Back to Journals » International Journal of Nanomedicine » Volume 16
Nucleus-Targeted Photosensitizer Nanoparticles for Photothermal and Photodynamic Therapy of Breast Carcinoma
Authors Liu J, Yin Y, Yang L, Lu B, Yang Z, Wang W, Li R
Received 29 September 2020
Accepted for publication 24 December 2020
Published 22 February 2021 Volume 2021:16 Pages 1473—1485
DOI https://doi.org/10.2147/IJN.S284518
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Thomas Webster
Jing Liu,1,* Yaru Yin,1,* Luxun Yang,1 Binghui Lu,1 Zhangyou Yang,2 Weidong Wang,3 Rong Li1
1Institute of Combined Injury, State Key Laboratory of Trauma, Burns and Combined Injury, Military Key Laboratory of Nanomedicine, Department of Military Preventive Medicine, Army Medical University, Chongqing, 400038, People’s Republic of China; 2Department of Pharmacy, Chongqing Medical University, Chongqing, 400010, People’s Republic of China; 3Department of Radiation Oncology, Sichuan Cancer Hospital, Chengdu, 610041, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Weidong Wang
Sichuan Cancer Hospital, Renmin South Road, Chengdu, 610041, Sichuan, People’s Republic of China
Email [email protected]
Rong Li
Army Medical University, No 30, Gaotanyan St., Chongqing, 400038, People’s Republic of China
Tel +86-13883996627
Fax +86023-68753127
Email [email protected]
Purpose: The near-infrared fluorescent dye indocyanine green (ICG) has shown great potential in the photodynamic therapy (PDT) and photothermal therapy (PTT) of cancer. However, its disadvantages of instability in aqueous solution, short half-life, and non-targeting accumulation limit the effectiveness of ICG PDT/PTT. To overcome the disadvantages of ICG in tumor treatment, we designed PEGylated-human serum albumin (PHSA)-ICG-TAT. In this nanoparticle, PEG4000, the HSA package, and nuclear targeting peptide TAT (human immunodeficiency virus 1 [HIV-1]-transactivator protein) were used to improve the water solubility of ICG, prolong the life span of ICG in vivo, and target the nuclei of tumor cells, respectively.
Methods: The PHSA-ICG-TAT was characterized in terms of morphology and size, ultraviolet spectrum, dispersion stability, singlet oxygen and cellular uptake, and colocalization using transmission electron microscopy and dynamic light scattering, and fluorescence assay, respectively. Subsequently, the anti-tumor effect of PHSA-ICG-TAT was investigated via in vitro and in vivo experiments, including cell viability, apoptosis, comet assays, histopathology, and inhibition curves.
Results: The designed ICG-loaded nanoparticle had a higher cell uptake rate and stronger PDT/PTT effect than free ICG. The metabolism of PHSA-ICG-TAT in normal mice revealed that there was no perceptible toxicity. In vivo imaging of mice showed that PHSA-ICG-TAT had a good targeting effect on tumors. PHSA-ICG-TAT was used for the phototherapy of tumors, and significantly suppressed the tumor growth. The tumor tissue sections showed that the cell gap and morphology of the tumor tissue had been obviously altered after treatment with PHSA-ICG-TAT.
Conclusion: These results indicate that the PHSA-ICG-TAT had a significant therapeutic effect against tumors.
Keywords: indocyanine green, human serum albumin, phototherapy, nuclear targeting
Introduction
Photodynamic therapy (PDT) and photothermal therapy (PTT) are established anti-cancer therapies. PDT kills cancer cells locally by using light to irradiate a photosensitizer, which leads to the generation of reactive oxygen species (ROS).1 PTT destroys biologically active molecules and induces the generation of ROS to kill cancer cells. This is achieved using light to irradiate photosensitizers, leading to the generation of thermal energy and increasing the temperature of cells.2 Most near-infrared (NIR) agents also have great potential for converting light energy into local hyperthermia (PTT) and/or generating ROS (PDT).3–5 Indocyanine green (ICG) is the only NIR reagent approved by the US Food and Drug Administration. It has been widely used in the clinical determination of cardiac output, liver function, blood flow, and ophthalmic angiography.6 In addition, ICG has strong NIR absorption, which can simultaneously achieve the PTT/PDT effect under the action of a single-wavelength NIR laser.7,8 However, ICG also has several intrinsic drawbacks, such as instability in aqueous solution, rapid body clearance, proneness to self-bleaching, and lack of targeting.9–11 These disadvantages significantly influence its imaging effect and efficacy of PTT/PDT.
In recent years, the rapid development of nanotechnology has provided technical support for overcoming these limitations. Moreover, it provides a platform for the wide application of ICG molecules.12,13 Studies on protein carriers have shown that the protein is a nanocarrier with great potential, which can be used for the sustained and controlled release of drugs, targeted delivery, bio-imaging, and tumor treatment. Through various methods, researchers combined different structural proteins (bovine serum albumin, human serum albumin [HSA], transferrin, etc) with biological antibodies, fluorescent molecules, metal ions, etc, and applied them to the diagnosis and treatment of tumors.14–19 This provides new ideas for the early diagnosis and effective treatment of tumors.
HSA, a single-chain glycosylated protein with many reticular gaps, is an ideal drug carrier. It can transport bile acid, steroid hormones, metal particles and many drug molecules, while maintaining the normal osmotic pressure of the blood.20 However, HSA has a fast degradation rate in the body, and is easily removed from the blood circulation. As a drug carrier alone, the sustained release time is short. At present, the methods commonly used to extend the half-life of drugs include poly ethylene glycol (PEG) modification and mutant construction.21 The shielding effect of PEG molecules may hinder the binding of vascular endothelial cell surface membrane scavenger receptors and HSA, thereby reducing HSA degradation and vascular permeability, and prolonging its vascular retention time.22
The nucleus is the control center of cell life activities. Using the nucleus as a target for the treatment of cancer will more effectively kill the tumor cells. Currently developed nano-carrier materials (eg, metals, mesoporous silica nanoparticles, lipids, and polymers) can target and deliver drug to the tumor microenvironment. However, the efficiency is low, and almost all nano-drugs cannot penetrate the nuclear membrane of cells.23–27 As a non-specific cell-penetrating peptide, TAT protein can carry various anti-cancer drugs, proteins, liposomes, and other substances of different sizes into the nucleus.28,29 Herein, we designed a nano-molecule PEGylated-HSA (PHSA)-ICG-TAT with HSA and PEG4000 as the backbone to load ICG and target the nucleus of tumor cells (Scheme 1). The anti-tumor mechanism of PHSA-ICG-TAT is based on the production of excessive 1O2 and heat in the nucleus, triggering a domino effect after irradiation with NIR laser light. Excessive accumulation of 1O2 and high temperature in the nucleus will cause DNA damage, destruction of the nucleus structure and function, and eventually lead to irreversible apoptosis of tumor cells. The nucleus-targeted PDT/PTT tumor therapy can effectively kill cancer cells and provide a more rapid and effective new approach to the treatment of cancer.
 |
Scheme 1 Schematic diagram of the anti-cancer therapy mechanism of PHSA-ICG-TAT under irradiation with light at a wavelength of 808 nm. Abbreviations: ICG, indocyanine green; PHSA, PEGylated human serum albumin. |
Materials and Methods
Materials
HSA, amino-polyethylene glycol-amino (Amine-PEG-Amine, MW 4K), ICG fluorescent dye, and nuclear targeting peptide TAT (human immunodeficiency virus 1 [HIV-1]-transactivator protein, TAT) were purchased from Sigma–Aldrich (St. Louis, MI, USA). N-hydroxysuccinimide (NHS) and N-(3-dimethyl aminopropyl)-N’-ethyl carbodiimide (EDC) were purchased from Aladdin reagent. Fetal bovine serum (FBS) was purchased from Hyclone. RPMI 1640 medium, phosphate-buffered saline (PBS), and 0.25% trypsin were purchased from Gibco (Grand Island, NY, USA). The Cell Counting Kit-8 (CCK-8) kit was purchased from the Tongren Institute of Chemistry (Japan); single-cell gel electrophoresis kit was purchased from Beijing Ximeijie Technology (Beijing, China); Annexin fluorescein isothiocyanate/propidium iodide (FITC/PI) apoptosis detection kit was purchased from BD Pharmingen (East Rutherford, NJ, USA,); Tris-HCl pH 8.8 buffer was purchased from Biyuntian Biotechnology Co., Ltd. (China).
Synthesis and Characterization of Materials
Synthesis of PHSA
HSA (50 mg) was accurately weighed, dissolved in 9 mL of pure water, and stirred magnetically in a 20 mL reaction flask. Approximately 20–40 mg of EDC and NHS were added into the reaction flask, and the duration of the activation reaction was 15 min. Next, NH2-PEG4000-NH2 (150 mg) and PBS (1 mL) were added to the reaction flask for 24 h. Following reaction, the material was placed in ultrapure water for dialysis and purification to obtain PHSA and stored in a refrigerator at 4°C.
Synthesis of PHSA-ICG
After adding 2 mL of PHSA liquid and 10 μL of DDT to the reaction flask, the reaction was stirred for 15 min at 25°C. Subsequently, 30 μL of ICG fluorescent dye was added, and the reaction was stirred for 12 h in the dark at 25°C. After the reaction, the pH of the purified water was adjusted to 7.0–7.5 to be used as dialysis solution with PBS, and the solution in the reaction bottle was dialyzed against the light for 24 h in the adjusted pH aqueous solution. The dialysis-purified PHSA-ICG was placed in a refrigerator at 4°C for subsequent use.
Synthesis of PHSA-ICG-TAT
After adding 1 mL of 2.5 mg/mL TAT into the reaction bottle, 5 mg of EDC and NHS were added, and the solution was stirred at 25°C for 10 min. At the same time, 1 mL of PHSA-ICG solution and 200 μL of Tris-HCl (pH 8.8) solution were added to a new reaction bottle, and the mixture was stirred for 15 min in the dark. Subsequently, 1 mL of the activated TAT solution was added to the PHSA-ICG solution and the reaction was continued for 12 h in the dark. After the reaction, the product was dialyzed in ultrapure water for 8 h to obtain purified PHSA-ICG-TAT and stored in a refrigerator at 4°C.
Characterization of PHSA-ICG and PHSA-ICG-TAT
The morphology of the nanoparticles was characterized using a transmission electron microscope (TEM; Tecnai G2 F20 S-TWIN TEM) with negative electron staining of the phosphotungstic acid protein. Furthermore, the size of the nanoparticles was tested by dynamic light scattering using a Brookhaven Zeta PALS analyzer. The ICG concentration of the nanomaterials was determined by the ultraviolet absorption method. Prior to the tests, different ICG standard concentration working solutions were prepared (0, 2.5, 5, 7.5, and 10 μM) to construct a standard curve and measure the content of ICG in PHSA-ICG-TAT and PHSA-ICG. In addition, 5 μM of ICG, PHSA, PHSA-ICG, and PHSA-ICG-TAT were dissolved in 50% ethanol for 200–900 nm ultraviolet spectrum analysis.
Detection of the Dispersion Stability of Materials
For the detection of dispersion stability, 10 μM PHSA-ICG-TAT and PHSA-ICG were added to 1 mL of PBS, RPMI 1640 medium, and FBS. After mixing, the solution was placed at 25°C in the dark for 24 h, centrifuged at 7,000 rpm for 10 min, and inspected for the presence of any precipitation.
Detection of Singlet Oxygen (1O2)
The photodynamic property of PHSA-ICG-TAT was evaluated by determining the generation of 1O2 according to the fluorescence signal of Singlet Oxygen Sensor Green (SOSG). SOSG was added to different solutions of ICG (5 μM), PHSA-ICG-TAT, and PHSA-ICG (PHSA-ICG and PHSA-ICG-TAT containing ICG equivalent to 5 μM) in PBS and irradiated with an 808 nm laser (power density: 2.0 W/cm2; irradiation power: 5 W; irradiation area: 2.5 cm2; 5 min). SOSG fluorescence was excited with a light source at a wavelength of 494 nm to a maximum of 525 nm following irradiation. The level of SOSG in the samples was evaluated by comparing the enhanced SOSG fluorescence measurement to that obtained for the control sample or background.
Cellular Uptake and Colocalization of Materials
The 4T1 cells (mouse breast carcinoma cells) were purchased from the cell bank of the Chinese Academy of Sciences (Beijing, China) and maintained in RPMI 1640 (Gibco) medium containing 10% FBS (Gibco). The cells were seeded in six-well plates at 2.0×105/well, and cultured at 37°C and 5% CO2 for 24 h until they adhered to the bottom of the plate. The ICG, PHSA-ICG, and PHSA-ICG-TAT (10 μM) were added to the cells, respectively, and the cell suspension was collected after 0, 1, 4, and 8 h. The cell suspension was filtered with a 70 μM filter, and the cells were resuspended in 200 μL of PBS in a 1.5 mL Eppendorf tube. Monochromatic flow fluorescence analysis was carried out at 640 nm excitation and 780 nm emission to determine the cell uptake of materials. The above cells cultured for 24 h were stained with Hoechst staining solution for 15 min and observed using a fluorescence microscope (DP80; Olympus, Tokyo, Japan) for the colocalization of materials.
Photothermal Heating Curve
The 4T1 cells were seeded in a 24-well plate at 1×105/well and cultured for 24 h. Subsequently, ICG, PHSA-ICG, and PHSA-ICG-TAT (50 μM) were added to cells, respectively, for 24 h. The cells were irradiated with light at a wavelength of 808 nm for 10 min (power density: 2.0 W/cm2), and the temperature was recorded at 0, 0.5, 1, 2, 3, 4, 5, 6, 7, 8, 9, and 10 min for each group.
In vitro PDT/PTT Anti-Tumor Effect
The 4T1 cells (4×103 cells/well) were seeded in 96-well plates and incubated overnight. The materials (2, 5 and 10 μM) were added to each group, and the cells were cultured for 24 h. The cell viability was measured at 24 h after irradiation with light at a wavelength of 808 nm (power density: 2.0 W/cm2, 5 min) using a CCK-8 kit. To compare the PDT and PTT effects of ICG alone and the prepared PHSA-ICG-TAT, the materials were stimulated under different conditions to assess their lethality to tumor cells. ICG and PHSA-ICG-TAT (10 μM) were added to the cells for 24 h. The cells were irradiated with light at a wavelength of 808 nm for 5 min (power density: 2.0 W/cm2) under different conditions (25°C, 4°C, addition of 4 μM NaN3 0.5 h before light irradiation). After 24 h of incubation, the cell viability was measured with a CCK-8 kit.
Cellular Mechanism of PDT/PTT Therapy
Cells and Treatments
The 4T1 cells (5×105 cells/well) were seeded in six-well plates and incubated overnight at 37°C in a humidified 5% CO2 atmosphere. ICG, PHSA-ICG, and PHSA-ICG-TAT (10 μM), as well as PBS (Control), were added to the cells, and the solutions were incubated for 24 h. The cells were irradiated with light at a wavelength of 808 nm (power density: 2.0 W/cm2) for 5 min and cultured further for 24 h.
Apoptosis
The cells in each group were washed with PBS, and the apoptosis was measured using an Annexin FITC/PI apoptosis detection kit and flow cytometer BD FACSCalibur (BD Pharmingen).
Comet Assay
The comet assay was performed as previously described.30 Briefly, molten 0.6% N-methyl-1,3-propanediamine was used to soak the frosted glass slide, which was left to dry naturally. Then, 70 μL of 0.7% LMPA at (37°C) was added to 10 μL of 4T1 cell suspension from each group to reach a cell number of 6,000. The solution was mixed and rapidly dripped on the frosted glass slide preheated at 37°C, immediately covered with a coverslip, and fixed at 4°C for 10 min. Subsequently, the coverslip was removed, and the cells were lysed in pre-cooled cell lysate at 4°C for 1 h in the dark. Electrophoresis was performed after unwinding for 30 min in alkaline electrophoresis buffer (electrophoresis parameters: 25 V, 300 mA, 25 min). After electrophoresis, the slides were rinsed and 20 ug/mL of EB 10 μL was added dropwise. The slides were covered with a coverslip and observed under a fluorescence microscope (DP80; Olympus). The extent of DNA damage was analyzed using the CASP software and evaluated as the percentage of DNA in the tail of the comet (Tail DNA%).
In vivo Toxicity of PHSA-ICG-TAT
Male BALB/c mice (eight weeks old, 20–22 g) were obtained from Animal Center of Army Medical University (Chongqing, China). The feeding conditions were as follows: ambient temperature 24 ± 1 °C, relative humidity 50 ± 5%, and light/dark cycle of 12/12 h. All the protocols were approved by the Animal Ethics Committee of the Army Medical University. The study design and all animal experimental procedures were performed in accordance with the Guide and Use of Laboratory Animals (Institute of Laboratory Animal Resources), and the Institutional Animal Care and Use Committee (IACUC) for Medical Research at Army Medical University. Mice were treated with disposable PHSA-ICG-TAT (10 mg/kg) or physiological saline solution through intraperitoneal injection. The tail vein blood was collected at 24 h after the injection of PHSA-ICG-TAT for the examination of blood biochemical parameters using a blood cell analyzer (Sysmex, Japan). The tissues were harvested at 24 h after the injection of PHSA-ICG-TAT to observe the hematoxylin and eosin (H&E) staining of pathological sections.
Anti-Tumor Effect of PHSA-ICG-TAT in vivo
Tumor Model
Female BALB/c mice (age: 8 weeks, weight: 20−22 g) were purchased from the Army Medical University Animal Center (Chongqing, China) under protocols approved by the Animal Ethics Committee of the Army Medical University. The study design and all animal experimental procedures were performed in accordance with the Guide and Use of Laboratory Animals (Institute of Laboratory Animal Resources), and the IACUC for Medical Research at Army Medical University. Tumor-bearing mice were prepared by subcutaneously injecting a suspension of 12×104 4T1 cells in PBS (100 μL) into the right lateral hind limbs of female mice.
In vivo Tumor Imaging
When the tumor size reached 2.5×2.5 mm, 30 μL of PHSA-ICG-TAT (equivalent to 15 μM of ICG) was injected into the tail vein of the mice for 24 h. Subsequently, in vivo fluorescence imaging was performed. The heart, liver, spleen, lung, and kidney tissues and tumor tissues of mice were dissected for ex vivo fluorescence imaging. The fluorescence was recorded using the Maestro all-optical imaging system (Caliper Life Sciences, Hopkinton, MA, USA).
Anti-Tumor Effect
When tumor sizes reached 2×2 mm, 60 μL of PHSA-ICG and PHSA-ICG-TAT (corresponding to 30 μM ICG) or 30 μM ICG was intratumorally injected into the tail veins of mice. After 24 h, the tumor of each mouse was exposed to a light source at a wavelength of 808 nm for 5 min (power density: 1 W/cm2; irradiation power: 2.5 W; irradiation area: 2.5 cm2). Images were captured to record changes in tumor size. Tumor sizes were measured using a caliper every day after the treatment. Tumor volumes (V) were calculated using the following equation: V = X × Y2/2, where X and Y are the longer and shorter diameters (mm) of the tumor, respectively. Seven days after laser irradiation, tumor tissues of mice were collected for the pathological observation of H&E staining.
Statistical Analysis
All the data presented in the study were derived from at least three independent experiments. We used the SPSS for Windows Version 18.0 (SPSS Inc., Chicago, IL, USA) software to analyze the data. Statistical analysis was performed using one-way analysis of variance, and variance between groups was determined using the least significant difference test. P<0.05 denoted statistically significant differences.
Results
Synthesis and Characterization
PHSA-ICG-TAT was synthesized through a three-step reaction. The morphology of nanoparticles was characterized by TEM to determine the particle size of the nanomaterials. As shown in Figure 1A, PHSA-ICG-TAT was slightly larger than PHSA-ICG, and both had a spherical shape. The results of the dynamic light scattering test also showed that the particle size of PHSA-ICG-TAT and PHSA-ICG was 22 and 18 nm, respectively (Figure 1B). Ultraviolet−vis absorbance spectra (Figure 1C) indicated that PHSA exhibited a high degree of Soret absorption at 280 nm (amino acid-conjugated double bond), whereas the ICG molecule had no such peak. After successive modification, the peak at 280 nm was gradually weakened. At the same time, the absorption peak gradually shifted to the right compared with the ICG peak, and PHSA-ICG-TAT exhibited the maximum absorption at 790 nm. The newly synthesized PHSA-ICG and PHSA-ICG-TAT materials were dispersed in PBS, RPMI 1640 medium, and FBS. After 24 h, they were centrifuged, and precipitation was not present. This shows that the chemical properties of PHSA-ICG and PHSA-ICG-TAT after PEG modification were relatively stable (Figure 1D). ICG, PHSA-ICG, and PHSA-ICG-TAT were added to 4T1 cells, and the fluorescence intensity in the cells was detected at different time points. The findings showed that ICG alone entered the cells slowly. After 8 h of drug treatment, the amount of ICG that entered the cells was significantly lower than that of PHSA-ICG and PHSA-ICG-TAT (Figure 1E). These results showed that the modified nanoparticles were more able to enter the cell and exert PTT and PDT effects.
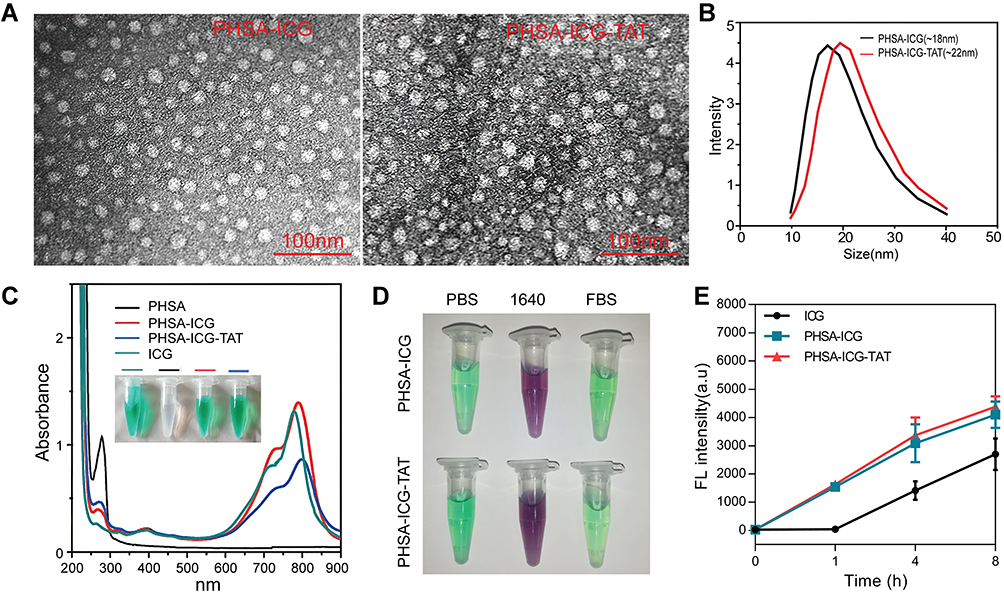 |
Figure 1 Characterization of nanomaterials. (A) TEM image of PHSA-ICG and PHSA-ICG-TAT negatively stained with phosphotungstic acid. (B) The average particle size of PHSA-ICG and PHSA-ICG-TAT. (C) UV−vis spectra of PHSA, PHSA-ICG, and PHSA-ICG-TAT (equivalent to 5 μM ICG), and ICG in PBS; the inset shows images of the nanomaterials. (D) The dispersion stability of PHSA-ICG and PHSA-ICG-TAT (equivalent to 10 μM ICG) in PBS, RPMI 1640 medium, and FBS. (E) The uptake capacity of 4T1 cells for ICG, PHSA-ICG, and PHSA-ICG-TAT (equivalent to 10 μM ICG). Abbreviations: FBS, fetal bovine serum; ICG, indocyanine green; PBS, phosphate-buffered saline; PHSA, PEGylated human serum albumin; TEM, transmission electron microscope; UV, ultraviolet. |
Detection of the PDT and PTT Effects of PHSA-ICG-TAT
To verify the targeting of the nucleus by PHSA-ICG-TAT, 4T1 cells were co-cultured with PHSA-ICG and PHSA-ICG-TAT for 24 h, and the nucleus was stained with Hoechst staining solution. The microscopic observation revealed that PHSA-ICG molecules mainly dispersed outside the nucleus. In contrast, PHSA-ICG-TAT molecules could penetrate and disperse in the nucleus (Figure 2A). SOSG was used to detect the ability of nanomaterials to generate 1O2 in PBS after being excited by light at a wavelength of 808 nm. The modified peaks of the ICG molecules were successively enhanced at 530 nm (Figure 2B). The results showed that, after the final modification, the compound molecule can enhance the generation of free radicals during the PTT process. After absorbing the light energy, ICG and PHSA-ICG-TAT increased the temperature to its maximum value within 5 min, which was subsequently gradually decreased. The temperature induced by ICG and PHSA-ICG-TAT at each time point after illumination was significantly higher than that measured in the PBS group. The photothermal conversion performance of PHSA-ICG-TAT was better than that of ICG. This is because ICG was encapsulated by protein to enhance the stability of light energy received by ICG, while reducing the quenching effect of ICG molecules (Figure 2C).
 |
Figure 2 (A) Subcellular localization of PHSA-ICG and PHSA-ICG-TAT (equivalent to 10 μM ICG). The cell nucleus was dyed blue by Hoechst, red by PHSA-ICG-TAT, and purple by merging blue and red, indicating that PHSA-ICG-TAT rather than PHSA-ICG entered the cell nucleus. (B) Singlet oxygen production and (C) temperature change induced by ICG and PHSA-ICG-TAT after light irradiation at a wavelength of 808 nm. Abbreviations: ICG, indocyanine green; PHSA, PEGylated human serum albumin. |
In vitro Photothermal Anti-Tumor Effect and Cellular Mechanism
To investigate the lethality of the nanomaterials against tumor cells, the 4T1 cells were treated with different concentrations of ICG, PHSA-ICG, and PHSA-ICG-TAT. After the nanomaterials were excited with light at a wavelength of 808 nm, the CCK-8 assay was used to detect the cell activity. It was found that, as the concentration of ICG increased, the lethality of nanomaterials on 4T1 cells also gradually increased. When the concentration of ICG is 10 μM, PHSA-ICG-TAT is significantly more lethal to 4T1 cells than PHSA-ICG and ICG alone (Figure 3A).
 |
Figure 3 In vitro photodynamic and photothermal killing of cancer cells. (A) Detection of the lethality of nanomaterials at different concentrations against 4T1 cells. Different concentrations of ICG, PHSA-ICG, and PHSA-ICG-TAT were applied to 4T1 cells, and the activity of the cells was tested using the CCK-8 assay after irradiation with light at a wavelength of 808 nm for 5 min. **P<0.01, *P<0.05, compared with the control group; ##P<0.01, comparison between PHSA-ICG and PHSA-ICG-TAT. (B) The photothermal and photodynamic capabilities of PHSA-ICG-TAT and ICG. The ICG and PHSA-ICG-TAT were irradiated with light at a wavelength of 808 nm for 5 min under the conditions of normal temperature, 4°C, and NaN3. Subsequently, the activity of 4T1 cells was tested with the CCK-8 assay. **P<0.01, compared with the control group; ##P<0.01, #P<0.05, comparison between ICG and PHSA-ICG-TAT. (C) The flow cytogram of the apoptosis of 4T1 cells after excitation of ICG, PHSA-ICG, and PHSA-ICG-TAT by light at a wavelength of 808 nm. (D) Statistics of transfer rate. **P<0.01, compared with the control group; ##P<0.01, comparison between PHSA-ICG and PHSA-ICG-TAT. (E) Comet images of DNA damage to 4T1 cells after stimulation of ICG, PHSA-ICG, and PHSA-ICG-TAT. (F) Statistics of Tail DNA% in comet experiments. **P<0.01, compared with the control group; ##P<0.01, comparison between PHSA-ICG and PHSA-ICG-TAT. Abbreviations: CCK-8, Cell Counting Kit-8; ICG, indocyanine green; PHSA, PEGylated human serum albumin. |
To further compare the lethality of ICG and PHSA-ICG-TAT against 4T1 cells, ICG and PHSA-ICG-TAT molecules were excited with light at a wavelength of 808 nm under different conditions, and cell viability was detected with the CCK-8 assay. Both ICG and PHSA-ICG-TAT were stimulated at room temperature and demonstrated the greatest lethality against tumor cells. Under these three excitation conditions, the PDT and PTT capabilities of PHSA-ICG-TAT were better than those of ICG alone (Figure 3B).
The mechanism through which PHSA-ICG-TAT nanoparticles killed tumor cells was investigated. It was found that PHSA-ICG-TAT significantly promoted the apoptosis of tumor cells compared with ICG and PHSA-ICG (Figure 3C and D). Moreover, the comet experiment showed that ICG, PHSA-ICG, and PHSA-ICG-TAT could significantly damage the DNA of tumor cells compared with the control group. Notably, PHSA-ICG-TAT caused the most significant damage to the DNA, as demonstrated by the tailing DNA which reached >80% (Figure 3E and F).
In vivo Toxicity of PHSA-ICG-TAT
Mice were intraperitoneally injected with PHSA-ICG-TAT and fed for 24 h to investigate the toxicity of the nuclear target material PHSA-ICG-TAT in vivo. The whole blood of the mice was obtained for the examination of blood biochemical parameters, and different tissues were used to observe the H&E staining. The results did not show a difference in blood biochemical parameters (Figure 4A–C). Observation of the tissue sections of the control group and the PHSA-ICG-TAT group revealed that there were no significant differences in the size and morphology of the heart, liver, spleen, lung, and kidney tissue cells (Figure 4D).
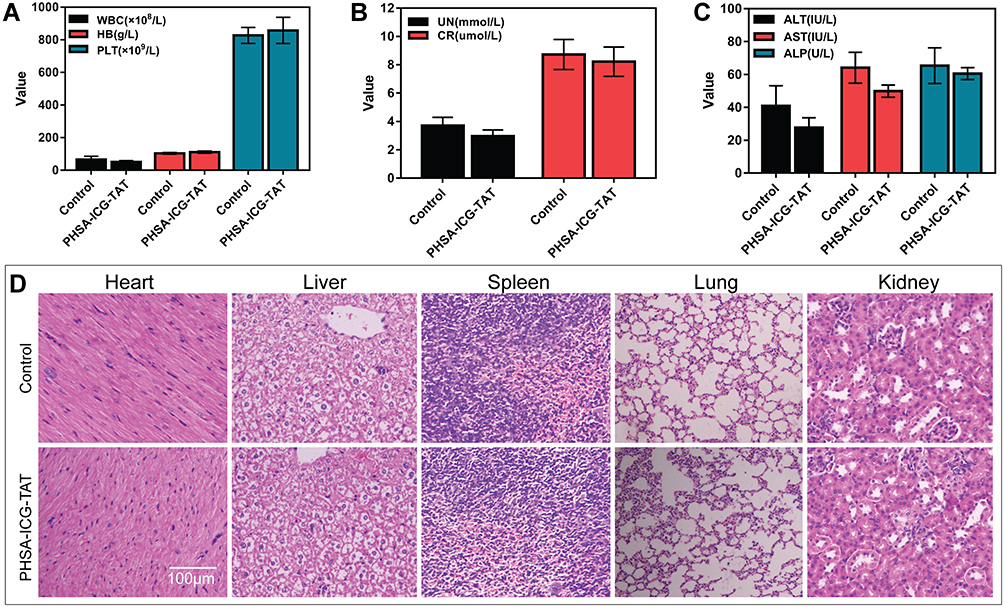 |
Figure 4 Toxicity of PHSA-ICG-TAT in mice. (A–C) The effect of PHSA-ICG-TAT on the blood biochemical parameters of mice. (D) The effect of PHSA-ICG-TAT on different tissues of mice. Abbreviations: ICG, indocyanine green; PHSA, PEGylated human serum albumin. |
Anti-Tumor Effect of PHSA-ICG-TAT in vivo
We investigated the feasibility of using PHSA-ICG-TAT for in vivo fluorescence imaging-guided PDT/PTT. Female BALB/c mice with subcutaneous 4T1 cell xenografts were used as the animal model. In vivo imaging of mice showed that PHSA-ICG-TAT only accumulated at the tumor site after injection through the tail vein. Fluorescence imaging of mouse visceral tissues and tumors further showed that PHSA-ICG-TAT only significantly accumulated in tumor tissues, but not in other tissues (Figure 5A). The digital photos of tumors treated with PDT/PTT in vivo (Figure 5B) showed that the tumor regression effect of the PHSA-ICG-TAT group was better than that of the free ICG and PBS groups. The change in tumor volume was measured daily following treatment for 16 days (Figure 5C). Compared with the control group, the ICG and PHSA-ICG groups showed non-significant tumor suppression after phototherapy. In the PHSA-ICG-TAT group, the tumor was gradually ablated after phototherapy. Body weight was monitored during the treatments, and the weights of mice in all treatment groups did not differ significantly from that measured in the control group (Figure 5D). These results indicated low levels of treatment-induced toxicity. To further evaluate the efficacy of PDT/PTT after treatment with PHSA-ICG-TAT, we observed the H&E staining of tumor sections 7 days post treatment (Figure 5E). The PBS group did not show obvious signs of tumor necrosis in histological sections. It was observed that the tumor cell gap was enlarged in the ICG and PHSA-ICG groups. In the PHSA-ICG-TAT group, the tumor cell gap was also enlarged and atrophied. These results indicated that PHSA-ICG-TAT induced minimal toxicity and high efficacy in the treatment of tumors.
 |
Figure 5 Anti-tumor effect of PHSA-ICG-TAT in vivo. (A) Fluorescence imaging of 4T1 tumor-bearing mice, tumors, heart, liver, spleen, lung, and kidney at 24 h after injection of PHSA-ICG-TAT. (B) Images of the 4T1 cell tumor xenograft-bearing mice captured before treatment (day 0), and at 7 and 15 days after treatment with PBS, free ICG, PHSA-ICG or PHSA-ICG-TAT, and laser exposure at 200 mW/cm2 for 5 min. (C) Tumor-growth curves of different groups of tumor-bearing mice after treatment. The error bars represent the standard deviations of six mice per group (**P<0.01, compared with the control group). (D) Body weights of different groups of tumor-bearing mice after treatment. (E) H&E-stained tumor sections collected from different groups of mice 7 days post treatment. Abbreviations: H&E, hematoxylin and eosin; ICG, indocyanine green; PHSA, PEGylated human serum albumin. |
Discussion
Breast cancer is a malignant tumor that seriously endangers women’s health. Currently, the main clinical treatment methods for this type of cancer include surgery, radiation therapy, and chemotherapy. Despite the continuous improvement of technology, the presence of residual cancer cells cannot be avoided, potentially leading to fatal recurrence. PDT/PTT is an established cancer therapy. Compared with traditional methods for the treatment of breast cancer, PDT/PTT has many advantages, such as precise light control of tumors, non-invasive penetration, and low toxicity to normal tissues.12,31 However, most of the currently reported photosensitizers are located in the cytoplasm, which may reduce the therapeutic effect of PDT/PTT. Considering that the nucleus is the most important organelle in the cell and contains most of the intracellular genetic material, the strategy of targeting the nucleus with a photosensitizer can greatly improve the therapeutic effect of tumors and further prevent recurrence. TAT is a regulator of HIV late transcription and a protein expressed by the long terminal repeat transactivated gene of HIV-1. The rich arginine and guanidine groups on TAT enable it to rapidly pass through the cell membrane. In addition, TAT is also a nuclear localization signal, which can effectively promote nuclear drug delivery.32,33 It was previously found that TAT can effectively mediate the entry of exogenous substances into cells; however, currently, it is incorporated into cell-penetrating peptides to transport various substances into cells and nuclei.34–36 It was found that the PDT of pancreatic cancer with chlorin e6 (Ce6)-binding metal ions under magnetic resonance was designed as follows: a kind of acid-sensitive material was used to temporarily block TAT and form a “masked” unit. After the unit reaches the tumor cells through blood, the acid-sensitive materials are removed due to the weak acid environment around the tumor tissue, exposing TAT. Because of different pH values, this process does not occur in normal cells. Next, TAT was exposed to induce the active materials to enter cancer cells and nuclei, and the PDT of cancer was realized under the action of NIR.37 To overcome the shortcomings of ICG in tumor treatment, we designed a nanoparticle based on an albumin and PEG4000 framework loaded with ICG and modified with TAT to target the nucleus. The PHSA-ICG-TAT has good stability and water solubility in PBS, RPMI 1640 medium, and FBS. As a non-specific cell-penetrating peptide, TAT can carry PHSA-ICG-TAT through the nuclear membrane and into the nucleus. The cell co-localization results showed that PHSA-ICG-TAT could accumulate in the nucleus, whereas PHSA-ICG could only accumulate outside the nucleus. Moreover, the PHSA-ICG-TAT is a spherical-like structure with a particle size of approximately 22 nm, which can be taken up into the cell by endocytosis. Hence, PHSA-ICG-TAT has a better cell uptake rate than ICG alone.
The therapeutic effect of ICG on tumors is mainly reflected in the effects of PDT and PTT. Under irradiation with NIR light, it can produce 1O2 to activate a series of immune responses, leading to apoptosis of tumor cells and the destruction of tumor tissue.38–40 At the same time, ICG can also generate thermal energy to increase the temperature of the nucleus. This effect destroys biologically active molecules (such as protein denaturation, enzyme inactivation) in the cell and induces ROS to cause tumor cell damage, necrosis, or thermal ablation of tumor tissue.2 It was found that the ability of ICG for 1O2 release after being loaded using the PHSA framework was stronger than that of ICG alone.The curve demonstrating the rise in temperature shows that the temperature of the PHSA-ICG group at each time point is higher than that of the ICG group. These two results indicate that the PHSA framework enhances the PDT/PTT effect of ICG. To verify this result, different concentrations of materials were co-cultured with 4T1 cells, and the cell activity was measured after irradiation with NIR light at a wavelength of 808 nm. The results showed that the lethality of ICG against 4T1 cells increased with the increase in the concentration of ICG. In addition, PHSA-ICG-TAT exhibited stronger lethality against 4T1 than ICG and PHSA-ICG, which shows that the damage to the nucleus is greater in tumor cells than in other intracellular regions. Apoptosis and DNA damage are the two most direct mechanisms of tumor death cell induced by the effects of ICG PDT/PTT. The results of flow cytometry and comet experiments showed that PHSA-ICG-TAT induced a higher apoptotic rate and Tail DNA% in 4T1 cells than ICG and PHSA-ICG alone. This further illustrates that the modification of TAT can guide ICG molecules to directly enter the nucleus of tumor cells and exert their PDT/PTT effect.
PHSA-ICG-TAT was injected into mice by intraperitoneal injection to investigate its toxicity. After 24 h of metabolism, the blood biochemical parameters and the morphology of the heart, liver, spleen, lung, and kidney tissue cells were not different between the PHSA-ICG-TAT and normal control groups. This shows that PHSA-ICG-TAT is safe for tissue treatment. The modification of photosensitive molecules with nanomaterials can enhance the enhanced permeability and retention effect on the tumor,41 so that the photosensitive molecules can more effectively reach and remain in the tumor tissue. Furthermore, nanomaterials can prevent the unintended release and inactivation of photosensitizers, thereby preventing the accumulation of photosensitizers in normal tissues.42 In the mouse in vivo experiment, the tumor was gradually ablated after PHSA-ICG-TAT phototherapy, and the tumor tissue section showed that the cell gap and morphology of the tumor tissue were significantly changed after treatment with PHSA-ICG-TAT. These findings demonstrated that PHSA-ICG-TAT had a significant therapeutic effect on tumors.
Conclusion
In this study, we obtained a novel targeted theranostic nanoparticle, termed PHSA-ICG-TAT, and demonstrated its excellent performance in PDT and PTT. When irradiated with NIR laser light, ICG can produce 1O2 in the nucleus, which in turn triggers a domino effect. At the same time, the PTT effect of ICG will increase the temperature of the nucleus and destroy the structure of biologically active molecules. Excessive accumulation of 1O2 in the nucleus and an increase in temperature will cause DNA damage and destruction of the nucleus structure and function, eventually leading to irreversible apoptosis. Cell colocalization experiments confirmed that PHSA-ICG-TAT targets the nucleus and can induce DNA damage and apoptosis in tumor cells. The eradication of tumors in vivo experiments confirmed that PHSA-ICG-TAT exerts a significant therapeutic effect.
Acknowledgment
This work was supported by the National Natural Science Foundation of China (No. 81773364, 11375124), Sichuan Province Natural Science Foundation (2017SZ0004), and Major Military Logistics Research Projects (AWS17J007).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Hwang HS, Shin H, Han J, et al. Combination of photodynamic therapy (PDT) and anti-tumor immunity in cancer therapy. J Pharm Investig. 2018;48(2):143–151. doi:10.1007/s40005-017-0377-x
2. Chen G, Roy I, Yang C, et al. Nanochemistry and nanomedicine for nanoparticle-based diagnostics and therapy. Chem Rev. 2016;116(5):2826–2885. doi:10.1021/acs.chemrev.5b00148
3. Zhang L, Wang D, Yang K, et al. Mitochondria-targeted artificial “Nano-RBCs” for amplified synergistic cancer phototherapy by a single NIR irradiation. Adv Sci (Weinh). 2018;5(8):1800049. doi:10.1002/advs.201800049
4. Liu J, Liang H, Li M, et al. Tumor acidity activating multifunctional nanoplatform for NIR-mediated multiple enhanced photodynamic and photothermal tumor therapy. Biomaterials. 2018;157:107–124. doi:10.1016/j.biomaterials.2017.12.003
5. Rajendrakumar SK, Chang NC, Mohapatra A, et al. A lipophilic IR-780 dye-encapsulated zwitterionic polymer-lipid micellar nanoparticle for enhanced photothermal therapy and NIR-based fluorescence imaging in a cervical tumor mouse model. Int J Mol Sci. 2018;19(4):1189. doi:10.3390/ijms19041189
6. Alander JT, Kaartinen I, Laakso A, et al. A review of indocyanine green fluorescent imaging in surgery. Int J Biomed Imaging. 2012;2012(1):940585. doi:10.1155/2012/940585
7. Sheng ZH, Hu DH, Zheng MB, et al. Smart human serum albumin-indocyanine green nanoparticles generated by programmed assembly for dual-modal imaging-guided cancer synergistic phototherapy. ACS Nano. 2014;8(12):12310–12322. doi:10.1021/nn5062386
8. Chen R, Wang X, Yao XK, et al. Near-IR-triggered photothermal/photodynamic dual-modality therapy system via chitosan hybrid nanospheres. Biomaterials. 2013;34(33):8314–8322. doi:10.1016/j.biomaterials.2013.07.034
9. Ott P. Hepatic elimination of indocyanine green with special reference to distribution kinetics and the influence of plasma protein binding. Pharmacol Toxicol. 1998;83(2):1–48. doi:10.1111/j.1600-0773.1998.tb01945.x
10. Ma Y, Tong S, Bao G, et al. Indocyanine green loaded SPIO nanoparticles with phospholipid-PEG coating for dual-modal imaging and photothermal therapy. Biomaterials. 2013;34(31):7706–7714. doi:10.1016/j.biomaterials.2013.07.007
11. Kirchherr AK, Briel A, Mäder K. Stabilization of indocyanine green by encapsulation within micellar systems. Mol Pharm. 2009;6(2):480–491. doi:10.1021/mp8001649
12. Zhang X, Li Y, Wei M, et al. Cetuximab-modified silica nanoparticle loaded with ICG for tumor-targeted combinational therapy of breast cancer. Drug Deliv. 2019;26(1):129–136. doi:10.1080/10717544.2018.1564403
13. Tang Y, Li Y, Li S, et al. Transformable nanotherapeutics enabled by ICG: towards enhanced tumor penetration under NIR light irradiation. Nanoscale. 2019;11(13):6217–6227. doi:10.1039/c9nr01049a
14. Xie J, Zheng Y, Ying JY. Protein-directed synthesis of highly fluorescent gold nanoclusters. J Am Chem Soc. 2009;131(3):888–889. doi:10.1021/ja806804u
15. Chen Q, Liang C, Wang C, et al. An imagable and photothermal ‘abraxane-like’ nanodrug for combination cancer therapy to treat subcutaneous and metastatic breast tumors. Adv Mater. 2015;27(5):903–910. doi:10.1002/adma.201404308
16. Yang JJ, Yang JH, Wei LX, et al. Rational design of protein-based MRI contrast agents. J Am Chem Soc. 2008;130(29):9260–9267. doi:10.1021/ja800736h
17. Huang P, Rong PF, Jin A, et al. Dye-loaded ferritin nanocages for multimodal imaging and photothermal therapy. Adv Mater. 2014;26(37):6401–6408. doi:10.1002/adma.201400914
18. Sun CJ, Yang H, Yuan Y, et al. Controlling assembly of paired gold clusters within apoferritin nanoreactor for in vivo kidney targeting and biomedical imaging. J Am Chem Soc. 2011;133(22):8617–8624. doi:10.1021/ja200746p
19. Jeong H, Huh M, Lee SJ, et al. Photosensitizer-conjugated human serum albumin nanoparticles for effective photodynamic therapy. Theranostics. 2011;6(1):230–239. doi:10.7150/thno/v01p0230
20. Otagiri M. A molecular functional study on the interactions of drugs with plasma proteins. Drug Metab Pharmacokinet. 2005;20(5):309–323. doi:10.2133/dmpk.20.309
21. Yin TJ, Cai H, Liu JY, et al. Biological evaluation of PEG modified nanosuspensions based on human serum albumin for tumor targeted delivery of paclitaxel. Eur J Pharm Sci. 2016;15(83):79–87. doi:10.1016/j.ejps.2015.12.019
22. Samkange T, D’Souza S, Obikeze K, et al. Influence of PEGylation on PLGA nanoparticle properties, hydrophobic drug release and interactions with human serum albumin. J Pharm Pharmacol. 2019;71(10):1497–1507. doi:10.1111/jphp.13147
23. Loomba L, Scarabelli T. Metallic nanoparticles and their medicinal potential. Part I: gold and silver colloids. Ther Deliv. 2013;4(7):859–873. doi:10.4155/tde.13.55
24. Ho D. Nanomaterial-based therapy: a new generation of cancer treatment. Therapy. 2009;6(1):99–103. doi:10.2217/14750708.6.1.99
25. Wang F, Wang YC, Dou S, et al. Doxorubicin-tethered responsive gold nanoparticles facilitate intracellular drug delivery for overcoming multidrug resistance in cancer cells. ACS Nano. 2011;5(5):3679–3692. doi:10.1021/nn200007z
26. Xiong XB, Lavasanifar A. Traceable multifunctional micellar nanocarriers for cancer-targeted co-delivery of MDR-1 siRNA and doxorubicin. ACS Nano. 2011;5(6):5202–5213. doi:10.1021/nn2013707
27. Meng H, Liong M, Xia T, et al. Engineered design of mesoporous silica nanoparticles to deliver doxorubicin and P-glycoprotein siRNA to overcome drug resistance in a cancer cell line. ACS Nano. 2010;4(8):4539–4550. doi:10.1021/nn100690m
28. Pan LM, He QJ, Liu JN, et al. Nuclear-targeted drug delivery of TAT peptide-conjugated monodisperse mesoporous silica nanoparticles. J Am Chem Soc. 2012;134(13):5722–5725. doi:10.1021/ja211035w
29. Copolovici DM, Langel K, Eriste E, et al. Cell-penetrating peptides: design, synthesis, and applications. ACS Nano. 2014;8(3):1972–1994. doi:10.1021/nn4057269
30. Singh NP. The comet assay: reflections on its development, evolution and applications. Mutat Res Rev Mutat Res. 2016;767:23–30. doi:10.1016/j.mrrev.2015.05.004
31. Hou XM, Wang XD, Liu R, et al. Facile synthesis of multifunctional Fe 3 O 4 @SiO 2 @Au magneto-plasmonic nanoparticles for MR/CT dual imaging and photothermal therapy. RSC Adv. 2017;7(31):18844–18850. doi:10.1039/C7RA00925A
32. Bechara C, Sagan S. Cell-penetrating peptides: 20 years later, where do we stand? FEBS Lett. 2013;587(12):1693–1702. doi:10.1016/j.febslet.2013.04.031
33. Borrelli A, Tornesello AL, Tornesello ML, et al. Cell penetrating peptides as molecular carriers for anti-cancer agents. Molecules. 2018;23(2):295. doi:10.3390/molecules23020295
34. Jin E, Zhang B, Sun X, et al. Acid-active cell-penetrating peptides for in vivo tumor-targeted drug delivery. J Am Chem Soc. 2013;135(2):933–940. doi:10.1021/ja311180x
35. Gao G, Jiang YW, Jia HR, et al. From perinuclear to intranuclear localization: a cell-penetrating peptide modification strategy to modulate cancer cell migration under mild laser irradiation and improve photothermal therapeutic performance. Biomaterials. 2019;223:119443. doi:10.1016/j.biomaterials.2019.119443
36. Bartomeu Garcia C, Shi D, Webster TJ. Tat-functionalized liposomes for the treatment of meningitis: an in vitro study. Int J Nanomedicine. 2017;12:3009–3021. doi:10.2147/IJN.S130125
37. Gao M, Fan F, Li D, et al. Tumor acidity-activatable TAT targeted nanomedicine for enlarged fluorescence/magnetic resonance imaging-guided photodynamic therapy. Biomaterials. 2017;133:165–175. doi:10.1016/j.biomaterials.2017.04.013
38. Chen Q, He S, Zhang FJ, et al. A versatile Pt-Ce6 nanoplatform as catalase nanozyme and NIR-II photothermal agent for enhanced PDT/PTT tumor therapy. Sci China Mater. 2020. doi:10.1007/s40843-020-1431-5
39. Fakhar-e-Alam M, Atif M, Alimgeer KS, et al. Synergistic effect of TEMPO-coated TiO2 nanorods for PDT applications in MCF-7 cell line model. Saudi J Biol Sci. 2020;27(12):3199–3207. doi:10.1016/j.sjbs.2020.09.027
40. Atif M, Iqbal S, Fakhar-E-Alam M, et al. Manganese-doped cerium oxide nanocomposite induced photodynamic therapy in MCF-7 cancer cells and antibacterial activity. Biomed Res Int. 2019;223:7156828. doi:10.1155/2019/7156828
41. Davis ME, Chen ZG, Shin DM. Nanoparticle therapeutics: an emerging treatment modality for cancer. Nat Rev Drug Discov. 2008;7(9):771–782. doi:10.1038/nrd2614
42. Master A, Livingston M, Sen Gupta A. Photodynamic nanomedicine in the treatment of solid tumors: perspectives and challenges. J Control Release. 2013;168(1):88–102. doi:10.1016/j.jconrel.2013.02.020
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.