Back to Journals » Nutrition and Dietary Supplements » Volume 11
Nutrition in sickle cell disease: recent insights
Authors Umeakunne K ![]() , Hibbert JM
, Hibbert JM ![]()
Received 18 December 2018
Accepted for publication 2 April 2019
Published 23 May 2019 Volume 2019:11 Pages 9—17
DOI https://doi.org/10.2147/NDS.S168257
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Chandrika Piyathilake
Video abstract presented by Kayellen Umeakunne and Jacqueline Hibbert.
Views: 3108
Kayellen Umeakunne,1 Jacqueline M Hibbert2
1Clinical Research Center, Bio-nutrition Core, Morehouse School of Medicine, Atlanta, GA, USA; 2Morehouse School of Medicine, Department of Microbiology, Biochemistry and Immunology, Atlanta, GA, USA
Abstract: A cure for sickle cell anemia (SCA) is not available to all who have inherited this devastating genetically inherited disease. However, increasing knowledge that nutritional problems are fundamental to the severity of the disease, has produced interest in promoting dietary supplementation for treating these patients. This review seeks to emphasize the understanding that both children and adults with sickle cell disease require much higher energy and protein consumption (more macronutrient intake) than healthy individuals and tend to suffer from undernutrition if energy intake is consistently low. Shortages may also exist for micronutrients, eg, Glutathione, which has both anti-inflammatory and anti-oxidant properties. Both chronic inflammation and oxidant stress are central issues for increased sickle cell disease severity. In conclusion, dedicating more effort and resources to establishing recommended dietary reference intakes (DRIs)/recommended dietary allowances (RDAs) for SCA patients is essential, and nutritional intervention should be included as an adjunct treatment in tandem with standard practice.
Keywords: macronutrients, micronutrients, inflammation, vaso-occlusive crisis (VOC)
Introduction
Sickle cell disease (SCD), involves widespread single-gene disorder hemoglobinopathies. The most common gene disorders are sickle cell anemia (HbSS or SCA), hemoglobin SC (HbSC) and hemoglobin Sβ thalassemia (HbSβthal). Patients with SCA suffer most severely, and these diseases represent a significant global public health concern, in endemic malaria environments. Over 100,000 people in the United States, are affected. Estimates recorded by the United Nations for the year 2008, were 20–25 million people worldwide living with SCD, with people of African descent primarily affected.1,2 This review addresses SCA with associated high severity, and regular costly hospital stays. In 2004 approximately 113,000 hospitalizations for SCD were recorded in the United States. The estimated hospital costs at that time were $488 million for the year.3 Earlier estimates for 1989–1993 were $475 million per year,4 indicating an approximate 3% increased hospitalization cost between 1993 and 2004.
Hydroxyurea (HU), the first drug approved by the FDA to treat SCD, has provided the ability to extend life and reduce morbidity and mortality in individuals affected by the disease.5 However, average life expectancy is still approximately 30 years lower for individuals with sickle cell anemia (SCA) than for the general population.6 Increased use of HU has resulted in decreased attention to new studies addressing nutritional deficiencies, which still exist and contribute to slowed growth, development, and reduced quality of life in this population. Nutrition is reported to impact many chronic health conditions associated with SCD, including chronic baseline inflammation,7 vaso-occlusive crisis (VOC), which is accompanied by frequent pain and greater occurrence of stroke, particularly in young children.8 Other severe manifestations of SCD are pulmonary hypertension, cardiovascular and renal disease.
Targeted evidence-based recommendations for nutrition therapy in sickle cell disease
It is becoming more apparent that current dietary recommendations for SCD should include more emphasis on adequate amounts of macronutrients.8 Traditional supplementation studies addressed only the association of SCA with a variety of micronutrient deficiencies, including zinc, copper, folic acid, pyridoxine, vitamin E, and more recently B6, B12, omega three fatty acids9 and vitamin D.10 The standard treatment protocol provides these supplements. One small study reported in 1985 demonstrated the efficacy of including macronutrient supplementation. Compared with only micronutrient supplements, intervention with macronutrients, (proteins carbohydrates and fats) showed measurable improvement in clinical condition, and reduced hospital admissions in growth delayed children with SCA.11 More recently, in a sickle cell mouse model, Manci et al, confirmed diminished organ damage and vascular leakage with a high protein to energy diet (35%) (Figure 1),12 based on a previous report by Archer et al, that macronutrient supplementation reduced chronic inflammation. Protein to energy ratio is a significant determinant of dietary adequacy, and for optimal growth control mice require 20% of energy from dietary protein. These studies showed a trending increased weight gain for S35 versus S20 mice (p<0.06). Also, inflammatory proteins C-reactive protein (CRP) and interleukin-6 (IL-6) decreased significantly in S35 versus S20 mice (P<0.05), suggesting that added macronutrient intervention could reduce sickle cell disease-associated inflammation, which drives disease severity and ultimate organ damage.13
 | Figure 1 High protein diet reduced organ damage in sickle cell mice. |
Energy and protein requirements
Adults and children with sickle cell anemia have a relative energy shortage. Hibbert et al, and others have shown that nutritional deficiency in SCA is secondary to a marked hypermetabolic state associated with higher energy requirements. Investigations demonstrated increased urea kinetics (a result of protein catabolism),14 erythropoiesis, myocardial energy expenditure,15 and proinflammatory cytokines,16 as significant contributors to the increased energy demands. In SCD patients, nutrients from the diet and amino acids from body protein catabolism channeled toward rapid red cell production, are replacing hemolyzed sickle red cells being constantly removed from the circulation. This metabolic irregularity drastically increases the energy requirement and reduces the availability of nutrients for growth and development in children and for maintaining adequate muscle mass in adults. The primary clinical manifestation of this relative nutrient deficiency is severe undernutrition. Singhal et al, measured the dietary intakes and resting metabolic rates (RMR) of 41 children with SCD and 31 control subjects with normal hemoglobin, aged three to six years. Dietary intake was assessed by weighed food consumption and RMR was determined by indirect calorimetry. The children with SCD had significantly elevated RMR compared with the healthy controls, after adjusting for gender and weight. Energy intake was similar for both groups, but the ratio of energy intake to RMR was significantly lower for the SCD compared with the control group. The authors concluded that this observation indicates a relative energy deficiency in SCD.17 These findings support a hypothesis for the increased need for energy from macronutrients (ie, proteins, carbohydrates, and fats), for pre-pubertal children with sickle cell disease. This condition is also likely to be the case for adults with SCD, particularly those that inherit the SCA genotype.
Macronutrients required to improve nutritional status in SCD – amino acids
Arginine
The amino acid arginine plays a vital role in the synthesis of nitric oxide in the endothelial cells. Nitric oxide stimulates muscle cells to relax thereby regulating blood flow and blood pressure through dilatation. Arginine metabolism is impaired in SCD and contributes to endothelial dysfunction, vaso-occlusive crises, and pulmonary hypertension. Arginine deficiency develops over time so that by adulthood it achieves a low steady state. Due to the interaction between arginine availability and nitric oxide levels, lack of adequate amounts of arginine leads to disruption of vascular homeostasis and oxidative stress. Therefore, in SCD arginine becomes an essential amino acid.18
Kehinde et al, investigated twenty (20) normal non-sickle cell anemia (NSCA) subjects and 20 SCA subjects receiving supplementation with L-arginine (1 gm/day for six weeks) to determine its effect on liver enzymes, lipid peroxidation, and nitric oxide metabolites. Plasma arginine, liver enzymes alanine aminotransferase (ALT), aspartate aminotransferase (AST), alkaline phosphatase (ALP), plasma total bilirubin concentration (TB), malondialdehyde concentration (MDA-a marker of oxidative stress) and nitric oxide (NOx) metabolite concentrations were estimated. ALT, AST, ALP (p<0.05 for all) and TB (p<0.001) were significantly higher for SCD subjects than for healthy controls at baseline. Plasma arginine and nitric oxide levels were significantly higher for the controls (p<0.001 and <0.05 respectively). Arginine supplementation caused a greater percentage increase in plasma arginine and nitric oxide in SCD than in non-SCD subjects (p<0.001).19 This research provides evidence that a chronic, oral low-dose supplementation with L-arginine improves liver function, increases plasma arginine concentration and nitric oxide metabolite levels in both non-SCD and SCD subjects, thereby reducing oxidative stress with greater sensitivity demonstrated in SCD subjects. A randomized, double-blind study in children showed that supplementing a ready-to-use food with arginine and citrulline resulted in increased bioavailability of arginine and improvement in endothelial function.20 Dietary supplementation with arginine and nitrates may help to alleviate endothelial dysfunction in SCD patients by maintaining ample substrate to generate nitric oxide. Arginine supplementation has also been shown to increase total antioxidant activity and erythrocyte integrity in SCA subjects.21 Foods with high nitrate and nitrite content include beets, spinach, radishes, celery, and mustard greens.22 Providing food sources of antioxidant Vitamins E, C and A (beta-carotene) can also boost the glutathione antioxidant defense system and work together with arginine-derived nitric oxide to combat oxidative stress.23,24 Dietary sources of bioactive food components that may be helpful for SCD associated chronic disease management, as well as energy and macronutrient requirements, are found in (Table 1).
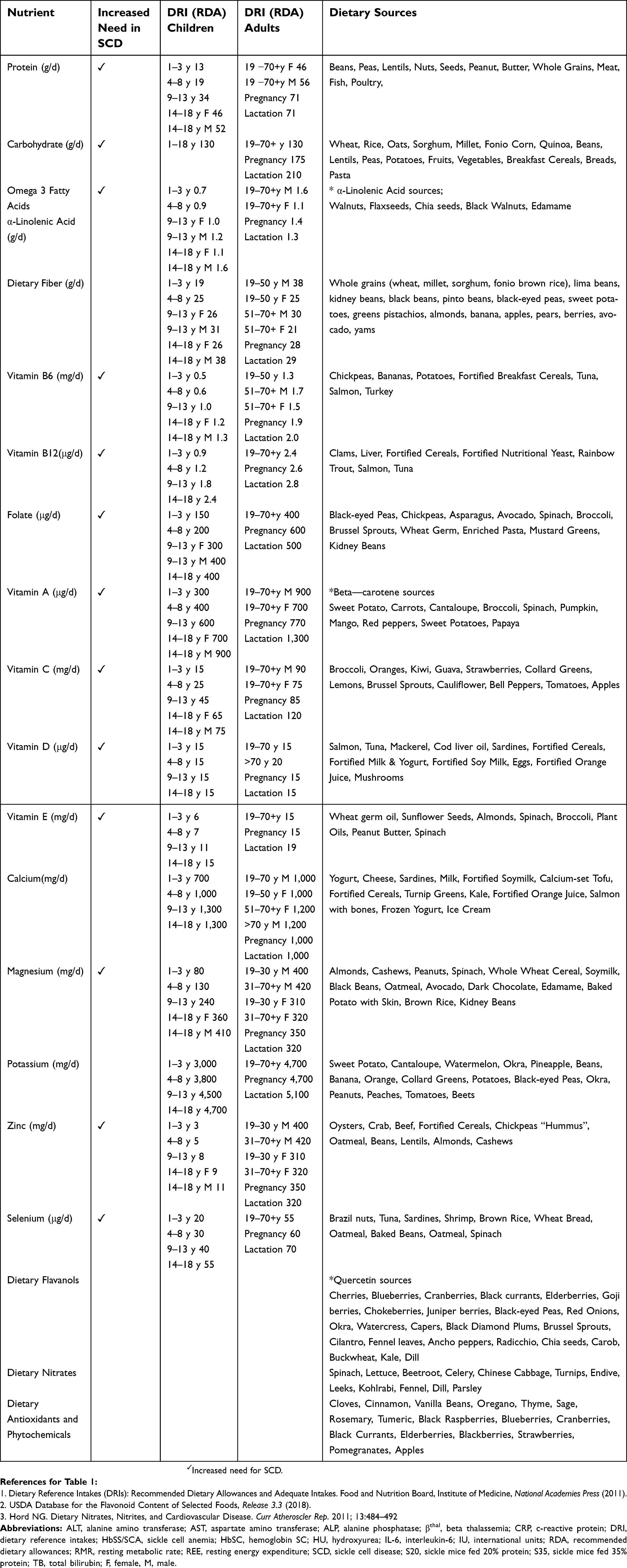 | Table 1 Dietary reference intakes for healthy individuals and dietary sources of nutrients suggested for nutrition management of SCD* |
Glutamine
Glutamine is a non-essential amino acid whose synthesis is ATP-dependent. Glutamine becomes conditionally essential in sickle cell disease due to its increased requirement. Deficiency for glutamine availability may result in metabolic stress, increased resting energy expenditure (REE), muscle wasting and decreased immune function. A study in 27 children and adolescents with SCD, supplemented with 600 mg/kg/day of oral glutamine resulted in decreased REE by 6 percent indicative of reduced protein turnover and improved glutamine nutritional status.25 Glutamine is also a precursor of NAD, and recent research indicates that glutamine may make sickle RBCs less adhesive.26 Evidence suggests that a pharmaceutical grade of L-glutamine is beneficial for decreasing the incidents of SCD-related vaso-occlusive (VOC) pain events without significant safety concerns.27 In 2017, the FDA approved pharmaceutical grade L-glutamine for children and adults with sickle cell disease. For individuals >5 years of age with repeated VOC pain events, oral L-glutamine at a dose of 0.3 g/kg twice per day is recommended, with a maximum daily dose of 30 g.28 L-Histidine, leucine, valine, and cysteine are also insufficient in SCD subjects.29
Vitamin D and SCD
Vitamin D is vital for calcium homeostasis and essential for bone mineralization. Deficiency of Vitamin D is common in sickle cell disease due to dark skin pigmentation, limited sun exposure, increased catabolism and decreased nutrient and energy intake. Deficiencies in Vitamin D contribute to osteopenia and osteoporosis which affect up to 80% of SCD patients. Patients with low serum Vitamin D (<14.1 ng/ml) have more crisis-related hospital visits per year than those with 25 (OH)D3 serum levels >34 ng/ml. Dietary intake of fish correlated with these findings.30 Vitamin D also functions to regulate immune responses and inflammation through its metabolite 1,25 dihydroxyvitamin D, which binds to the vitamin D receptor to serve as a transcription factor, inducing vitamin D-responsive genes present in cells of the immune system.31,32 A two year randomized clinical trial investigating the effect of a high dose of 100,000 International units (IU) (equivalent to 3,333 IU/day) versus the standard treatment 12,000 IU (equivalent to 400 IU/day) of oral vitamin D3 supplements for reducing risk of respiratory infections, was studied in 62 children and adolescents with SCD, aged 3-20 years. The results showed a significant reduction in respiratory events for both groups during the two years. The group receiving 3,333 IU/day administered as 100,000 IU once per month showed a decrease in annual respiratory events from 4.34±0.35 at baseline to 1.49±0.37 at year 2. Similarly, the group receiving 400 IU/day administered as 12,000 once monthly showed a decrease in annual respiratory events from 3.91±0.35 to 1.54±0.37. Ninety-eight percent of the high-dose group also stabilized at a mean serum 25-hydroxyvitamin D concentration of 37.0 ng/ml. This study showed a protective effect of Vitamin D supplementation against respiratory infections commonly found in children with sickle cell disease.33 Sources of dietary Vitamin D are limited and include ergocalciferol (Vit. D2) from mushrooms, fortified milk, plant milk and yogurt, fortified orange juice, fortified breakfast cereals, and as cholecalciferol (Vit. D3) in fatty fish, cheese, egg yolks, and liver.
Importance of hydration and SCD
Hydration plays an essential role in sickle cell anemia. Cells become sickled due to reduced hydration status and hemolytic anemia. Poorly hydrated erythrocytes lead to increased viscosity and may contribute to the vaso-occlusive crisis in SCD.34 It is crucial to promote proper hydration by frequent intake of water and other fluids, and to avoid physical activity and extreme weather that result in excessive sweating.35 Even sickle cell trait carriers can experience increased blood viscosity during strenuous sports.36 Avoiding dietary sodium intake can help to maintain appropriate hydration status by preventing water from leaving the erythrocytes.37 Dietary recommendations for maintaining good hydration status include limiting high sodium, processed foods, and snacks while consuming water and fluids throughout the day.
The gut microbiome: considerations in SCD
The gut microbiome contains trillions of bacteria, collectively termed microbiota, that play a significant role in host immunity. While a balance of commensal and pathogenic bacteria maintains the gut homeostasis, a predominance of pathogenic bacteria in the gut may arise from inadequate intake of the dietary substrate for gut microbiota, physical damage, and antibiotic use. If the prevalence of pathogenic bacteria compromises the intestinal barrier, disruption of the mucosal T-cell homeostasis and inflammation may result. There is a link between the gut microbiome and many inflammatory diseases. These include type 2 diabetes, allergies and colorectal cancer.38 Dietary intake of prebiotic substrates from legumes, grains, fruit, and vegetables, as well as probiotics from fermented dairy, soy, and grains, provide optimal substrates for the gut microbiota that promotes the predominance of protective commensal bacteria such as Lactobacillus and Bifidobacterium. These foods also provide vitamins, minerals, phytochemicals and antioxidants for the host.39
Few studies have determined whether the gut microbiota of SCD patients differs from those without SCD. In mouse models, the gut microbiota has been shown to regulate neutrophil aging via Toll-like receptors (TLRs) and myeloid differentiation factor 88 (Myd88) mediated signaling pathways, leading to TNFα-induced VOC.40 There is a notion that VOC contributes to dysbiosis through the subclinical intestinal ischemia it causes from its presence in the splanchnic vasculature. Further exacerbation of this compromise may arise from dietary factors or medications. The intestinal microbiome facilitates the synthesis of nitric oxide (Figure 2)41 in the gut through the arginine citrulline pathway and in human endothelial cells from dietary flavanol derived gut metabolites. The impact of nutritional arginine deficiency in preventing adequate nitric oxide production and resultant oxidative stress and inflammation may be due to both alterations of gut bacteria and impairment of the gut microbial pathway. Gut modulation of bioactive components of food sources of dietary phytochemicals, flavanols and amino acids like arginine, may aid in decreasing oxidative stress and improving both endothelial function and blood pressure in SCD. Also, studies through the Minority Coalition for Precision Medicine and National Microbiome Initiative are currently exploring the relationship between gut microbiota, circulating activated neutrophils and VOC.42 Using 16SrRNA sequencing studies individuals with SCD and no VOC or antibiotic usage, are compared with sickle cell trait (AS) carriers. Compared with AS, SCD had a lower relative abundance of three species from Firmicutes phylum (Pseudobutyrivibrio, Faecalibacterium, Subdoligranulum) and two from Bacteriodetes phylum (Prevotella, Alistipes). Relative abundance of Escherichia-Shigella from Proteobacteria phylum was higher in SCD. When further correlated with clinical parameters, lactate dehydrogenase (LDH) correlated positively with the genera from Firmicutes phylum. LDH is associated with hemolysis in SCD patients.43 Maintaining a healthy gut microbiota through adequate dietary intake of fruits, vegetables, whole grains, legumes, and fermented foods may aid the SCD patient in optimizing host immunity.
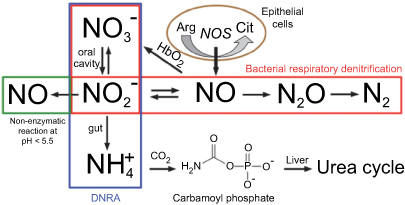 | Figure 2 Nitrogen pathways in the human gut. |
Emerging medicinal plant derived nutrients for SCD
Interest in natural products is gaining attention as an integrative approach to management of sickle cell disease. Many of these tropical plants are native to the countries where high rates of sickle cell disease exist. Derivatives from plants have been shown to contain antioxidant properties from bioactive components such as phytochemicals and flavanols.44 Exploration for the use of extracts from the tropical plant Moringa oleifera is in progress, to determine the antioxidant capacity in the treatment of oxidative stress in sickle cell disease. Ethanol extracts of Moringa oleifera showed antioxidant values between 77 and 4,458 μg/ml.45 Other plant leaves known to contain phytochemicals, include Cajanus cajan, Zanthoxylum zanthoxyloides, and Carica papaya. Experiments using 2% sodium metabisulfite to induce red cell sickling in an in vitro model, demonstrated that these plants could aid in the resistance of hemolysis and reduce the number of sickled red blood cells.46 Results in animal models did not show acute toxicity of the Cajanus cajan leaf.47
Conclusion
Management of SCD is complex and multifactorial. Nutritional risks are high, and after more than 100 years of following this disease, investigating the use of nutrition as adjuvant therapy for addressing multiple diet-related chronic disorders associated with SCD is still not a priority for providing adequate treatment. The focus of much of the research has been about increasing the red cell count by various methods, without considering that the changes in the form and function of the sickle red cells may be associated with developing a nutrient deficiency. For example, individuals with sickle cell, experience varied levels of hemolytic anemia, which reduces the oxygen-carrying capacity of the blood. This is associated with increased rates or red cell production and therefore increasing amounts of young red cells (reticulocytes) in the blood. Red cell production requires many substrates, not the least of which is protein. Protein synthesis is associated a with high energy cost and limits nutrient availability for growth and maintenance of body mass. The reticulocytes also readily stick to the blood vessel endothelial cells due to increased availability of adhesion sites. Therefore, the flow of nutrients for other essential metabolic needs is limited. So, it is not surprising that a sufficient diet for a healthy age, gender, and body-mass matched individual will not cover the nutritional needs of the person grappling with SCD. There has been no attempt to calculate dietary requirements for these individuals, as has been done for healthy people without the disease. Frankly, developing recommended dietary intakes will be a daunting task, as this involves gathering clinical information (ie, body composition, anthropometry, energy metabolism, measuring circulating nutrients by venipuncture and food intakes by self-reported diet diaries, in tandem with on-site compliance measurements, and more). These investigations will require recruiting many participants of different ages, followed prospectively for at least six months. These data would then be used to calculate the optimal nutritional requirements for different age groups of individuals with varying types of sickle cell disease, including HbSS, β-thallasemia and HbSC, the most abundant and severe of this group of hemoglobinopathies.
The protocol for this type of investigation has already been developed for healthy individuals and is worth repeating for this health challenged group. Estimating dietary needs for those affected by SCD can pave the way toward macronutrient sufficiency, ie, required energy and protein, which are most deficient in patients with SCD. Identifying and recommending foods needed to supplement the elevated metabolism of individuals with SCD will improve growth and development, promote weight maintenance, conserve muscle mass, and reduce inflammation for these patients. Finally, this paper addresses the need for the development of a comprehensive Medical Nutrition Therapy approach to treating SCD. This approach should include an emphasis on high dietary requirements for macronutrients (protein carbohydrate and fat) while incorporating evidence-based research supporting the use of food sources of polyphenolic phytochemicals, flavanols and gut microbial required prebiotics. We suggest that these components in combination with the vitamins, minerals, and omega-3 fatty acids routinely used in standard treatment, may provide adjuvant therapy for the SCD-associated chronic disease burden, and promote sustainable health, quality of life and increased longevity for this patient population.
Acknowledgments
Umeakunne K reports support from the Academy of Nutrition and Dietetics Foundation and the National Institute on Minority Health and Health Disparities (NIMHD) and National Institute of Allergy and Infectious Diseases (NIAID) Grant Number U54MD007588. Hibbert JM reports support from the Georgia Research Alliance (GRA) Phase I funding grant number GRA.VL18.F1, the Department of Microbiology/Biochemistry/Immunology microgrant fund #500061, MSM, and Grant Number 8G12MD007602 from the National Institute of Minority Health and Health Disparities (NIMHD). The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIMHD or the NIH.
Disclosure
Jacqueline M Hibbert and Kayellen Umeakunne report a patent US20110294727A1 issued to Hibbert JM, Stiles JK, Hyacinth HI, Umeakunne K. The authors report no other conflicts of interest in this work.
References
1. Hassell KL. Population estimates of sickle cell disease in the U.S. Am J Prev Med. 2010;38(Suppl):S512–S521. doi:10.1016/j.amepre.2009.12.022
2. Weatherall DJ. Hemoglobinopathies worldwide: present and future. Curr Mol Med. 2008;8:592–599. doi:10.2174/156652408786241375
3. Steiner CA, Miller JL. Sickle cell disease patients in U.S. hospitals, 2004. Statistical brief. No. 21. Rockville (MD): Agency for Healthcare Research and Quality; December 2006.
4. Davis H, Moore RM
5. Platt OS. Hydroxyurea for the treatment of sickle cell anemia. N Engl J Med. 2008;358:1362–1369. doi:10.1056/NEJMct0708272
6. Platt OS, Brambilla DJ, Rosse WF, et al. Mortality in sickle cell disease. Life expectancy and risk factors for early death. N Engl J Med. 1994;330:1639–1644.
7. Egberg MD. Progressive loss of brain volume in children with sickle cell anemia and silent cerebral infarct: a report from the silent cerebral infarct transfusion trial. Correspondence. Am J Hematol. 2018:E406.
8. Hyacinth HI. Sickle-cell anaemia needs more food? Lancet Haematol. 2018;5(4):e130. doi:10.1016/S2352-3026(18)30032-2
9. Daak A, Rabinowicz A, Ghebremeskel K. Omega three fatty acids are a potential therapy for patients with sickle cell disease. Nat Rev Dis Primers. 2018;4, Article number:15. doi:10.1038/s41572-018-0012-9
10. Adegoke SA, Smith OS, Adekile AD, Figueiredo MS. Relationship between serum 25-hydroxyvitamin D and inflammatory cytokines in paediatric sickle cell disease. Cytokine. 2017;96:87–93. Epub 2017 Apr 5. doi:10.1016/j.cyto.2017.03.010
11. Heyman MB, Katz R, Hurst D, et al. Growth retardation in sickle-cell disease treated by nutritional support. Lancet. 1985;325:903–906. doi:10.1016/S0140-6736(85)91677-0
12. Manci EA, Hyacinth HI, Capers PL, et al. High protein diet attenuates histopathologic organ damage and vascular leakage in transgenic murine model of sickle cell anemia. Exp Biol Med (Maywood). 2014;239(8):966–974. Epub 2014 May 19. doi:10.1177/1535370214531863
13. Archer DR, Stiles JK, Newman GW, et al. C-reactive protein and Interleukin-6 are decreased in transgenic sickle cell mice fed a high protein diet. J Nutr. 2008;138:1148–1152. doi:10.1093/jn/138.6.1148
14. Hibbert JM, Forrester T, Jackson AA. Urea kinetics: comparison of oral and intravenous dose regimens. Eur J Clin Nutr. 1992;46:405–409.
15. Hibbert JM, Creary MS, Gee BE, et al. Erythropoiesis and myocardial oxygen consumption contribute significantly to hypermetabolism of childhood sickle cell anemia. J Pediatr Gastroenterol Nutr. 2006;43:680–687.
16. Hibbert JM, Hsu LL, Bhathena SJ, et al. Proinflammatory cytokines and the hypermetabolism of children with sickle cell disease. Exp Biol Med. 2005;230:68–74. doi:10.1177/153537020523000109
17. Singhal A, Parker S, Linsell L, Serjeant G. Energy intake and resting metabolic rate in preschool Jamaican children with homozygous sickle cell disease. Am J Clin Nutr. 2002;75:1093–1097. doi:10.1093/ajcn/75.6.1093
18. Morris CR. Alterations of the arginine metabolome in sickle cell disease. A growing rationale for arginine therapy. Hematol Oncol Clin N Am. 2014;28:301–321. doi:10.1016/j.hoc.2013.11.008
19. Kehinde MO, Ogungbemi SI, Anigbogu CN, Jaja SI. L-Arginine supplementation enhances antioxidant activity and erythrocyte integrity in SCA subjects. Pathophysiology. 2015;22:137–142. doi:10.1016/j.pathophys.2015.05.001
20. Cox SE, Ellins EA, Marealle AI, et al. Ready-to-use- food supplement, with or without arginine and citrulline, with daily chloroquine in Tanzanian children with sickle-cell disease: a double-blind, random order crossover trial. Lancet. 2018;5:e147–e160. doi:10.1016/S2352-3026(18)30020-6
21. Bondonno CP, Blekkenhorst LC, Liu AH, et al. Vegetable-derived bioactive nitrate and cardiovascular health. Mol Aspects Med. 2018;61:83–91. doi:10.1016/j.mam.2017.08.001
22. Kobayashi J, Ohtake K, Uchida H. NO-rich diet for lifestyle-related disease. Nutrients. 2015;7:4911–4937. doi:10.3390/nu7064911
23. Little JA, Hauser KP, Martyr SE, et al. Hematologic, biochemical, and cardiopulmonary effects of L-arginine supplementation or phosphodiesterase 5 inhibition in patients with sickle cell disease who are on hydroxyurea therapy. Eur J Haematol. 2009;82:315–321. doi:10.1111/j.1600-0609.2009.01210.x
24. Gizi A, Papassotiriou I, Apostolakou F, et al. Assessment of oxidative stress in patients with sickle cell disease: the glutathione system and the oxidant-antioxidant status. Blood Cells Mol Dis. 2011;46:220–225. doi:10.1016/j.bcmd.2011.01.002
25. Williams R, Olivi S, Li CS, et al. Oral glutamine supplementation decreases resting energy expenditure in children and adolescents with sickle cell anemia. J Pediatr Hematol Oncol. 2004;26(10):619–625. doi:10.1097/01.mph.0000140651.65591.b8
26. Niihara Y, Matsui NM, Shen YM, et al. L-Glutamine therapy reduces endothelial adhesion of sickle red blood cells to human umbilical vein endothelial cells. BMC Blood Disord. 2005;5:4.
27. DeBaun MR, Vichinsky P. Vaso-occlusive pain management in sickle cell disease. UpToDate. 2018;1–52.
28. Quinn CT. L-gutamine for sickle cell anemia: more questions than answers. Blood. 2018;132(7):689. doi:10.1182/blood-2018-03-834440
29. Minniti CP. L-glutamine and the dawn of combination therapy for sickle cell disease. New Egl J Med. 2018;379(3):292–294. doi:10.1056/NEJMe1800976
30. McCaskill ML, Ogunsakin O, Hottor T, Harville E, Kruse-Jarres R. Serum 25-hydroxyvitamin D and diet mediates vaso-occlusive related hospitalizations in sickle cell disease patients. Nutrients. 2018;10(10):1–15. doi:10.3390/nu10101384
31. Bikle DD. Extraskeletal actions of vitamin D. Ann NY Acad Sci. 2016;1376(1):29–52. doi:10.1111/nyas.13219
32. Wei R, Christakos S. Mechanisms underlying the regulation of innate and adaptive immunity by vitamin D. Nutrients. 2015;7(10):8251–8260. doi:10.3390/nu7105392
33. Lee MT, Kattan M, Fennoy I, et al. Randomized phase 2 trial of monthly vitamin D to prevent respiratory complications in children with sickle cell disease. Blood Adv. 2018;2(9):969–978. doi:10.1182/bloodadvances.2017013979
34. Rinehart J, Gulcicek EE, Joiner CH, Lifton RP, Gallagher PG. Determinants of erythrocyte hydration: current opinion in hematogy. Curr Opin Hematol. 2010;17(3):191–197. doi:10.1097/MOH.0b013e32833800d0
35. Piel FB, Steinberg MH, Rees DC. Sickle cell disease. N Engl J Med. 2017;376(16):1561–1573. doi:10.1056/NEJMra1510865
36. Diaw M, Samb A, Diop S, et al. Effects of hydration and water deprivation on blood viscosity during a soccer game in sickle cell trait carriers. British J of Sports Med. 2014;48(4):326–331. doi:10.1136/bjsports-2012-091038
37. Williams-Hooker R, Hankins J, Ringwald-Smith K, et al. Evaluation of hydration status, sodium and fluid intake in children with sick cell anaemia. J Blood Disord Transf. 2013;4(3):1–4. doi:10.4172/2155-9864.1000143
38. Flint HJ, Scott KP, Louis P, Duncan SH. The role of the gut microbiota in nutrition and health. Nat Rev Gastroenterol Hepatol. 2012;9(10):577–589. doi:10.1038/nrgastro.2012.156
39. Bayram B, Gonzalez-Sarrias A, Istas G, et al. Breakthrough in the health effects of plant food bioactives; A perspective on microbiomics, Nutri(epi)genomics, and metabolomics. J Agric Food Chem. 2018;66(41):10686–10692. doi:10.1021/acs.jafc.8b03385
40. Zhang D, Chen G, Manwani D, et al. Neutrophil ageing is regulated by the microbiome. Nature. 2015;525(7570):528–532. doi:10.1038/nature15367
41. Tiso M, Schechter AN. Nitrate reduction to nitrite, nitric oxide and ammonia by gut bacteria under physiological conditions. PLoS One. 2015. doi:10.1371/journal.pone.0119712
42. Lim SH, Fast L, Morris A. Sickle cell vaso-occlusive crisis: it’s a gut feeling. J Transl Med. 2016;14(1):334–336. doi:10.1186/s12967-016-1092-5
43. Lim SH, Morris A, Li K, et al. Intestinal microbiome analysis revealed dysbiosis in sickle cell disease. Am J Hematology. 2018;93(4):E91–E93. doi:10.1002/ajh.25019
44. Khan SA, Damanhouri G, Ali A, et al. Precipitating factors and targeted therapies in combating the perils of sickle cell disease – A special nutritional consideration. Nutr Metab (Lond). 2016;13(50):1–12. doi:10.1186/s12986-016-0109-7
45. Wright RJ, Lee KS, Hyacinth HI, et al. An investigation of the antioxidant capacity in extracts from moringa oleifera plants grown in jamaica. Plants. 2017;6(48):1–8. doi:10.3390/plants6040048
46. Nurain IO, Bewaji CO, Johnson JS, et al. Potential of three ethnomedicinal plants as anti-sickling agents. Mol Pharm. 2017;14(1):172–182. doi:10.1021/acs.molpharmaceut.6b00767
47. Tang R, Tian R, Cai J, et al. Acute and sub-chronic toxicity of Cajanus cajan leaf extracts. Pharm Biol. 2017;55(91):1740–1746. doi:10.1080/13880209.2017.1309556
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.