Back to Journals » OncoTargets and Therapy » Volume 10
NRAS-mutant melanoma: current challenges and future prospect
Authors Muñoz-Couselo E ![]() , Zamora Adelantado E, Ortiz C, Soberino García J, Perez-Garcia J
, Zamora Adelantado E, Ortiz C, Soberino García J, Perez-Garcia J
Received 28 March 2017
Accepted for publication 6 June 2017
Published 8 August 2017 Volume 2017:10 Pages 3941—3947
DOI https://doi.org/10.2147/OTT.S117121
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Ingrid Espinoza
Eva Muñoz-Couselo,1,2 Ester Zamora Adelantado,1,2 Carolina Ortiz,1,2 Jesús Soberino García,3 José Perez-Garcia3
1Medical Oncology Department, Vall d’Hebron Hospital, Barcelona, Spain; 2Vall d’Hebron Institute of Oncology (VHIO), Barcelona, Spain; 3Baselga Institute of Oncology, Hospital Quirón, Barcelona, Spain
Abstract: Melanoma is one of the most common cutaneous cancers worldwide. Activating mutations in RAS oncogenes are found in a third of all human cancers and NRAS mutations are found in 15%–20% of melanomas. The NRAS-mutant subset of melanoma is more aggressive and associated with poorer outcomes, compared to non-NRAS-mutant melanoma. Although immune checkpoint inhibitors and targeted therapies for BRAF-mutant melanoma are transforming the treatment of metastatic melanoma, the ideal treatment for NRAS-mutant melanoma remains unknown. Despite promising preclinical data, current therapies for NRAS-mutant melanoma remain limited, showing a modest increase in progression-free survival but without any benefit in overall survival. Combining MEK inhibitors with agents inhibiting cell cycling and the PI3K–AKT pathway appears to provide additional benefit; in particular, a strategy of MEK inhibition and CDK4/6 inhibition is likely to be a viable treatment option in the future. Patients whose tumors had NRAS mutations had better response to immunotherapy and better outcomes than patients whose tumors had other genetic subtypes, suggesting that immune therapies – especially immune checkpoint inhibitors – may be particularly effective as treatment options for NRAS-mutant melanoma. Improved understanding of NRAS-mutant melanoma will be essential to develop new treatment strategies for this subset of patients with melanoma.
Keywords: metastatic melanoma, NRAS mutation, MEK inhibitor, immunotherapy, trametinib, binimetinib
Introduction
Throughout the recent 5 years, there has been a significant shift in the therapeutic management of disseminated melanoma. The clinical success of BRAF-targeted therapy and immunotherapy with high responders and long survivors suggests that short- and long-term disease control can be a reality for the unclearly defined subgroups of patients with melanoma. BRAF and NRAS co-mutations are not mutually exclusive; however, the sole finding of double-mutated cells in a resistant tumor is insufficient to determine follow-up therapy, and combinational therapy targeting different pathways will be necessary.1–4
Some specific driver mutations, each with well-known unique clinical and genetic features, have been described; these driver mutations occur in multiple oncogenes including BRAF, NRAS, and CKIT, which are the most commonly described, and may serve as potential therapeutic targets. Despite this progress, few advances have been made in developing targeted therapeutic strategies for the 50% of patients whose melanomas are BRAF wild-type (WT). The best-characterized subgroup of BRAF-WT tumors comprises 15%–20% of all melanomas that harbor activating NRAS mutations. The small GTPase, NRAS, was the first oncogene identified in melanoma and other mutational subtypes of melanoma; patients with mutant NRAS tumors tend to be older and have a history of chronic ultraviolet (UV) exposure.5–7 Histologically, mutant NRAS tumors are more aggressive than other subtypes and have thicker lesions, elevated mitotic activity, and higher rates of lymph node metastasis.8,9 Given the more aggressive disease seen with mutant NRAS patients who have not received any specific therapy for the disease, it is not surprising that NRAS mutation status is a predictor of poorer outcomes in patients with melanoma who harbor this mutation, with lower median overall survival (OS) compared to non-NRAS-mutant melanoma.6–8 Moreover, and in contrast with BRAF-mutant melanoma, little progress has been made in developing targeted therapeutic strategies for NRAS-mutant melanoma; no effective small-molecule inhibitors have been approved that specifically target NRAS, although MEK inhibitors have demonstrated modest clinical activity in a phase II trial, with clinical benefit in progression-free survival (PFS), but without a clear benefit in OS.3 A better understanding of the biological and signaling characteristics of the NRAS-mutant melanoma will outline some effective therapeutic strategies for its therapeutic management that are now urgently needed.
Ras proteins as an oncogene
Despite the attention focused upon BRAF-mutant melanoma, NRAS was the first melanoma oncogene to be identified. It is known that approximately one-third of all human cancers have oncogenic mutations in the small GTPase RAS family.10 The RAS family of GTPases consists of KRAS, HRAS, and NRAS. Although NRAS, KRAS, and HRAS share structural and functional similarities, mutations in KRAS are the most frequent RAS mutations in human malignant disease; moreover, in melanoma, the most commonly mutated isoform of RAS mutation typically occur at codons 12, 61, or, less frequently, 13, with 15% of cases harboring point mutations.11 Whereas mutant NRAS(Q61) disrupts the GTPase activity of RAS, locking it in its active conformation, NRAS(G12) and NRAS(G13) mutations affect the Walker A-motif (p-loop) of the protein, thus decreasing its sensitivity to GTPase-accelerating proteins.12,13 Mutations in G12/13 and Q61 can all be described as activating; yet, they affect the NRAS protein in a distinct way as they favor the formation of GTP-bound, active RAS proteins.
Genetic evidence in experimental systems provides strong evidence that the RAF–MEK–ERK pathway is critical to the ability of RAS to induce cell proliferation, migration, and survival – highlighting the functional and biochemical relationship between RAS and this pathway in cancer.14 Whereas KRAS mutations are frequent in colorectal, lung, and pancreatic cancers, NRAS mutations are, by far, the predominant alteration among RAS isoforms in melanoma.
Mutations in NRAS constitutively activate intracellular signaling through a variety of pathways – most notably, the RAS–RAF–MAPK and PI3K–AKT pathways. These mutations activate MAPK signaling to a similar degree as BRAF mutations and rarely co-occur with mutations in the PI3K–AKT pathways, suggesting that mutant NRAS drives this pathway as well. These activated signaling pathways induce cell-cycle dysregulation, pro-survival pathways, and cellular proliferation.15
NRAS-mutant melanoma
NRAS was the first melanoma oncogene to be identified in 1984 in a screen of melanoma cell lines for genes that possessed transforming properties and were identified as activating mutations in NRAS in 4/30 samples.16 Currently, mutations of NRAS, KRAS, or HRAS are known to be present in 20%, 2%, and 1% of all melanomas tested, respectively.17 The most common oncogenic change present in >80% of all NRAS mutations is a point mutation leading to the substitution of glutamine to leucine at position 61, with mutations at positions 12 and 13 occurring with less frequency.18
NRAS mutations occur at a fairly consistent rate of 15%–20% at all non-uveal sites of melanoma, including sun-exposed and sun-unexposed skin, mucosal, and acral sites of origin. This distribution contrasts with BRAF mutations, which are more common in intermittently sun-exposed skin, and with KIT mutations, which are present predominantly in mucosal and acral melanomas. Furthermore, in contrast to BRAF, NRAS mutations are rarely present in benign melanocytic nevi – with the exception of congenital nevi.9 The presence of NRAS mutations in melanoma has prognostic significance.12,19 Typical patients harboring NRAS mutations tend to be older (>55 years) than patients with BRAF mutations, with a chronic pattern of UV exposure and lesions are usually located at the extremities and have greater levels of mitosis than BRAF-mutant melanomas. Moreover, NRAS mutations are associated with lower rates of ulceration and thicker primary tumors, with the presence of NRAS mutations an adverse prognostic factor leading to shorter MSS. Several studies examining the effect of NRAS mutations on OS have found different results; when OS was measured from the time of primary disease, NRAS mutations were found to have no impact on OS.20,21 However, in two other studies where OS was measured from the time of biopsy of advanced disease, NRAS mutations were associated with improved OS when compared to tumors with BRAF mutations or those WT for both.22,23
Current management strategies
Improved understanding of genetic and molecular basis of melanoma has revolutionized treatment options for this disease. Genetic profiling of melanomas varies widely between institutions and practices and, in spite of these advances, the treatment of melanoma remains challenging in terms of therapy selection. Because no therapeutic agents have been approved specifically for NRAS-mutant melanoma, due to the fact that several different strategies of directly targeting RAS have not resulted in effective therapeutics, mutational profiling of NRAS is not performed routinely by many clinicians although identifying NRAS mutations may have prognostic implications and facilitate clinical trial enrolment.
Farnesyl transferase inhibitors
Farnesyl transferase inhibitors are a class of drugs designed to prevent the posttranslational modification of Ras and its insertion into the plasma membrane. Although this mechanism of inhibition showed promising preclinical activity, the clinical experience in different clinical trials with these inhibitors has been very disappointing – with several serious side effects and very few responses observed.24–26 In melanoma, farnesyl transferase inhibitors were evaluated in a small phase II trial with 14 patients whose NRAS mutation status was unknown; in this trial, none of the patients experienced a clinical response.27 The lack of success observed with these drugs is attributable to the fact that many critical cellular proteins are farnesylated, in addition to Ras. Currently, there are no further ongoing clinical trials with farnesyl transferase inhibitors.
MEK inhibitors
After the lack of success in directly targeting NRAS was objectivized, focus developed toward targeting the critical signal-transduction pathways of the MAPK pathway, using MEK inhibitors. First-generation MEK inhibitors (PD 098059, U0126) showed promising inhibition in preclinical models of melanoma, but they did not progress to clinical trials.28 CI-1040 (PD 184352) and its derivate PD-0325901 (PD-901) were the first MEK inhibitors tested in a clinical trial. CI-1040 was identified as a drug with a favorable safety profile, but with low oral bioavailability and high metabolism that led to plasma drug levels insufficient for antitumor activity.29 PD-901 is a second-generation MEK inhibitor with some clinical response, as demonstrated in a clinical trial with 48 patients with melanoma (three patients experienced partial responses and 10 had temporary stable disease); however, the high incidence of adverse events – particularly ocular and neurologic – were observed, and limited further development of this drug.30 Overall, the results of early clinical trials with early-generation MEK inhibitors were disappointing (10% objective response rate) and retrospective genotyping for the NRAS/BRAF mutations did predict for clinical benefit.
Newer MEK inhibitors have been developed with a better safety profile and antitumor activity. The first of these newer generation MEK inhibitors to be developed was selumetinib (AZD-6244; ARRY-142886) – another second-generation inhibitor of MEK1/2 with potent inhibition of cell lines in both RAS- and RAF-mutant cancers.31 Initially, it was tested in a phase I trial with 11 patients with melanoma, where activity was observed with one partial response (in a NRAS-mutant patient) and seven patients had stable disease; two other clinical trials were performed, but they did not show clear benefit with selumetinib.32 In a phase II trial study with BRAF-WT and NRAS-unselected patients with melanoma, selumetinib was compared with temozolomide; the study reported equivalent/inferior response rates for selumetinib (5.8% vs 9.7%) and no difference in PFS between the two groups [hazard ratio (HR) 1.07].33 In another phase II trial of docetaxel with or without selumetinib in patients with BRAF-WT advanced melanoma, no difference in OS was noted between the two groups.34 This agent was subsequently evaluated only in BRAF V600-mutant melanoma in combination with dacarbazine as compared to dacarbazine alone; although PFS was extended (median 5.6 vs 3.0 months), no improvement in OS was identified (13.9 vs 10.5 months).35 Selumetinib has, moreover, been tested for uveal melanoma in two phase II trials with a PFS advantage, as compared to chemotherapy.36,37 To date, no trials have been specifically conducted for NRAS-mutant melanoma.
Clinical development of third-generation MEK inhibitors led to focused attention in targeting MEK in patients with NRAS-mutant melanoma. Both trametinib (GSK1120212) and MEK 162 (ARRY-438162) are potent inhibitors of MEK1/2, with sustained MAPK pathway inhibition at clinically achievable doses.38,39 Trametinib is an allosteric inhibitor of MEK1/2 that was first tested in a phase I trial among patients with advanced melanoma; efficacy was demonstrated in BRAF-mutant patients, but no responses were seen in NRAS-mutant patients.40 Trametinib has received FDA approval for use in the treatment of BRAF V600-mutant melanoma as a single agent and in combination with dabrafenib. This approval was based on improved OS in a phase III trial for trametinib compared with dacarbazine in BRAF-mutant melanoma,38 and the strong activity demonstrated in another phase III trial among patients with BRAF-mutant melanoma treated in the METRIC study; an overall response rate of 24% and a median PFS of 4.8 months, both significantly better than with standard chemotherapy, were observed.38 Additionally, a phase III trial analyzing the efficacy of BRAF inhibition with dabrafenib alone compared to a combination of dabrafenib and trametinib demonstrated clear benefit with the combination treatment in terms of response rate, PFS, and OS.41 Potential combination strategies of trametinib combined with other agents may play a role in the future of NRAS-mutant patients. Binimetinib (MEK 162) is the MEK1/2 inhibitor that was the first to show significant activity in NRAS-mutant melanoma. In a phase I trial with patients who had advanced solid tumors, MEK 162 showed promising signs of clinical activity.42 In a phase II study of binimetinib in patients with advanced NRAS-mutant melanoma, 20% showed partial responses and 43.3% stable disease, with a median PFS of 3.7 months.43 Based on these promising results, a randomized phase III trial, the NEMO trial was performed. In this trial, binimetinib improved PFS compared with dacarbazine (2.8 months vs 1.5 months, HR 0.62 [95% confidence interval 0.47–0.80]) and was tolerable. Grade 3–4 adverse events seen in at least 5% of patients in either group were increased creatine phosphokinase (19% vs 0%), hypertension (7% vs 2%), anemia (2% vs 5%), and neutropenia (1% vs 9%) in binimetinib group versus dacarbazine group, respectively; serious adverse events (all grades) occurred in 34% patients in the binimetinib group and in 22% patients in the dacarbazine group. Binimetinib might represent a new treatment option for patients with NRAS-mutant melanoma after failed immunotherapy.44
Other MEK inhibitors such as pimasertib (AS703026), cobimetinib (GDC-0973), TAK-733, and RO4987655 have been tested for efficacy in different clinical trials with patients with melanoma. Pimasertib has been evaluated in a phase I trial with 17 NRAS-mutant patients, with two partial and two complete responses.45 Cobimetinib has not been evaluated specifically for the NRAS cohort but, in combination with vemurafenib, has demonstrated a promising activity for patients with BRAF-mutant melanoma46 (Table 1).
 | Table 1 Phase I/II/III trials of MEK inhibitors in NRAS-mutant melanoma |
MEK inhibitor combinations
Although the next generation of MEK inhibitors are showing promising clinical efficacy, the relatively suboptimal response rate and PFS has led to interest in different MEK inhibitor-based combinations. The combination of MEK inhibition with RAF, EGFR–PI3K–AKT, and CDK4/6, which are the two particular pathways of interest, are currently being evaluated in clinical trials.
MEK + CDK4-6 inhibitors, which are regulators of the G1/S cell-cycle checkpoint inhibiting cancer cell growth, are being tested in phase I/II trials. Early results for the combination of ribociclib (LEE001) with binimetinib in patients with NRAS-mutant melanoma have shown a partial response in 33% and stable disease in 52% of patients.47 Another phase I/II trial with a combination of trametinib and palbociclib in patients with solid tumors and with a specific cohort for NRAS-mutant melanoma is ongoing.
The combination of MEK + PI3K/AKT inhibitors has been shown to synergistically inhibit the growth of NRAS-mutant melanoma cell lines. In melanoma cell lines where BRAF inhibitor resistance is mediated through an acquired NRAS mutation, the combination of a MEK inhibitor plus a PI3K/mTOR inhibitor was noted to overcome drug resistance and inhibit cell survival.48 Several early-phase studies in solid tumors have been performed, but it is not yet clear what the optimal combination of signal transduction inhibitors will be for NRAS-mutant melanoma. There are multiple phase I/II studies examining these combinations in patients with melanoma.
Other combinations for NRAS-mutant melanoma
Other targets such as polo-like kinase 1 (PLK1), TANK-binding kinase 1 (TBK1), and ROCK 1/2 are overexpressed in NRAS-mutant melanoma and the combination of MEK inhibitors plus specific inhibitors in these targets may show promising results in the different phase I/II trials that are ongoing.49–51
Immune-based therapies
Immune therapies are playing an increasing role in the treatment of patients with metastatic melanoma, regardless of its BRAF or NRAS mutation status, particularly when there is no specific targeted therapy available.52
Despite the current lack of effective and specifically approved targeted therapies for NRAS-mutant melanoma, there is some evidence that NRAS mutational status may predict for response to other therapies. Immune-based therapies are the standard of care in melanoma therapeutics, mainly used as first-line therapy particularly in patients with BRAF-WT melanoma, and are used regardless of tumor genotype. Some retrospective data suggest that patients with NRAS-mutant melanoma may have higher response rates to immunotherapies.
A retrospective analysis of patients treated with high-dose interleukin-2 demonstrated that the majority of the responders were patients with NRAS-mutant melanoma, and that those patients with either BRAF-mutant or BRAF/NRAS-WT melanoma were less likely to respond.53 It is not yet clear whether BRAF or NRAS mutational status predicts for better responses in patients with melanoma receiving the antiCTLA-4 antibody ipilimumab or anti-PD-1/PD-L1 antibodies; however, some publications suggest that patients with NRAS mutations respond better to these agents.54,55 In the only published analysis to date, the disease control rate of patients with melanoma on ipilimumab therapy was noted to be 30% and 33% for those with and without BRAF mutations, respectively55 (Table 2).
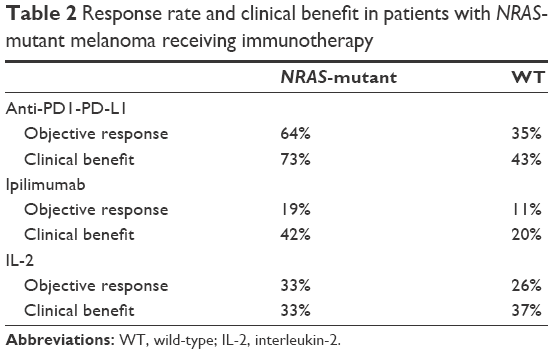 | Table 2 Response rate and clinical benefit in patients with NRAS-mutant melanoma receiving immunotherapy |
Future perspectives
Although the current target and immunotherapeutic agents may offer some hope to patients with NRAS-mutant melanoma, none of these therapies are mutation specific and have shown modest response rates and carry risk of significant toxicities. In contrast with BRAF-mutant melanoma, to date, no effective molecularly targeted therapeutic strategies have been approved for NRAS-mutant or -WT melanoma.
Many targeted strategies are now being evaluated for NRAS-mutant melanoma, although these tumors appear to be more heterogeneous than those with BRAF mutations. The most promising data from clinical investigations are with regard to MEK inhibition; however, the relatively short PFS indicates that either combination strategies or other targeted approaches will be necessary to achieve more clinically important disease responses. Both combinations for pathway interference (MEK + PI3K/mTOR and MEK + CDK 4,6) as well as combination of targeted therapy with immunotherapy are, to date, the most promising strategy to interfere with current targets refractory to chemotherapy, such as NRAS-mutant melanoma.
Conclusion
NRAS-mutant melanoma is a relatively common subtype of this disease (15%–20% of patients harbor this mutation) with a known poor prognosis. Although, currently, there are no targeted therapies that directly target NRAS, a high number of newer targeted therapeutic strategies, particularly mono- and combination therapy with MEK inhibitors, hold promise of being effective treatment strategies in the near future in the several clinical trials that are being conducted. Immune-based therapies are not genotype-specific but appear to be at least as or even more effective in the NRAS-mutant population compared to other melanoma subtypes.
Acknowledgment
The authors thank all patients with melanoma and their families for their help in participating in the investigations into malignant melanoma.
Author contributions
All authors contributed toward data analysis, drafting and revising of the paper, and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
Robert C, Schachter J, Long GV, et al; KEYNOTE-006 investigators. Pembrolizumab versus ipilimumab in advanced melanoma. N Engl J Med. 2015;372(26):2521–2532. | ||
Ascierto PA, McArthur GA, Dréno B, et al. Cobimetinib combined with vemurafenib in advanced BRAF(V600)-mutant melanoma (coBRIM): updated efficacy results from a randomised, double-blind, phase 3 trial. Lancet Oncol. 2016;17(9):1248–1260. | ||
Dhillon S. Dabrafenib plus trametinib: a review in advanced melanoma with a BRAF (V600) mutation. Target Oncol. 2016;11(3):417–428. | ||
Larkin J, Chiarion-Sileni V, Gonzalez R, et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med. 2015;373(1):23–34. | ||
Curtin JA, Fridlyand J, Kageshita T, et al. Distinct sets of genetic alterations in melanoma. N Engl J Med. 2005;353(20):2135–2147. | ||
Jakob JA, Bassett RL Jr, Ng CS, et al. NRAS mutation status is an independent prognostic factor in metastatic melanoma. Cancer. 2012;118(16):4014–4023. | ||
Lee JH, Choi JW, Kim YS. Frequencies of BRAF and NRAS mutations are different in histological types and sites of origin of cutaneous melanoma: a meta-analysis. Br J Dermatol. 2011;164(4):776–784. | ||
Thumar J, Shahbazian D, Aziz SA, Jilaveanu LB, Kluger HM. MEK targeting in N-RAS mutated metastatic melanoma. Mol Cancer. 2014;13:45. | ||
Devitt B, Liu W, Salemi R, et al. Clinical outcome and pathological features associated with NRAS mutation in cutaneous melanoma. Pigment Cell Melanoma Res. 2011;24(4):666–672. | ||
Diaz-Flores E, Shannon K. Targeting oncogenic Ras. Genes Dev. 2007;21(16):1989–1992. | ||
Malumbres M, Barbacid M. RAS oncogenes: the first 30 years. Nat Rev Cancer. 2003;3(6):459–465. | ||
Daud A, Bastian BC. Beyond BRAF in melanoma. Curr Top Microbiol Immunol. 2012;355:99–117. | ||
Fedorenko IV, Gibney GT, Smalley KS. NRAS mutant melanoma: biological behavior and future strategies for therapeutic management. Oncogene. 2013;32(25):3009–3018. | ||
Drosten M, Dhawahir A, Sum EY, et al. Genetic analysis of Ras signalling pathways in cell proliferation, migration and survival. EMBO J. 2010;29(6):1091–1104. | ||
Hodis E, Watson IR, Kryukov GV, et al. A landscape of driver mutations in melanoma. Cell. 2012;150(2):251–263. | ||
Albino AP, Le Strange R, Oliff AI, Furth ME, Old LJ. Transforming ras genes from human melanoma: a manifestation of tumour heterogeneity? Nature. 1984;308(5954):69–72. | ||
Milagre C, Dhomen N, Geyer FC, et al. A mouse model of melanoma driven by oncogenic KRAS. Cancer Res. 2010;70(13):5549–5557. | ||
Bos JL. Ras oncogenes in human cancer: a review. Cancer Res. 1989;49(17):4682–4689. | ||
Charbel C, Fontaine RH, Malouf GG, et al. NRAS mutation is the sole recurrent somatic mutation in large congenital melanocytic nevi. J Invest Dermatol. 2014;134(4):1067–1074. | ||
Akslen LA, Angelini S, Straume O, et al. BRAF and NRAS mutations are frequent in nodular melanoma but are not associated with tumor cell proliferation or patient survival. J Invest Dermatol. 2005;125(2):312–317. | ||
Ellerhorst JA, Greene VR, Ekmekcioglu S, et al. Clinical correlates of NRAS and BRAF mutations in primary human melanoma. Clin Cancer Res. 2011;17(2):229–235. | ||
Omholt K, Platz A, Kanter L, Ringborg U, Hansson J. NRAS and BRAF mutations arise early during melanoma pathogenesis and are preserved throughout tumor progression. Clin Cancer Res. 2003;9(17):6483–6488. | ||
Platz A, Egyhazi S, Ringborg U, Hansson J. Human cutaneous melanoma; a review of NRAS and BRAF mutation frequencies in relation to histogenetic subclass and body site. Mol Oncol. 2008;1(4):395–405. | ||
Konstantinopoulos PA, Karamouzis MV, Papavassiliou AG. Post-translational modifications and regulation of the RAS superfamily of GTPases as anticancer targets. Nat Rev Drug Discov. 2007;6(7):541–555. | ||
Smalley KS, Eisen TG. Farnesyl transferase inhibitor SCH66336 is cytostatic, pro-apoptotic and enhances chemosensitivity to cisplatin in melanoma cells. Int J Cancer. 2003;105(2):165–175. | ||
Niessner H, Beck D, Sinnberg T, et al. The farnesyl transferase inhibitor lonafarnib inhibits mTOR signaling and enforces sorafenib-induced apoptosis in melanoma cells. J Invest Dermatol. 2011;131(2):468–479. | ||
Gajewski TF, Salama AK, Niedzwiecki D, et al; Cancer and Leukemia Group B. Phase II study of the farnesyltransferase inhibitor R115777 in advanced melanoma (CALGB 500104). J Transl Med. 2012;10:246. | ||
Friday BB, Adjei AA. Advances in targeting the Ras/Raf/MEK/Erk mitogen-activated protein kinase cascade with MEK inhibitors for cancer therapy. Clin Cancer Res. 2008;14(2):342–346. | ||
Rinehart J, Adjei AA, Lorusso PM, et al. Multicenter phase II study of the oral MEK inhibitor, CI-1040, in patients with advanced non-small-cell lung, breast, colon, and pancreatic cancer. J Clin Oncol. 2004;22(22):4456–4462. | ||
LoRusso PM, Krishnamurthi SS, Rinehart JJ, et al. Phase I pharmacokinetic and pharmacodynamic study of the oral MAPK/ERK kinase inhibitor PD-0325901 in patients with advanced cancers. Clin Cancer Res. 2010;16(6):1924–1937. | ||
Yeh TC, Marsh V, Bernat BA, et al. Biological characterization of ARRY-142886 (AZD6244), a potent, highly selective mitogen-activated protein kinase kinase 1/2 inhibitor. Clin Cancer Res. 2007;13(5):1576–1583. | ||
Adjei AA, Cohen RB, Franklin W, et al. Phase I pharmacokinetic and pharmacodynamic study of the oral, small-molecule mitogen-activated protein kinase kinase 1/2 inhibitor AZD6244 (ARRY-142886) in patients with advanced cancers. J Clin Oncol. 2008;26(13):2139–2146. | ||
Kirkwood JM, Bastholt L, Robert C, et al. Phase II, open-label, randomized trial of the MEK1/2 inhibitor selumetinib as monotherapy versus temozolomide in patients with advanced melanoma. Clin Cancer Res. 2012;18(2):555–567. | ||
Gupta A, Love S, Schuh A, et al. DOC-MEK: a double-blind randomized phase II trial of docetaxel with or without selumetinib in wild-type BRAF advanced melanoma. Ann Oncol. 2014;25(5):968–974. | ||
Robert C, Dummer R, Gutzmer R, et al. Selumetinib plus dacarbazine versus placebo plus dacarbazine as first-line treatment for BRAF-mutant metastatic melanoma: a phase 2 double-blind randomised study. Lancet Oncol. 2013;14(8):733–740. | ||
Carvajal RD, Schwartz GK, Mann H, Smith I, Nathan PD. Study design and rationale for a randomised, placebo-controlled, double-blind study to assess the efficacy of selumetinib (AZD6244; ARRY-142886) in combination with dacarbazine in patients with metastatic uveal melanoma (SUMIT). BMC Cancer. 2015;15:467. | ||
Carvajal RD, Sosman JA, Quevedo JF, et al. Effect of selumetinib vs chemotherapy on progression-free survival in uveal melanoma: a randomized clinical trial. JAMA. 2014;311(23):2397–2405. | ||
Flaherty KT, Robert C, Hersey P, et al; METRIC Study Group. Improved survival with MEK inhibition in BRAF-mutated melanoma. N Engl J Med. 2012;367(2):107–114. | ||
Gilmartin AG, Bleam MR, Groy A, et al. GSK1120212 (JTP-74057) is an inhibitor of MEK activity and activation with favorable pharmacokinetic properties for sustained in vivo pathway inhibition. Clin Cancer Res. 2011;17(5):989–1000. | ||
Falchook GS, Lewis KD, Infante JR, et al. Activity of the oral MEK inhibitor trametinib in patients with advanced melanoma: a phase 1 dose-escalation trial. Lancet Oncol. 2012;13(8):782–789. | ||
Long GV, Stroyakovskiy D, Gogas H, et al. Combined BRAF and MEK inhibition versus BRAF inhibition alone in melanoma. N Engl J Med. 2014;371(20):1877–1888. | ||
Bendell JC, Javle M, Bekaii-Saab TS, et al. A phase 1 dose-escalation and expansion study of binimetinib (MEK162), a potent and selective oral MEK1/2 inhibitor. Br J Cancer. 2017;116(5):575–583. | ||
Ascierto PA, Schadendorf D, Berking C, et al. MEK162 for patients with advanced melanoma harbouring NRAS or Val600 BRAF mutations: a non-randomised, open-label phase 2 study. Lancet Oncol. 2013;14(3):249–256. | ||
Dummer R, Schadendorf D, Ascierto PA, et al. Binimetinib versus dacarbazine in patients with advanced NRAS-mutant melanoma (NEMO): a multicentre, open-label, randomised, phase 3 trial. Lancet Oncol. 2017;18(4):435–445. | ||
Le A, et al. Pimasertib, a selective oral MEK 1/2 inhibitor, shows clinical activity in cutaneous and uveal metastatic melanoma in the phase I program. EADO congress. 2012; abstract C06. | ||
Larkin J, Ascierto PA, Dréno B, et al. Combined vemurafenib and cobimetinib in BRAF-mutated melanoma. N Engl J Med. 2014;371(20):1867–1876. | ||
Sosman JA, Kittaneh M, Lolkema MPJ, et al. A phase 1b/2 study of LEE011 in combination with binimetinib (MEK162) in patients with NRAS-mutant melanoma: early encouraging clinical activity. J Clin Oncol. 2014;32(Suppl 15):9009. | ||
Greger JG, Eastman SD, Zhang V, et al. Combinations of BRAF, MEK, and PI3K/mTOR inhibitors overcome acquired resistance to the BRAF inhibitor GSK2118436 dabrafenib, mediated by NRAS or MEK mutations. Mol Cancer Ther. 2012;11(4):909–920. | ||
Lin CC, Su WC, Yen CJ, et al. A phase I study of two dosing schedules of volasertib (BI 6727), an intravenous polo-like kinase inhibitor, in patients with advanced solid malignancies. Br J Cancer. 2014;110(10):2434–2440. | ||
Vogel CJ, Smit MA, Maddalo G, et al. Cooperative induction of apoptosis in NRAS mutant melanoma by inhibition of MEK and ROCK. Pigment Cell Melanoma Res. 2015;28(3):307–317. | ||
Vu HL, Aplin AE. Targeting TBK1 inhibits migration and resistance to MEK inhibitors in mutant NRAS melanoma. Mol Cancer Res. 2014;12(10):1509–1519. | ||
Johnson DB, Lovly CM, Flavin M, et al. Impact of NRAS mutations for patients with advanced melanoma treated with immune therapies. Cancer Immunol Res. 2015;3(3):288–295. | ||
Joseph RW, Sullivan RJ, Harrell R, et al. Correlation of NRAS mutations with clinical response to high-dose IL-2 in patients with advanced melanoma. J Immunother. 2012;35(1):66–72. | ||
Johnson DB, Puzanov I. Treatment of NRAS-mutant melanoma. Curr Treat Options Oncol. 2015;16(4):15. | ||
Shahabi V, Whitney G, Hamid O, et al. Assessment of association between BRAF-V600E mutation status in melanomas and clinical response to ipilimumab. Cancer Immunol Immunother. 2012;61(5):733–737. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.