Back to Journals » Cancer Management and Research » Volume 11
Nuclear GAPDH is vital for hypoxia-induced hepatic stellate cell apoptosis and is indicative of aggressive hepatocellular carcinoma behavior
Authors Gong Y, Zou B ![]() , Peng S, Li P, Zhu G, Chen J, Chen J, Liu X, Zhou W, Ding L, Chen Y, Zeng L, Zhang B, Cai C, Li J
, Peng S, Li P, Zhu G, Chen J, Chen J, Liu X, Zhou W, Ding L, Chen Y, Zeng L, Zhang B, Cai C, Li J ![]()
Received 20 January 2019
Accepted for publication 6 April 2019
Published 30 May 2019 Volume 2019:11 Pages 4947—4956
DOI https://doi.org/10.2147/CMAR.S202268
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Chien-Feng Li
Yihang Gong,1,* Baojia Zou,1,* Siqi Peng,2 Peiping Li,1 Genglong Zhu,1 Jiafan Chen,1 Jianxu Chen,1 Xialei Liu,1 Wenying Zhou,3 Lei Ding,1 Yutong Chen,2 Linjuan Zeng,2 Baimeng Zhang,1 Chaonong Cai,1 Jian Li1
1Department of Hepatobiliary Surgery; 2Department of Oncology; 3Central Laboratory, The Fifth Affiliated Hospital of Sun Yat-sen University, Zhuhai, Guangdong Province 519000, People’s Republic of China
*These authors contributed equally to this work
Background/aim: Hepatic stellate cells (HSCs) are critical determinants of liver tumor behavior such as vascular invasion, cell proliferation and migration. The apoptosis of HSCs can inhibit tumor growth and contribute to repressing hepatocellular carcinoma (HCC) progression. Our study aims to investigate the impact of nuclear glyceraldehyde-3-phosphate dehydrogenase (GAPDH) on HSCs under hypoxic conditions and the association of nuclear GAPDH with HCC patient outcomes and tumor progression.
Patients and methods: Following stable cell passage, 0.3% O2 was used to induce hypoxia. Cell proliferation and apoptosis were analyzed using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-tetrazolium bromide (MTT) assays and flow cytometry, respectively. Proteins expression were detected by extracting nuclear and cytoplasmic proteins and performing Western blots. GAPDH nuclear translocation was blocked by the agent deprenyl. Immunohistochemical staining for GAPDH was investigated in 137 HCC tissue samples from our center. An analysis of the clinicopathological features, Kaplan-Meier analysis and Cox proportional hazards regression analysis were applied.
Results: MTT assays and flow cytometry analyses showed that the nuclear accumulation of GAPDH led to the apoptotic death of HSCs, while blockade of this process with deprenyl significantly decreased apoptosis. Western blots revealed that deprenyl inhibited the nuclear translocation of GAPDH. An analysis of the immunohistochemical staining of HSCs in HCC tissue samples (137) revealed that nuclear GAPDH expression was significantly positively correlated with HIF-1α expression. Overall survival (OS) and time-to-recurrence (TTR) estimated by Kaplan-Meier analyses showed that patients with high HIF-1α or low nuclear GAPDH levels in HSCs had significantly poorer prognosis compared with patients with low HIF-1α or high nuclear GAPDH expression in HSCs. Moreover, patients with combined high HIF-1α/low nuclear GAPDH expression in HSCs had the worst prognosis. The Cox regression analysis revealed that the combination of nuclear GAPDH/HIF-1α expression in HSCs was an independent prognostic factor for OS and TTR in HCC patients.
Conclusions: These findings provide a novel mechanism underlying the involvement of intranuclear GAPDH in hypoxia-induced HSCs apoptosis and a correlation between nuclear GAPDH levels and the clinical prognosis, which may prompt the development of a novel therapeutic strategy for HCC.
Keywords: apoptosis, GAPDH, HSC, HCC, hypoxia
Introduction
Hepatocellular carcinoma (HCC) is one of the most fatal solid tumors worldwide.1 Hepatic stellate cells (HSCs) are essential in the development of HCC. Upon exposure to hypoxia, HSCs undergo either apoptosis or activation and the balance of this process is vital in the progression of HCC.2–4 Activated HSCs, which are characterized by excessive α-smooth muscle actin (α-SMA) expression,5 in HCC are critical determinants of malignant tumor behavior such as vascular invasion and tumor cell proliferation and migration.6,7 In contrast, the apoptosis of HSCs can suppress tumor cell proliferation and invasion.8 For example, the selective apoptosis of activated HSCs effectively restores tumor growth and attenuates HCC progression.9 However, the mechanisms of HSCs apoptosis remain poorly understood.
Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) is a pivotal glycolytic enzyme that is commonly expressed in all tissues and plays a critical role in regulating the cellular energy supply.10 This molecule activates the apoptosis pathway after translocating to the nucleus, leading to cell death under hypoxic conditions.11 Moreover, deprenyl (selegiline) is an effective agent that prevents hypoxia-induced GAPDH nuclear translocation and cell death in cancer.12,13 In addition, hypoxia is considered a key event underlying HCC progression. However, the function of nuclear GAPDH and the relationship between hypoxia and nuclear GAPDH in HSCs remain unknown.
Therefore, we hypothesized that nuclear GAPDH played a role in promoting HSCs apoptosis under hypoxic conditions and affected patient prognosis. In the current study, we investigated whether nuclear GAPDH translocation was induced by hypoxia in HSCs and determined how it was associated with HCC patient outcomes and tumor progression.
Materials and methods
Cell culture and chemical treatment
The LX2 cell line (Merck Millipore, Germany) was obtained from Fudan University, Shanghai, People’s Republic of China, and was cultured in normal glucose (5 mM) 1,640 basic medium supplemented with 10% fetal bovine serum and 1% penicillin at 37 °C in a humidified incubator with a 5% CO2 atmosphere. Cells were cultured without serum for 24 h before exposure to hypoxia. The hypoxic environment was created in a hypoxia glove box (Coy) with a calibrated gas containing 0.3% O2 at 37 °C, and the CO2 concentration was maintained at 5% under both conditions. The cells were maintained in the incubator at 37 °C for different durations. When appropriate, cells were preincubated with different concentrations of deprenyl (S3740, Selleck Chemicals, Houston, TX, USA) for 2 hrs prior to exposure to hypoxia, and unexposed cells were used as controls.
Western blot analysis and preparation of nuclear and cytoplasmic fractions
Western blot analyses were performed using previously described methods.14 Target proteins were detected by incubating the membranes with the following primary antibodies: GAPDH (ab8245; Abcam, USA), α-SMA (ab32575; Abcam, USA), hypoxia-inducible factor-1α (HIF-1α) (A11945; ABclonal, USA), Bcl-2 (catalog no. 2872; CST, USA), Bax (catalog no. 2772; CST, USA), p53 (catalog no. 2524; CST, USA), α-Tubulin (ab4074; Abcam, USA), Histone H3 (BM4855; Boster, China) and β-Actin (ac004; ABclonal, USA). Nuclear and cytoplasmic proteins were extracted using a subcellular fractionation kit (P0027; Beyotime, Shanghai, China) according to the manufacturer’s protocol. The results were captured using an automatic fluorescence/chemiluminescence image analysis system (Tanon, Shanghai, China).
Flow cytometry analysis of cellular apoptosis
All apoptosis measurements were performed after cells received the intervention for different times. Briefly, LX2 cells (1×106 cells/ml) were collected, centrifuged at 1,000×g for three minutes at room temperature, washed twice with PBS (1×) and resuspended in 100 µl of binding buffer (Annexin V-FITC Staining kit, DOJINDO, Shanghai, China) containing 5 µl of annexin-V-FITC and 5 µl of PI solution according to the manufacturer’s instructions. After 20 mins of incubation at room temperature in the dark, 400 µl of fresh binding buffer was added to the mixture. Samples were immediately analyzed.
MTT proliferation assay
MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-tetrazolium bromide) was added to the 96-well plates, which were placed at 37 °C. After incubation for 4 hrs, the medium was discarded and the precipitates (formazan) were dissolved with dimethyl sulfoxide (DMSO, MP, 196,055, Santa Ana, USA). The absorbance was detected at a wavelength of 490 nm. To subtract the background, a blank control was included that contained only culture medium and MTT.
Tissue samples
A retrospective study of patients who were pathologically diagnosed with HCC from 2011 to 2016 at our center was performed, and 137 HCC tumor specimens were used for immunohistochemical staining. All patients were pathologically confirmed to have HCC and underwent primary surgical resection. Samples were formalin-fixed and paraffin-embedded histological sections from the Department of Pathology at our center. TNM staging was performed according to the 2010 American Joint Committee on Cancer (AJCC) staging system. The follow-up time ranged from 1 to 2,440 days (median, 909 days). Written informed consent was obtained from the patients. The study was approved by the Institutional Ethics Review Board of the 5th Affiliated Hospital, Sun Yat-sen University.
Immunohistochemical staining and analysis
Paraffin-embedded HCC tissues were sectioned at a thickness of 4 µm and then dewaxed in xylene after heating to 70 °C for 70 mins. Rehydration was performed in a gradient of ethanol solutions. A sodium citrate solution was applied for antigen retrieval under high pressure and temperature. Then, the sections were incubated with primary antibody overnight at 4 °C. After three washes with PBS, sections were incubated with HRP-labeled goat anti-mouse secondary antibody for 60 mins at room temperature. Finally, the slides were counterstained with hematoxylin. The same dilution of nonimmune rabbit serum was used as the negative control. The immunohistochemistry kit was obtained from MaiXin (KIT-9921, Fujian, China). In addition, the 3ʹ-diaminobenzidine tetrahydrochloride (DAB, 1:1:1, DAB-0031, MaiXin, Fujian, China) reagent was used to develop the staining.
Immunostaining results were independently analyzed by two pathologists. Nuclear protein expression was measured by multiplying the stained area by the intensity score in 4 random areas of each section at 400× magnification under a microscope (Axio Observer, ZEISS, Germany). The percentage of positive cells was scored as 1 (1–25%), 2 (26–50%), 3 (51–75%), or 4 (76–100%), and the staining intensity was scored as 0+, 1+, 2+, or 3+. In accordance with the literature,15,16 staining scores ≤4 and ≥6 were regarded as low expression and high expression, respectively.
Statistical analysis
The statistical and graphing software programs used in this study were SPSS 20.0 (SPSS, Inc., Chicago, IL, USA) and GraphPad Prism 7.0 (GraphPad Software, La Jolla, CA, USA), and each experiment was repeated at least three times. Data are presented as the mean ± standard deviation (SD). The correlation analysis was performed using Spearman’s correlation rank test. Overall survival (OS) and time-to-recurrence (TTR) were determined using the Kaplan-Meier survival analysis and the log-rank test. The univariate survival analysis, Cox proportional hazards regression analysis, and correlations between different factors were analyzed using SPSS 20.0 software. p≤0.05 was considered statistically significant in all analyses.
Results
Hypoxia affects the activation of LX2 cells and promotes apoptosis and the nuclear translocation of GAPDH
The effects of exposure to hypoxia for different periods on LX-2 cell apoptosis were investigated using flow cytometry. Hypoxia significantly increased annexin V-FITC/PI incorporation in a dose-dependent manner (Figure 1A and B). Cells were also analyzed using MTT assays to confirm the increase in apoptosis. Hypoxia exposure (24, 48 and 72 h) decreased cell viability, similar to the findings obtained using flow cytometry (Figure 1C). The activation and apoptosis of LX2 cells depended on the degree of hypoxia exposure. Western blot analyses revealed an increasing and then decreasing trend in α-SMA expression along with an increase in HIF-1α levels (Figure 1D). Nuclear and cytoplasmic protein extracts were obtained to determine if nuclear GAPDH levels were affected by hypoxia. An increased time of exposure to hypoxia significantly increased the translocation of cytoplasmic GAPDH into the nucleus (Figure 1E).
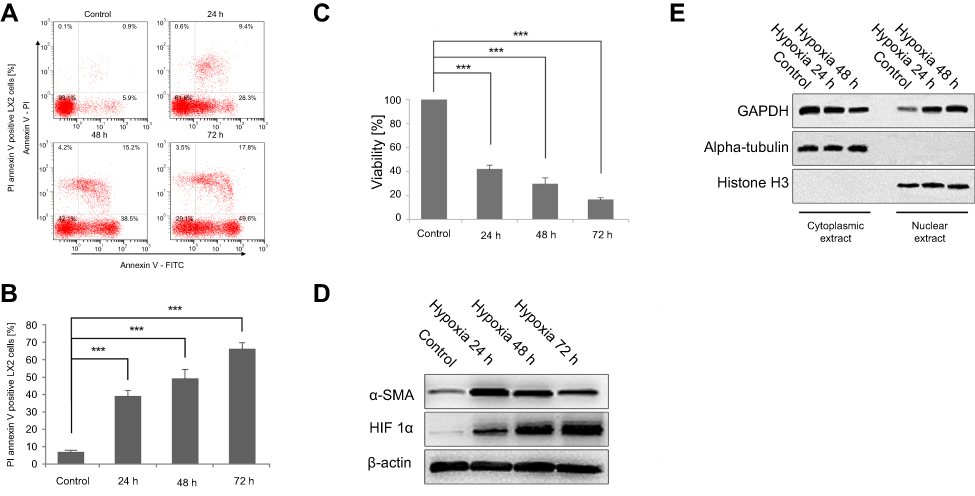 | Figure 1 Hypoxia exposure affects the activation of LX2 cells and increases cell apoptosis and GAPDH nuclear translocation. (A and B) Cell apoptosis was detected using annexin V-FITC/PI double staining and flow cytometry (A), and bar graphs (B) show the effect of hypoxia on apoptosis. (C) Cell viability was measured using the MTT assay after hypoxia. (D and E) Western blots showing the effects of hypoxia on the activation of LX2 cells (D) and the accumulation of nuclear GAPDH (E). Untreated samples served as controls.Note: Data are presented as the mean ± SEM. n.s., not significant, *p<0.05, **p<0.01, and ***p<0.001, n=3.Abbreviations: GAPDH, glyceraldehyde-3-phosphate dehydrogenase; α-SMA, α-smooth muscle actin; HIF-1α, hypoxia-inducible factor 1α. |
Deprenyl restored hypoxia-induced GAPDH nuclear translocation and apoptosis in LX2 cells
Next, the specific inhibitor deprenyl was applied to further confirm whether nuclear GAPDH contributed to apoptosis under hypoxic conditions. Deprenyl toxicity was detected after exposing LX2 cells to 0.001 to 400 µM deprenyl for 0–5 days. The toxicity of 400 µM deprenyl was negligible in LX2 cells (Figure 2A and B). Deprenyl efficiently inhibited the nuclear translocation of GAPDH but did not alter HIF-1α expression in hypoxia-treated LX2 cells (Figure 2C). Cell viability was measured after treatment with a single concentration of deprenyl (10 µM) and exposure to hypoxia for different periods (24 and 48 h). As shown in Figure 2D, preincubation with deprenyl efficiently prevented the loss of cell viability induced by hypoxia. Meanwhile, annexin V-FITC/PI staining and subsequent flow cytometry analyses showed that the apoptosis of LX2 cells under hypoxia exposure was restored by deprenyl (Figure 2E and F). The effect of GAPDH after nuclear blockade was further confirmed by Western blots of apoptosis-related proteins. Deprenyl can reverse hypoxia-induced higher Bax/Bcl-2 ratio. Accordingly, the increase in the level of the p53 protein was also inhibited by the deprenyl treatment (Figure 2G).
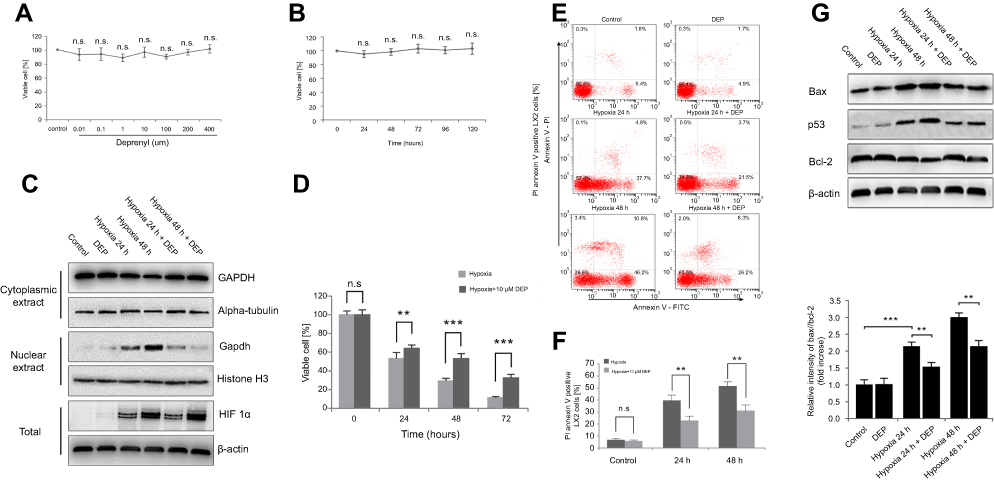 | Figure 2 Deprenyl inhibits the hypoxia-induced nuclear translocation of GAPDH and protects LX2 cells from apoptosis. (A and B) Deprenyl toxicity was detected using the MTT assay. LX2 cells were exposed to different concentrations of deprenyl for 48 hrs (A) or to 10 µM deprenyl for different times (B). LX2 cells were exposed to hypoxia for different periods after preincubation with 10 µM deprenyl. (C) The effect of deprenyl on hypoxia-induced nuclear GAPDH accumulation. (D) Cell viability was measured using the MTT assay. (E and F) Cell apoptosis was detected using annexin V-FITC/PI double staining and flow cytometry (E), and bar graphs (F) show the effect of deprenyl on apoptosis. (G) The effect of deprenyl on the levels of apoptotic proteins in LX2 cells. Untreated samples served as controls.Note: Data are presented as the mean ± SEM. n.s., not significant, *p<0.05, **p<0.01, ***p<0.001, n=3.Abbreviations: GAPDH, glyceraldehyde-3-phosphate dehydrogenase; DEP, deprenyl. |
Nuclear GAPDH and HIF-1α expression in HSCs in the context of HCC
α-SMA levels were first detected to identify the presence of HSCs. Negative and positive controls are shown in Figure 3A and B, respectively. We examined the expression of nuclear GAPDH and HIF-1α, an indicator of hypoxia. Representative images with low and high nuclear GAPDH and HIF-1α expression are shown (Figure 3C–J). Low and high nuclear GAPDH levels were observed in 62% (85/137) and 38% (52/137) of HCC samples, respectively. Low and high HIF-1α expression was observed in 51.1% (70/137) and 48.9% (67/137) of all cases, respectively.
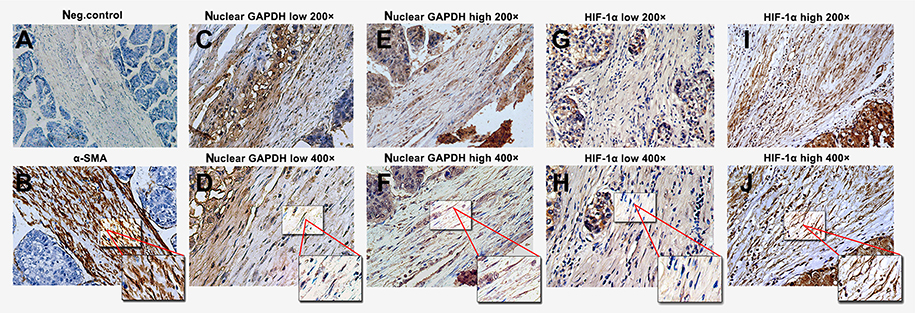 | Figure 3 Immunohistochemical staining for α-SMA, nuclear GAPDH, and HIF-1α in HCC tissues containing HSCs. (A) α-SMA staining in the negative control and HSCs (B) in HCC tissues. (C–F) Different nuclear GAPDH expression patterns in HSCs within HCC tissues. (G–J) Different HIF-1α expression patterns in HSCs within HCC tissues. (A, C, E, G, and I: 200× magnification; B, D, F, H, and J: 400× magnification).Abbreviations: GAPDH, glyceraldehyde-3-phosphate dehydrogenase; α-SMA, α-smooth muscle actin; HIF-1α, hypoxia-inducible factor 1α; HCC, hepatocellular carcinoma; HSCs, Hepatic stellate cells. |
The correlation between clinicopathological features and nuclear GAPDH and hif-1α expression in hscs
We further explored the correlations between clinicopathological features and the IHC results for nuclear GAPDH and HIF-1α in the cohort of 137 HCC patients (Table 1). Nuclear GAPDH expression was negatively correlated with the TNM stage (p=0.026) and vascular invasion (p=0.007). Meanwhile, HIF-1α expression was positively correlated with the TNM stage (p=0.002), tumor size (p<0.001) and vascular invasion (p<0.001). Furthermore, Spearman’s correlation analysis revealed a significant positive correlation between nuclear GAPDH expression and HIF-1α expression (rs=0.326, p<0.001, Table 2).
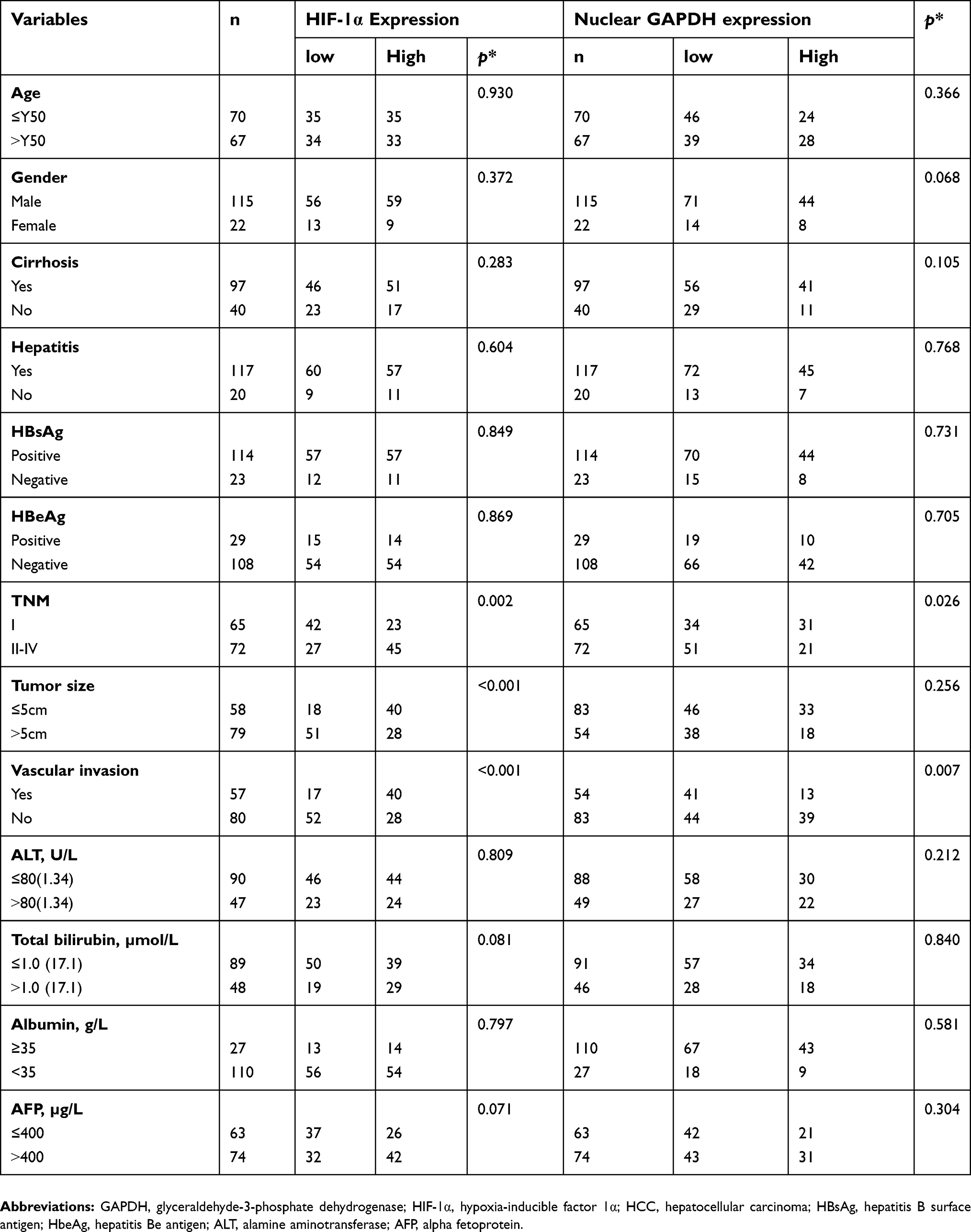 | Table 1 Association of nuclear GAPDH and HIF-1α expression with clinicopathologic characteristics in 137 HCC patients |
 | Table 2 Spearman correlation analysis between nuclear GAPDH and HIF-1α proteins expression in HCC |
Association of nuclear GAPDH and HIF-1α levels in HSCs with the clinical outcomes in HCC patients
The Kaplan-Meier analysis and log-rank test were used to examine the relationship between nuclear GAPDH and HIF-1α levels in HSCs and the clinical outcomes of patients with HCC. The mean OS and TTR were 768 days and 629 days in HCC patients with low nuclear GAPDH expression and 1,140 days (p=0.012; Figure 4A) and 982 days (p=0.001; Figure 4B) in those with high levels of nuclear GAPDH. For HIF-1α, the mean OS and TTR were 1,040 days and 920 days in HCC patients with low expression and 776 days (p<0.001, Figure 4C) and 604 days (p<0.001, Figure 4D) in those with high expression.
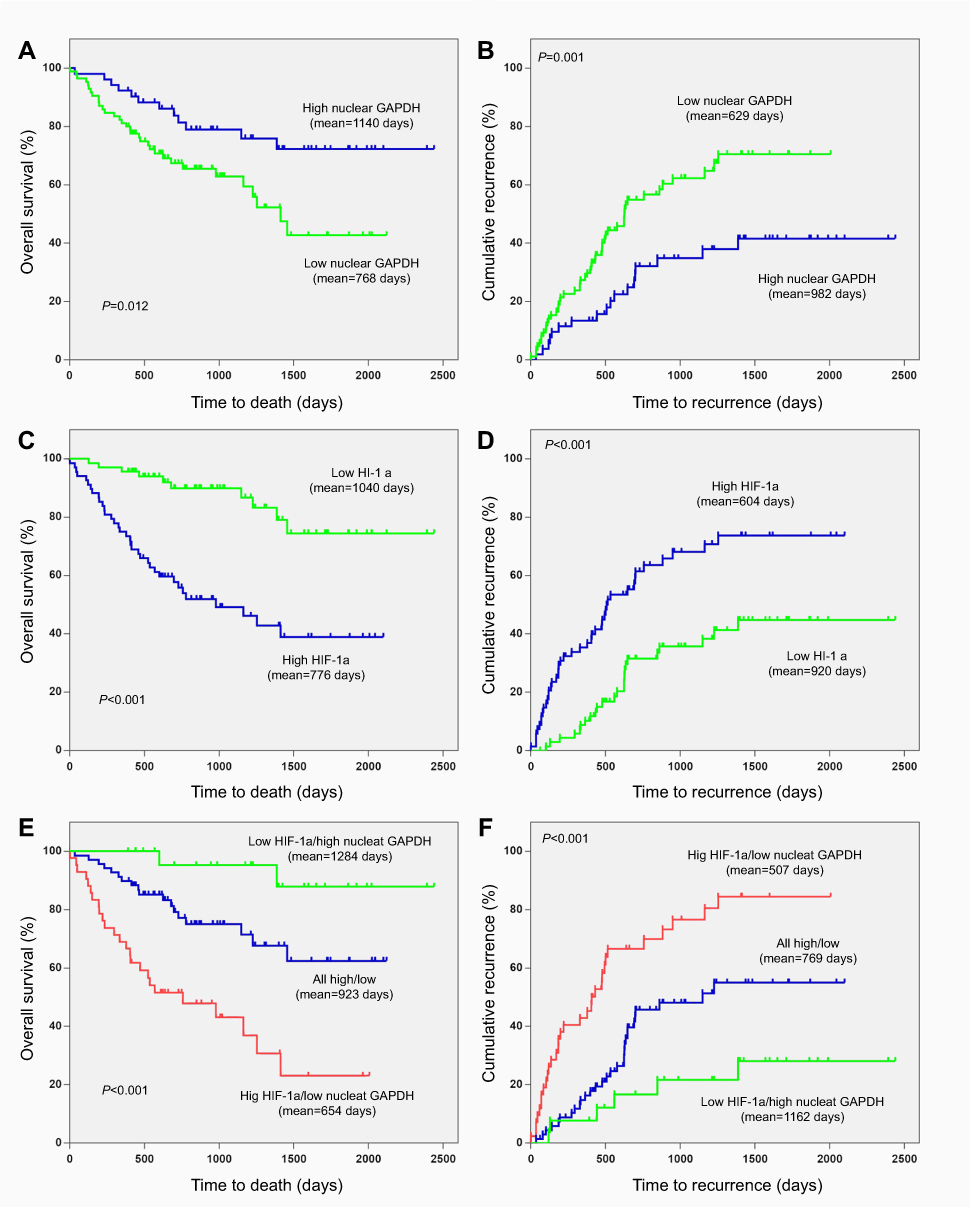 | Figure 4 Kaplan–Meier survival analyses based on nuclear GAPDH and HIF-1α expression in HCC patients. (A) The OS of postoperative HCC patients with low/high nuclear GAPDH expression. (B) The TTR of postoperative HCC patients with low/high nuclear GAPDH expression. (C) The OS of postoperative HCC patients with low/high HIF-1α expression. (D) The TTR of postoperative HCC patients with low/high HIF-1α expression. (E) The OS of postoperative HCC patients based on both nuclear GAPDH and HIF-1α expression. (F) The TTR of postoperative HCC patients based on both nuclear GAPDH and HIF-1α expression. |
We next divided the 137 HCC patients into 3 subgroups according to nuclear GAPDH and HIF-1α expression in HSCs. Then, we performed Kaplan–Meier survival analysis to examine correlations with clinical outcomes in the various subgroups. As shown in Figure 4E and F, HCC patients with low nuclear GAPDH/high HIF-1α expression (group 1) exhibited the shortest OS (mean, 654 days) and TTR (mean, 507 days). In contrast, patients with high nuclear GAPDH/low HIF-1α expression (group 3) displayed the longest OS (mean, 1,284 days) and TTR (mean, 1,162 days). Moreover, patients with low or high levels of both nuclear GAPDH and HIF-1α expression (group 2) displayed intermediate OS (mean, 923 days) and TTR (mean, 769 days).
Association of nuclear GAPDH and HIF-1α expression with patient survival
Univariate analysis indicated that tumor size, vascular invasion, TNM stage, alpha-fetoprotein (AFP), nuclear GAPDH, HIF-1α, and the combined nuclear GAPDH/HIF-1α levels were significantly correlated with OS and TTR (p-values for OS: <0.001, 0.017, 0.014, 0.010, 0.012, <0.001, and <0.001, respectively; p-values for TTR: 0.046, <0.001, <0.001, 0.039, 0.001, <0.001, and <0.001, respectively) (Table 3). Cox multivariate proportional hazard regression analysis was performed by incorporating the significant factors in the univariate analysis. Briefly, the nuclear GAPDH/HIF-1α combination as well as AFP were independent prognostic factors for OS. Moreover, TNM stage, AFP and the nuclear GAPDH/HIF-1α combination were independent prognostic factors for TTR (Table 3).
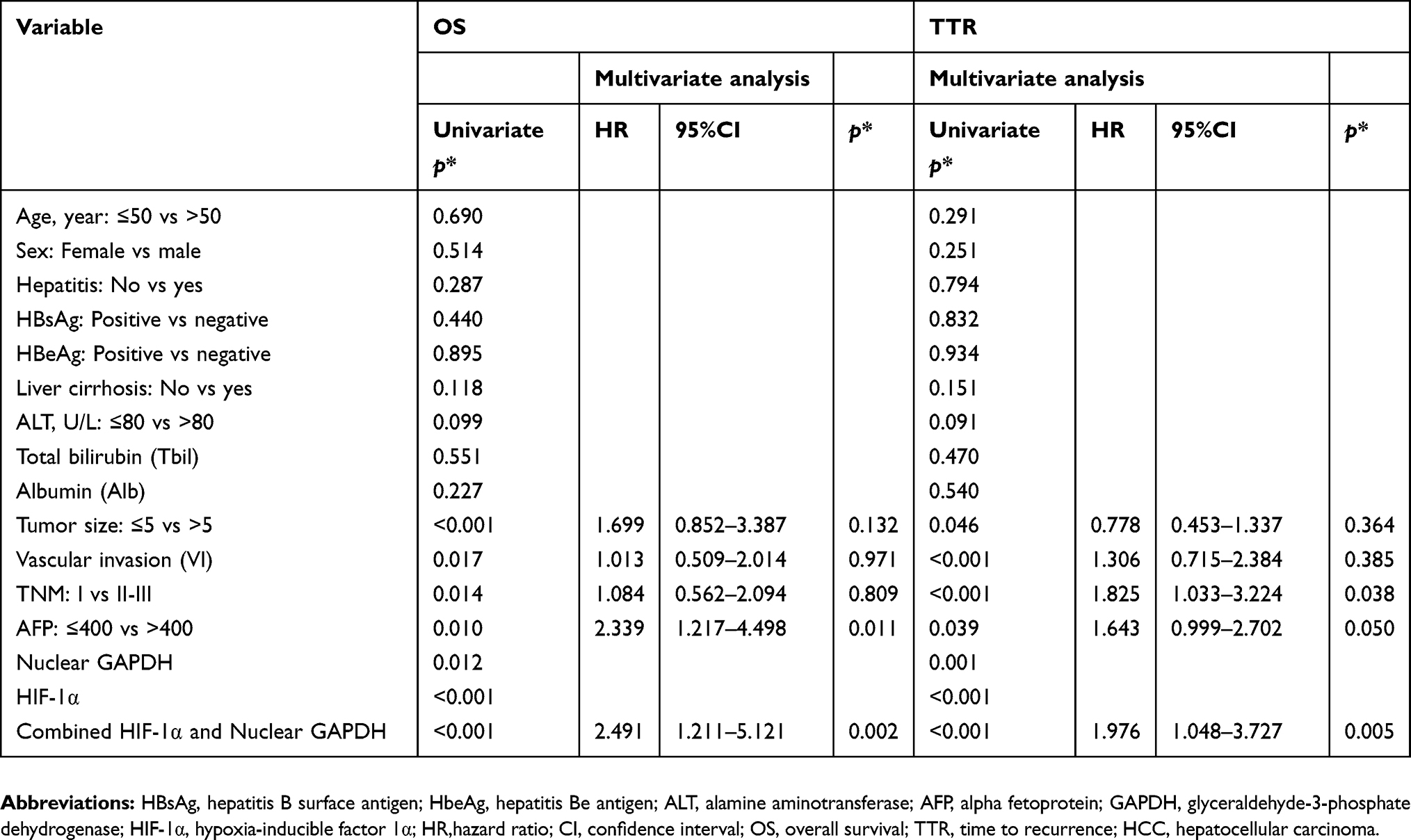 | Table 3 Univariate and multivariate analyses of factors associated with OS and TTR in 137 cases of HCC patients |
Discussion
In the present study, we reported that GAPDH accumulated in the nucleus and induced apoptosis in HSCs under hypoxic conditions. Furthermore, nuclear GAPDH expression was correlated with tissue hypoxia in postsurgical samples. Nuclear GAPDH, HIF-1α and the nuclear GAPDH/HIF-1α combination are independent prognostic factors for the clinical outcomes of patients with HCC.
A hypoxic microenvironment is a typical feature of HCC,17 and exposure to hypoxia can either induce apoptosis or promote HSC activation.4,5 The balance of HSC activation and apoptosis is a controversial topic in the field of cirrhosis and HCC development, particularly in the context of hypoxia.18 The mechanisms underlying both processes remain poorly understood. HSCs are activated by exposure to certain levels of hypoxia. When hypoxia becomes more intense, oxidative stress becomes so severe that it induces cellular apoptosis, as has been shown in many cancer cell lines in previous studies and the present study.19
GAPDH has been reported to actively participate in apoptosis-related processes in the nucleus, and this function is independent of its enzymatic activity in the process of glycolysis in the cytoplasm.20 Several studies have revealed a role for GAPDH nuclear translocation in cell death in ovarian cancer, pancreatic cancer and neuroblastoma.21–23 Consistent with previous studies, nuclear GAPDH plays a significant role in HSC apoptosis in the present study.
The upregulation of nuclear GAPDH in HSCs was also correlated with a better prognosis of patients with HCC. Data from the present study suggest that this correlation is probably due to the nuclear translocation of GAPDH in HSCs, which causes apoptosis and thus serves as a protective factor against tumor growth and metabolism. In contrast, low levels of nuclear GAPDH in HSCs were correlated with poor prognosis. Similarly, activated HSCs were shown to inhibit tumor cell death, induce blood vessel formation, enhance immune cell apoptosis, and promote tumor growth in previous studies.3,24,25 The apoptosis of HSCs has been reported to reduce the secretion of biological factors, including TGF-β1 and PDGF-BB, which protect cancer cells from cell death via the cAMP/PKA/SMO pathway by activating GLI2.4,26 In addition, TGF-β1 promotes HCC invasion and metastasis through the ERK pathway by upregulating FGFR4.27 These mechanisms may contribute to repressing HCC progression. However, these previous studies specifically focused on the effects of apoptotic HSCs on tumor cell lines, and the potential relationship between apoptotic HSCs and the clinicopathological features of HCC patients has not been explored. Our study confirmed that nuclear GAPDH induces HSC apoptosis, particularly under hypoxic conditions, and nuclear GAPDH levels correlate closely with patient outcomes and may be a valuable predictor of prognosis.
The results from the present study suggest a strong correlation between nuclear GAPDH and HIF-1α. Previous studies have shown that GAPDH, a key glycolytic enzyme, contains a hypoxia responsive element (HRE) in its promoter which has been identified as a target of HIF-1α. Therefore, HIF-1α can modulate the expression of GAPDH through HRE.28,29 In turn, GAPDH promotes expression of HIF-1α by binding to TRAF2 and enhancing NF-κB activity. There is a positive feedback loop between HIF-1α and GAPDH.30,31 However, these studies did not focus on GAPDH in the nucleus, and the interaction between nuclear GAPDH and HIF-1α remained largely unknown. In our future research, we will study these issues in more depth.
In summary, our data suggest for the first time that nuclear GAPDH plays a pivotal role in promoting HSCs apoptosis under hypoxic conditions. These findings were further confirmed in vivo using postsurgical HCC samples. Therefore, nuclear GAPDH in activated HSCs is a protective factor against HCC growth and should be considered in future therapeutic strategies for patients with HCC.
Acknowledgments
This work was supported by grants from the National Natural Science Foundation of China (two projects, Grant Nos. 81502503 and 81872348), the Natural Science Foundation of Guangdong Province (Grant No. 2016A030310191), Project of Administration of Traditional Chinese Medicine of Guangdong Province of China (Grant No. 20181062), the Fundamental Research Funds for the Central Universities (Sun Yat-sen University, Grant No. 17ykpy60), and the open project of the Key Laboratory of Tropical Disease Control (Sun Yat-sen University), the Ministry of Education (Grant No. 2018kfkt04). In addition, Yihang Gong particularly wishes to thank Danyang Pan, whose company provided him with powerful spiritual support over the past two years.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Altekruse SF, McGlynn KA, Reichman ME. Hepatocellular carcinoma incidence, mortality, and survival trends in the United States from 1975 to 2005. J Clin Oncol. 2009;27(9):1485–1491. doi:10.1200/JCO.2008.20.7753
2. Ji Q, Yang L, Zhou J, et al. Protective effects of paeoniflorin against cobalt chloride-induced apoptosis of endothelial cells via HIF-1alpha pathway. Toxicol In Vitro. 2012;26(3):455–461. doi:10.1016/j.tiv.2012.01.016
3. Ma H, Xie L, Zhang L, et al. Activated hepatic stellate cells promote epithelial-to-mesenchymal transition in hepatocellular carcinoma through transglutaminase 2-induced pseudohypoxia. Commun bio. 2018;1:168. doi:10.1038/s42003-018-0052-4
4. Geng ZM, Jha RK, Li B, et al. Sorafenib inhibition of hepatic stellate cell proliferation in tumor microenvironment of hepatocellular carcinoma: a study of the sorafenib mechanisms. Cell Biochem Biophys. 2014;69(3):717–724. doi:10.1007/s12013-014-9858-y
5. Deng J, Huang Q, Wang Y, et al. Hypoxia-inducible factor-1alpha regulates autophagy to activate hepatic stellate cells. Biochem Biophys Res Commun. 2014;454(2):328–334. doi:10.1016/j.bbrc.2014.10.076
6. Carloni V, Luong TV, Rombouts K. Hepatic stellate cells and extracellular matrix in hepatocellular carcinoma: more complicated than ever. Liver Int. 2014;34(6):834–843. doi:10.1111/liv.12465
7. Amann T, Bataille F, Spruss T, et al. Activated hepatic stellate cells promote tumorigenicity of hepatocellular carcinoma. Cancer Sci. 2009;100(4):646–653. doi:10.1111/j.1349-7006.2009.01087.x
8. Liu CH, Chern GJ, Hsu FF, et al. A multifunctional nanocarrier for efficient TRAIL-based gene therapy against hepatocellular carcinoma with desmoplasia in mice. Hepatology. 2018;67(3):899–913. doi:10.1002/hep.29513
9. Sun B, Zhang X, Cheng X, et al. Intratumoral hepatic stellate cells as a poor prognostic marker and a new treatment target for hepatocellular carcinoma. PLoS One. 2013;8(11):e80212. doi:10.1371/journal.pone.0080212
10. Nicholls C, Li H, Liu JP. GAPDH: a common enzyme with uncommon functions. Clin Exp Pharmacol Physiol. 2012;39(8):674–679. doi:10.1111/j.1440-1681.2011.05599.x
11. Hara MR, Agrawal N, Kim SF, et al. S-nitrosylated GAPDH initiates apoptotic cell death by nuclear translocation following Siah1 binding. Nat Cell Biol. 2005;7(7):665–674. doi:10.1038/ncb1268
12. Hara MR, Thomas B, Cascio MB, et al. Neuroprotection by pharmacologic blockade of the GAPDH death cascade. Proc Natl Acad Sci U S A. 2006;103(10):3887–3889. doi:10.1073/pnas.0511321103
13. Oakes D. Antiparkinson efficacy of deprenyl. DATATOP steering committee of Parkinson Study Group. Ann Neurol. 1993;34(4):634. doi:10.1002/(ISSN)1531-8249
14. Zou B, Liu X, Zhang B, et al. The expression of FAP in hepatocellular carcinoma cells is induced by hypoxia and correlates with poor clinical outcomes. J Cancer. 2018;9(18):3278–3286. doi:10.7150/jca.25775
15. Rotterud R, Fossa SD, Nesland JM. Protein networking in bladder cancer: immunoreactivity for FGFR3, EGFR, ERBB2, KAI1, PTEN, and RAS in normal and malignant urothelium. Histol Histopathol. 2007;22(4):349–363. doi:10.14670/HH-22.349
16. Zhou L, Yao LT, Liang ZY, et al. Nuclear translocation of fibroblast growth factor receptor 3 and its significance in pancreatic cancer. Int J Clin Exp Pathol. 2015;8(11):14640–14648.
17. Luo D, Wang Z, Wu J, Jiang C, Wu J. The role of hypoxia inducible factor-1 in hepatocellular carcinoma. Biomed Res Int. 2014;2014:409272. doi:10.1155/2014/409272
18. Tsuchida T, Friedman SL. Mechanisms of hepatic stellate cell activation. Nat Rev Gastroenterol Hepatol. 2017;14(7):397–411. doi:10.1038/nrgastro.2017.38
19. Wanka C, Brucker DP, Bahr O, et al. Synthesis of cytochrome C oxidase 2: a p53-dependent metabolic regulator that promotes respiratory function and protects glioma and colon cancer cells from hypoxia-induced cell death. Oncogene. 2012;31(33):3764–3776.
20. Barbini L, Rodriguez J, Dominguez F, Vega F. Glyceraldehyde-3-phosphate dehydrogenase exerts different biologic activities in apoptotic and proliferating hepatocytes according to its subcellular localization. Mol Cell Biochem. 2007;300(1–2):19–28. doi:10.1007/s11010-006-9341-1
21. Butera G, Pacchiana R, Mullappilly N, et al. Mutant p53 prevents GAPDH nuclear translocation in pancreatic cancer cells favoring glycolysis and 2-deoxyglucose sensitivity. Biochim Et Biophys Acta Mol Cell Res. 2018;1865(12):1914–1923. doi:10.1016/j.bbamcr.2018.10.005
22. Dando I, Pacchiana R, Pozza ED, et al. UCP2 inhibition induces ROS/Akt/mTOR axis: role of GAPDH nuclear translocation in genipin/everolimus anticancer synergism. Free Radic Biol Med. 2017;113:176–189. doi:10.1016/j.freeradbiomed.2017.09.022
23. Leisner TM, Moran C, Holly SP, Parise LV. CIB1 prevents nuclear GAPDH accumulation and non-apoptotic tumor cell death via AKT and ERK signaling. Oncogene. 2013;32(34):4017–4027. doi:10.1038/onc.2012.408
24. Xia YH, Wang ZM, Chen RX, et al. T-cell apoptosis induced by intratumoral activated hepatic stellate cells is associated with lung metastasis in hepatocellular carcinoma. Oncol Rep. 2013;30(3):1175–1184. doi:10.3892/or.2013.2571
25. Zhang L, Li Y, Qiao L, Zhao Y, Wei Y, Li Y. Protective effects of hepatic stellate cells against cisplatin-induced apoptosis in human hepatoma G2 cells. Int J Oncol. 2015;47(2):632–640. doi:10.3892/ijo.2015.3024
26. Fingas CD, Bronk SF, Werneburg NW, et al. Myofibroblast-derived PDGF-BB promotes Hedgehog survival signaling in cholangiocarcinoma cells. Hepatology. 2011;54(6):2076–2088. doi:10.1002/hep.24588
27. Huang J, Qiu M, Wan L, et al. TGF-beta1 promotes hepatocellular carcinoma invasion and metastasis via ERK pathway-mediated FGFR4 expression. Cellular Physiol Biochem. 2018;45(4):1690–1699. doi:10.1159/000487737
28. Zhang JY, Zhang F, Hong CQ, et al. Critical protein GAPDH and its regulatory mechanisms in cancer cells. Cancer Biol Med. 2015;12(1):10–22.
29. Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell. 2007;129(7):1261–1274. doi:10.1016/j.cell.2007.06.009
30. Chiche J, Pommier S, Beneteau M, et al. GAPDH enhances the aggressiveness and the vascularization of non-Hodgkin’s B lymphomas via NF-kappaB-dependent induction of HIF-1alpha. Leukemia. 2015;29(5):1163–1176. doi:10.1038/leu.2014.324
31. Gao X, Wang X, Pham TH, et al. NleB, a bacterial effector with glycosyltransferase activity, targets GAPDH function to inhibit NF-kappaB activation. Cell Host Microbe. 2013;13(1):87–99. doi:10.1016/j.chom.2012.11.010
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.