Back to Journals » Journal of Inflammation Research » Volume 19
NRG1 Suppresses NLRP3 Inflammasome Activation and Endothelial-Mesenchymal Transition in Cerebral Ischemia-Reperfusion Injury: Association with the AKT/NF-κB Pathway
Authors Cai Y, Wang Y ![]() , Wang D
, Wang D ![]() , Chen H
, Chen H ![]() , Zhu K
, Zhu K ![]() , Cai X
, Cai X ![]() , Sun J
, Sun J ![]()
Received 4 December 2025
Accepted for publication 24 February 2026
Published 28 February 2026 Volume 2026:19 584926
DOI https://doi.org/10.2147/JIR.S584926
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Ning Quan
Yaozhuo Cai,1,2,* Yuzhen Wang,1,2,* Dandan Wang,1,2 Hao Chen,1,2 Kaiqi Zhu,1,2 Xueli Cai,1,2 Jingping Sun1,2
1Department of Neurology, the Municipal Central Hospital of Lishui, the Fifth Affiliated Hospital of Wenzhou Medical University, Lishui, Zhejiang, People’s Republic of China; 2Lishui Clinical Research Center for Neurological Diseases, Lishui, Zhejiang, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Jingping Sun, Department of Neurology, the Municipal Central Hospital of Lishui, the Fifth Affiliated Hospital of Wenzhou Medical University, No. 289, Kuocang Street, Lishui, Zhejiang, People’s Republic of China, 323000, Email [email protected] Xueli Cai, Department of Neurology, the Municipal Central Hospital of Lishui, the Fifth Affiliated Hospital of Wenzhou Medical University, No. 289, Kuocang Street, Lishui, Zhejiang, People’s Republic of China, 323000, Email [email protected]
Background: Endothelial damage and NLRP3 inflammasome activation are key mechanisms underlying cerebral ischemia-reperfusion (I/R) injury. Neuregulin-1 (NRG1) alleviates endothelial damage, blood-brain barrier (BBB) disruption, and neurological deficits following acute ischemic stroke (AIS). This study investigates whether NRG1 attenuates endothelial-mesenchymal transition (EndMT) during endothelial injury by suppressing NLRP3 inflammasome activation, potentially through the AKT/NF-κB pathway, in cerebral I/R injury.
Methods: Middle cerebral artery occlusion/reperfusion (MCAO/R) mouse models and oxygen-glucose deprivation/reoxygenation (OGD/R)-treated immortalized human cerebral microvascular endothelial cells (hCMEC/D3) were established and treated with NRG1. Neurological deficits were assessed using the modified Neurological Severity Score (mNSS), while cerebral infarct volume and caspase-1 activity were quantified. Cell viability and death were evaluated by the Cell Counting Kit-8 (CCK-8), lactate dehydrogenase (LDH) assays, and TUNEL staining. Protein expression was analyzed using Western blotting and immunofluorescence staining.
Results: NRG1 intervention significantly improved mNSS, reduced cerebral infarct volume, and decreased caspase-1 activity in MCAO/R mice. In OGD/R-treated hCMEC/D3, NRG1 enhanced cell viability while reducing cell death, as indicated by decreased LDH release and TUNEL-positive cells. Western blotting and immunofluorescence staining revealed that NRG1 enhanced AKT phosphorylation, inhibited NF-κB p65 phosphorylation, and downregulated NLRP3 and IL-1β expression. These effects were associated with the amelioration of occludin reduction and suppression of α-smooth muscle actin (α-SMA) increase during EndMT progression.
Conclusion: The results demonstrate that NRG1 reduces cerebral infarct volume, alleviates neurological deficits, and suppresses NLRP3 inflammasome activation in association with modulation of the AKT/NF-κB pathway, thereby attenuating EndMT-associated proteins following cerebral I/R injury.
Keywords: NRG1, ischemia/reperfusion injury, NLRP3 inflammasome, endothelial-mesenchymal transition
Introduction
Acute ischemic stroke (AIS) is the most common stroke subtype, which requires early restoration of blood flow to ischemic regions through recanalization therapies.1 However, the acute reperfusion of the ischemic brain areas may further induce cerebral tissue damage, termed ischemia-reperfusion (I/R) injury. Furthermore, the damage caused by I/R injury is not confined to the specific ischemic tissue but can also exert systemic effects that impact remote organs.2 The underlying pathophysiology is multifaceted, encompassing critical events such as oxidative stress, inflammatory cascades, diverse modes of cell death, and mitochondrial impairment.3
As a cytosolic multiprotein complex central to innate immunity, the NLRP3 inflammasome orchestrates the activation of inflammatory caspases, leading to pyroptosis and the maturation of pro-inflammatory cytokines.4 It has been established that NLRP3 is activated in the brain following I/R and that its inhibition can mitigate endothelial damage, BBB disruption, and cellular pyroptosis.5–7 Endothelial cells, central to BBB integrity, can undergo a pathological phenotypic shift known as endothelial-mesenchymal transition (EndMT) following cerebral I/R injury, characterized by loss of tight junctions and acquisition of mesenchymal features.8,9 While inflammatory signaling is a known trigger for EndMT in other contexts, the NLRP3 inflammasome, which is expressed in endothelial cells, and its specific role in this process after cerebral I/R remains to be elucidated.10
Neuregulin 1 (NRG1), an epidermal growth factor (EGF) family member, is a signaling protein mediating intercellular interactions. It is predominantly expressed in the brain, heart, mammary glands, and other tissues, and regulates tissue development and maturation.11 Current studies suggest therapeutic applications of NRG1 in diabetic gastroparesis, post-myocardial infarction inflammation/fibrosis reduction, heart failure suppression, type 2 diabetes management, and tumor-targeted therapy.12–16 Notably, NRG1 exerts cardioprotective effects against myocardial I/R injury, with studies suggesting the involvement of the PI3K/AKT pathway, which can suppress NF-κB-mediated pro-inflammatory responses.17,18 Previous studies have demonstrated that NRG1 significantly attenuates endothelial damage, BBB disruption, cerebral edema, and neurological deficits following reperfusion.19,20 These effects are initiated by NRG1 binding to ErbB receptors, triggering their phosphorylation and the subsequent activation of downstream signaling cascades, such as MAPK and PI3K/AKT.21 However, whether NRG1’s endothelial protection in cerebral I/R involves the modulation of the NLRP3 inflammasome and, consequently, the inhibition of EndMT, is unknown. Furthermore, the potential involvement of the AKT/NF-κB axis in this putative mechanism has not been explored.
Therefore, this study aims to elucidate the protective role of NRG1 and its underlying mechanisms in cerebral I/R injury. We hypothesize that NRG1 attenuates EndMT by suppressing NLRP3 inflammasome activation, potentially through the modulation of the AKT/NF-κB pathway in endothelial cells.
Materials and Methods
Animals and Treatment
The specific pathogen-free (SPF) male C57BL/6J mice (9–10 weeks old, 25–30 g; n = 45) were purchased from PIZHOU Oriental & Xiaohe Technology (Production License No. SCXK (su) 2017–0003). The animals were group-housed and acclimated for one week under standardized conditions (25 ± 1°C, 65 ± 5% humidity, 12/12 h light/dark cycle) with ad libitum access to food and water. The mice were randomly divided into three groups (n = 11 per group) using a random number table: (1) sham; (2) middle cerebral artery occlusion/reperfusion (MCAO/R) + vehicle treatment (vehicle); (3) MCAO/R + NRG1 (NRG1). During surgery, 8 mice died, and 4 were excluded due to unsuccessful MCAO/R modeling. NRG-1β (100 μg/kg; EGF-like domain, R&D Systems, Minneapolis, MN) or vehicle (0.1% BSA/PBS) was administered via tail vein at 1 hour post-ischemia and 24 hours post-reperfusion (Figure 1A). The dose of NRG1 was selected based on previous studies demonstrating its efficacy in neuroprotection in stroke models.22 All experimental protocols complied with the ethical guidelines approved by the Institutional Animal Care and Use Committee (IACUC) of Lishui University (Ethical Approval No. 2024YD0159).
 |
Figure 1 Effects of NRG1 on neurological deficits and cerebral infarct volume in MCAO/R mice. (A) In vivo experimental protocol. (B) Statistical analysis of neurological scores (n = 11). (C) TTC staining and statistical analysis of cerebral infarct volume (n = 4). Data are expressed as mean ± SD, with individual data points overlaid on the bar graphs. ## p < 0.01 vs sham; * p < 0.05, ** p < 0.01 vs vehicle. |
MCAO/R Model
The MCAO/R model was employed to induce focal cerebral ischemia. Mice were anesthetized via intraperitoneal injection of a 1.25% (w/v) solution of avertin (30 μL/g; BR4108423, Bioleaper, China). The left common carotid artery (CCA) was exposed, and a poly-L-lysine-coated nylon monofilament (Cinontech, China) with a rounded tip was inserted from the CCA into the left middle cerebral artery (MCA). The filament was left in place for 1 hour to induce ischemia, removed for reperfusion, and mice were euthanized after 48 hours of reperfusion. Sham-operated mice underwent vascular isolation without filament insertion. Throughout the procedure, body temperature was maintained at 36.5 ± 0.5°C using a thermostatic heating pad.
Infarct Volume Measurement and Neurological Evaluation
The modified Neurological Severity Score (mNSS) was assessed at 24 and 48 hours post-ischemia by an experimenter blinded to the group allocation to evaluate sensorimotor function, reflexes, balance, and abnormal movements. Each failed task or absent response scored 1 point, with total scores ranging from 0 for normal to 18 for severe impairment. Mice with mNSS scores below 5 at 24 hours post-ischemia were considered to have an unsuccessful MCAO/R model. At 48 hours post-surgery, mice were euthanized by cervical dislocation. Brains were collected, frozen at −80°C, and sectioned into 5–7 consecutive coronal slices. Sections were incubated in 2% 2,3,5-triphenyltetrazolium chloride (TTC; Sigma-Aldrich, USA) at room temperature for 15 minutes in the dark, followed by fixation in 4% paraformaldehyde (4% PFA). Infarct volume percentage was quantified by a blinded investigator using ImageJ (version 1.61, NIH, Bethesda, USA), where the infarct area was delineated manually after adjusting image threshold to consistently distinguish stained from unstained tissue, and the percentage was calculated with the formula: (infarct volume / hemispheric volume) × 100%.
Caspase-1 Activity Assay
Caspase-1 activity in brain tissue homogenates was measured using a Caspase-1 Activity Assay Kit (Beyotime, China). Caspase-1 catalyzes the substrate Ac-YVAD-pNA to generate p-nitroaniline (pNA), which exhibits an absorption peak at 405 nm. Activity was quantified by comparing absorbance values to a standard curve.
Cell Culture
The immortalized human cerebral microvascular endothelial cell line (hCMEC/D3) was provided by Henan Engineering Research Center of Industrial Microbiology. hCMEC/D3 cells were cultured in Endothelial Cell Medium (ECM, ScienCell, USA) supplemented with 5% fetal bovine serum (FBS), 1% Endothelial Cell Growth Supplement (ECGS), and 1% penicillin/streptomycin solution (P/S). Cells were maintained at 37°C in a humidified incubator with 95% air and 5% CO2. hCMEC/D3 cells were seeded in 25 cm2 cell culture flasks and subcultured at 80% confluence. The cells within 15 passages were selected for experiments.
Oxygen-Glucose Deprivation/Reoxygenation (OGD/R) Model and Intervention
In vitro, ischemia was simulated using the OGD/R model. MCC950 (S7809; Selleck, USA), a selective NLRP3 inflammasome inhibitor, and Nigericin sodium salt (Nigericin) (S6653; Selleck), an NLRP3 inflammasome activator, were employed. Cells were divided into six groups: (1) control; (2) OGD/R - 0h (OGD); (3) OGD/R - 4h (vehicle); (4) OGD/R + NRG1 (NRG1); (5) OGD/R + NRG1 + MCC950 (NRG1 + MCC950); (6) OGD/R + NRG1 + Nigericin (NRG1 + Nig). The cells were first exposed to glucose-free DMEM (Gibco, USA) in an anaerobic environment with 94% N2, 5% CO2, and 1% O2 for 4 hours. Subsequently, cells were returned to normoxic conditions for 4 hours of reoxygenation. At the onset of reoxygenation, NRG1 (100 ng/mL), MCC950 (10 μmol/L), and Nigericin (100 ng/mL) were administered, with all concentrations selected based on concentrations established in previous studies.20,23,24 Throughout the process, the control group was cultured in ECM under normoxic conditions for the same duration, while the OGD group underwent OGD without subsequent reoxygenation.
Cell Viability and Death Measurement
Following OGD/R and drug administration, cell viability was evaluated using the Cell Counting Kit-8 (CCK-8) cytotoxicity assay (Beyotime, China). At 2 hours post-reoxygenation, 10 μL of CCK-8 reagent was added to each well, followed by a 2-hour incubation. The optical density (OD) value was measured at 450 nm to calculate cell viability as follows: Cell viability (%) = [(OD of treated group – OD of blank control) / (OD of control group – OD of blank control)] × 100. Additionally, cell death was estimated by measuring the absorbance at 490 nm using the lactate dehydrogenase (LDH) cytotoxicity assay kit (Beyotime, China). Cell death rate was calculated as: Cell death (%) = [(OD of treated group – OD of control group) / (OD of maximum enzyme activity – OD of control group)] × 100.
Terminal Deoxynucleotidyl Transferase-Mediated dUTP Nick End Labeling (TUNEL) Staining
At 48 hours after MCAO/R, saline and 4% PFA were sequentially perfused through the heart. Brain tissues were rapidly removed, fixed in 4% PFA, then paraffin-embedded for sectioning. In vitro, after OGD/R treatment, hCMEC/D3 cells were fixed with 4% PFA for 30 minutes. The brain sections or cells were then processed using the One Step TUNEL Apoptosis Assay Kit (Beyotime, China). Subsequently, the samples were stained with 4′,6-diamidino-2-phenylindole (DAPI; Thermo Fisher Scientific, USA) for 5 minutes. Images were captured using an inverted fluorescence microscope (DMi8, Leica, Germany). For quantification, TUNEL-positive and DAPI-positive cells were counted in three non-overlapping random fields per sample by a blinded observer. The percentage of TUNEL-positive cells was calculated as: TUNEL positive nuclei (%) = (TUNEL+ cells / total DAPI+ cells) × 100%.
Immunofluorescence Staining
As described previously, brain sections or cells were fixed with 4% PFA. The samples were permeabilized with Enhanced Immunostaining Permeabilization Buffer (Beyotime, China) for 10 minutes and blocked at room temperature for 1 hour. Subsequently, sections or cells were incubated overnight at 4°C with the following primary antibodies: anti-occludin (1:500; ab216327, Abcam, UK) and anti-α-smooth muscle actin (α-SMA; 1:1000; ab7817, Abcam). After washing with PBS, samples were incubated in the dark for 1 hour at room temperature with AlexaFluor 647 goat anti-mouse IgG (1:1000 for α-SMA; A21235, Thermo Fisher Scientific) and AlexaFluor 488 goat anti-rabbit IgG (1:500 for occludin; A11008, Thermo Fisher Scientific). Then, nuclei were counterstained with DAPI. Finally, images were acquired using a laser scanning confocal microscope (STELLARIS 5, Leica). Quantitative analysis of mean fluorescence intensity for in vitro samples was performed using ImageJ after applying a consistent threshold and subtracting background signal; all images for comparison were acquired and processed under identical settings. Images of in vivo brain sections are presented as representative micrographs to illustrate protein localization.
Western Blotting Analysis
Protein concentrations in extracts from brain tissues and hCMEC/D3 cells were measured by the Enhanced BCA Protein Assay Kit (Beyotime, China) following the manufacturer’s protocol. Protein samples were separated by electrophoresis and transferred onto polyvinylidene fluoride (PVDF) membranes (Millipore, USA). After blocking for 1.5 hours, membranes were incubated overnight at 4°C with the following primary antibodies: anti-occludin (1:1000; ab216327, Abcam), anti-α-SMA (1:1000; ab7817, Abcam), anti-NLRP3 (1:1000; ab263899, Abcam), anti-NRG1 (1:5000; ab53104, Abcam), anti-IL-1β (1:1000; ab254360, Abcam), anti-phospho-AKT (p-AKT; 1:5000; 66,444-1-Ig, Proteintech, China), anti-AKT (1:5000; 60,203-2-Ig, Proteintech), anti-phospho-NF-κB p65 (p-NF-κB p65; 1:1000; 3033S, CST, USA), anti-NF-κB p65 (1:1000; 8242S, CST), anti-GAPDH (1:1000; 5174S, CST), anti-β-Tubulin (1:1000; 2128S, CST). Membranes were then incubated with either Anti-Mouse-HRP secondary antibody (1:10,000; 7076S, CST) or Anti-Rabbit-HRP secondary antibody (1:10,000; 7074S, CST) at room temperature for 1 hour. Protein bands were visualized using the Invitrogen iBright FL1500 Imaging System (Thermo Fisher Scientific) with consistent acquisition settings across compared lanes. Band intensities from independent biological replicates were measured after background subtraction and normalized to the corresponding loading control.
Statistical Analysis
The data were expressed as mean ± standard deviation (SD). The sample size (n) represents the number of independent biological replicates. Image analysis and cell counting were performed using Adobe Photoshop 2023 and ImageJ. Statistical analysis was conducted with GraphPad Prism software (version 8.0.2; GraphPad Software, USA). Comparisons between two groups and among multiple groups were analyzed using unpaired t-test and one-way analysis of variance (ANOVA), respectively. Statistical significance was defined as p < 0.05. All data presented herein are original and have not been published previously.
Results
NRG1 Reduces Cerebral Infarct Volume and Neurological Deficits in MCAO/R Mice
The results demonstrated that compared to the vehicle group (n = 10), NRG1 group (n = 10) exhibited significantly lower mNSS at both 24 and 48 hours (Figure 1B). TTC-stained coronal sections revealed a marked increase in infarct area in the vehicle group (44.98 ± 4.46% of hemispheric area, p < 0.01 vs sham), confirming successful model establishment. The NRG1 group significantly reduced infarct size (29.90 ± 6.58%, p < 0.05 vs vehicle) (Figure 1C).
NRG1 Attenuates NLRP3 Inflammasome Activation in MCAO/R Mice
Western Blotting analysis confirmed upregulated protein levels of NLRP3 (1.51 ± 0.13, p < 0.01 vs sham) and IL-1β (1.45 ± 0.03, p < 0.01 vs sham) in the vehicle group (Figure 2A). TUNEL staining revealed a significant increase in the percentage of TUNEL-positive cells in the vehicle group (61.08 ± 3.42%, p<0.01 vs sham) (Figure 2B). Furthermore, caspase-1 activity was markedly elevated in the vehicle group (157.82 ± 10.60, p < 0.01 vs sham) (Figure 2C). The NRG1 treatment attenuated NLRP3 inflammasome activation during MCAO/R, as evidenced by reduced caspase-1 activity (130.38 ± 6.14, p < 0.01 vs vehicle), decreased NLRP3 (1.18 ± 0.04, p < 0.01) and IL-1β (1.24 ± 0.04, p < 0.01) protein levels, and lower proportions of TUNEL-positive cells in the NRG1 group (25.35 ± 1.96%, p<0.01 vs vehicle) (Figure 2A–C). Notably, NRG1 protein expression was elevated in the vehicle group (1.46 ± 0.13, p < 0.01 vs sham) (Figure 2A).
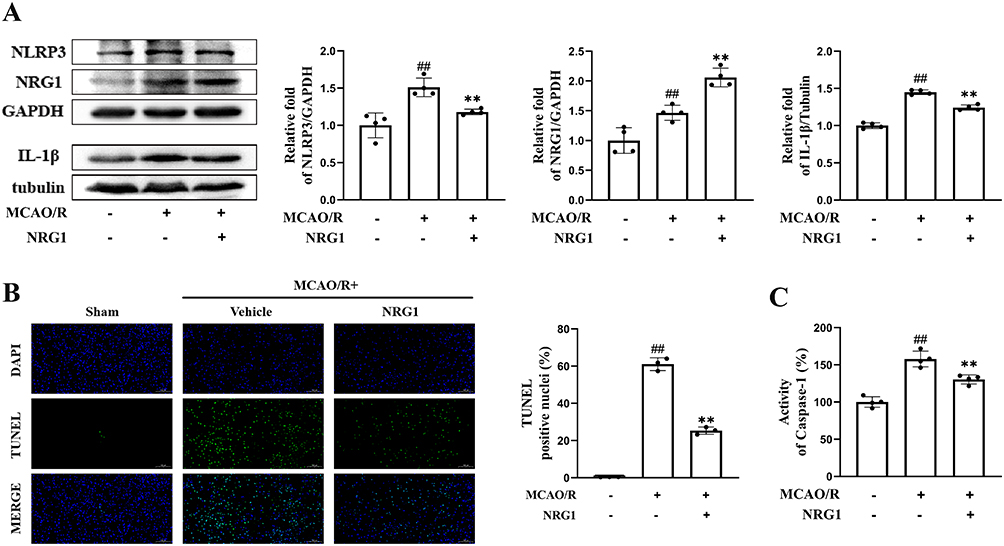 |
Figure 2 Effects of NRG1 on pyroptosis-related components in MCAO/R mice. (A) Western blotting analysis of NRG1, NLRP3, and IL-1β expression (n = 4). (B) Percentage of TUNEL-positive cells in ipsilateral brain tissue post-MCAO/R (scale bar = 100 μm) (n = 3). (C) Caspase-1 activity in brain tissue homogenates (n = 4). Data are expressed as mean ± SD, with individual data points overlaid on the bar graphs. ## p < 0.01 vs sham; ** p < 0.01 vs vehicle. |
NRG1 Attenuated OGD/R-Induced Injury in hCMEC/D3
To investigate the protective effects of NRG1 on hCMEC/D3 cells following OGD/R, cell viability, LDH release, and TUNEL staining were assessed at 4 hours post‑reoxygenation (Figure 3A). Compared to the control group, the vehicle group significantly reduced cell viability (73.27 ± 1.04%, p < 0.01 vs control). This effect was attenuated by NRG1 administration during reoxygenation (78.69 ± 1.12%, p < 0.01 vs vehicle) and further ameliorated by NRG1 combined with the MCC950 (86.74 ± 1.69%, p < 0.01 vs vehicle) (Figure 3B). Additionally, LDH release was markedly reduced in the NRG1 group (13.54 ± 1.26%, p < 0.01 vs vehicle) and the NRG1 + MCC950 group (9.81 ± 1.68%, p < 0.01 vs vehicle), whereas the NRG1 + Nig. group showed the attenuation of this protective effect (16.67 ± 1.35%, p < 0.05) (Figure 3C). TUNEL staining analysis showed a reduction in TUNEL-positive cells across all intervention groups (p < 0.01 vs vehicle) (Figure 3D).
 |
Figure 3 Effects of NRG1 on OGD/R-induced injury in hCMEC/D3. (A) In vitro experimental protocol. (B) Cell viability assessed by the Cell Counting Kit-8 (CCK-8) assay. (C) LDH release in the culture medium to evaluate cell death. (D) Percentage of TUNEL-positive endothelial cells post-OGD/R (scale bar = 100 μm) (n = 3). Data are expressed as mean ± SD, with individual data points overlaid on the bar graphs. ## p < 0.01 vs control; ** p < 0.01 vs vehicle; $ p < 0.05, $$ p < 0.01 vs NRG1. |
NRG1 Inhibits NLRP3 Inflammasome Activation Through AKT/NF-κB Pathway in OGD/R-Injured hCMEC/D3
As previously observed in MCAO/R mice, OGD/R-injured hCMEC/D3 exhibited significantly elevated protein levels of NLRP3 (2.49 ± 0.15, p < 0.01 vs control) and IL-1β (2.06 ± 0.09, p < 0.01 vs control), whereas the OGD group showed no significant differences compared to controls (Figure 4A). However, NRG1 markedly reduced these levels (p < 0.01 vs vehicle). Subsequently, we investigated whether the AKT/NF-κB pathway plays a critical role in NRG1-mediated NLRP3 inflammasome suppression. Western blotting analysis revealed that the vehicle group exhibited elevated levels of p-AKT (2.16 ± 0.06, p < 0.01 vs control) and p-NF-κB p65 (2.25 ± 0.08, p < 0.01 vs control), indicating pathway activation (Figure 4B and C). NRG1 treatment further enhancing p-AKT levels (2.40 ± 0.08, p < 0.05 vs vehicle) while significantly downregulated p-NF-κB p65 expression (1.92 ± 0.21, p < 0.05 vs vehicle).
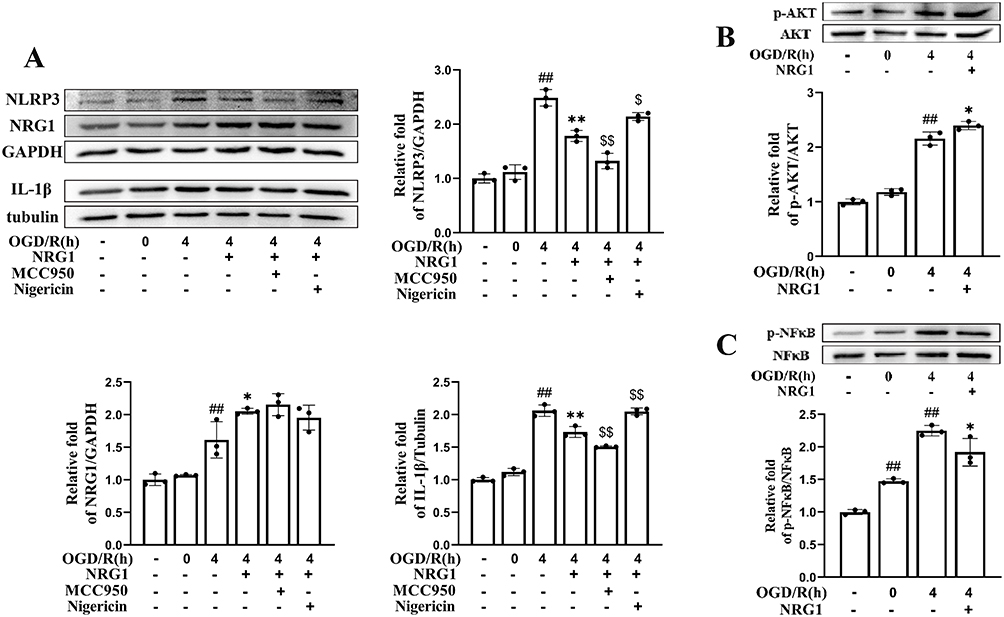 |
Figure 4 NRG1 inhibits NLRP3 inflammasome activation, which is associated with modulation of the AKT/NF-κB Pathway in OGD/R-injured hCMEC/D3. (A) Western blotting analysis of NRG1, NLRP3, and IL-1β expression (n = 3). (B and C) NRG1 increased phosphorylated AKT levels and decreased phosphorylated NF-κB p65 levels (n = 3). Data are expressed as mean ± SD, with individual data points overlaid on the bar graphs. ## p < 0.01 vs control; * p < 0.05, ** p < 0.01 vs vehicle; $ p < 0.05, $$ p < 0.01 vs NRG1. |
NRG1 Inhibits EndMT Progression in Endothelial Cells Both in vivo and in vitro
Representative immunofluorescence images demonstrated that MCAO/R significantly increased the signal for α-SMA and reduced that for occludin in brain sections, changes that were reversed by NRG1 treatment (Figure 5A). Consistently, quantitative Western blotting analysis revealed decreased occludin expression (0.81 ± 0.02, p < 0.01 vs sham) and elevated α-SMA levels (1.95 ± 0.12, p < 0.01 vs sham) in the vehicle group, with NRG1 attenuating these changes (p < 0.05 vs vehicle) (Figure 5B). Similar trends were observed in vitro (Figure 6A and B). Notably, MCC950 enhanced NRG1’s protective effect on OGD/R-induced endothelial barrier dysfunction in hCMEC/D3, whereas Nigericin suppressed this effect, as validated by immunofluorescence and Western blotting (Figure 6B).
 |
Figure 5 Effects of NRG1 on EndMT in the infarct area of the MCAO/R mice. (A) Immunofluorescence staining of occludin and α-SMA in the ipsilateral ischemic cerebral hemisphere (scale bar = 50 μm). (B) Western blot analysis of occludin and α-SMA expression (n = 4). Data are expressed as mean ± SD, with individual data points overlaid on the bar graphs. ## p < 0.01 vs sham; * p < 0.05, ** p < 0.01 vs vehicle. |
 |
Figure 6 Effects of NRG1 on EndMT in hCMEC/D3 cells following OGD/R injury. (A) Western blotting analysis of occludin and α-SMA expression (n = 3). (B) Immunofluorescence staining of occludin and α-SMA in hCMEC/D3 post-OGD/R (scale bar = 10 μm) and quantitative analysis of the mean fluorescence intensity (n = 3). Data are expressed as mean ± SD, with individual data points overlaid on the bar graphs. ## p < 0.01 vs control; * p < 0.05, ** p < 0.01 vs vehicle; $ p < 0.05, $$ p < 0.01 vs NRG1. |
Discussion
The primary therapeutic strategy for AIS involves achieving early reperfusion within the therapeutic time window through intravenous thrombolysis and intra-arterial endovascular therapy.1 However, cerebrovascular recanalization may paradoxically induce I/R injury, adversely affecting patient prognosis. Recent studies have demonstrated that NLRP3-mediated pyroptosis plays a pivotal role in cerebral I/R injury, highlighting the therapeutic potential of targeting the NLRP3 inflammasome or its regulatory pathways.6,25 This study investigated the protective effects of NRG1 against I/R injury with a specific focus on cerebrovascular endothelial cells. We demonstrated that NRG1 attenuates I/R injury by suppressing NLRP3 inflammasome activation, an effect associated with modulation of the AKT/NF-κB pathway. Furthermore, we provide the first evidence linking NLRP3 inflammasome activation to EndMT in this context, showing that NRG1 mitigates both processes.
As a pleiotropic signaling molecule in the EGF family, NRG1 is critical for nervous system development and homeostasis, with its signaling output tightly regulated by specific isoforms, cell-specific receptors, and the local microenvironment.21,26 Studies have demonstrated that NRG1 exerts neuroprotective and reparative effects in diverse models of neurological disorders, including schizophrenia, multiple sclerosis, and peripheral nerve injury, supporting its recognition as a biologically rational target for cerebral I/R injury.26,27 To support this hypothesis, NRG1 has been demonstrated to exert neuroprotective and anti-inflammatory effects in neuroinflammatory and ischemic stroke models. In this study, NRG1 treatment significantly reduced cerebral infarct volume and alleviated neurological deficits compared to the vehicle group, confirming its neuroprotective effects against I/R injury.
The NLRP3 inflammasome serves as a pivotal cytosolic platform for caspase-1 activation.28 I/R-induced pathological stimuli, including oxidative stress, lactic acidosis, and ion imbalance, lead to NLRP3 inflammasome assembly and the consequent caspase-1-dependent cleavage of pro-IL-1β and pro-IL-18.29 The mature cytokines are then released through GSDMD-formed membrane pores, inducing pyroptosis.29 In this study, NRG1 treatment in the MCAO/R mice significantly reduced NLRP3 and IL-1β protein levels, suppressed caspase-1 activity, and decreased TUNEL-positive cell counts. These findings demonstrate that NRG1 exerts neuroprotective effects against cerebral I/R injury by inhibiting NLRP3 inflammasome-mediated inflammatory responses. Consistent with the observed suppression of NLRP3 inflammasome activation, the concomitant reduction in TUNEL-positive cells suggests a decrease in inflammasome-associated cell death.
In vitro studies on macrophages and microglia have demonstrated that NRG1 binding to the ErbB4 receptor activates AKT phosphorylation, which in turn suppresses the canonical NF-κB pathway by inhibiting NF-κB p65 phosphorylation and nuclear translocation, thereby inhibiting the expression of NLRP3 and pro-inflammatory cytokines.20,30 While NRG1 also demonstrates endothelial protective and BBB-stabilizing effects in experimental injury and cerebral malaria models, its role in modulating the endothelial NLRP3 inflammasome during cerebral I/R was unclear.31,32 Building on the detection of NLRP3 in stroke-affected endothelium, we found that NRG1 enhanced endothelial cell viability, reduced LDH release, and decreased TUNEL-positive cell ratios after OGD/R.6 Furthermore, NRG1 promoted AKT phosphorylation, suppressed p-NF-κB p65 expression, and significantly reduced NLRP3 and IL-1β protein levels compared to the OGD/R group. These findings further demonstrate that NRG1 reduces endothelial cell death by inhibiting the NLRP3 inflammasome activation, an effect concurrent with modulation of the AKT/NF-κB pathway.
EndMT is a process where endothelial cells transform into mesenchymal-like cells under various stimuli.9 Studies have shown that cerebrovascular endothelial cells in mouse stroke models partially lose their tight junctions and adopt a mesenchymal phenotype.8 Additionally, inflammatory cytokines, including TNF-α, IL-1β, and IL-6, are known to promote EndMT progression.33,34 In this study, compared to the vehicle group, the NRG1 group exhibited reduced NLRP3 and IL-1β protein expression, increased the tight junction protein occludin levels, and suppressed the mesenchymal marker α-SMA expression. Furthermore, the NLRP3 inhibitor MCC950 enhanced NRG1-mediated inhibition of OGD/R-induced EndMT, whereas the NLRP3 activator Nigericin attenuated this protection. These results indicate that NRG1 mitigates EndMT progression by suppressing NLRP3 inflammasome activation and inflammatory cytokine expression.
Translating these preclinical findings into clinical benefit requires addressing several key challenges. Although clinical studies in heart failure have established preliminary safety for recombinant human NRG1, its application in stroke faces specific hurdles. First, its short plasma half-life highlights the need for brain-targeted delivery strategies, such as nanocarriers or intranasal delivery.21,35 Second, future efficacy studies should be conducted in clinically relevant models that incorporate aging and comorbidities.36 Finally, defining the optimal therapeutic window and dosing regimen for NRG1, particularly when combined with recanalization therapies, will be a crucial focus for future research.
This study has limitations that should be considered. One key limitation is that the mechanistic link between NRG1 and NLRP3 suppression is primarily associative, based on correlative changes in protein phosphorylation and downstream markers. Future studies employing pathway-specific inhibitors or genetic manipulations are needed to establish causality within the AKT/NF-κB pathway. Additionally, our findings are derived from an acute observation window, underscoring the need for longer-term studies to assess chronic EndMT progression and the durability of NRG1’s effects. Finally, our characterization of EndMT, while focused on the established core indicators occludin and α-SMA, was constrained by practical limitations from encompassing a broader panel of markers, including VE-cadherin, vimentin, that would have offered further mechanistic insight.
In summary, our findings demonstrate that NRG1 reduces cerebral infarct volume, alleviates neurological deficits, and inhibits NLRP3 inflammasome activation in association with modulation of the AKT/NF-κB pathway, thereby decreasing inflammatory cytokine release and attenuating the acute-phase changes in EndMT-associated proteins following cerebral I/R injury (Figure 7). These results provide a theoretical foundation for developing NRG1 as a promising candidate for alleviating cerebral I/R injury after ischemic stroke.
 |
Figure 7 Proposed mechanism of NRG1-mediated neuroprotection and attenuation of EndMT-associated proteins expression. In this model, NRG1 is associated with increased AKT phosphorylation, which correlates with reduced NF-κB nuclear translocation and decreased expression of inflammasome-associated components, including NLRP3, IL-1β, and IL-18. These changes are concurrent with the suppression of EndMT-associated proteins expression following cerebral I/R injury. |
ARRIVE Guidelines Compliance Statement
The authors confirm that they have adhered to the ARRIVE (Animal Research: Reporting of In Vivo Experiments) guidelines 2.0 in the design, conduct, analysis, and reporting of this study. A completed ARRIVE checklist is available as Supplementary Material 1.
Data Sharing Statement
The original contributions presented in the study are included in the article/supplementary materials. Further inquiries can be directed to the corresponding author.
Author Contributions
Yaozhuo Cai: Writing – review & editing, Writing – original draft, Validation, Visualization, Methodology, Investigation, Formal analysis, Data curation, Conceptualization. Yuzhen Wang: Writing – review & editing, Writing – original draft, Validation, Resources, Methodology, Conceptualization. Dandan Wang: Writing – review & editing, Investigation. Hao Chen: Writing – review & editing, Visualization, Formal analysis. Kaiqi Zhu: Writing – review & editing, Formal analysis. Xueli Cai: Writing – review & editing, Writing – original draft, Supervision, Resources, Project administration, Funding acquisition, Conceptualization. Jingping Sun: Writing – review & editing, Writing – original draft, Validation, Supervision, Resources, Project administration, Methodology, Data curation, Conceptualization.
All authors gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This study was supported by the Basic Public Welfare Research Program of Zhejiang Province (Grant No. 2022SJGYZC02) and the Medical Science and Technology Project of Zhejiang Province (Grant No. 2022RC301).
Disclosure
The authors declare no conflict of interest.
References
1. Peng B, Wang Y. Chinese guidelines for diagnosis and treatment of acute ischemic stroke 2023. Chin J Neurol. 2024;57(6):523–13. doi:10.3760/cma.j.cn113694-20240410-00221
2. Kashef SM, Abo Elnasr SE. Effect of peripheral blood mononuclear cells on ischemia-reperfusion injury of sciatic nerve of adult male albino rat: histological, immunohistochemical, and ultrastructural study. Ultrastruct Pathol. 2024;48(3):172–191. doi:10.1080/01913123.2024.2321144
3. Zhang A, Wang S, Wang P, Wang Y. Progress in pathological mechanism of ischemic stroke and prevention and treatment of traditional chinese medicine. Chin J Exp Traditional Med Formulae. 2020;26(5):227–240. doi:10.13422/j.cnki.syfjx.20200538
4. Gao L, Dong Q, Song Z, Shen F, Shi J, Li Y. NLRP3 inflammasome: a promising target in ischemic stroke. Inflamm Res. 2017;66(1):17–24. doi:10.1007/s00011-016-0981-7
5. Xu Q, Zhao B, Ye Y, et al. Relevant mediators involved in and therapies targeting the inflammatory response induced by activation of the NLRP3 inflammasome in ischemic stroke. J Neuroinflammation. 2021;18(1):123–145. doi:10.1186/s12974-021-02137-8
6. Yang F, Wang Z, Wei X, et al. NLRP3 deficiency ameliorates neurovascular damage in experimental ischemic stroke. J Cereb Blood Flow Metab. 2014;34(4):660–667. doi:10.1038/jcbfm.2013.242
7. Ward R, Li W, Abdul Y, et al. NLRP3 inflammasome inhibition with MCC950 improves diabetes-mediated cognitive impairment and vasoneuronal remodeling after ischemia. Pharmacol Res. 2019;142:237–250. doi:10.1016/j.phrs.2019.01.035
8. Bai Y, Zhang Y, Han B, et al. Circular RNA DLGAP4 ameliorates ischemic stroke outcomes by targeting miR-143 to regulate endothelial-mesenchymal transition associated with blood–brain barrier integrity. J Neurosci. 2018;38(1):32–50. doi:10.1523/jneurosci.1348-17.2017
9. Jiang H, Zhou Y, Zhang W, et al. Molecular mechanisms of endothelial–mesenchymal transition and its pathophysiological feature in cerebrovascular disease. Cell Biosci. 2025;15(1). doi:10.1186/s13578-025-01393-y
10. Maleszewska M, Moonen J-RAJ, Huijkman N, van de Sluis B, Krenning G, Harmsen MC. IL-1β and TGFβ2 synergistically induce endothelial to mesenchymal transition in an NFκB-dependent manner. Immunobiology. 2013;218(4):443–454. doi:10.1016/j.imbio.2012.05.026
11. Rösler TW, Depboylu C, Arias-Carrión O, et al. Biodistribution and brain permeability of the extracellular domain of neuregulin-1-β1. Neuropharmacology. 2011;61(8):1413–1418. doi:10.1016/j.neuropharm.2011.08.033
12. Cui W, Ma Y, Zhang L, et al. Neuregulin 1 improved gastric motility and reduced gastric inflammation by activating the α7nAChR through the cholinergic anti-inflammatory pathway in diabetic rats. Toxicol Appl Pharmacol. 2025;495:117205. doi:10.1016/j.taap.2024.117205
13. Xiao J, Li B, Zheng Z, et al. Therapeutic effects of neuregulin-1 gene transduction in rats with myocardial infarction. Coron Artery Dis. 2012;23(7):460–468. doi:10.1097/MCA.0b013e32835877da
14. Jabbour A, Hayward CS, Keogh AM, et al. Parenteral administration of recombinant human neuregulin-1 to patients with stable chronic heart failure produces favourable acute and chronic haemodynamic responses. Eur J Heart Fail. 2011;13(1):83–92. doi:10.1093/eurjhf/hfq152
15. Arai T, Hayashi E, Maeda S, et al. Liver-derived neuregulin1α stimulates compensatory pancreatic β cell hyperplasia in insulin resistance. Nat Commun. 2025;16(1):1950–1961. doi:10.1038/s41467-025-57167-0
16. Jonna S, Feldman RA, Swensen J, et al. Detection of NRG1 gene fusions in solid tumors. Clin Cancer Res. 2019;25(16):4966–4972. doi:10.1158/1078-0432.Ccr-19-0160
17. Ebner B, Lange SA, Hollenbach D, et al. In situ postconditioning with neuregulin-1β is mediated by a PI3K/Akt-dependent pathway. Can J Cardiol. 2015;31(1):76–83. doi:10.1016/j.cjca.2014.10.035
18. Lin Y, Liu H, Wang X. Neuregulin‑1, a microvascular endothelial‑derived protein, protects against myocardial ischemia‑reperfusion injury. Int J Mol Med. 2020;46(3):925–935. doi:10.3892/ijmm.2020.4662
19. Wu L, Walas S, Leung W, et al. Neuregulin1-β decreases IL-1β-induced neutrophil adhesion to human brain microvascular endothelial cells. Transl Stroke Res. 2015;6(2):116–124. doi:10.1007/s12975-014-0347-9
20. Simmons LJ, Surles-Zeigler MC, Li Y, Ford GD, Newman GD, Ford BD. Regulation of inflammatory responses by neuregulin-1 in brain ischemia and microglial cells in vitro involves the NF-kappa B pathway. J Neuroinflammation. 2016;13(1):237–251. doi:10.1186/s12974-016-0703-7
21. Noll JM, Sherafat AA, Ford GD, Ford BD. The case for neuregulin-1 as a clinical treatment for stroke. Front Cell Neurosci. 2024;18:1325630. doi:10.3389/fncel.2024.1325630
22. Noll JM, Li Y, Distel TJ, Ford GD, Ford BD. Neuroprotection by exogenous and endogenous neuregulin-1 in mouse models of focal ischemic stroke. J Mol Neurosci. 2019;69(2):333–342. doi:10.1007/s12031-019-01362-4
23. Wu X, Wang B, Zhou Y, et al. NLRP3 inflammasome inhibitor MCC950 reduces cerebral ischemia/reperfusion induced neuronal ferroptosis. Neurosci Lett. 2023;795:137032. doi:10.1016/j.neulet.2022.137032
24. Xin Q, Xu F, Ma Z, Wu J. β-caryophyllene mitigates ischemic stroke-induced white matter lesions by inhibiting pyroptosis. Exp Cell Res. 2024;442(1):114214. doi:10.1016/j.yexcr.2024.114214
25. Franke M, Bieber M, Kraft P, Weber ANR, Stoll G, Schuhmann MK. The NLRP3 inflammasome drives inflammation in ischemia/reperfusion injury after transient middle cerebral artery occlusion in mice. Brain Behav Immun. 2021;92:221–231. doi:10.1016/j.bbi.2020.12.009
26. Kataria H, Alizadeh A, Karimi-Abdolrezaee S. Neuregulin-1/ErbB network: an emerging modulator of nervous system injury and repair. Prog Neurobiol. 2019;180:101643. doi:10.1016/j.pneurobio.2019.101643
27. Ding Y, Zhang Y, Zhang L. The role of the neuregulin 1-ErbB4 signaling pathway in neurological disorders. J Physiol Pharmacol. 2024;75(6):579–594. doi:10.26402/jpp.2024.6.01
28. She Y, Shao L, Zhang Y, et al. Neuroprotective effect of glycosides in Buyang Huanwu decoction on pyroptosis following cerebral ischemia-reperfusion injury in rats. J Ethnopharmacol. 2019;242:112051. doi:10.1016/j.jep.2019.112051
29. Panbhare K, Pandey R, Chauhan C, Sinha A, Shukla R, Kaundal RK. Role of NLRP3 inflammasome in stroke pathobiology: current therapeutic avenues and future perspective. ACS Chem Neurosci. 2024;15(1):31–55. doi:10.1021/acschemneuro.3c00536
30. Alizadeh A, Dyck SM, Kataria H, et al. Neuregulin-1 positively modulates glial response and improves neurological recovery following traumatic spinal cord injury. Glia. 2017;65(7):1152–1175. doi:10.1002/glia.23150
31. Lok J, Zhao S, Leung W, et al. Neuregulin-1 effects on endothelial and blood–brain barrier permeability after experimental injury. Transl Stroke Res. 2012;3(S1):119–124. doi:10.1007/s12975-012-0157-x
32. Liu M, Solomon W, Cespedes JC, Wilson NO, Ford B, Stiles JK. Neuregulin-1 attenuates experimental cerebral malaria (ECM) pathogenesis by regulating ErbB4/AKT/STAT3 signaling. J Neuroinflammation. 2018;15(1):104–118. doi:10.1186/s12974-018-1147-z
33. Mahler GJ, Farrar EJ, Butcher JT. Inflammatory cytokines promote mesenchymal transformation in embryonic and adult valve endothelial cells. Arterioscler Thromb Vasc Biol. 2013;33(1):121–130. doi:10.1161/atvbaha.112.300504
34. Lee JG, Kay EP. NF-κB is the transcription factor for FGF-2 that causes endothelial mesenchymal transformation in cornea. Invest Ophthalmol Vis Sci. 2012;53(3):1530–1538. doi:10.1167/iovs.11-9102
35. Gao R, Zhang J, Cheng L, et al. A Phase II, randomized, double-blind, multicenter, based on standard therapy, placebo-controlled study of the efficacy and safety of recombinant human neuregulin-1 in patients with chronic heart failure. J Am Coll Cardiol. 2010;55(18):1907–1914. doi:10.1016/j.jacc.2009.12.044
36. Ergul A, Hafez S, Fouda A, Fagan SC. Impact of comorbidities on acute injury and recovery in preclinical stroke research: focus on hypertension and diabetes. Transl Stroke Res. 2016;7(4):248–260. doi:10.1007/s12975-016-0464-8
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.