with more hypertension, proteinuria, and a trend for more severe renal pathology. The ARR for dialysis and death was significantly worse in the overweight/obese group compared with the normal weight group. IgA nephropathy patients in the overweight/obese group had lower GFRs, more CKD stage III or worse renal dysfunction, and more patients required dialysis or died compared with normal weight IgA nephropathy patients. Being overweight is also an independent risk factor for development of hypertension in patients with IgA nephropathy (Figure 2).19,20 Moderate weight loss in overweight patients with chronic nondiabetic proteinuric nephropathies induced a marked reduction in proteinuria, whereas in overweight subjects who did not lose weight, renal function worsened with time.21
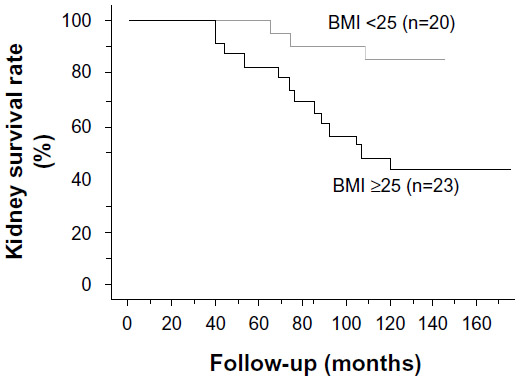 | Figure 2 Kidney survival rate of IgA nephropathy patients in the group with a BMI <25 kg/m2 and the group with a BMI>25 kg/m2. |
Obesity may also amplify the effects of other renal insults, even those that are usually considered to be relatively benign, such as unilateral nephrectomy. Retrospective analysis of patients who underwent unilateral nephrectomy (13.6±8.6 years previously) revealed that 62% of those with a BMI>30 kg/m2 developed proteinuria or renal insufficiency, whereas only 12% with a BMI <30 kg/m2 developed these disorders.22 Thus, obesity may have additive or synergistic effects to worsen renal function in patients with preexisting glomerulopathies or even more subtle renal injury, and weight loss may attenuate the impact of renal injury from other causes.
Mechanisms of obesity-associated kidney disease
Type II diabetes and hypertension together account for more than 75% of ESRD.8 Although much of the increasing prevalence of ESRD has been attributed to increasing type II diabetes, most of these patients are also hypertensive, making it difficult to separate the impact of increased blood pressure and diabetes in contributing to CKD. However, as discussed previously, there is evidence that obesity may contribute to kidney injury through additional mechanisms besides hypertension and type II diabetes.
Early functional and structural effects of obesity on the kidneys
Rapid weight gain in experimental animals as well as in humans initially causes increased renal tubular sodium reabsorption which, in turn, leads to compensatory renal vasodilation and increased GFR.23 The glomerular hyperfiltration that occurs after weight gain eventually subsides and may be replaced by a gradual decrease in GFR as renal injury and nephron loss occur in association with prolonged obesity-induced hypertension and diabetes.
The mechanisms responsible for the early rise in GFR in obesity are not entirely clear but may be mediated, in part, via a macula densa feedback mechanism.24 As discussed later, visceral obesity and associated compression of the kidneys may increase loop of Henle sodium chloride reabsorption, reducing sodium chloride delivery to the macula densa and causing, via tubuloglomerular feedback, reductions in afferent arteriolar resistance and increases in renal blood flow, GFR, and renin secretion. The increased GFR and elevated blood pressure would tend to return distal sodium chloride delivery toward normal in the face of increase loop reabsorption and therefore help to restore sodium balance. However, these compensatory mechanisms also lead to increased glomerular wall tension and glomerular hypertrophy, changes that may lead to renal injury, glomerulosclerosis, and ultimately nephron loss.
Structural changes in the kidneys occur within a few weeks after rapid weight gain. For example, in dogs placed on a high fat diet for only 7–9 weeks, there was enlargement of Bowman’s space, increased glomerular cell proliferation, increased mesangial matrix, thicker basement membranes, and increased expression of glomerular transforming growth factor β.25 These early renal changes occurred with only modest hypertension, no evidence of diabetes, and only mild metabolic abnormalities. Deposition of mesangial matrix, glomerular basement membrane thickening, and fibrotic changes in obesity may initially protect against glomerular capillary overstretching despite increased glomerular hydrostatic pressure due to renal vasodilation and elevated arterial pressures. However, these glomerular changes, if progressive, could eventually impinge on the glomerular lumen and reduce filtration surface area, initiating a positive feedback that further increases blood pressure and causes additional kidney injury. This slowly developing vicious cycle may be accelerated in the setting of metabolic derangements induced by obesity, such as hyperglycemia, inflammation, oxidative stress, and dyslipidemia.
Obese humans often develop proteinuria, frequently in the nephrotic range, that may be followed by progressive loss of kidney function even in the absence of diabetes or severe hypertension.26 The most common types of renal lesions observed in renal biopsies of obese subjects are focal and segmental glomerulosclerosis and glomerulomegaly. A review of 6,818 biopsies indicated that the incidence of obesity-related glomerulopathy, defined as combined focal glomerulosclerosis and glomerulomegaly, rose ten-fold from 1990 to 2000, coincident with the rapid increase in the prevalence of obesity during this period.27
Renal “lipotoxicity”
Ectopic deposition of lipids into nonadipose tissues, such as the kidney, often occurs in obesity. Excessive lipid deposition into nonadipose organs can lead to accumulation of toxic metabolites, such as diacylglycerols and ceramides, derived from metabolism of fats/fatty acids and sphingolipids.28 These metabolites may lead to mitochondrial dysfunction, endoplasmic reticulum stress, apoptosis, and eventually renal dysfunction and injury. Supporting this hypothesis is the finding that treatment with 3-hydroxy-3-methylglutaryl-coenzyme A reductase inhibitors may improve proteinuria and preserve renal function.29
The mechanisms by which obesity causes nephron injury are not fully understood but likely involve a combination of hemodynamic, metabolic, and inflammatory changes. Activation of the sympathetic nervous system (SNS) and renin–angiotensin–aldosterone system (RAAS) and physical compression may all contribute to hypertension, which along with metabolic disorders (eg, diabetes), glomerular hyperfiltration and inflammation, may cause renal injury, although the quantitative importance of these stimuli and how they interact to cause CKD are poorly understood.
Obesity and hypertension
The importance of obesity as a cause of hypertension is supported by experimental studies showing that weight gain raises blood pressure; clinical studies showing that weight loss lowers blood pressure in normotensive or hypertensive obese subjects; and epidemiological studies showing that excess weight gain is a good predictor for development of hypertension.5,30,31
Experimental animal studies have demonstrated a consistent rise in blood pressure with excess weight gain induced by prolonged high fat diet.23,32,33 These animal models exhibit many of the clinical manifestations of the metabolic syndrome often observed in obese humans. Although the mechanisms responsible for obesity-induced hypertension are not fully understood, there is considerable evidence that abnormal kidney function plays a key role. Obesity causes excess renal sodium reabsorption, impaired renal-pressure natriuresis, and expansion of extracellular fluid volume, which lead to increased arterial blood pressure.5,31 Increases in blood pressure and GFR offset increased renal sodium reabsorption so that sodium balance is maintained despite impaired renal-pressure natriuresis, albeit at a higher arterial pressure (Figure 3). However, chronically elevated blood pressure coupled with renal vasodilation and glomerular hyperfiltration, SNS and RAAS activation, inflammation and metabolic derangements eventually causes renal injury which, in turn, further impairs renal-pressure natriuresis and exacerbates hypertension and kidney injury as discussed previously (Figure 3).
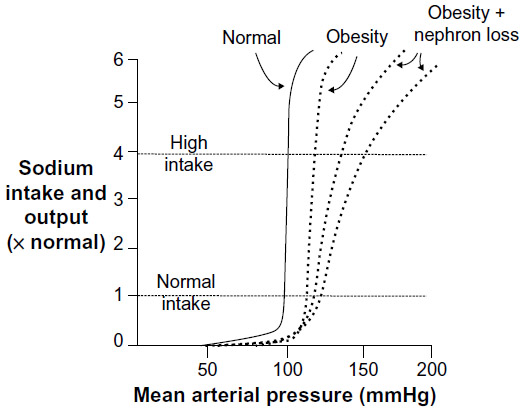 | Figure 3 Effect of obesity to shift the renal-pressure natriuresis curve to higher arterial pressure. |
Clinical studies also suggest that excess weight gain is a key contributor to elevated blood pressure in most patients with primary hypertension. Weight loss of only 5%–10% lowers blood pressure in normotensive as well as in hypertensive obese subjects and reduces the need for antihypertensive medications.34 Clinical trials have also demonstrated the effectiveness of weight loss in primary prevention of hypertension.35 Although weight loss does not completely normalize blood pressure in all obese patients, this is perhaps not surprising since long-term obesity-induced hypertension may also cause pathological cardiovascular and renal changes that are not always reversible, such as glomerular injury and loss of functional nephrons. These changes can make the hypertension more severe and more difficult to control, even with weight loss and antihypertensive medications.
Population studies have also shown that blood pressure is closely correlated with anthropometric indices of obesity, such as BMI (Figure 4), waist circumference, or waist-to-hip ratio.9,36,37 The strong association between excess weight and hypertension has been observed in diverse populations throughout the world and in populations of similar origins living in different locations.37,38 Thus, obesity appears to be a major cause of primary hypertension, and excess weight gain is a good predictor for future development of hypertension.
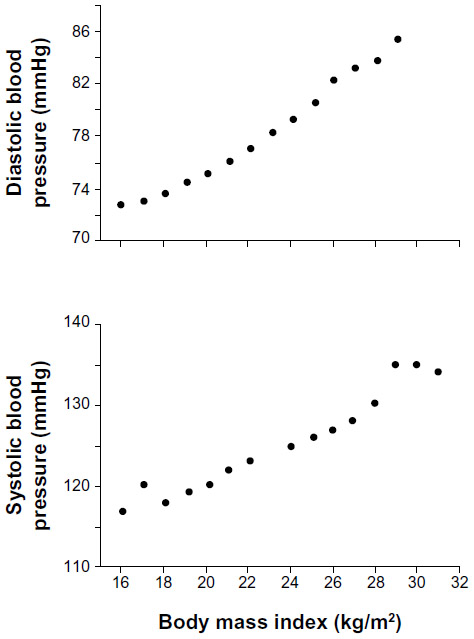 | Figure 4 Relationship between body mass index and systolic and diastolic blood pressures in 22,354 Korean subjects. |
Role of the sympathetic nervous system
Obese subjects have elevated SNS activity as assessed by plasma norepinephrine, urinary norepinephrine excretion, tissue norepinephrine spillover, or microneurography techniques.5,31 SNS activation is mild and occurs in specific tissues such as kidneys and skeletal muscle, whereas cardiac sympathetic activity may actually be reduced due to baroreflex inhibition.39 Pharmacologic blockade of adrenergic receptors reduces blood pressure to a greater extent in obese patients compared with lean hypertensive patients.40 The chronic effects of SNS activation on blood pressure appear to be mediated mainly by the renal nerves since renal denervation attenuates sodium reabsorption and development of hypertension associated with obesity in experimental animals.41 Renal denervation also reduces blood pressure in obese patients whose hypertension is resistant to the usual pharmacological therapy.42 These observations implicate activation of the SNS, especially the renal sympathetic nerves, as a major contributor to the development and maintenance of obesity hypertension.
The mechanisms of renal SNS activation in obesity have not been fully elucidated but several factors have been suggested: 1) hyperleptinemia, 2) angiotensin II (ANG II), 3) hyperinsulinemia, 4) impaired baroreflex sensitivity, and 5) physical compression of the kidneys due to visceral fat and intrarenal or perirenal fat. Because these factors have been previously reviewed,43–45 we will provide only a brief discussion of three mechanisms that appear to be especially important in obesity-induced hypertension.
Role of leptin-melanocortin system in obesity-induced SNS activation
Leptin is an adipokine that has powerful central nervous system effects, regulating energy balance by reducing appetite and stimulating energy expenditure via activation of the SNS. Mice lacking the ability to synthesize leptin due to a nonsense mutation in the leptin gene (ob/ob mice) or mice with nonfunctional leptin receptors (db/db mice) develop severe obesity. However, these mice which lack a functional leptin axis are usually not hypertensive despite profound adiposity and associated metabolic derangements.46 Similar findings have been reported in humans with leptin gene mutations who, despite severe obesity and most features of the metabolic syndrome, have normal blood pressure and reduced, rather than increased, SNS activity.47
Experimental studies have also shown that intracerebroventricular or intravenous infusions of leptin to produce blood concentrations similar to those found in obesity, increase renal sympathetic nervous activity and blood pressure.5,31,48,49 This rise in arterial pressure develops despite reductions in food intake and body mass. Leptin’s chronic effects to raise blood pressure appear to be mediated through the SNS, as combined alpha- and beta-adrenergic blockade completely abolished the hypertensive response.5,31
Leptin’s effects to activate the SNS and raise blood pressure appear to be mediated through activation of the hypothalamic proopiomelanocortin (POMC) pathway (Figure 5). POMC neurons send projections to second order neurons in the paraventricular nucleus and brainstem, where they release α-melanocyte stimulating hormone, which in turn, is the primary agonist for melanocortin 3 and 4 receptors (MC3R and MC4R, respectively). Deletion of leptin receptors specifically on POMC neurons completely abolished the blood pressure effects of leptin.50 Also, pharmacological blockade of central nervous system MC3R and MC4R completely abolished leptin’s acute effects on renal SNS activation51 as well as the chronic hypertensive effects of leptin.52 The blood pressure effects of leptin-mediated POMC activation are mediated largely by the MC4R rather than MC3R since mice with MC4R deficiency have no increase in SNS activity or rise in blood pressure during chronic hyperleptinemia.53
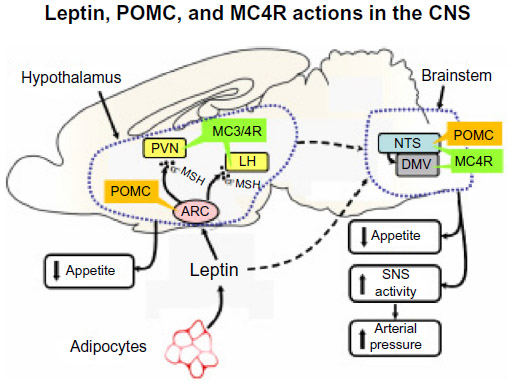 | Figure 5 Possible links between leptin and its effects on the hypothalamus, sympathetic nervous system (SNS) activity, and hypertension. |
Further support for the concept that the POMC-MC4R pathway may be an important link between obesity, SNS activation, and hypertension comes from the finding that humans and rodents with POMC or MC4R mutations have early-onset morbid obesity and many characteristics of the metabolic syndrome, including insulin resistance, hyperinsulinemia, and hyperlipidemia, but do not have increased SNS activity or hypertension.31,53,54 Moreover, chronic administration of an MC4R agonist caused significant increases in blood pressure in humans as well as in rodents.31,55 Thus, in humans and rodents, chronic activation of the POMC-MC4R pathway raises blood pressure, and the presence of a functional MC4R system appears to be necessary for obesity and hyperleptinemia to increase SNS activity and BP.
Role of renin–angiotensin–aldosterone system
Multiple studies in experimental animals and in humans suggest that the RAAS is mildly activated in obesity and contributes to increased blood pressure.5 Angiotensin converting enzyme inhibitors (ACEIs) or angiotensin receptor blockers (ARBs) blunt sodium reabsorption, extracellular volume expansion, and increased arterial pressure in obese dogs.56,57 In obese Zucker rats, there is increased sensitivity to the blood pressure effects of ANG II since blockade of the RAAS lowers blood pressure more than in lean rats despite comparable plasma renin activity.58
The direct effects of ANG II to stimulate renal sodium retention and aldosterone secretion are well known.59 However, whether the effects of ANG II to increase blood pressure in obesity are primarily mediated via direct actions on the kidneys or stimulation of aldosterone is still unclear.
ANG II also constricts efferent arterioles, which not only contributes to increased peritubular capillary reabsorption and therefore increased tubular sodium reabsorption, but also increases glomerular hydrostatic pressure.59 Therefore, activation of the RAAS may contribute to glomerular injury observed in obesity by exacerbating increased glomerular pressure caused by increased arterial blood pressure and vasodilation of afferent arterioles. Blockade of the RAAS with ACEIs or ARBs reduces proteinuria in diabetic and nondiabetic CKD patients.60–62 In the Ramipril in Non-diabetic Renal Failure Trial, obesity predicted an increased incidence of adverse renal outcomes including ESRD, but treatment with the ACEI ramipril attenuated this increased risk (Figure 6).63 Clinical studies comparing the effectiveness of RAAS antagonism in obese versus lean hypertensive patients would provide insight into the management of obesity hypertension and renal disease.
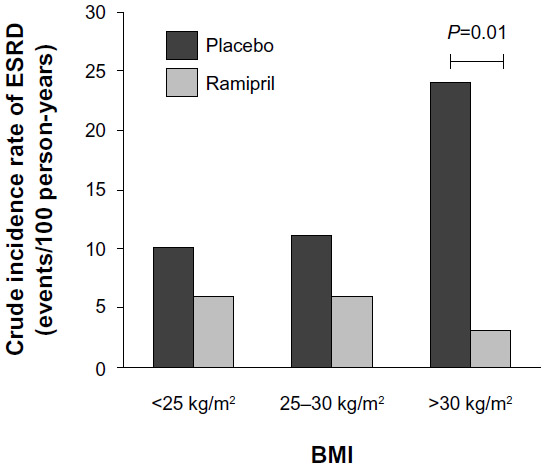 | Figure 6 The incidence of ESRD for obese, normal, and overweight patients and the effect of ramipril treatment. |
Plasma aldosterone levels are often mildly elevated in obese hypertensive patients, and a relative hyperaldosteronism may be partly responsible for “resistant” hypertension that is often observed in obese individuals. In a small study of 21 obese patients (mean BMI 32.4 kg/m2) with essential hypertension and preexisting target organ injury, low dose spironolactone (12.5 mg) was added to chronic ACEI therapy for 4 weeks and then withdrawn.64 During spironolactone treatment, mean office blood pressure, 24-hour ambulatory blood pressure, nocturnal blood pressures, and urinary albumin:creatinine ratio were all significantly reduced. Multivariate analyses suggested the improvement in proteinuria was related mainly to changes in GFR, although blood pressure effects could not be ruled out. In patients with type II diabetic nephropathy, addition of spironolactone, but not losartan, to a regimen including maximal angiotensin converting enzyme inhibition affords greater renoprotection despite similar reductions in blood pressure.65
Although the mechanisms responsible for the renoprotective effects of mineralocorticoid receptor (MR) antagonism in obesity have not been fully elucidated, MR antagonism in obese dogs markedly attenuated sodium retention, hypertension, and glomerular hyperfiltration.66 This protection occurred despite marked activation of the renin-angiotensin system levels, suggesting that combined blockade of MR and ACEI or ARB might be especially effective in preventing obesity-induced sodium retention and hypertension. The observation that MR antagonism attenuated glomerular hyperfiltration may also have important implications for renal protection in obesity, although there are no studies, to our knowledge, which have tested this directly in obese humans.
Role of visceral obesity and renal sinus fat in obesity hypertension
Visceral obesity may increase blood pressure, in part, through physical compression of the kidneys.67 In obese dogs and rabbits fed a high fat diet for several weeks, there was almost complete encapsulation of the kidneys and penetration of fat into the renal sinuses. These changes were associated with marked increases in renal interstitial hydrostatic pressure, which may act as a compressive force on the loop of Henle, reducing tubular flow rate and increasing fractional sodium reabsorption.43,68
High intra-abdominal pressure, secondary to increased visceral fat accumulation, may also contribute to renal compression. Intra-abdominal pressure is highly correlated with sagittal abdominal diameter, with some obese patients having intra-abdominal pressures as high as 40 mmHg.69 High intrarenal pressures and reduced flow rate into the loop of Henle would, in turn, reduce sodium chloride delivery to the macula densa, increasing renin secretion and ANG II formation, which would lead to further sodium reabsorption. Small increases (3–4 mmHg) in renal interstitial hydrostatic pressure may inhibit renal tubular sodium reabsorption, however, large increases of the magnitude observed in obese dogs (to around 19 mmHg) would increase sodium reabsorption. Hence, increases in intrarenal pressures caused by fat around and within the kidneys may be an important mechanism by which obesity causes hypertension.24,25,70
In addition to the potential compressive force of visceral and perinephritic fat on the kidneys, accumulation of fat in the renal sinuses has been associated with stage II hypertension in an elderly population and the number of antihypertensive drugs required to control blood pressure independent of other fat depots, including subcutaneous, intra-abdominal, and retroperitoneal fat.16 Additionally, renal sinus fat is independently associated with CKD.17 Although fat accumulation around and within the kidneys cannot account for the initial increase in blood pressure that occurs with rapid weight gain, it may partly explain why abdominal obesity is much more closely associated with hypertension than subcutaneous obesity.
Therapeutic interventions for obesity-induced renal dysfunction
Current therapeutic strategies for treating obesity-associated kidney disease rely largely on treating associated risk factors, including hypertension, diabetes, and hyperlipidemia. Unfortunately, few randomized clinical trials have evaluated the effectiveness of specific antihypertensive agents in obese individuals. In a multicenter placebo-controlled study of 232 obese patients with hypertension, the Treatment in Obese Patients with Hypertension (TROPHY) study compared the efficacy of the ACEI lisinopril to the diuretic hydrochlorothiazide over a 12-week period.71 Blood pressure reductions were similar in the two treatment groups, although hydrochlorothiazide elevated glucose levels compared to lisinopril. In the Candesartan Role on Obesity and on Sympathetic System (CROSS) study, the antihypertensive, neuroadrenergic, and metabolic effects of the ARB candesartan were compared to hydrochlorothiazide in 127 obese hypertensive patients.72 Blood pressure was reduced to similar levels by both medications. Candesartan treatment reduced muscle sympathetic nerve activity (measured by microneurography of the peroneal nerve) and improved insulin sensitivity, whereas hydrochlorothiazide had no effect on muscle sympathetic nerve activity and actually worsened insulin sensitivity.
Because of the lack of large prospective clinical trials evaluating treatment strategies in obese hypertensive patients, there are very few specific treatment recommendations for obesity hypertension. This is surprising given that obesity accounts for a large portion of the patients with hypertension. In a German survey, 75% of hypertensive patients treated by primary care physicians were overweight or obese.73 In 2012, the European Association for the Study of Obesity and the European Society of Hypertension published a consensus document discussing the pathophysiologic mechanisms and epidemiology of obesity hypertension, although no recommendations for specific antihypertensive treatments were made.74 There are specific blood pressure targets for patients with diabetes (<130/80 mmHg), and it seems reasonable for similar blood pressure targets to be recommended in obese individuals. However, due to the paucity of data from large clinical trials, no specific guidelines for treating these patients have been published. Some inferences may be drawn from randomized, controlled clinical trials of drug therapy in essential hypertension patients since most of these trials have included many subjects who were overweight or obese. Because hypertension is more difficult to control in obese subjects, especially when there is significant renal injury, combination drug therapy is often required to achieve goal blood pressures.
Obesity-induced renal dysfunction will become an increasingly difficult clinical dilemma as the prevalence of obesity continues to increase. Ideally, strategies to prevent weight gain and to induce weight loss in patients who are already obese would ultimately reduce the incidence of obesity-mediated renal disease. Weight loss through nonsurgical (lifestyle modification) or surgical interventions can reduce blood pressure and proteinuria and potentially decrease the incidence of advanced renal dysfunction in overweight and obese individuals. Even modest weight loss of 5%–10% has been shown to produce substantial benefits, including reductions in blood pressure and risk of diabetes.75
A systematic review by Afshinnia et al of 522 subjects from five controlled trials and eight uncontrolled trials demonstrated an association of weight loss with decreased proteinuria and microalbuminuria.76 Dietary restriction, exercise, pharmacological therapy, and bariatric surgery were included in these trials as weight loss interventions. Each 1 kg weight loss was associated with 110 mg decrease in proteinuria, independent of reduction in mean arterial pressure.
Lifestyle modification
Caloric restriction and increased physical activity are first-line therapy recommendations for obese individuals. This strategy is based on animal and clinical data showing that intentional weight loss with diet and exercise improves most of the metabolic, cardiovascular, and renal derangements associated with obesity. Several clinical studies have shown specific renal benefits of reducing body mass on blood pressure, proteinuria, and renal function.
Sixty-three patients with biopsy-proven, obesity-related glomerulopathy underwent physician-supervised weight loss programs including diet and exercise. These patients were divided into three groups: 1) >3% reduction in BMI, 2) stable weight, or 3) significant weight gain (>3% increase in BMI).77 Metabolic parameters and renal function were assessed over a 2-year period. Over the first 6 months, 27 patients had an average drop in BMI of 8.3% and a mean decrease in proteinuria of 35%. After 2 years, these patients achieved a 9.2% reduction in BMI, a 51% decrease in proteinuria, and concomitant reductions in blood pressure and dyslipidemia. These findings are consistent with previously published studies. Morales et al randomly assigned 30 overweight patients (BMI >27 kg/m2) with either diabetic or nondiabetic proteinuric nephropathies to either receive a low-calorie normal protein diet or to maintain their usual diet for 5 months.26 As expected, subjects in the low-calorie diet group showed a significant reduction in body weight and BMI compared to those in the usual diet group, who showed increased body weight and BMI. Mean weight loss in the diet group was 4.1% and this was associated with a 31.2% reduction in proteinuria, whereas proteinuria increased in the usual diet control group. There were no significant differences in renal function assessed by serum creatinine or estimated GFR between the two groups, although renal function remained stable in the diet group and showed significant worsening in the control group. This illustrates that a moderate weight loss (in this case just 4%) can significantly reduce proteinuria.
Exercise is an important component of sustained weight loss programs. In a study of patients with metabolic syndrome, weight loss strategies of diet only versus diet plus aerobic exercise were compared.26 Serum creatinine decreased more in the diet plus exercise group compared to the diet group (−14.9% versus −8.1%, respectively), and maximal oxygen consumption and baroreflex sensitivity were improved in only the diet plus exercise group. While albuminuria and muscle sympathetic nerve activity were reduced in both the diet and diet plus exercise groups, markers of SNS activation, fitness, inflammation (C-reactive protein), and activation of the RAAS (plasma renin activity) were improved by exercise in addition to dietary therapy.
There has been concern that certain diets, such as high-protein diets, may have adverse renal consequences. Brinkworth et al assessed renal function over a 1-year period in 68 individuals with abdominal obesity and without renal dysfunction. Individuals were randomized to either an isocaloric, low-carbohydrate diet (4% carbohydrate, 35% protein, 61% fat) or high-carbohydrate diet (46% carbohydrate, 24% protein, 30% fat).79 After 1 year, there were no significant changes in serum creatinine or estimated GFR in either group, demonstrating no significant adverse effects of weight reduction strategies with these diets. Whether longer periods of time on a high protein diet would have adverse renal consequences is unclear.
Surgical interventions
Nonsurgical interventions such as diet, exercise, and appetite-suppressing medications are often ineffective in producing long-term weight loss lasting more than 1 year. Therefore, surgical interventions such as bariatric surgery have been evaluated as a strategy to achieve long-term weight reduction and improve kidney function. Bariatric surgery attenuates obesity-mediated dyslipidemia and insulin resistance and reduces blood pressure, and several clinical studies have shown improvements in serum creatinine and proteinuria with surgical weight loss.80,81
In a 1-year prospective study, morbidly obese patients underwent Roux-en-Y gastric bypass (RYGB) surgery, laparoscopic adjustable gastric banding, or laparoscopic sleeve gastrectomy.82 One year after surgery, patients in all three surgical groups showed reductions in mean BMI, mean arterial pressure, and urinary and serum inflammatory markers. Patients with impaired renal function (defined as serum cystatin C >0.8 mg/L) at baseline had significant improvement in renal function 1 year after surgery. Reductions in serum and urine inflammatory markers (macrophage migration inhibitory factor, monocyte chemotactic protein-1, and chemokine ligand-18) were directly correlated with weight loss.
A recent randomized 1-year study compared the likelihood of achieving target blood pressure (<130 mmHg), hemoglobin A1c (<7.0%), and low-density lipoprotein cholesterol (<100 mg/dL) with RYGB surgery and lifestyle and intensive medical management.83 Forty-nine percent of the gastric bypass patients achieved the target endpoints compared to only 19% of the lifestyle and medical management arm. Patients in the gastric bypass group lost more weight and subsequently required three fewer medications compared to the lifestyle-medical management group.
A systematic review and meta-analysis by Navaneethan et al assessed 13 studies (eleven observational and two randomized controlled trials) to determine the potential benefits of weight loss in patients with CKD.84 Weight loss interventions included nonsurgical (diet, exercise, and pharmacologic agents) and surgical strategies. Nonsurgical interventions were associated with a decrease in BMI (weighted mean difference of −3.67 kg/m2), which significantly reduced systolic blood pressure and proteinuria while halting the decline in GFR over the mean follow-up period of over 7 months. Surgical interventions decreased BMI in morbidly obese individuals (BMI>40 kg/m2) with glomerular hyperfiltration (GFR>125 mL/min). Subsequently, these individuals experienced a reduction in GFR as well as systolic blood pressure and albuminuria, although this was over a short follow-up time period. In contrast, while nonsurgical interventions were also associated with reductions in BMI, there were no significant changes observed in GFR. The authors concluded that the “no treatment benefit” of the nonsurgical group could potentially be viewed as a treatment success because the treated group experienced no change in the GFR over a 7-month time period, while the untreated control group experienced a decline in GFR. They concluded that while weight loss reduces the risks of CKD in these patients, the overall evidence supporting intentional weight loss in those with mild to moderate CKD to slow progression of renal disease is “modest at best” and warrants further study.
The mechanisms by which bariatric surgery ameliorates renal dysfunction are only partially known and are largely inferred from studies on the mechanism of obesity-induced hypertension and renal injury. Attenuated SNS activity and reduction in sodium reabsorption are two potential beneficial effects of bariatric surgery since bariatric surgery decreases markers of overall renal sympathetic activity.85
Potential negative effects of gastric bypass surgery, particularly RYGB, are malabsorption and hyperoxaluria, which can exacerbate renal dysfunction. In one study, 7.7% of patients undergoing RYGB developed hyperoxaluria compared with only 4.6% of controls.86 This effect was not observed in gastric banding or sleeve gastrectomy procedures, which do not increase urinary oxalate excretion.
Although bariatric surgery has significant benefits for patients with severe obesity and underlying renal dysfunction, there are important risks associated with these procedures. However, in a retrospective analysis of over 27,000 patients undergoing bariatric surgery from 2006–2008, the absolute incidence of complication rates from these procedures remained below 10%.87 A risk-benefit assessment in this patient population must be carefully evaluated. Bariatric surgical procedures will be refined, safer, and more efficacious as they are more widely employed as a strategy to reduce renal and cardiovascular risks.
The “obesity paradox”
As discussed previously, overweight and obesity are important risk factors for hypertension and cardiovascular and renal disease. Being overweight or obese also increases the risk for other chronic diseases, including several types of cancer as well as all-cause mortality. However, there is a reverse epidemiology, known as the “obesity paradox”, in overweight and obese patients with ESRD, myocardial infarction, heart failure, stroke, cancer, diabetes, and other debilitating diseases.88–95 For example, several epidemiologic studies have shown that higher BMI in ESRD or dialysis patients is associated with a lower rather than higher mortality.88,89,91,92 Compared to patients with normal BMI (23.0–24.9 kg/m2), those who were overweight (BMI 25.0–29.9 kg/m2) or obese (BMI>30 kg/m2) had a significantly lower relative risk of death.88,89,91 Thus, being overweight or obese appeared to be associated with a survival benefit in dialysis patients, a benefit that was observed in “healthier” as well as the “sicker” subgroups of patients.91 Molnar et al92 studied mortality and BMI changes of 14,632 dialysis patients during the transplant waitlist period and also observed that patients with lower BMI, reduced muscle mass, or unintentional weight loss or muscle loss had a higher mortality.
The obesity paradox has also been observed in other diseases associated with high mortality rates, such as myocardial infarction, heart failure, and diabetes.90,93–98 Several potential explanations have been proposed for the obesity paradox. One possibility is that heavier patients may be presenting earlier with less advanced disease compared to those with lower BMI. Fonarow et al98 reviewed data from over 108,927 hospitalized patients in the Acute Decompensated Heart Failure National Registry and found that mortality in patients with a BMI of 27.8–33.3 kg/m2 was significantly lower than in patients with a BMI between 23.7–27.7 kg/m2, and that patients with BMI between 16–23.6 kg/m2 had the highest inpatient mortality. However, patients with higher BMI were younger and were less likely to have systolic failure than patients with lower BMI. Cachexia associated with heart failure and other debilitating chronic diseases, including ESRD and cancer, may also contribute to higher mortality in lean compared to obese patients. Being underweight or experiencing unintentional weight loss may reflect a general poor health status associated with high mortality in these patients. Overweight and obese patients may be more hemodynamically stable93 and less likely to develop protein-energy malnutrition than lean patients with critical illnesses.94
The apparent obesity paradox has been suggested to be simply the result of selection bias or methodological errors associated with epidemiological studies. For example, “collider stratification” is a source of selection bias that is relatively common in epidemiological research.95 This selection bias is caused by conditioning on a variable (eg, ESRD) that is affected by an exposure (eg, obesity) and shares common causes with the outcome, known as a collider (eg, mortality). A schematic of this potential source of selection bias is shown in Figure 7. Banack and Kaufman,95 using the National Health and Nutrition Examination Survey and the National Death Index, found that the apparent obesity paradox for heart failure disappeared after correction for selection bias. Evidence for selection bias as an explanation for the apparent paradoxical relationship of BMI, diabetes, and mortality has also been recently found.96,97 Although we are not aware of similar analyses for ESRD, this example illustrates the need to consider and correct for potential selection or treatment biases in epidemiological studies that can lead to counterintuitive conclusions. Unmeasured risk factors and biases related to selection and treatment of patients with complex chronic disease should be considered as possible sources of the apparent “obesity paradox”.
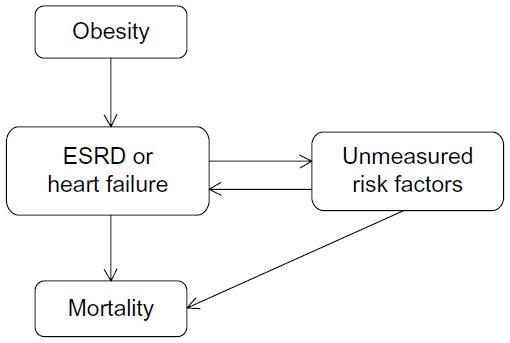 | Figure 7 Schematic of potential effects of obesity on debilitating diseases, such as heart failure and ESRD, and impact on mortality of unmeasured risk factors such as poor nutrition, cachexia, unstable hemodynamic status, genetics, and others. |
Summary
Data from experimental, clinical, and population studies clearly demonstrate that obesity is a driving force for development of CKD. Obesity-associated hypertension, diabetes, and dyslipidemia may act synergistically to increase the risk of renal dysfunction. Obesity triggers a cascade of intrarenal and neurohormonal changes that ultimately cause impaired renal-pressure natriuresis, increased sodium reabsorption, hypertension, and kidney dysfunction (Figure 8). Increased visceral adiposity activates the SNS and RAAS systems in addition to physically compressing the kidneys, leading to altered intrarenal hemodynamics and impaired sodium excretion. Surgical and nonsurgical weight loss strategies ameliorate several obesity-mediated risk factors, such as hypertension and diabetes, which have adverse consequences on the kidneys. However, additional clinical trials are needed to evaluate the long-term effects of such therapies.
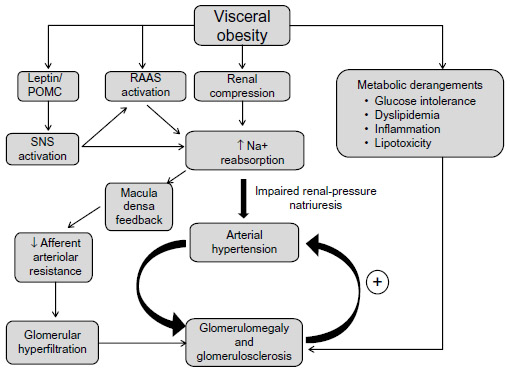 | Figure 8 Summary of potential mechanisms by which obesity causes hypertension and renal injury. |
Acknowledgments
This research was supported by grants from the National Heart, Lung, and Blood Institute (P01 HL 51971) and the National Institute of General Medical Sciences (P20 GM104357).
Disclosure
The authors report no conflicts of interest in this work.
References
Obesity and overweight. Fact Sheet N°311 [webpage on the Internet]. Geneva: World Health Organization. [updated Mar 2013; cited October 24, 2013]. Available from: http://www.who.int/mediacentre/factsheets/fs311/en/index.html. Accessed October 18, 2013. | |
Obesity and overweight trends in the U.S., 2011. Adult obesity facts [webpage on the Internet]. Atlanta: Center for Disease Control and Prevention. [updated Aug 2013; cited October 24, 2013]. Available from: http://www.cdc.gov/nccdphp/dnpa/obesity/trend/index.htm. Accessed on September 14, 2013. | |
Hall JE, Henegar JR, Dwyer TM, et al. Is obesity a major cause of chronic renal disease? Adv Ren Replace Ther. 2004;11(1):41–54. | |
Tchernof A, Després JP. Pathophysiology of human visceral obesity: an update. Physiol Rev. 2013;93(1):359–404. | |
Hall JE, Crook ED, Jones DW, Wofford MR, Dubbert PM. Mechanisms of obesity-associated cardiovascular and renal disease. Am J Med Sci. 2002;324(3):127–137. | |
National chronic kidney disease fact sheet 2010 [webpage on the Internet]. Atlanta: Center for Disease Control and Prevention. [updated November 5, 2012; cited October 24, 2013]. Available from: http://www.cdc.gov/diabetes/pubs/factsheets/kidney.htm. Accessed on September 14, 2013. | |
Kidney disease statistics for the United States. NIH publication No 12-3895 [webpage on the Internet]. Bethesda: U.S. Department of Health and Human Services – National Kidney and Urologic Diseases Clearinghouse. [updated November 15, 2012; cited October 24, 2013]. Available from: http://kidney.niddk.nih.gov/kudiseases/pubs/kustats/. Accessed on September 14, 2013. | |
U.S. Renal Data System, 2012 annual data report: atlas of chronic kidney disease and end-stage renal disease in the United States [webpage on the Internet]. Bethesda: National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases; 2012. Available from: http://www.usrds.org/atlas12.aspx. | |
Garrison RJ, Kannel WB, Stokes J, Castelli WP. Incidence and precursors of hypertension in young adults: The Framingham Offspring Study. Prev Med. 1987;16(2):235–251. | |
Ejerblad E, Fored CM, Lindblad P, Fryzek J, McLaughlin JK, Nyrén O. Obesity and risk for chronic renal failure. J Am Soc Nephrol. 2006; 17(6):1695–1702. | |
Hsu CY, McCulloch CE, Iribarren C, Darbinian J, Go AS. Body mass index and risk for end-stage renal disease. Ann Intern Med. 2006;144(1):21–28. | |
Foster MC, Hwang SJ, Larson MG, et al. Overweight, obesity, and the development of stage 3 CKD: the Framingham Heart Study. Am J Kidney Dis. 2008;52(1):39–48. | |
Gelber RP, Kurth T, Kausz AT, et al. Association between body mass index and CKD in apparently healthy men. Am J Kidney Dis. 2005;46(5):871–880. | |
Burton JO, Gray LJ, Webb DR, et al. Association of anthropometric obesity measures with chronic kidney disease risk in a non-diabetic patient population. Nephrol Dial Transplant. 2012;27(5):1860–1866. | |
Gomez P, Ruilope LM, Barrios V, et al; FATH Study Group. Prevalence of renal insufficiency in individuals with hypertension and obesity/overweight: the FATH study. J Am Soc Nephrol. 2006;17(12 Suppl 3):S194–S200. | |
Chughtai HL, Morgan TM, Rocco M, et al. Renal sinus fat and poor blood pressure control in middle-aged and elderly individuals at risk for cardiovascular events. Hypertension. 2010;56(5):901–906. | |
Foster MC, Hwang SJ, Porter SA, Massaro JM, Hoffmann U, Fox CS. Fatty kidney, hypertension, and chronic kidney disease: the Framingham Heart Study. Hypertension. 2011;58(5):784–790. | |
Berthoux F, Mariat C, Maillard N. Overweight/obesity revisited as a predictive risk factor in primary IgA nephropathy. Nephrol Dial Transplant. 2013;(28 Suppl 4):iv160–iv166. | |
Bonnet F, Deprele C, Sassolas A, et al. Excessive body weight as a new independent risk factor for clinical and pathological progression in primary IgA nephritis. Am J Kidney Dis. 2001;37(4):720–727. | |
Kataoka H, Ohara M, Shibui K, et al. Overweight and obesity accelerate the progression of IgA nephropathy: prognostic utility of a combination of BMI and histopathological parameters. Clin Exp Nephrol. 2012;16(5):706–712. | |
Chagnac A, Weinstein T, Korzets A, Ramadan E, Hirsch J, Gafter U. Glomerular hemodynamics in severe obesity. Am J Physiol Renal Physiol. 2000;278(5):F817–F822. | |
Praga M, Hernández E, Herrero JC, et al. Influence of obesity on the appearance of proteinuria and renal insufficiency after unilateral nephrectomy. Kidney Int. 2000;58(5):2111–2118. | |
Hall JE, Brands MW, Dixon WN, Smith MJ. Obesity-induced hypertension. Renal function and systemic hemodynamics. Hypertension. 1993;22(3):292–299. | |
Hall JE, Louis K. Dahl Memorial Lecture. Renal and cardiovascular mechanisms of hypertension in obesity. Hypertension. 1994;23(3):381–394. | |
Henegar JR, Bigler SA, Henegar LK, Tyagi SC, Hall JE. Functional and structural changes in the kidney in the early stages of obesity. J Am Soc Nephrol. 2001;12(6):1211–1217. | |
Morales E, Valero MA, León M, Hernández E, Praga M. Beneficial effects of weight loss in overweight patients with chronic proteinuric nephropathies. Am J Kidney Dis. 2003;41(2):319–327. | |
Kambham N, Markowitz GS, Valeri AM, Lin J, D’Agati VD. Obesity-related glomerulopathy: an emerging epidemic. Kidney Int. 2001;59(4):1498–1509. | |
Unger RH, Scherer PE. Gluttony, sloth and the metabolic syndrome: a roadmap to lipotoxicity. Trends Endocrinol Metab. 2010;21(6):345–352. | |
Tonelli M, Moyé L, Sacks FM, Cole T, Curhan GC; Cholesterol and Recurrent Events Trial Investigators. Effect of pravastatin on loss of renal function in people with moderate chronic renal insufficiency and cardiovascular disease. J Am Soc Nephrol. 2003;14(6):1605–1613. | |
Jones A, Charakida M, Falaschetti E, et al. Adipose and height growth through childhood and blood pressure status in a large prospective cohort study. Hypertension. 2012;59(5):919–925. | |
Hall JE, da Silva AA, do Carmo JM, et al. Obesity-induced hypertension: role of sympathetic nervous system, leptin, and melanocortins. J Biol Chem. 2010;285(23):17271–17276. | |
Carroll JF, Huang M, Hester RL, Cockrell K, Mizelle HL. Hemodynamic alterations in hypertensive obese rabbits. Hypertension. 1995;26(3):465–470. | |
Dobrian AD, Davies MJ, Prewitt RL, Lauterio TJ. Development of hypertension in a rat model of diet-induced obesity. Hypertension. 2000;35(4):1009–1015. | |
Jones DW, Miller ME, Wofford MR, et al. The effect of weight loss intervention on antihypertensive medication requirements in the hypertension Optimal Treatment (HOT) study. Am J Hypertens. 1999;12(12 Pt 1–2):1175–1180. | |
Stevens VJ, Obarzanek E, Cook NR, et al; Trials for the Hypertension Prevention Research Group. Long-term weight loss and changes in blood pressure: results of the Trials of Hypertension Prevention, phase II. Ann Intern Med. 2001;134(1):1–11. | |
Alexander J, Dustan HP, Sims EAH, et al. Report of the Hypertension Task Force. Department of Health Education and Welfare publication no (DHEW) 70-1631. Washington, DC: US Department of Health, Education, and Welfare. 1979:61–77. | |
Jones DW, Kim JS, Andrew ME, Kim SJ, Hong YP. Body mass index and blood pressures in Korean men and women: the Korean National Blood Pressure Survey. J Hypertens. 1994;12(12):1433–1437. | |
Cooper RS, Rotimi CN, Ward R. The puzzle of hypertension in African-Americans. Sci Am. 1999;280(2):56–63. | |
Vaz M, Jennings G, Turner A, Cox H, Lambert G, Esler M. Regional sympathetic nervous activity and oxygen consumption in obese normotensive human subjects. Circulation. 1997;96(10):3423–3429. | |
Wofford MR, Anderson DC, Brown CA, Jones DW, Miller ME, Hall JE. Antihypertensive effect of alpha- and beta-adrenergic blockade in obese and lean hypertensive subjects. Am J Hypertens. 2001;14(7 Pt 1):694–698. | |
Kassab S, Kato T, Wilkins FC, Chen R, Hall JE, Granger JP. Renal denervation attenuates the sodium retention and hypertension associated with obesity. Hypertension. 1995;25(4 Pt 2):893–897. | |
Esler MD, Krum H, Schlaich M, Schmieder RE, Böhm M, Sobotka PA; Symplicity HTN-2 Investigators. Renal sympathetic denervation for treatment of drug-resistant hypertension: one-year results from the Symplicity HTN-2 randomized, controlled trial. Circulation. 2012;126(25):2976–2982. | |
Hall JE. The kidney, hypertension, and obesity. Hypertension. 2003;41(3 Pt 2):625–633. | |
Davy KP, Hall JE. Obesity and hypertension: two epidemics or one? Am J Physiol Regul Integr Comp Physiol. 2004;286(5):R803–R813. | |
da Silva AA, do Carmo J, Dubinion J, Hall JE. The role of the sympathetic nervous system in obesity-related hypertension. Curr Hypertens Rep. 2009;11(3):206–211. | |
do Carmo JM, da Silva AA, Dubinion J, et al. Control of metabolic and cardiovascular function by the leptin-brain melanocortin pathway. IUBMB Life. 2013;65(8):692–698. | |
Ozata M, Ozdemir IC, Licinio J. Human leptin deficiency caused by a missense mutation: multiple endocrine defects, decreased sympathetic tone, and immune system dysfunction indicate new targets for leptin action, greater central than peripheral resistance to the effects of leptin, and spontaneous correction of leptin-mediated defects. J Clin Endocrinol Metab. 1999;84(10):3686–3695. | |
Shek EW, Brands MW, Hall JE. Chronic leptin infusion increases arterial pressure. Hypertension. 1998;31(1 Pt 2):409–414. | |
Dubinion JH, da Silva AA, Hall JE. Chronic blood pressure and appetite responses to central leptin infusion in rats fed a high fat diet. J Hypertens. 2011;29(4):758–762. | |
do Carmo JM, da Silva AA, Cai Z, Lin S, Dubinion JH, Hall JE. Control of blood pressure, appetite, and glucose by leptin in mice lacking leptin receptors in proopiomelanocortin neurons. Hypertension. 2011;57(5):918–926. | |
Haynes WG, Morgan DA, Djalali A, Sivitz WI, Mark AL. Interactions between the melanocortin system and leptin in control of sympathetic nerve traffic. Hypertension. 1999;33(1 Pt 2):542–547. | |
da Silva AA, Kuo JJ, Hall JE. Role of hypothalamic melanocortin 3/4-receptors in mediating chronic cardiovascular, renal, and metabolic actions of leptin. Hypertension. 2004;43(6):1312–1317. | |
Tallam LS, da Silva AA, Hall JE. Melanocortin-4 receptor mediates chronic cardiovascular and metabolic actions of leptin. Hypertension. 2006;48(1):58–64. | |
Greenfield JR. Melanocortin signalling and the regulation of blood pressure in human obesity. J Neuroendocrinol. 2011;23(2):186–193. | |
Greenfield JR, Miller JW, Keogh JM, et al. Modulation of blood pressure by central melanocortinergic pathways. N Engl J Med. 2009;360(1):44–52. | |
Hall JE, Henegar JR, Shek EW, Brands MW. Role of renin-angiotensin system in obesity hypertension. Circulation. 1997;96:1–33. | |
Robles RG, Villa E, Santirso R, et al. Effects of captopril on sympathetic activity, lipid and carbohydrate metabolism in a model of obesity-induced hypertension in dogs. Am J Hypertens. 1993;6(12):1009–1015. | |
Alonso-Galicia M, Brands MW, Zappe DH, Hall JE. Hypertension in obese Zucker rats. Role of angiotensin II and adrenergic activity. Hypertension. 1996;28(6):1047–1054. | |
Hall JE, Brands MW, Henegar JR. Angiotensin II and long-term arterial pressure regulation: the overriding dominance of the kidney. J Am Soc Nephrol. 1999;(10 Suppl 12):S258–S265. | |
Kunz R, Friedrich C, Wolbers M, Mann JF. Meta-analysis: effect of monotherapy and combination therapy with inhibitors of the renin angiotensin system on proteinuria in renal disease. Ann Intern Med. 2008;148(1):30–48. | |
Gansevoort RT, Sluiter WJ, Hemmelder MH, de Zeeuw D, de Jong PE. Antiproteinuric effect of blood-pressure-lowering agents: a meta-analysis of comparative trials. Nephrol Dial Transplant. 1995;10(11):1963–1974. | |
Heeg JE, de Jong PE, van der Hem GK, de Zeeuw D. Efficacy and variability of the antiproteinuric effect of ACE inhibition by lisinopril. Kidney Int. 1989;36(2):272–279. | |
Mallamaci F, Ruggenenti P, Perna A, et al; REIN Study Group. ACE inhibition is renoprotective among obese patients with proteinuria. J Am Soc Nephrol. 2011;22(6):1122–1128. | |
Bomback AS, Muskala P, Bald E, Chwatko G, Nowicki M. Low-dose spironolactone, added to long-term ACE inhibitor therapy, reduces blood pressure and urinary albumin excretion in obese patients with hypertensive target organ damage. Clin Nephrol. 2009;72(6):449–456. | |
Mehdi UF, Adams-Huet B, Raskin P, Vega GL, Toto RD. Addition of angiotensin receptor blockade or mineralocorticoid antagonism to maximal angiotensin-converting enzyme inhibition in diabetic nephropathy. J Am Soc Nephrol. 2009;20(12):2641–2650. | |
de Paula RB, da Silva AA, Hall JE. Aldosterone antagonism attenuates obesity-induced hypertension and glomerular hyperfiltration. Hypertension. 2004;43(1):41–47. | |
Hall JE, Brands MW, Henegar JR. Mechanisms of hypertension and kidney disease in obesity. Ann N Y Acad Sci. 1999;892:91–107. | |
Hall JE, Jones DW, Kuo JJ, da Silva A, Tallam LS, Liu J. Impact of obesity on hypertension and renal disease. Curr Hypertens Rep. 2003;5(5):386–392. | |
Sugerman H, Windsor A, Bessos M, Wolfe L. Intra-abdominal pressure, sagittal abdominal diameter and obesity comorbidity. J Intern Med. 1997;241(1):71–79. | |
Alonso-Galicia M, Dwyer TM, Herrera GA, Hall JE. Increased hyaluronic acid in the inner renal medulla of obese dogs. Hypertension. 1995;25(4 Pt 2):888–892. | |
Reisin E, Weir MR, Falkner B, Hutchinson HG, Anzalone DA, Tuck ML. Lisinopril versus hydrochlorothiazide in obese hypertensive patients: a multicenter placebo-controlled trial. Treatment in Obese Patients With Hypertension (TROPHY) Study Group. Hypertension. 1997;30(1 Pt 1):140–145. | |
Grassi G, Seravalle G, Dell’Oro R, et al; CROSS Study. Comparative effects of candesartan and hydrochlorothiazide on blood pressure, insulin sensitivity, and sympathetic drive in obese hypertensive individuals: results of the CROSS study. J Hypertens. 2003;21(9):1761–1769. | |
Bramlage P, Pittrow D, Wittchen HU, et al. Hypertension in overweight and obese primary care patients is highly prevalent and poorly controlled. Am J Hypertens. 2004;17(10):904–910. | |
Jordan J, Yumuk V, Schlaich M, et al. Joint statement of the European Association for the Study of Obesity and the European Society of Hypertension: obesity and difficult to treat arterial hypertension. J Hypertens. 2012;30(6):1047–1055. | |
Wing RR, Lang W, Wadden TA, et al; Look AHEAD Research Group. Benefits of modest weight loss in improving cardiovascular risk factors in overweight and obese individuals with type 2 diabetes. Diabetes Care. 2011;34(7):1481–1486. | |
Afshinnia F, Wilt TJ, Duval S, Esmaeili A, Ibrahim HN. Weight loss and proteinuria: systematic review of clinical trials and comparative cohorts. Nephrol Dial Transplant. 2010;25(4):1173–1183. | |
Shen WW, Chen HM, Chen H, Xu F, Li LS, Liu ZH. Obesity-related glomerulopathy: body mass index and proteinuria. Clin J Am Soc Nephrol. 2010;5(8):1401–1409. | |
Straznicky NE, Grima MT, Lambert EA, et al. Exercise augments weight loss induced improvement in renal function in obese metabolic syndrome individuals. J Hypertens. 2011;29(3):553–564. | |
Brinkworth GD, Buckley JD, Noakes M, Clifton PM. Renal function following long-term weight loss in individuals with abdominal obesity on a very-low-carbohydrate diet vs high-carbohydrate diet. J Am Diet Assoc. 2010;110(4):633–638. | |
Chagnac A, Weinstein T, Herman M, Hirsh J, Gafter U, Ori Y. The effects of weight loss on renal function in patients with severe obesity. J Am Soc Nephrol. 2003;14(6):1480–1486. | |
Schuster DP, Teodorescu M, Mikami D, Foreman K, Rogers P, Needleman BJ. Effect of bariatric surgery on normal and abnormal renal function. Surg Obes Relat Dis. 2011;7(4):459–464. | |
Fenske WK, Dubb S, Bueter M, et al. Effect of bariatric surgery-induced weight loss on renal and systemic inflammation and blood pressure: a 12-month prospective study. Surg Obes Relat Dis. 2013;9(4):559–568. | |
Ikramuddin S, Korner J, Lee WJ, et al. Roux-en-Y gastric bypass vs intensive medical management for the control of type 2 diabetes, hypertension, and hyperlipidemia: the Diabetes Surgery Study randomized clinical trial. JAMA. 2013;309(21):2240–2249. | |
Navaneethan SD, Yehnert H, Moustarah F, Schreiber MJ, Schauer PR, Beddhu S. Weight loss interventions in chronic kidney disease: a systematic review and meta-analysis. Clin J Am Soc Nephrol. 2009;4(10):1565–1574. | |
van de Borne P, Watrin I, Bouquegneau M, et al. Ambulatory blood pressure and neuroendocrine control after diet-assisted gastric restrictive surgery. J Hypertens. 2000;18(3):301–306. | |
Matlaga BR, Shore AD, Magnuson T, Clark JM, Johns R, Makary MA. Effect of gastric bypass surgery on kidney stone disease. J Urol. 2009;181(6):2573–2577. | |
Turgeon NA, Perez S, Mondestin M, et al. The impact of renal function on outcomes of bariatric surgery. J Am Soc Nephrol. 2012;23(5):885–894. | |
Lowrie EG, Lew NL. Death risk in hemodialysis patients: the predictive value of commonly measured variables and an evaluation of death rate differences between facilities. Am J Kidney Dis. 1990;15(5):458–482. | |
Kalantar-Zadeh K, Block G, Humphreys MH, Kopple JD. Reverse epidemiology of cardiovascular risk factors in maintenance dialysis patients. Kidney Int. 2003;63:793–808. | |
Kalantar-Zadeh K, Block G, Horwich T, Fonarow GC. Reverse epidemiology of conventional cardiovascular risk factors in patients with chronic heart failure. J Am Coll Cardiol. 2004;43(8):1439–1444. | |
Leavey SF, McCullough K, Hecking E, Goodkin D, Port FK, Young EW. Body mass index and mortality in ‘healthier’ as compared with ‘sicker’ haemodialysis patients: results from the Dialysis Outcomes and Practice Patterns Study (DOPPS). Nephrol Dial Transplant. 2001;16(12):2386–2394. | |
Molnar MZ, Streja E, Kovesdy CP, et al. Associations of body mass index and weight loss with mortality in transplant-waitlisted maintenance hemodialysis patients. Am J Transplant. 2011;11(4):725–736. | |
Horwich TB, Fonarow GC, Hamilton MA, MacLellan WR, Woo MA, Tillisch JH. The relationship between obesity and mortality in patients with heart failure. J Am Coll Cardiol. 2001;38(3):789–795. | |
Fung F, Sherrard DJ, Gillen DL, et al. Increased risk for cardiovascular mortality among malnourished end-stage renal disease patients. Am J Kidney Dis. 2002;40(2):307–314. | |
Banack HR, Kaufman JS. The “obesity paradox” explained. Epidemiology. 2013;24(3):461–462. | |
Lajous M, Bijon A, Fagherazzi G, et al. Body mass index, diabetes, and mortality in French women: explaining away a “paradox”. Epidemiology. 2014;25(1):10–14. | |
Standl E, Erbach M, Schnell O. Defending the con side: obesity paradox does not exist. Diabetes Care. 2013;36(Suppl 2):S282–S286. | |
Fonarow GC, Srikanthan P, Costanzo MR, Cintron GB, Lopatin M; ADHERE Scientific Advisory Committee and Investigators. An obesity paradox in acute heart failure: analysis of body mass index and inhospital mortality for 108,927 patients in the Acute Decompensated Heart Failure National Registry. Am Heart J. 2007;153(1):74–81. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.